

Summary
Type 1 diabetes (T1D) is one of the most common and severe chronic diseases affecting both children and adults. The aetiology of the disease remains unknown, and thus far no ‘true’ cure for those affected is available. Indeed, exogenous insulin replacement therapy to manage glucose metabolism to the best degree possible remains the current standard of care. However, despite a recent array of truly impressive improvements designed to enhance disease management (e.g. insulin analogues, continuous glucose monitoring, insulin pumps), it is still difficult for the vast majority of patients to reach recommended target HbA1C levels (< 7·0%). As a result of suboptimal disease management, far too many patients with T1D have an increased risk for disease‐associated complications such as nephropathy, neuropathy and retinopathy, as well as hypoglycaemia. New treatment modalities are therefore needed urgently to bring a ‘true’ cure (disease prevention/disease reversal) to patients with T1D. Here we consider issues that collectively pose a major stumbling block in T1D research with respect to identifying a means to prevent and/or cure the disease. We begin this Perspective by discussing new insights emanating from studies of the pancreas in human T1D; findings which may, at least in part, explain why previous interventions seeking disease prevention/reversal have yielded insufficient benefit. We then turn to suggestions that could optimise the outcome of future clinical trials. Finally, we direct attention to recommendations for the global T1D research community; messages we deem to have the potential to improve our chances of finding the elusive T1D ‘cure’.
Keywords: autoimmune disease, beta cells, immune therapy, insulin, type 1 diabetes
Abbreviations
- T1D
type 1 diabetes
- MHC
major histocompatibility complex
Why have previous interventions seeking disease prevention/reversal been unsuccessful?
The concept of seeking to reverse autoimmunity by altering the self‐destructive immune response against insulin‐secreting pancreatic beta cells is not new. Indeed, since the advent of immunosuppression trials with cyclosporin and azathioprine in the early 1980s, much effort and money, and the time of many physicians, scientists and patients, has been devoted to this cause 1. Unfortunately, despite considerable investment into this line of research, the results have, to a large extent, been considered ‘disappointing’ in terms of their ability to provide durable preservation of insulin production in patients with new‐onset disease.
We are not suggesting that there is no glimmer of hope, nor that all efforts have proved ineffective in every subject; far from it. Indeed, a limited number of intervention trials have shown protection of beta cell function in a subpopulation of subjects, although the results have been viewed (by some) as suboptimal. As a case in point, serum C‐peptide production (a marker of insulin production and surviving beta cells) could be preserved temporarily in certain patients, while in other patients insulin requirements were reduced 2, 3, 4, 5, 6. Significant heterogeneity in the preservation and decline of C‐peptide between individuals in clinical trials is a major issue that may be circumvented by developing other end‐points and biomarkers to predict disease heterogeneity. However, for the vast majority of patients with T1D type 1 diabetes (T1D) durable effects have not, so far, been achieved 2, 3, 4, 5, 6. This represents an important predicament, as concerns continue to exist regarding potential adverse events (e.g. viral reactivation and oncological events) that are often associated with the use of immunosuppression and immunomodulation 7, 8. As a result, it is vital for the T1D research community to question whether short‐term preservation of serum C‐peptide production is worth this risk and decide what level of C‐peptide production and for what time‐period is, in reality, significant to the life of the patient.
Given the unfortunate observation that immune intervention (alone) has shown limited success, it is even more concerning that a facet in research potentially capable of dramatically improving this situation, namely understanding the aetiology of T1D, remains unidentified. If an inciting or exacerbating agent for T1D were indeed known, be it dietary or infectious, our approach to this disease, and perhaps our success rate with treatments, might be quite different. Currently, we are acting in somewhat of an intellectual vacuum and stumbling in the dark in terms of our knowledge of the pathogenesis of this disease 9. Hopefully, given the recent well‐funded and highly organised multi‐centre, multi‐cultural and geographically diverse efforts designed to identify the aetiology of T1D [e.g. The Environmental Determinants of Diabetes in the Young (TEDDY), TrialNet, Daisy, Network for Pancreatic Organ Donors with Diabetes (nPOD), Persistent Virus Infection in Type 1 Diabetes Network (PevNet), Diabetes Virus Detection Study (DiViD)], the detailed pathogenesis and natural history of this disease will not remain unknown for much longer.
