

Summary
Sequence‐specific RNA binding proteins (RBP) are important regulators of the immune response. RBP modulate gene expression by regulating splicing, polyadenylation, localization, translation and decay of target mRNAs. Increasing evidence suggests that RBP play critical roles in the development, activation and function of lymphocyte populations in the immune system. This review will discuss the post‐transcriptional regulation of gene expression by RBP during lymphocyte development, with particular focus on the Tristetraprolin family of RBP.
Keywords: cell activation, gene regulation, inflammation
Other Articles Published in this Review Series Lessons from The ageing human B cell repertoire: a failure of selection? Clinical and Experimental Immunology 2016, 183: 50–56. Eosinophils: important players in humoral immunity. Clinical and Experimental Immunology 2016, 183: 57–64. Transcription factors regulating B cell fate in the germinal centre. Clinical and Experimental Immunology 2016, 183: 65–75.
Introduction
Regulation of gene expression can occur at every step during the transfer of genetic information from DNA to protein. Transcript abundance is regulated by transcription factors, epigenetic marks such as DNA methylation and by RNA decay, which is an important mechanism for the dynamic control of gene expression. In order to translate mRNA efficiently into protein, localization and availability are as important as transcript abundance. Moreover, qualitative changes in the RNA complement of the cell, which can include alternative splicing, polyadenylation, RNA methylation and RNA editing, have important consequences for the fate of RNA and for the makeup of the proteome. These processes are regulated dynamically by RNA binding proteins (RBP), which have gained increasing prominence as important post‐transcriptional regulators of gene expression. Remarkably, it has been estimated that more than 1500 RBP are encoded by the mammalian genome 1, 2, 3, 4.
RBP carry out their functions by binding to RNA sequences or secondary structures via diverse RNA binding motifs. Some RBP bind short, single‐stranded sequences within mRNA, including AU‐rich elements (AREs), GU‐rich elements or polypyrimidine tracts 5. Specific RNA sequences may interact with a variety of RBP, sometimes with opposing functions, and therefore can result in alternative RNA fates such as stabilization or destabilization of target transcripts (Fig. 1) 6. The ARE is a sequence motif often found in 3′ untranslated regions (3′UTR) of mRNA. It is associated primarily with RNA decay and can interact with multiple proteins with different affinities in the nanoMolar range (Table 1). While the Tristetraprolin (ZFP36) family of RBP bind AREs via tandem CCCH zinc finger domains, HuR (human antigen R) encoded by ELAV1 (embryonic lethal abnormal vision like 1), CUGBP1 (CUG triplet repeat RNA binding protein 1), AUF1 (AU‐rich element binding factor 1, also known as hnRNP D; heterogeneous nuclear ribonucleoprotein D), nucleolin and TIA1 (T cell intracellular antigen 1) and their paralogues bind through RNA recognition motifs (RRM). By contrast, KSRP (KH type splicing regulatory protein), Mex3D (Tino) and Fragile X mental retardation‐related protein 1 (FXR1), bind AREs via K‐homology (KH) domains, a protein domain that was first recognized in in the human hnRNP‐K protein 14, 15.
Figure 1.
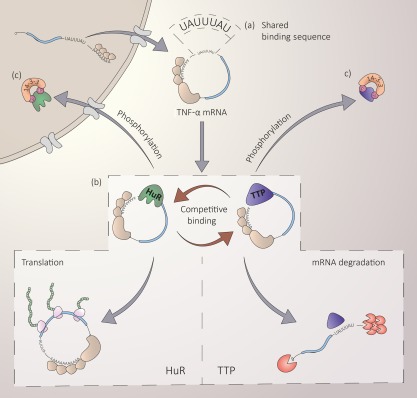
Competitive binding by human antigen R (HuR) and Tis11 can direct alternative target mRNA fates. HuR and Tis11 can compete for the same binding sequences, resulting in opposing mRNA fates. (a) Competition for binding to target mRNA is determined by the affinity each RNA binding proteins (RBP) has to shared binding sequences. (b) The relative expression of these RBP will affect competitive binding. (c) Post‐translational regulation of RBP, for example via phosphorylation, affects their ability to bind to target mRNAs.
Table 1.
Binding affinity of tristetraprolin (TTP) and human antigen R (HuR) to synthetic RNA fragments
| RNA | ARE sequence | nM binding affinity to TTP | nM binding affinity to HuR | Reference |
|---|---|---|---|---|
| AUUUAUUUAUUUA | 3·6 | 7, 8 | ||
| AUUUAUUUAUUUAUUUAUUUAUUUA | 0·5 | 0·50 | 9 | |
| UUAUUUAUU | 3·0 | 7, 8 | ||
| UAUUUAU | 19·0 | 7, 8 | ||
| UUUUUUUU | Not bound | 10 | ||
| UUUUUUUUU | 0·97 | 10 | ||
| UUUUUUUUUUUUU | 280·0 | 7, 8 | ||
| UUUUAUUUAUUUU | 3·2 | 7, 8 | ||
| UUUUAUUUUUUUU | 56·0 | 7, 8 | ||
| UUUUAUUUCUUUU | 83·0 | 7, 8 | ||
| UUUUAUUUGUUUU | 28·0 | 7, 8 | ||
| UUUUUUUUAUUUU | 54·0 | 7, 8 | ||
| UUUUCUUUAUUUU | 93·0 | 7, 8 | ||
| UUUUGUUUAUUUU | 28·0 | 7, 8 | ||
| UUAUUUUUU | 130·0 | 7, 8 | ||
| UUUUUUAUU | 120·0 | 7, 8 | ||
| UUUUAUAUUUU | 160·0 | 7, 8 | ||
| UUUUAUUAUUUU | 18·0 | 7, 8 | ||
| UUUUAUUUAUUUU | 3·2 | 7, 8 | ||
| UUUUAUUUUAUUUU | 6·4 | 7, 8 | ||
| UUUUAUUUUUAUUUU | 17·0 | 7, 8 | ||
| NNUUNNUUU | 0·96 | 10 | ||
| UAUUAUUUU | 1·14 | 10 | ||
| A AUUUAUUU | 1·01 | 10 | ||
| CUUUCUUUCUUUCUUUC | 0·96 | 10 | ||
| cFos | UAUUUAUAUUUUUAUUUUAUUU | 34 | 11 | |
| c‐Myc | UUA C C AUCUUUUUUUUUCUUUA | >1000 | 11 | |
| Cox‐2 | UAUUA AUUUA AUUAUUUA AUA AUAUUUAUAUUA A A | 13·6 | 10 | |
| GM‐CSF | AUUUAUUUAUUUAUUUAUUUA | 21 | 11 | |
| IL‐1β | UAUUUAUUUAUUUAUUUGUUUGUUUGUUUUAUU | 0·12 | 10, 12 | |
| IL‐2 | UAUUUAUUUA A AUAUUUA A AUUUUAUAUUUAUU | 44 | 9·5 | 10, 11, 12 |
| IL‐3 | UAUUUAUUUAUGUAUUUAUGUAUUUAUUUAUUUAU | 4 | 11 | |
| IL‐8 | UAUUUAUUAUUUAUGUAUUUAUUUA A | 1·09 | 10, 12 | |
| TNF‐α | AUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUA | 2 | 0·35 | 10, 11, 12 |
| cFos | 3′UTR | 10 | 13 | |
| IL‐2 | 3′UTR | 32·77 | 10 | |
| TNF‐α | 3′UTR | 3·87 | 10 |
Dissociation constants (Kd) for TTP and HuR ARE interactions at 25°C. Data taken from published fluorescence anisotropy data. Adenosine residues are shown in red, cytosine and guanine residues are shown in blue and N (any) residues are shown in purple. ARE = adenosine–uridine‐rich element; GM‐CSF = granulocyte–macrophage colony‐stimulating factor; TNF‐α = tumour necrosis factor‐alpha; IL = interleukin.
