

Abstract
Aggregation of the microtubule‐associated protein tau is a key feature of Alzheimer's disease and other so‐called tauopathies, yet what causes this protein to aggregate and what renders it toxic is only slowly being revealed. Because tau spreads in a stereotypical pattern through the diseased brain, it has been proposed that it possesses prion‐like properties, with aggregation‐prone tau facilitating the conversion of “naïve” tau into “toxic” forms. The current study by Wegmann et al (2015) addresses whether tau fulfils classical “prion criteria” by assessing its spreading and toxicity in the absence of endogenous tau. Using different transgenic and viral paradigms, the authors demonstrate that, although tau still propagates in this scenario, there is a decrease in its misfolding and neurotoxicity. They therefore conclude that tau is not a genuine prion, at least when the current definition of these infectious proteins is applied.
Subject Categories: Neuroscience
Widespread protein aggregation is an inherent part of ageing (David et al, 2010), yet only a few proteins are regarded as signature proteins in neurodegenerative diseases where they are believed not only to aggregate but also to have a critical role in neuronal demise. One such example is tau, a scaffolding protein that in mature neurons is mainly concentrated in the axons, where it binds to microtubules. Under neurodegenerative conditions such as Alzheimer's disease (AD), tau accumulates in a hyperphosphorylated, fibrillar form as neurofibrillary tangles (NFTs) that can be visualized with specific antibodies or by silver impregnation techniques. Insight into the underlying pathomechanisms has been facilitated by the identification of pathogenic mutations in the tau‐encoding MAPT gene in familial forms of frontotemporal dementia (FTD). This in turn has led to the generation of transgenic mouse models that reproduce key features of the tau pathology of AD such as hyperphosphorylation, NFT formation and neuronal loss. In the current study, Wegmann et al (2015) made use of such models that express FTD mutant tau (P301L tau).
In post‐mortem AD tissue, NFTs have been shown to appear in a stereotypic pattern that is initiated in the entorhinal cortex and sequentially appears in additional brain areas suggestive of spreading along neuronal connections. An increasing body of evidence has shown that aggregated tau can form a misfolded tau “seed” which can behave like a prion. In this prion‐like state, tau can recruit monomeric endogenous tau and induce its aggregation. Furthermore, tau seeds can be released into the extracellular space, gain entry into neighbouring or synaptically connected cells and trigger further misfolding and aggregation of physiological tau (Clavaguera et al, 2009; Frost et al, 2009; Guo & Lee, 2011; Asai et al, 2015). However, whether all these propagation characteristics are sufficient for tau to be considered a genuine prion remains a matter of debate.
Wegmann et al (2015) tested known characteristics of “true” prions in the context of tau spreading in mice. For instance, it is established that accepted prions such as PrPSc induce the misfolding of endogenous PrPC, which becomes corrupted and converted into PrPSc, with subsequent accumulation of PrPSc causing neurodegeneration. Interestingly, simply eliminating naïve PrPC from mice renders neuronal tissue resistant to both the propagation and infectivity of PrPSc (Brandner et al, 1996). In other words, the corruptor PrPSc requires physiological (naïve) PrPC to spread along neural connections. Wegmann et al (2015) equated P301L mutant tau to the corruptor and endogenous mouse tau to the naïve form of tau, a protein that has been shown to co‐aggregate with human tau in tau transgenic mouse models (de Calignon et al, 2012; Van der Jeugd et al, 2012), although older studies indicated that endogenous murine tau prevents a tau pathology in mice overexpressing a human tau‐containing artificial chromosome (Andorfer et al, 2003). In analogy to what is happening in “the prion world,” one would have expected that the ablation of mouse tau would abolish the propagation of mutant P301L tau, if this protein were to behave like the true prion PrPSc. Surprisingly, human P301L tau was propagated on a mouse tau‐null background; however, there was reduced neurotoxicity, suggesting a role for mouse tau in the neurotoxic events seen in tau transgenic models (Fig 1). As stated by Wegmann et al (2015), these results indicate that tau cannot strictly be classified as a prion protein under current definitions.
Figure 1. Comparison of the toxic principles of PrPS c and tau.
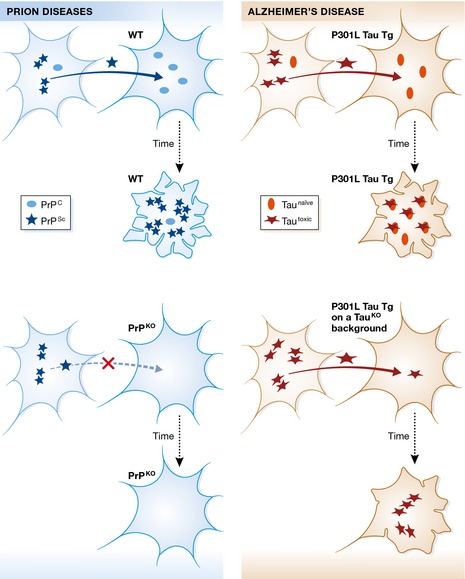
The present report addresses what is known about the propagation and toxicity of prions (shown on the left) and asks the question whether tau in Alzheimer's disease (on the right) displays the same characteristics. In the case of prion diseases, a form of the prion protein known as PrPS c (Scrapie) propagates from cell to cell and, upon uptake by a recipient cell, leads to an infectious conversion of the endogenous, cellular form of prion (PrPC) into the toxic PrPS c form, ultimately leading to neuronal demise (as graphically illustrated by the extremely ruffled cell membrane). Importantly, both propagation and toxicity are halted, when the recipient cells lacks endogenous PrPC expression (PrP knockout, PrPKO). In their study, Wegmann et al (2015) use different transgenic (P301L Tau Tg) and viral paradigms, to demonstrate that, although P301L tau still propagates, there is a decrease in its misfolding and neurotoxicity in the absence of endogenous mouse tau (as visualized by the different extent of membrane ruffling under the two conditions). The assumption is that P301L tau is synonymous with tautoxic and that endogenous mouse tau exists in a naïve form. As a possible model to explain the reduced toxicity in the absence of endogenous (naïve) tau, the authors suggest that co‐aggregation of mouse tau with human P301L tau may create more toxic “strains” of tau aggregates, with stronger effects on tau pathology in mice. The authors conclude that P301L mutant tau may not be a genuine prion.