How can we optimise the outcomes of future research and clinical trials?
If we are to yield improved results despite the aforementioned gaps in our knowledge regarding disease aetiology, as well as seemingly ‘overdependence’ on interventions seeking immunosuppression, we need to find different ways of approaching T1D research. Among the first of the concepts to consider here is one difficult to convey to the T1D research community without stirring emotions. In short, perhaps past research into new treatments has relied too heavily on studies of animal models, in‐vitro systems and, in the case of human research, scrutiny of peripheral blood rather than the human pancreas. Clearly, this statement has the potential to be considered inflammatory as so much, indeed the vast majority, of T1D research over previous decades has been directed at these efforts.
If one agrees with this premise (and clearly not all will), it is also wise to suggest that we must now place more emphasis on studies of human patients throughout the T1D disease course (i.e. before disease onset, at or near diagnosis and post‐onset), highlighting analyses of the pancreas combined with lifestyle/environmental encounters. This notion is a key message of this Perspective. While we might be tempted to end our argument there, we deem it vital to explain our rationale further to emphasize this point, because evidence for this concept has grown dramatically in the last 18 months.
Are autoreactive T cells really the main drivers of disease?
Recent insights into the immunopathology of T1D at the level of the pancreas should make us pause and reconsider the conventional view that this disease is primarily, if not solely, caused by autoreactive T cells. It is possible that this assumption may have contributed at least in part to the aforementioned suboptimal clinical trial results. To be clear from the beginning, we are not suggesting in any way that T cells are not of importance to the pathogenesis of the disease; in fact, genetic studies strongly suggest a role for the major histocompatibility complex (MHC) class II locus 10, but rather that other constituents such as beta cells and exocrine pancreas may also be contributing, perhaps equally, to disease progression 11.
What draws us to make such declarations? In short, a series of emerging curiosities regarding the disease. First among these is the observation that beta cell loss is often curiously lobular (Fig. 1a,b), suggesting that an external trigger may render distinct pancreatic lobes susceptible to immune infiltration and beta cell loss 12. Beyond this is the issue of lobular area. Insulitis is discrete, affecting only a portion of islets. Indeed, in newly diagnosed T1D (i.e. less than 1 month after onset), a lesion is only present in approximately half of patients (more in individuals with young age at disease onset and less in those with adult onset T1D) 13. It might therefore be possible that an external trigger(s) could lead to a relapsing–remitting disease course, literally flaring up and burning through groups of islets at a time (Fig. 2).
Figure 1.
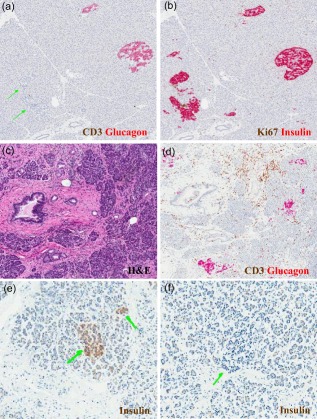
Largely unrecognized pathological features of the endocrine and exocrine pancreas in human type 1 diabetes (T1D). A common but widely underappreciated feature of endocrine pancreatic pathology in T1D involves lobular variance in the presence of (a) glucagon‐positive islets (i.e. devoid of insulin; indicated by double green arrows) versus (b) islets that contain both insulin‐ and glucagon‐positive cells. In this case (nPOD 6195), regions in close proximity clearly demonstrate such marked differences, as determined by the immunohistochemistry of adjacent pancreatic sections with anti‐glucagon and anti‐insulin antibodies, as described 23. A second characteristic involves features of the exocrine pancreas. This case (nPOD 6240) demonstrates, via standard haematoxylin and eosin (H&E), (c) fibrosis as well as (d) focal pancreatitis and peri‐ductal inflammation (determined by immunohistochemisty with anti‐glucagon and anti‐CD3 antibodies, as described 23). Finally, for many years it was presumed that the pancreas of patients with T1D with long‐standing disease was devoid of beta cells, but recent evidence suggests otherwise. The pancreas of many patients contain (e) insulin‐positive cells observed as small clusters or single cells (double green arrows), as well as (f) islets devoid of insulin (single green arrow) for years to decades following the diagnosis of T1D. Shown herein is a pancreatic section of a 60 year‐old patient with T1D (nPOD 6042) with disease duration of 59 years. Once again, expression was determined by immunohistochemistry 23. Photomicrographs courtesy of Dr Martha Campbell‐Thompson.