Regulation of RNA stability provides an important mechanism by which the abundance of different classes of RNA, including mRNA, can be controlled. The first evidence for a role of AREs in mRNA stability came in 1986, when they were identified as conserved sequences in the 3′UTR of transcripts encoding cytokines and some proto‐oncogenes. These proteins were known to be encoded by mRNAs with short half‐lives, and it was demonstrated that the ARE from the 3′UTR of granulocyte–macrophage colony‐stimulating (GM‐CSF) mRNA, when incorporated into a reporter construct, was a destabilizing element 16. At around the same time, it was recognized that a consensus sequence (TTATTTAT) was present in the 3′UTRs of human and mouse tumour necrosis factor (TNF) and interleukin (IL)‐1 mRNAs as well as the human lymphotoxin, colony‐stimulating factor, human and rat fibronectin, and almost all of the then sequenced human and mouse interferons 17. As this consensus sequence appeared at a higher rate than would be dictated by chance alone, it was suggested that it may have some regulatory capacity in controlling the magnitude of the inflammatory response 17. Two years later, this hypothesis was confirmed when it was shown that these identified AREs confer instability to a number of cytokine mRNAs 18. In the same year, AREs contained within the c‐Fos 3′UTR were shown to destabilize the transcript, and play a pivotal role in the process of deadenylation 19. It has been shown since that ARE‐containing mRNAs are recruited selectively to the deadenylase machinery 20, 21. Furthermore, exosome mediated 3′‐5′ mRNA decay has been shown to be an important mechanism of ARE‐containing mRNA decay 22, 23. AU‐rich elements are found in mRNAs from a diverse range of species, including yeast 24, drosophila 25, sea urchin 26 and mouse, indicating that the control of mRNA stability via AREs is a conserved regulatory process that has been selected for during evolution 6.
Some RBP recognize secondary RNA structures, such as the constitutive decay element (CDE); a stem‐loop sequence found in more than 100 transcripts 5. The functional relationship between CDEs and AREs in controlling RNA stability remains to be elucidated fully; however, it is likely that each motif promotes RNA decay via independent mechanisms. A β‐globin reporter transcript, carrying the 3′UTR of TNF, remained labile in mutant cell lines defective for ARE‐mediated mRNA decay, indicating that the TNF 3′UTR contains a further destabilizing element 27. To analyse this further, RAW 264·7 macrophages were transfected stably with a GFP reporter comprising the β‐globin 3′UTR, into which TNF AREs or a section of the TNF 3′UTR thought to contain a CDE were inserted 27. Treatment of these stably transfected cell lines with lipopolysaccharide (LPS), or activation of p38 or PI3K signalling in NIH 3T3 cells, inhibited ARE‐mediated but not CDE‐mediated TNF‐α mRNA destabilization 27. The CDE is bound by the RBP Roquin‐1 and Roquin‐2, which have overlapping functions in the regulation of T cell activation 28, 29, 30, 31.