However, if P301L tau is not a prion, what is it?—On the one hand, P301L tau propagation in the absence of naïve mouse tau excludes it from the club of “true” prions. However, the data also indicate that P301L tau might behave even worse than true prions, the propagation of which can be prevented just by eliminating the endo‐genous protein in its naïve form. Human P301L tau demonstrated unique characteristics and independence in propagating along neuronal networks. It appears that the only way to eliminate the toxic effects of aggregated tau is by eliminating or reducing this toxic form of tau. It is possible that P301L tau does not require naïve mouse tau to propagate in mouse brains because it co‐exists in two different states: a soluble state that could even functionally replace mouse tau, and an aggregated state that corrupts and absorbs naïve forms of tau, eventually leading to the formation of NFTs. The balance between these two conformations of tau could determine the progression of tau pathology, with pro‐aggregation factors accelerating the process and anti‐aggregation factors reducing the advance of the pathology. Taking into account these considerations, the reduced neurotoxicity and the delayed tau pathology observed in the absence of mouse tau could be caused by a reduction in the availability of soluble, naïve P301L tau, kidnapped in assuming functions proper of mouse tau, such as binding and stabilizing microtubules. Another possibility, as stated by Wegmann et al (2015), is that co‐aggregation of mouse tau with human P301L tau may create more toxic “strains” of tau aggregates, with stronger effects on tau pathology (Fig 1). In any case, the results provide a clear message that pure distinctive human tau effects are best analysed on a mouse tau‐null background. What is unexplored and may be worth further investigation is the role of the different isoforms of tau, whereas transgenic mice generally overexpress just one tau isoform, endogenous tau exists as three isoforms in normal adult mice, with different functions and differences in their subcellular localization.
Apart from potential artefacts generated by the expression of endogenous mouse tau, it is also necessary to consider the possibility of potential artefacts due to overexpression of human tau in mice. Wegmann et al (2015) dealt with this by working with two different mouse models: one with threefold overexpression of P301L mutant tau specifically in the entorhinal cortex (ECrTgTau) and the other with 10‐fold overexpression under the control of a “pan‐neuronal” promoter (rTg4510). The consistency of the data in both models leads to increased confidence in the conclusions that are drawn. It appears that the different levels of overexpression only affected the speed of development of tau pathology, being faster at higher levels of human tau expression. Nevertheless, future P301L tau knock‐in experiments might provide a definitive answer.
In conclusion, Wegmann, Hyman and colleagues provide conclusive evidence that human mutant tau may not be a genuine prion. However, there are more similarities than differences between “true” prions and self‐aggregating proteins involved in the pathogenesis of AD and other neurodegenerative diseases. Expanding the prion concept to include a larger group of “potential club members” appears to be in the mind of many researchers in the field. Future work building on this and other studies will determine whether human tau will be accepted in the exclusive club of prions or a novel club will be founded to include proteinaceous nucleating particles such as tau.
See also: S Wegmann et al (December 2015)
References
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86: 582–590 [DOI] [PubMed] [Google Scholar]
- Asai H, Ikezu S, Tsunoda S, Medalla M, Luebke J, Haydar T, Wolozin B, Butovsky O, Kugler S, Ikezu T (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: 1584–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996) Normal host prion protein necessary for scrapie‐induced neurotoxicity. Nature 379: 339–343 [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez‐Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires‐Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73: 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Bioly 11: 909–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C (2010) Widespread protein aggregation as an inherent part of aging in C. elegans . PLoS Biol 8: e1000450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI (2009) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284: 12845–12852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM (2011) Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer‐like tangles. J Biol Chem 286: 15317–15331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D'Hooge R, Mandelkow EM (2012) Cognitive defects are reversible in inducible mice expressing pro‐aggregant full‐length human Tau. Acta Neuropathol 123: 787–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S, Maury EA, Kirk MJ, Saqran L, Roe A, DeVos SL, Nicholls S, Fan Z, Takeda S, Cagsal‐Getkin O, William CM, Spires‐Jones TL, Pitstick R, Carlson GA, Pooler AM, Hyman BT (2015) Removing endogenous tau does not prevent tau propagation yet reduces its neurotoxicity. EMBO J 34: 3028–3041 [DOI] [PMC free article] [PubMed] [Google Scholar]