Figure 2.
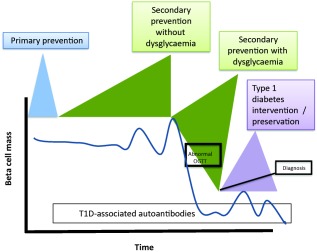
Model showing potential intervention points and hypothesised decline of beta‐cells as currently seen in type 1 diabetes (T1D) 16. Four key therapeutic windows of opportunity exist in the natural history of T1D – :primary prevention (prior to formation of any T1D‐associated autoantibodies); secondary prevention (post‐autoantibody development); secondary prevention with dysglycaemia (post‐autoantibody development with abnormal glucose tolerance); and finally T1D intervention/preservation (post‐hyperglycaemic onset).
This finding raises a series of pathogenic curiosities. Autoantibodies predict the risk for T1D reliably 14, so it may be that there are periods when there is little or no insulitis, as well as those when immune‐based therapies would be expected to be ineffective, as depicted in Fig. 2. Certainly for the prediabetic phase this is not an unreasonable notion, because after disease onset insulitis is present more consistently in discrete areas 15. Given this evidence from pancreatic pathology, prediabetic interventions should therefore be focused on the phase directly before diagnosis, when oral glucose tolerance tests become abnormal and one can be sure that immune reactivity is present (Fig. 2). To use common vernacular, ‘timing is everything’, and perhaps part of our frustration in seeking to prevent/reverse the disease is that timings have not been optimal. Lobular disease could also be anatomical in origin. Islet blood supply is often described as portal, and immune cells may move from islet to islet via blood (or lymphatics) within a lobule before resolution within that lobule. A better understanding of migration and lymphatic drainage within these areas is needed before we can fully interpret lobularity. Hence, there exists a great need for further research in this area.
Given that we do not understand the primary causes and natural history of T1D, one must consider that the hypothesis ‘T1D is an exclusively T cell‐driven autoimmune disease’ might not be correct. Innate inflammatory factors may be instrumental for pathogenesis, meaning that the disease could be a result of infections or a genetically based dysregulation of the immune system, or a combination of the two. There is also a growing body of evidence highlighting the importance of beta cells themselves to disease pathogenesis, which will be discussed in more detail below.
Now for a novel thought: is T1D definitely antigen‐specific?
While not widely considered in discussions of the pathogenesis of T1D, there is an early and substantial effect on the exocrine pancreas in those with this disease, as evidenced by pancreatic inflammation 16, as well as a 30% reduction in total pancreatic volume present at the time of diagnosis when compared with non‐diabetic individuals 17. Morphological examinations often show considerable engagement of the exocrine pancreas with pancreatitis (sometimes patchy), and focal periductal inflammation and fibrosis (Fig. 1c,d) 18.
These observations of general inflammation should perhaps direct us towards the use of non‐specific anti‐inflammatory therapies, and may suggest a possible reason why antigen‐specific interventions have been historically less successful. Collectively, the available evidence suggests that T1D in humans is a pancreatic disease in toto, perhaps progressing in a relapsing–remitting fashion, with its main clinical manifestations emanating from the loss of the insulin‐producing cells. However, even if T1D is not predominantly antigen‐specific, induction of tissue‐specific immune regulation (which would be best achieved by antigen) could still be a great benefit, and a useful way to combat any focal immune response, whatever its driver.