Post‐translational regulation of RBP
The activity of RBP is governed by the same signalling pathways that control lymphocyte development and activation in response to antigen and cytokine receptor signalling 32. Phosphorylation of RBP controls their function and localization, and it is likely that RBP may act as a point of convergence for the activity of numerous kinases 32. This includes the PI3K pathway, protein kinase B (PKB), mTOR (mammalian target of rapamycin) and p38 mitogen‐activated protein kinase (MAPK). Phosphoproteomic analysis of exogenous Tis11, transfected stably into RAW264·7 macrophages, identified 14 sites of phosphorylation which include 11 serine residues, two threonine residues and one tyrosine reside 33. Phosphorylation of Tis11 and Tis11b at specific serine residues, by p38 MAPK/MK2 and MK2 and PKB, respectively, results in their sequestration by 14–3–3 proteins, and diminished mRNA decay activity due in part to decreased affinity for AREs 9, 34, 35, 36, 37, 38. Additionally, phosphorylated Tis11 is impaired in its ability to recruit mRNA decay enzymes, resulting in decreased deadenylation of target transcripts 20, 21. While association with 14–3–3 proteins is sufficient to prevent effective deadenylase recruitment, this impairment is worsened when Tis11 is phosphorylated 20. This suggests that phosphorylation of Tis11 inhibits its ability to promote mRNA decay in both a 14–3–3‐dependent and ‐independent manner. Furthermore, phosphorylation induced assembly of Tis11/14–3–3 protein complexes result in the exclusion of Tis11 from stress granules, a further mechanism by which phosphorylation impairs Tis11‐mediated mRNA decay (Fig. 2) 36. Recent evidence suggests that MK2‐mediated phosphorylation of serine 52 and serine 178 of Tis11 protect the protein from proteasomal degradation, and may inhibit Tis11 from autoregulating its expression by destabilization of its own mRNA 33. Despite this, replacing serine 52 and 178 codons with non‐phosphorylatable alanine codons in the endogenous murine Tis11 locus, resulted in the production of a mutant form of Tis11, which is highly active and promotes destabilization of target mRNAs 33. Indeed, this mutated Tis11 can compete effectively with an excess of wild‐type Tis11 to promote more rapid mRNA degradation, further suggesting that phosphorylation of Tis11 decreases its affinity for RNA 33. In addition, the dual‐specificity phosphatase 1 (DUSP1), which antagonizes the MAPK signalling pathway, has been shown to regulate Tis11 phosphorylation status 39. Targeted deletion of Dusp1 gives rise to prolonged activation of p38 MAPK signalling, which promotes phosphorylation of serine 52 and serine 178 of Tis11, resulting in the accumulation of inactive Tis11, and subsequent stabilization of its target mRNAs following LPS treatment of bone marrow macrophages (BMMs) 39. Tis11 can also be ubiquitinated by tumour necrosis factor receptor‐associated factor 2 (TRAF2) in a phosphorylation‐dependent manner 40, resulting in protein stabilization 41. It is likely that Tis11 degradation by the proteasome is ubiquitin‐independent, and instead is dependent upon intrinsically disordered N‐ and C‐terminals of the protein, a mechanism which is conserved in the invertebrate drosophila 42. Phosphorylation of HuR at serine residues 202 and 242, which reside within the hinge region of the protein, and serine 158 and 221 by PKCα, affect the subcellular localization of the protein, due to increased binding of 14–3–3 proteins, resulting in nuclear accumulation of HuR 43, 44, 45. Furthermore, stress‐inducing arsenite treatment of HeLa cells resulted in Janus kinase 3 (JAK3) phosphorylation of HuR at tyrosine 200, preventing its localization to stress granules, resulting in accelerated decay of HuR target mRNAs 46.
Figure 2.
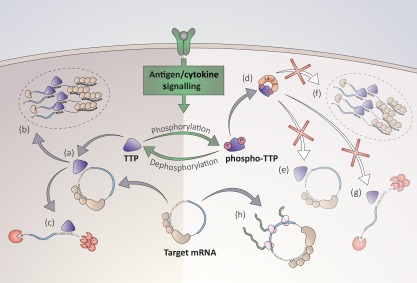
Phosphorylation‐mediated regulation of Tis11 downstream of antigen/cytokine receptor signalling. Tis11 is regulated downstream of antigen/cytokine signalling which culminates in phosphorylation of specific serine residues on the protein. (a) Dephosphorylated Tis11 regulates target mRNA expression negatively through binding specific adenosine–uridine‐rich elements (AREs) in the 3′UTR. (b) Tis11 targets mRNA to sites of degradation such as processing bodies. (c) Tis11 recruits mRNA degradation machinery. (d) Phosphorylation of Tis11 results in 14‐3‐3 binding and regulates its function negatively, allowing Tis11 target mRNAs to be translated and expressed. (e) Phosphorylated Tis11 has reduced binding affinity for target mRNAs. (f) Phosphorylation may result in the exclusion Tis11 from sites of degradation such as procession bodies. (g) Phosphorylated Tis11 is deficient in its ability to recruit mRNA decay machinery. (h) As a result of this there may be increased translation of Tis11b target mRNAs.
RBP have also been reported to regulate kinase expression through conserved AREs in the 3′UTRs of PKBα, β and γ and p38α 47. It is likely that RBP regulate the activity of a number of phosphatases, including members of the DUSP family. Under conditions of oxidative stress, DUSP1 mRNA was stabilized in HeLa cells by the binding of HuR and NF90 48, 49. HuR further promotes DUSP1 expression by facilitating recruitment of the DUSP1 transcript to the translation machinery, and enhancing translation efficiency 50. DUSP1 is also a target of Tis11, which plays a role in the degradation of its mRNA 51, 52. Indeed, DUSP1 transcript levels were shown to be elevated in Tis11–/– dendritic cells 52. This constitutes a potential feedback loop preventing excessive attenuation of Tis11 signalling, thus limiting inflammatory responses. Therefore, DUSP1 mRNA is a target of numerous RBP, indicating that its expression is regulated tightly both at the transcriptional and post‐transcriptional levels, to prevent aberrant MAPK signalling.
Competitive binding of RBP alters the fate of mRNA targets
Binding sequences within target mRNAs interact frequently with a number of different RBPs, with differing functions (Fig. 1) 6, 53, therefore the same sequences within mRNAs can direct alternative fates including stabilization or destabilization of target RNAs 53. In macrophages and T lymphocytes, translation of TNF mRNA is dependent upon binding of HuR to AU‐rich elements within its 3′UTR in the absence of Tis11 9, 54. In a model of the dynamic regulation of TNF mRNA, the phosphorylation of Tis11 by the p38‐MK2 pathway reduces its affinity for the TNF AREs, allowing HuR to bind and promote stabilization and translation of the transcript (Fig. 1) 9. The phosphorylation‐regulated exchange between Tis11 and HuR may allow for a rapid and reversible switch between unstable, non‐translated mRNA and stable, translatable mRNA; however, the extent to which this competitive mechanism occurs in vivo remains to be elucidated. Indeed, a myeloid‐specific deletion of HuR results in an exacerbated inflammatory profile, including enhanced levels of TNF resulting from decreased transcriptional silencing 55. KSRP has been shown to compete for binding of inducible nitric oxide synthase (iNOS) mRNA with HuR in human epithelial colon carcinoma DLD‐1 cells 56. Like Tis11, KSRP has been shown to destabilize target mRNAs. Upon activation, of DLD‐1 cells with a cytokine mixture containing IL‐1β, interferon (IFN)‐γ and TNF, KSRP RNA binding affinity was decreased while HuR affinity for AREs was increased 56. Although Tis11 has not been shown to bind to human iNOS transcripts, it was shown to interact with KSRP in the exosome, indicating a complex interplay of RBP in the regulation of mRNA stability.
RBP in lymphocyte development
A number of roles for RBP in key immunological processes have already been established; however, their role in lymphocyte development remains less well defined 5, 57. Much of the work characterizing the involvement of RBP in lymphocyte development has made use of conditional gene targeting studies in mice 57.