It is possible that different immunopathological processes may contribute to the development of T1D, carrying important implications for treatment and prevention strategies. Potentially related islet and blood autoimmune response phenotypes may coincide with and precede disease. A study by Arif et al. 19 revealed two distinct types of insulitic lesions in children/adolescents with newly diagnosed diabetes distinguishable by the degree of cellular infiltrate and presence of B cells (termed ‘hyper‐immune CD20hi’ and ‘pauci‐immune CD20lo’). Subjects had only one infiltration phenotype and were partitioned by this into two equal‐sized groups that differed significantly by age at diagnosis, with hyperimmune CD20hi subjects being 5 years younger.
Interestingly, up‐regulation of MHC class I has been reported to be associated with T1D 20, 21. However, the issue is by no means straightforward, as expression can be heterogeneous 20 and up‐regulation of MHC RNA is not necessarily associated with protein up‐regulation 22, leaving the possibility that there may be incomplete turnover of MHC class I molecules on the cell surface. No intervention to date has been able to reverse this key pathogenic feature, and more attention should be given to this issue. Resolution may be brought forth most expeditiously by a highly collaborative approach such as has been spearheaded by the nPOD consortium 23. nPOD is an innovative Juvenile Diabetes Research Foundation (JDRF)‐funded project that provides donated organs from people with T1D to leading diabetes scientists all over the world for use in their research studies. It provides researchers with a unique opportunity to literally see and understand more clearly the initial steps of T1D and their physiological impact on the pancreas and immune system.
Is there time to intervene after disease onset?
The T1D research community has, for several decades, communicated the message that as most beta cells have already been destroyed by the time of diagnosis, there is no time to intervene in disease progression. However, we now know that insulin‐positive (albeit perhaps dysfunctional) beta cells can often be observed many years post‐disease onset (Fig. 1e,f). Thus, if there is sufficient beta cell mass left after onset in at least some individuals, it might be conceivable to consider later intervention trials in an attempt to change the disease course for some of those already affected. While evidence for this is limited, at least one recent study 24 noted the ability to intervene positively in subjects with T1D duration up to 2 years post‐disease onset. Clearly, our views on this subject require re‐evaluation and titration.
Is C‐peptide a relevant marker for disease progression?
Which end‐points should be used in clinical trials for T1D? The end‐points currently used in nearly all intervention trials have been focused upon preservation of C‐peptide and secondary mechanistic immunological end‐points. If, however, interventional trials are started later in the disease course we would have to establish more sensitive outcome measures than C‐peptide, because C‐peptide declines more slowly in adults with established diabetes than in young children 25. As definite specific biomarkers for disease progression are still lacking, a range of alternative analyses should be considered and developed further, such as pancreas size and composition, methylated insulin assays and beta cell imaging. Such biomarkers should be validated fully, perhaps utilizing the resources of international workshops. In the meantime, perhaps C‐peptide is the most clinically important outcome that we can measure. Indeed, a recent study examining the association of quantitative C‐peptide concentration with outcomes in the Diabetes Control and Complications Trial found that across the range of values, higher amounts of secreted C‐peptide were associated with lower glycated haemoglobin (HbA1c), lower daily insulin dose, less severe hypoglycaemia and less risk of retinopathy 26.
Should we enroll children and young adults into clinical trials?
Because a large fraction of subjects affected by T1D are children and young adults, careful consideration must be given to these cohorts in intervention trials. Under what circumstances is it ethical to include children and young adults? The declaration of Helsinki and other country‐specific guidelines provide ethical regulations on the execution of clinical trials in children. However, failures of some recent investigative trials such as those involving interleukin (IL)−1 blockade have led to rebuttal from parents that children should only be treated with drugs that have a proven effect in adults. This is a complicated dilemma, because there is evidence (for example from studies of anti‐CD3) that some drugs will work in children and young adults but not in older adults 27. The reason for this divergence is unknown, and could point towards different mechanisms of T1D in childhood or simply the fact that the effect of the drug is smaller because the disease is less aggressive in adults.