HuR
As germline deletion of HuR is lethal at the embryonic stage, HuR function has been studied in mice using a variety of conditional gene deletion systems, as well as a transgenic system targeted to macrophages 58, 59, 60, 61. Inducible cre‐mediated deletion of HuR using tamoxifen‐regulatable cre resulted in reduced numbers of common lymphoid‐ and B cell‐progenitors 62. Loss of lymphocyte progenitors was linked to increased apoptosis resulting from excessive p53 activity, an effect that was attributed to a requirement for HuR to stabilize mouse double minute 2 homologue (Mdm2) mRNA, which encodes an important negative regulator of p53 57, 62. Inducible cre systems are, however, challenging to use as the tamoxifen‐induced activation of creERT2 itself can result in increased apoptosis of haematopoietic cells in vivo 63, 64. Furthermore, the link to DNA damage/p53 response has not been widely reproducible; indeed, HuR has been shown to increase expression of p53 65, 66, 67, 68. It also remains unclear why HuR function was attributed to a single RNA–protein interaction when a prevailing view is that HuR, like many RBP, acts to control multiple targets 1. HuR can control additional post‐transcriptional processes such as polyadenylation 69 and splicing 59, 70, although it remains unknown what influence this might have on early lymphocyte development.
Conditional deletion of HuR at the pro‐B cell stage using mb1‐cre indicated that HuR appears to be dispensable for the majority of B cell development and homeostasis. However, the B1 B cell subset was depleted, suggesting an intrinsic requirement for HuR in the development or maintenance of this population 59. The same study found an important role for HuR in mediating the germinal centre (GC) reaction and for the production of class‐switched antibodies in response to thymus‐independent (TI) antigens 59. In addition, it highlights a role for HuR in the quality control of the transcriptome and in regulating mitochondrial metabolism. In the absence of HuR, excessive production of reactive oxygen species led to B cell death 59. Additionally, there was alternative splicing of DLST, a subunit of the α‐KDGH complex which is essential for the maintenance of tricarboxylic acid (TCA)‐cycle flux and cell energy supply, resulting in reduced transcription of enzymatically active protein 59. Conditional deletion of HuR in early thymocyte development, using lck‐cre, results in defective cell cycle progression of DN thymocytes, T cell receptor (TCR) signalling and T cell egress from the thymus 60. These mice had enlarged thymi, but suffer substantial losses of peripheral T cell subsets due to decreased positive selection of HuR‐deficient thymocytes, and an inability of single‐positive T cells to respond to chemotactic signals 60. HuR has been shown to regulate the stability of a number of proinflammatory cytokine mRNAs, including IL‐4, IL‐13, TNF‐α and IL‐17 71, 72, 73. Conditional deletion of HuR in activated T cells under the control of OX40‐cre indicated a gene dosage effect of HuR in the control of cytokine production 74. While GATA binding protein 3 (GATA3), IL‐4 and IL‐13 mRNA was reduced in heterozygous conditional knock‐outs, IL‐2, IL‐4 and IL‐13 mRNA and protein expression was increased in homozygous conditional knock‐outs 74. These apparently discordant results reflect potentially disparate roles for HuR in mRNA metabolism and translation.
AUF1
Recent PAR‐CLIP (photoactivatable‐ribonucleoside‐enhanced cross‐linking and immunoprecipitation) analysis to identify RBP binding sites indicated that AU‐binding factor 1 (AUF1) acts globally to destabilize RNA targets; however, it may also stabilize a discrete subset of RNAs 75. Additionally, AUF1 plays an important role in promoting translation through interactions with HuR 75. AUF1–/– mice have a small (two‐fold) reduction in the number of splenocytes due to reduced numbers of B and T lymphocytes compared to wild‐type control mice 76. Lymphocyte populations are normal in the bone marrow and thymus, suggesting a predominant role for AUF1 in peripheral lymphocyte homeostasis. The deficit in splenocyte numbers was attributed to a slight decrease in follicular B cell numbers, suggesting that AUF1 may play a role in maintaining the splenic follicular B cell compartment; however, the total numbers of splenic B cells remain unchanged, as the reduction in follicular B cell numbers is counterbalanced by the increase in absolute numbers of marginal zone (MZ) and newly formed B cells 76. In the absence of AUF1, splenic follicular B cells undergo increased apoptosis, resulting from reduced expression of the anti‐apoptotic proteins Bcl2, A1 and Bcl‐XL 76. The transcripts encoding these proteins contain potential binding sites for AUF1 57. While thymus‐dependent (TD) immune responses are normal in AUF1–/– mice, TI humoral responses are attenuated despite no impairment in GC formation or class switch recombination (CSR), suggesting that AUF1 may also be important for the functionality of MZ B cells in the spleen 76. The prosurvival protein Bcl2 is required to maintain mature B cells in healthy mice 77, 78. AUF1 has been shown to bind to the AU‐rich sequence in the 3′UTR of Bcl2 mRNA in mature primary B cells 79. Deletion of these AU‐rich elements reduces Bcl2 protein expression 79. This is due in part to the lack of AUF1 binding to the 3′UTR of Bcl2, which is required for the stabilization and subsequent translation of the Bcl2 transcript, promoting survival of mature B cells 79.
Roquin‐1 and Roquin‐2
Roquin‐1 and Roquin‐2 bind to secondary RNA structures known as CDE. These proteins are functionally redundant in their capacity to maintain T cell homeostasis 29. In the absence of Roquin‐1 and ‐2 in T cells, or in mice expressing a dominant negative Roquin‐1 (sanroque), T cells differentiate spontaneously into T follicular helper cells (Tfh) 28, 29, 30, 31. This is associated with a lupus‐like autoimmune phenotype in the sanroque mice 31. Roquin‐1 and ‐2 redundantly control the expression of ICOS (inducible co‐stimulator) and OX40 (tumour necrosis factor superfamily member 4) mRNAs, both of which are important in the regulation of effector T cell differentiation and function 29. In addition, the Roquin proteins act in concert with the endoribonuclease Regnase‐1 to inhibit T helper type 17 (Th17) differentiation by co‐operatively binding and promoting degradation of the RNA of Th17‐promoting factors, which include IL‐6, ICOS, c‐Rel and IFN regulatory factor 4 (IRF‐4) 80. Indeed, in the absence of Roquin‐1 and ‐2, Th17 cells were seen to accumulate in the lungs of mice 80. Both Roquin and Regnase‐1 are cleaved downstream of TCR signalling by the paracaspase mucosa‐associated lymphoid tissue lymphoma translocation protein 1 (MALT‐1), resulting in their inactivation, releasing Roquin and Regnase‐1 mRNA targets from repression, providing a mechanism by which TCR signal strength is translated into T cell differentiation 80, 81.
Tis11 family
Tristetraprolin (TTP, Tis11, ZFP36) is the founding member of a small family of RNA binding proteins. The Tis11 family consists of four members in mammals: Tis11, Tis11b (BRF1, ZFP36l1, BERG36, ERF‐1), Tis11d (BRF2, ZFP36l2, ERF‐2), and a fourth protein, ZFP36l3, expressed only in mouse placenta and yolk sac 82, 83. Tis11 orthologues have been found in most vertebrates studied, including lizards and alligators; however, no sequences corresponding to Tis11 have been found in any bird species 84. Despite this, orthologous protein sequences for Tis11b and Tis11d were discovered in chickens, with high sequence conservation to alligator and lizard 84. In addition to vertebrate orthologues, MEX‐5 and MEX‐6 proteins, which have important roles in determining embryonic asymmetry in Caenorhabditis elegans, share the conserved CCCH zinc finger domains with Tis11, but with slightly different spacing of the zinc fingers 85. Tis11 was described originally in 1991 as an immediate early gene induced by the phorbol ester tetradecanoyl phorbol acetate (TPA) and by growth factors 86; however, it is now known that Tis11 family members are up‐regulated downstream of antigen receptor signalling and in response to LPS, insulin and serum stimulation 87, 88, 89, 90, 91, 92, 93, 94. The Tis11 knock‐out mouse exhibited a severe syndrome characterized by growth retardation, cachexia, arthritis, chronic inflammation and autoimmunity that was caused by excess TNF 95. Tis11 was shown subsequently to bind the ARE of TNF mRNA directly, resulting in the destabilization and degradation of the mRNA 91. Moreover, deletion of the TNF ARE in mice showed that it was a dominant inhibitory sequence that limited TNF production and its associated pathology 96. As macrophages were shown to be an important cellular source of TNF in Tis11–/– mice 97, a myeloid‐specific deletion of Tis11 was generated to confirm whether the excess of TNF in Tis11 germline knock‐outs was produced by myeloid cells 98. However, mice with a LysMcre‐mediated myeloid‐specific knock‐out of Tis11 did not recapitulate the phenotype of the germline knock‐outs, providing evidence that Tis11 has important functional roles outside the myeloid lineage to inhibit inflammation and autoimmunity 97, 99. Tis11 family members share two highly conserved CCCH tandem zinc fingers preceded by a YKTEL motif 100. Mutation of any of the eight amino acids responsible for co‐ordinating the zinc ions within the zinc finger domains completely disrupts RNA binding 101. Tis11 family proteins interact with AREs in the 3′UTR of target mRNAs, where each zinc finger domain binds a separate 5′‐UAUU‐3′ subsite, thus the Tis11 family members bind preferentially a nonameric consensus sequence: UUAUUUAUU with 3·6 nM binding affinity (Table 1) 7, 100. This nonameric sequence is found several times in the 3′UTR of TNF‐α, a well‐characterized target of Tis11 102. Investigations using RNA SELEX selected the same consensus binding site for the Tis11 protein 103. Optimal Tis11 binding requires an RNA substrate greater than 7 nt in length, which must contain two adenylate residues within a U‐rich sequence, the spacing of which contribute to binding affinity of Tis11 7. Thus, Tis11 still has strong (6·4 nM) binding affinity for substrates containing AUUUUA motifs, but only intermediate affinity for AUUA and AUUUUUA binding sequences (18 and 17 nM, respectively) (Table 1) 7.The RNA binds to the zinc fingers in a linear fashion, free from secondary structure, indicating that RNA folding may actually inhibit RNA binding 100. As the presence of AU‐rich sequences alone does not define an RNA as a Tis11 family target, secondary structure may constitute a key factor which distinguishes a functional from a non‐functional ARE. This presents an intriguing possibility whereby binding affinity of Tis11 family proteins may be regulated dynamically by changes in RNA structure.
Once bound to the RNA, the Tis11 family proteins can interact with a number of different effector molecules to bring about degradation or translational arrest of the target transcript. Tis11 associates indirectly with poly(A)‐specific ribonuclease (PARN), resulting in de‐adenylation and destabilization of target mRNAs 104. It has also been shown that Tis11 and Tis11b associate with the decapping enzymes Dcp1 and Dcp2, the 5′‐3′ exonuclease Xrn1 via an N‐terminal activation domain, which acts as a binding platform for mRNA decay enzymes 82, 105, 106. Tis11 has also been shown to co‐exist with the CCR4–CAF‐1 deadenylase complex, which may interact with Tis11 via the largest protein in the complex, Not1 21, 107, 108. This suggests either that RNA decay mediated by Tis11 occurs via a number of different mechanisms, or that Tis11 acts to co‐ordinate RNA decay pathways. AREs are found in the 3′UTRs of a range of mRNAs encoding cytokines, transcription factors, cell cycle regulators and regulators of apoptosis. However, as not all predicted targets have been shown to be physiologically relevant, data from conditional knock‐out models should be considered.
Tis11b‐deficient mice die from a failure of chorioallantoic fusion during embryogenesis, and manifest vascularization defects in the embryo 109, 110. This phenotype is associated with elevated levels of vascular endothelial growth factor‐A (VEGF‐A) in the embryo and in cultured embryonic fibroblasts. In transformed Tis11b‐deficient fibroblasts, increases in VEGF‐A protein are not the result of increased Vegfa mRNA stability, but arise from the increased association of Vegfa mRNA with ribosomes, indicating that Tis11b can regulate translation efficiency independently of mRNA stability 109. This is potentially an important observation because AREs are considered primarily to confer instability on mRNA targets of Tis11 family members. While it has already been recognized that RBP including HuR, T cell intracellular antigen (TIA) and TIA‐1‐related protein (TIAR) are involved in the translational control of RNA targets, it is important to consider the numerous levels of regulation at which RBP can act when defining target transcripts 111. The mechanism of translational repression, mediated by the Tis11 family of RNA binding proteins, remains to be elucidated fully. Like Tis11b, Tis11 has also been shown to shift target transcripts to lighter polysome fractions, resulting in decreased or repressed translation 112. This translational repression of target transcripts is dependent upon the interaction of Tis11 with the DEAD‐box helicase RCK (DDX6), which localizes to processing‐bodies (P‐bodies), sites of cellular mRNA turnover 112. The Tis11 family of proteins promote localization of ARE‐containing transcripts into P‐bodies. This is augmented when the availability of mRNA decay enzymes is limiting, suggesting that Tis11, Tis11b and Tis11d may sequester target mRNAs in P bodies when RNA decay is inefficient 113.