Moving forward, therapeutic interventions in T1D should consider the notion of age as a facet for agent selection. There is a limited but convincing body of data which suggests that T1D heterogeneity might be influenced by age and will, in turn, be associated with variations in pathogenic features of disease development. In the case of the anti‐CD3 trials, perhaps there was less dependence on T cells with increasing age. In any case, we should consider including individuals with rapidly progressive disease in trials of immunomodulatory therapies, because otherwise drugs might be missed that could have benefit solely in paediatric T1D. As patients with rapid disease progression tend to be younger children, safety considerations will be paramount. Safety studies should therefore be carried out in adults, followed by efficacy studies in children. In cases where it is known that there are no side effects [for example, as expected for antigenic and some glucagon‐like peptide 1 (GLP1)‐based therapies], paediatric trials could perhaps be considered before completion of studies in adults.
Should we stratify subjects for clinical trials?
At present, patients are enrolled preferentially into intervention studies as soon as possible after the onset of T1D (i.e. < 3 months/100 days after onset). Inherent with this approach, however, is a vast variation in clinical appearance. Some patients will rapidly lose their entire C‐peptide production within a few months, whereas others will exhibit what is known as an extended ‘honeymoon’ phase and have preserved C‐peptide levels and low requirement of insulin even at 1 year after diagnosis (Fig. 3). Composite T1D TrialNet data from an international network of researchers who are exploring ways to prevent, delay and reverse the progression of T1D indicate a mean decrease of only 15% in the area under the curve response to a mixed meal tolerance test during the first year of T1D in 21–46‐year‐old patients 25. This variation in clinical outcome increases the power requirement for any interventional trial, so in the absence of well‐established biomarkers much larger studies are needed. Although partial loss of beta cell function is the common denominator, it is possible that the underlying aetiology or severity of immune attack may vary considerably between individuals. Analysis of other factors [e.g. insulin resistance, body mass index (BMI), C‐peptide at trial onset and age] could reveal mechanisms that will impact significantly the progression of beta cell loss 25. It may be possible that enhanced imaging techniques such as non‐invasive detection of insulitis and beta cell mass may uncover suitable biomarkers to predict the speed of disease progression 28. Patients with predicted rapid progression of T1D could be allocated to interventional trials focusing preferentially on the inhibition of immune‐mediated aetiology. Patients with predicted slow progression could be invited to participate in trials at 6 months to 1 year after disease onset, which would focus on beta cell protective agents, substances triggering beta cell proliferation or substances preventing superimposed gluco‐lipotoxicity. Careful stratification of subjects should therefore enhance the likelihood of finding an effective means to intervene and halt disease progression.
Figure 3.
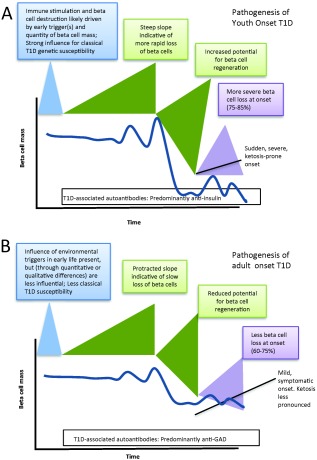
Variability of beta cell decline following diagnosis of type 1 diabetes (T1D) as a function of age. T1D is often discussed as if it were a singular disease. However, many pathogenic features differ between its diagnosis in (a) youth‐ versus (b) adult‐onset T1D.
What steps do the global T1D research community need to take to achieve the goal of pharmacological ‘cure’?
Why have numerous intervention studies often applying intensive immunosuppression failed to durably preserve beta cell function? It could be possible that the level of immune suppression needed to inhibit the progression of T1D is simply not ethically justifiable, because it would lead to extremely undesirable side effects. In addition, the aetiology of the disease, for example a recurrent infectious trigger, might erode therapeutic benefits and prevent us from durably saving any remaining beta cells. One way to circumvent this issue may be to start combination therapy with an induction agent that modulates the immune system more strongly, followed by a maintenance therapy consisting of antigenic tolerance induction and, for example, anti‐inflammatory drugs or metabolic stabilization, e.g. drugs that act on GLP‐1 29, 30. This approach may enhance efficacy while minimizing side effects because of the lower dose of immune‐suppression required. Last, but not least, adaptive immune memory cells are present in a certain fraction of patients with T1D, and alefacept 31 (LFA3 anti‐CD2 fusion protein) is one drug which inhibits these cells while leaving regulatory mechanisms intact.