Tis11d knock‐out mice are viable for approximately 2 weeks after birth; however, they have significant defects in haematopoiesis, resulting in anaemia and thrombocytopenia 114. The phenotypical differences observed in the germline ablation of Tis11 family members suggests that although these proteins have highly conserved RNA binding domains, they have some non‐redundant functions during development. This difference could result from tissue specific expression or interactions with different effector proteins, although there is a multitude of overlapping functions between family members and clear evidence for redundancy in T cell development 115.
Young mice which have a double conditional deletion of Tis11b and Tis11d in lymphocytes using CD2cre have reduced thymic cellularity and an increased proportion of double‐negative (DN) and CD8 single‐positive thymocytes. Subsequently, they develop T cell acute lymphoblastic leukaemia (T‐ALL) 115. Tis11b‐ and Tis11d‐deficient DN thymocytes are able to bypass the β‐selection check‐point, which ensures that all T cells express the TCR‐β chain, resulting in the inappropriate proliferative expansion of TCR‐β‐negative T cells 115. The emergence of this aberrant population of TCR‐β‐negative cells requires significantly up‐regulated Notch‐1 protein, which is encoded by an mRNA that is a direct target of Tis11b and Tis11d 115. A role for Tis11 as a negative regulator of B cell development in ageing mice was proposed to occur via negative regulation of E2A mRNA 116. However, it is likely that the Tis11 family proteins play important roles in both early and late B cell development and function (unpublished data from R.N.).
RBP in disease
Disrupted ARE‐mediated processes can arise from a number of different mechanisms, including mutations in AREs, chromosomal deletions of AREs, changes in the abundance of differentially expressing ARE mRNA isoforms and changes in the function, localization or binding affinity of RBP 117. The majority of known RBP defects manifest as neurodegenerative disorders due to the high prevalence of alternative splicing in the brain, a major function of RBP 118, 119. However, RBP dysfunction can also result in a range of human cancers, as cell growth and proliferation is known to be modulated by a variety of RBP. The immune response involves a variety of inflammatory responses, which must be regulated tightly in part by RBP to prevent chronic inflammatory disease and cancer.
RBP in cancer
Genetic translocation of proto‐oncogenes, including Bcl2, Bcl6 and c‐Myc, are found in a variety of B cell tumours, including Burkitt lymphoma, follicular B cell lymphoma and ‘double‐hit’ (DH) mature B cell lymphomas that specifically involve the Myc and Bcl2 loci 120, 121. Enhanced expression of Bcl2 drives B cell survival and has been shown to be important for the survival and progression of B cell leukaemia 78, 122. AREs present within the Bcl2 3′UTR are required to promote Bcl2 mRNA stability and protein expression in mature B cells through binding of HuR 79. In addition to B cell leukaemias, enhanced expression of HuR has been associated with high‐grade malignant brain tumours, including glioblastoma multiforme and medulloblastoma, where it contributes to tumour progression by stabilizing various angiogenic factors such as VEGF 123. HuR is also implicated in colon cancer, where stabilization of the proinflammatory enzyme COX‐2 (cyclooxygenase‐2) results in enhanced growth and tumourogenicity of human colon cancer cells 124. Thus, therapeutically targeting HuR could be considered as a component of new cancer treatments, for example resulting in reduced expression of proto‐oncogenes such as Bcl2 and COX‐2. ARE dysregulation can also result in carcinogenesis. Removing AREs in the 3′UTR of fos mRNA resulted in increased oncogenic potential of this proto‐oncogene 125. Furthermore, genomic rearrangement of the CCND1 (cyclinD1) 3′UTR resulted in over‐expression of CCND1 mRNA in mantle cell lymphoma 126. The resulting perturbation of the G1/S‐phase transition in the cell cycle contributed to augmented tumour development 126. AUF1 has been shown to decrease carcinogenesis in a number of malignancies by destabilizing the prosurvival factor Bcl2, or cyclinD1 127. However, it has also been shown to be elevated in various cancers, including lymphomas, sarcomas and carcinomas, where it may play a pathological role 127. Alternative splicing and splice factor mutations are found consistently in haematopoietic malignancies, implicating the importance of studying this RBP function in cancer 128. The Tis11 family proteins have been implicated as important tumour suppressors by destabilizing transcripts whose over‐expression is related to malignancy 82. Indeed, tumour formation induced by injection of v‐H‐Ras‐transformed mast cells was delayed when cells additionally over‐expressed Tis11 129. Recent evidence also suggests that Tis11 expression is decreased in human invasive breast cancer cell lines when compared to healthy breast cell lines 130. Tis11 has been shown further to regulate a subset of transcripts which may be involved in the growth, invasion and metastasis of cancer cells 130, 131. Deletion of Tis11b and Tis11d in lymphocytes results in the development of T‐ALL in young mice, in a Notch‐1 dependent manner 115. Despite this, over‐expression of Tis11b has been shown to contribute to leukaemogenesis in primary cells and cell lines, mediated by the fusion transcription factor AML1–MTG8 132. AML1–MTG8 is generated by a genetic translocation, resulting in the dysregulation of genes in haematopoietic progenitors, found in 40% of acute myeloid leukaemia subtype M2 132. Furthermore, increased expression of Tis11b has been found in primary breast cancer 133 and Tis11, Tis11b and Tis11d were found to be elevated in a number of human cancer cell lines when compared to healthy tissues 134. This suggests that the Tis11 family play complex and varied roles in a range of human cancers.
RBP in inflammatory pathology
Post‐transcriptional regulation of mRNA turnover plays an important role in cytokine expression, and many cytokine mRNAs contain AREs in their 3′UTRs. Deleting AREs in the 3′UTR of TNF‐α results in a spontaneous inflammatory syndrome which comprises chronic arthritis and Crohn's like intestinal inflammation 96. A similar autoimmune phenotype is exhibited by Tis11‐deficient mice, demonstrating a key role for Tis11 in restraining chronic TNF signalling 91. HuR has also been shown to stabilize proinflammatory cytokines in activated T cells 74. This results in augmented responses to inflammatory diseases such as experimental autoimmune encephalomyelitis (EAE) 72. These studies indicate that targeted ablation of HuR activity may help to resolve chronic inflammatory conditions by reducing the stabilization of a number of proinflammatory proteins. Despite this, HuR over‐expression in myeloid lineage cells has been shown to dampen inflammatory responses resulting from post‐transcriptional silencing of proinflammatory effectors 55. Indeed, mice lacking HuR in myeloid cells exhibited increased susceptibility to chemical‐induced colitis and colitis‐associated cancer 55. These seemingly contradictory results, between innate and adaptive cells, should be considered prior to investigating the therapeutic targeting of HuR in inflammatory pathologies.
Concluding remarks
The central dogma of molecular biology, a term first coined by Francis Crick, describes the transfer of genetic information from DNA to messenger RNA (mRNA), and from mRNA copies to produce proteins, which constitute the functional machinery of the cell 135. Gene expression, when it refers to the production of proteins, can be regulated at every stage in this process. This review has focused on the post‐transcriptional regulation of gene expression by RNA binding proteins (RBP). While gene expression can be regulated positively and negatively by transcription factor binding, mRNA decay represents an important mechanism by which the abundance of mRNA is controlled in the cytoplasm. The roles of some RBP as limiting factors for immune cell activation and inflammation are becoming clearer, but their role in lymphocyte development is less well characterized 5, 57. The most insightful results have come from the characterization of transgenic mice 57. In order to progress, the field must consider not only the target transcripts of RBP but how RBP are regulated, and their co‐operative interactions with each other and with microRNAs in the regulation of mRNA stability.
The immune response involves a variety of inflammatory responses, which must be regulated tightly to prevent chronic inflammatory disease and cancer. Post‐transcriptional mechanisms that control the stability of mRNA and/or transcription are particularly important in the resolution of inflammation. The half‐life of mRNAs encoding pro‐ or anti‐inflammatory proteins can be highly variable; therefore, blocking transcription may not provide sufficiently rapid changes in transcript abundance to alter the magnitude of inflammation 136. As such, regulation of mRNA stability constitutes an important regulatory mechanism by which inflammatory mediators are controlled. Evidence for the post‐transcriptional regulation of proinflammatory effector proteins by RBP demonstrates their critical involvement in mediating inflammatory homeostasis and cancer. Given the importance of RBP‐mediated regulation of a range of cellular processes, remarkably few RBP have been identified as oncogenes or tumour‐suppressors 118. While HuR has been implicated in a number of human malignancies, caution should be exercised when considering RBP as future therapeutic targets. RBP regulate diverse RNA targets, and few are specific to one tissue. It is therefore imperative to have a greater understanding of basic RBP biology before we can target them for therapeutic benefit.
The roles of RBP in inflammatory disease and cancer are becoming increasingly well characterized; however, little has been done to investigate their roles during ageing. As the process of ageing is associated commonly with an increasing inflammatory phenotype (inflammaging), it would be interesting to see whether some of this is due to a loss of the post‐transcriptional regulation of inflammatory mediators such as cytokines and chemokines.
Disclosure
The authors have no conflicting financial interests.
Acknowledgements
We would like to thank Elisa Monzon‐Casanova for critical reading of the manuscript.
References
- 1. Keene JD. RNA regulons: coordination of post‐transcriptional events. Nat Rev Genet 2007; 8:533–43. [DOI] [PubMed] [Google Scholar]
- 2. Araujo PR, Yoon K, Ko D, et al. Before it gets started: regulating translation at the 5′UTR. Comp Funct Genomics 2012; 2012:475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA‐binding proteins. Cell 2012; 149:1393–406. [DOI] [PubMed] [Google Scholar]
- 4. Gerstberger S, Hafner M, Tuschl T. A census of human RNA‐binding proteins. Nat Rev Genet 2014; 15:829–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol 2014; 15:484–91. [DOI] [PubMed] [Google Scholar]
- 6. Turner M, Katsikis PD. A new mechanism of gene regulation mediated by noncoding RNA. J Immunol 2012; 189:3–4. [DOI] [PubMed] [Google Scholar]
- 7. Brewer BY. RNA sequence elements required for high affinity binding by the zinc finger domain of tristetraprolin: conformational changes coupled to the bipartite nature of AU‐rich mRNA‐destabilizing motifs. J Biol Chem 2004; 279:27870–7. [DOI] [PubMed] [Google Scholar]
- 8. Brooks SA, Blackshear PJ. Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochim Biophys Acta 2013; 1829:666–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tiedje C, Ronkina N, Tehrani M, et al. The p38/MK2‐driven exchange between tristetraprolin and HuR regulates AU‐rich element‐dependent translation. PLOS Genet 2012; 8:e1002977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meisner NC, Hackermuller J, Uhl V, Aszodi A, Jaritz M, Auer M. mRNA openers and closers: modulating AU‐rich element‐controlled mRNA stability by a molecular switch in mRNA secondary structure. Chembiochem 2004; 5:1432–47. [DOI] [PubMed] [Google Scholar]
- 11. Hau HH, Walsh RJ, Ogilvie RL, Williams DA, Reilly CS, Bohjanen PR. Tristetraprolin recruits functional mRNA decay complexes to ARE sequences. J Cell Biochem 2007; 100:1477–92. [DOI] [PubMed] [Google Scholar]
- 12. Ray D, Kazan H, Chan ET, et al. Rapid and systematic analysis of the RNA recognition specificities of RNA‐binding proteins. Nat Biotechnol 2009; 27:667–70. [DOI] [PubMed] [Google Scholar]