In conclusion, we should consider the possibility that T1D is caused by multiple and interlaced mechanisms. It is not likely to be cured using a single mode of intervention, and a combinatorial approach is therefore required a priori. Redirection of the focus of global T1D research from predominantly T cell‐mediated autoimmunity studies to more comprehensive and wide‐ranging strategies may uncover agents that are able to preserve beta cell mass and reduce beta cell stress in addition to modulating the adaptive and innate immune mechanisms (Table 1). Combinations of therapies may also be required to allow the use of lower doses of immunomodulatory agents which would limit toxicity while achieving additive or synergistic effects on the disease process.
Table 1.
Recommendations for type 1 diabetes (T1D) research
| Research stage | Recommendation |
|---|---|
| Preclinical research | • More detailed studies of human T1D pathology are needed |
| • Intensive investigation should be carried out into the disease cause/driver (other than autoimmunity) such as aberrant sensitivity of beta cells to metabolic stress, viruses, bacteria, etc. | |
| • Biomarkers should be developed which could be used to map the natural history of T1D and therefore establish whether the disease follows a relapsing/remitting, primary progressive or secondary progressive time–course (or indeed whether the time course depends on the individual) | |
| Phase 1 studies | • Well‐powered studies are required to determine the optimal and rational combinations of therapies for treatment |
| • Surrogate markers of beta cell stress/loss need to be developed and tested (e.g. methylated insulin, non‐invasive imaging, immune biomarkers, etc.) | |
| Phase 2 studies | • Appropriately‐powered Phase 2 studies are required to account for T1D heterogeneity |
| • Biomarkers should be developed to enable the stratification of susceptible patient subsets | |
| • Children should be enrolled unless there is any compelling argument (safety/mechanism) not to |
Disclosure
None declared.
Acknowledgements
The authors declare that no conflicts of interest relevant to this paper exist. M. G. V. H. is an employee of Novo Nordisk in addition to his academic appointment at La Jolla Institute. Editorial assistance for the preparation of the manuscript was funded by Novo Nordisk.
References
- 1. Skyler JS. Update on worldwide efforts to prevent type 1 diabetes. Ann NY Acad Sci 2008; 1150:190–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herold KC, Hagopian W, Auger JA et al. Anti‐CD3 monoclonal antibody in new‐onset type 1 diabetes mellitus. N Engl J Med 2002; 346:1692–8. [DOI] [PubMed] [Google Scholar]
- 3. Keymeulen B, Vandemeulebroucke E, Ziegler AG et al. Insulin needs after CD3‐antibody therapy in new‐onset type 1 diabetes. N Engl J Med 2005; 352:2598–608. [DOI] [PubMed] [Google Scholar]
- 4. Keymeulen B, Walter M, Mathieu C et al. Four‐year metabolic outcome of a randomised controlled CD3‐antibody trial in recent‐onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010; 53:614–23. [DOI] [PubMed] [Google Scholar]
- 5. Bach JF. Anti‐CD3 antibodies for type 1 diabetes: beyond expectations. Lancet 2011; 378:459–60. [DOI] [PubMed] [Google Scholar]
- 6. Pescovitz MD, Greenbaum CJ, Krause‐Steinrauf H et al. Rituximab, B‐lymphocyte depletion, and preservation of beta‐cell function. N Engl J Med 2009; 361:2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dantal J, Soulillou JP. Immunosuppressive drugs and the risk of cancer after organ transplantation. N Engl J Med 2005; 352:1371–3. [DOI] [PubMed] [Google Scholar]
- 8. Di Bisceglie AM, Lok AS, Martin P, Terrault N, Perrillo RP, Hoofnagle JH. Recent US Food and Drug Administration warnings on hepatitis B reactivation with immune‐suppressing and anticancer drugs: just the tip of the iceberg? Hepatology 2015; 61:703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Battaglia M, Atkinson MA. The streetlight effect in type 1 diabetes. Diabetes 2015; 64:1081–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Todd JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity 2001; 15:387–95. [DOI] [PubMed] [Google Scholar]
- 11. Atkinson MA. Losing a grip on the notion of beta‐cell specificity for immune responses in type 1 diabetes: can we handle the truth? Diabetes 2014; 63:3572–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eisenbarth GS. Banting lecture 2009: an unfinished journey: molecular pathogenesis to prevention of type 1A diabetes. Diabetes 2010; 59:759–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. In't VP. Insulitis in human type 1 diabetes: the quest for an elusive lesion. Islets 2011; 3:131–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eisenbarth GS. Insulin autoimmunity: immunogenetics/immunopathogenesis of type 1A diabetes. Ann NY Acad Sci 2003; 1005:109–18. [DOI] [PubMed] [Google Scholar]
- 15. Krogvold L, Skog O, Sundstrom G et al. Function of isolated pancreatic islets from patients at onset of type 1 diabetes; Insulin secretion can be restored after some days in a non‐diabetogenic environment in vitro. Results from the DiViD study. Diabetes 2015; 64:2506–12. [DOI] [PubMed] [Google Scholar]
- 16. Rodriguez‐Calvo T, Ekwall O, Amirian N, Zapardiel‐Gonzalo J, von Herrath MG. Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 2014; 63:3880–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gaglia JL, Guimaraes AR, Harisinghani M et al. Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J Clin Invest 2011; 121:442–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gepts W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes 1965; 14:619–33. [DOI] [PubMed] [Google Scholar]
- 19. Arif S, Leete P, Nguyen V et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 2014; 63:3835–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foulis AK, Farquharson MA, Hardman R. Aberrant expression of class II major histocompatibility complex molecules by B cells and hyperexpression of class I major histocompatibility complex molecules by insulin containing islets in type 1 (insulin‐dependent) diabetes mellitus. Diabetologia 1987; 30:333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bottazzo GF, Dean BM, McNally JM, MacKay EH, Swift PG, Gamble DR. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med 1985; 313:353–60. [DOI] [PubMed] [Google Scholar]
- 22. Skog O, Korsgren S, Wiberg A et al. Expression of human leukocyte antigen class I in endocrine and exocrine pancreatic tissue at onset of type 1 diabetes. Am J Pathol 2015; 185:1129–138. [DOI] [PubMed] [Google Scholar]
- 23. Pugliese A, Yang M, Kusmarteva I et al. The juvenile diabetes research foundation network for pancreatic organ donors with diabetes (nPOD) program: goals, operational model and emerging findings. Pediatr Diabetes 2014; 15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haller MJ, Gitelman SE, Gottlieb PA et al. Anti‐thymocyte globulin/G‐CSF treatment preserves beta cell function in patients with established type 1 diabetes. J Clin Invest 2015; 125:448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Greenbaum CJ, Beam CA, Boulware D et al. Fall in C‐peptide during first 2 years from diagnosis: evidence of at least two distinct phases from composite Type 1 Diabetes TrialNet data. Diabetes 2012; 61:2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lachin JM, McGee P, Palmer JP. Impact of C‐peptide preservation on metabolic and clinical outcomes in the diabetes control and complications trial. Diabetes 2014; 63:739–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hagopian W, Ferry RJ Jr, Sherry N et al. Teplizumab preserves C‐peptide in recent‐onset type 1 diabetes: two‐year results from the randomized, placebo‐controlled Protege trial. Diabetes 2013; 62:3901–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trudeau JD, Kelly‐Smith C, Verchere CB et al. Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 2003; 111:217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Skyler JS. Immune therapy for treating type 1 diabetes: challenging existing paradigms. J Clin Invest 2015; 125:94–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von HM. Combination therapies for type 1 diabetes: why not now? Immunotherapy 2010; 2:289–91. [DOI] [PubMed] [Google Scholar]
- 31. Rigby MR, DiMeglio LA, Rendell MS et al. Targeting of memory T cells with alefacept in new‐onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Diabetes Endocrinol 2013; 1:284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]