- 13. Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, Hla T. The RNA‐binding protein HuR regulates the expression of cyclooxygenase‐2. J Biol Chem 2003; 278:25227–33. [DOI] [PubMed] [Google Scholar]
- 14. Vasudevan S, Steitz JA. AU‐rich‐element‐mediated upregulation of translation by FXR1 and Argonaute 2. Cell 2007; 128:1105–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baber JL, Libutti D, Levens D, Tjandra N. High precision solution structure of the C‐terminal KH domain of heterogeneous nuclear ribonucleoprotein K, a c‐myc transcription factor. J Mol Biol 1999; 289:949–62. [DOI] [PubMed] [Google Scholar]
- 16. Shaw G, Kamen R. A conserved AU sequence from the 3′ untranslated region of GM‐CSF mRNA mediates selective mRNA degradation. Cell 1986; 46:659–67. [DOI] [PubMed] [Google Scholar]
- 17. Caput D, Beutler B, Hartog K, Thayer R, Brown‐Shimer S, Cerami A. Identification of a common nucleotide sequence in the 3′‐untranslated region of mRNA molecules specifying inflammatory mediators. Proc Natl Acad Sci USA 1986; 83:1670–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Beutler B, Thompson P, Keyes J, Hagerty K, Crawford D. Assay of a ribonuclease that preferentially hydrolyses mRNAs containing cytokine‐derived UA‐rich instability sequences. Biochem Biophys Res Commun 1988; 152:973–80. [DOI] [PubMed] [Google Scholar]
- 19. Wilson T, Treisman R. Removal of poly(A) and consequent degradation of c‐fos mRNA facilitated by 3′ AU‐rich sequences. Nature 1988; 336:396–9. [DOI] [PubMed] [Google Scholar]
- 20. Clement SL, Scheckel C, Stoecklin G, Lykke‐Andersen J. Phosphorylation of tristetraprolin by MK2 impairs AU‐rich element mRNA decay by preventing deadenylase recruitment. Mol Cell Biol 2011; 31:256–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marchese FP, Aubareda A, Tudor C, Saklatvala J, Clark AR, Dean JL. MAPKAP kinase 2 blocks tristetraprolin‐directed mRNA decay by inhibiting CAF1 deadenylase recruitment. J Biol Chem 2010; 285:27590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee D, Gao M, O'Connor JP, et al. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU‐rich elements. EMBO J 2002; 21:165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen CY, Gherzi R, Ong SE, et al. AU binding proteins recruit the exosome to degrade ARE‐containing mRNAs. Cell 2001; 107:451–64. [DOI] [PubMed] [Google Scholar]
- 24. Fernandez MR, Porte S, Crosas E, et al. Human and yeast zeta‐crystallins bind AU‐rich elements in RNA. Cell Mol Life Sci 2007; 64:1419–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cairrao F, Halees AS, Khabar KS, Morello D, Vanzo N. AU‐rich elements regulate Drosophila gene expression. Mol Cell Biol 2009; 29:2636–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Asson‐Batres MA, Spurgeon SL, Diaz J, DeLoughery TG, Bagby GC, Jr. Evolutionary conservation of the AU‐rich 3′ untranslated region of messenger RNA. Proc Natl Acad Sci USA 1994; 91:1318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stoecklin G, Lu M, Rattenbacher B, Moroni C. A constitutive decay element promotes tumor necrosis factor alpha mRNA degradation via an AU‐rich element‐independent pathway. Mol Cell Biol 2003; 23:3506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pratama A, Ramiscal RR, Silva DG, et al. Roquin‐2 shares functions with its paralog Roquin‐1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity 2013; 38:669–80. [DOI] [PubMed] [Google Scholar]
- 29. Vogel KU, Edelmann SL, Jeltsch KM, et al. Roquin paralogs 1 and 2 redundantly repress the ICOS and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity 2013; 38:655–68. [DOI] [PubMed] [Google Scholar]
- 30. Heissmeyer V, Vogel KU. Molecular control of Tfh‐cell differentiation by Roquin family proteins. Immunol Rev 2013; 253:273–89. [DOI] [PubMed] [Google Scholar]
- 31. Vinuesa CG, Cook MC, Angelucci C, et al. A RING‐type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005; 435:452–8. [DOI] [PubMed] [Google Scholar]
- 32. Venigalla RK, Turner M. RNA‐binding proteins as a point of convergence of the PI3K and p38 MAPK pathways. Front Immunol 2012; 3:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ross EA, Smallie T, Ding Q, et al. Dominant suppression of inflammation via targeted mutation of the mRNA destabilizing protein tristetraprolin. J Immunol 2015; 195:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benjamin D, Schmidlin M, Min L, Gross B, Moroni C. BRF1 protein turnover and mRNA decay activity are regulated by protein kinase B at the same phosphorylation sites. Mol Cell Biol 2006; 26:9497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidlin M, Lu M, Leuenberger SA, et al. The ARE‐dependent mRNA‐destabilizing activity of BRF1 is regulated by protein kinase B. EMBO J 2004; 23:4760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stoecklin G, Stubbs T, Kedersha N, et al. MK2‐induced tristetraprolin:14‐3‐3 complexes prevent stress granule association and ARE‐mRNA decay. EMBO J 2004; 23:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson BA, Stehn JR, Yaffe MB, Blackwell TK. Cytoplasmic localization of tristetraprolin involves 14‐3‐3‐dependent and ‐independent mechanisms. J Biol Chem 2002; 277:18029–36. [DOI] [PubMed] [Google Scholar]
- 38. Maitra S, Chou CF, Luber CA, Lee KY, Mann M, Chen CY. The AU‐rich element mRNA decay‐promoting activity of BRF1 is regulated by mitogen‐activated protein kinase‐activated protein kinase 2. RNA 2008; 14:950–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smallie T, Ross EA, Ammit AJ, et al. Dual‐specificity phosphatase 1 and tristetraprolin cooperate to regulate macrophage responses to lipopolysaccharide. J Immunol 2015; 195:277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schichl YM, Resch U, Lemberger CE, Stichlberger D, de Martin R. Novel phosphorylation‐dependent ubiquitination of tristetraprolin by mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase kinase 1 (MEKK1) and tumor necrosis factor receptor‐associated factor 2 (TRAF2). J Biol Chem 2011; 286:38466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Resch U, Cuapio A, Sturtzel C, Hofer E, de Martin R, Holper‐Schichl YM. Polyubiquitinated tristetraprolin protects from TNF‐induced, caspase‐mediated apoptosis. J Biol Chem 2014; 289:25088–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ngoc LV, Wauquier C, Soin R, et al. Rapid proteasomal degradation of posttranscriptional regulators of the TIS11/tristetraprolin family is induced by an intrinsically unstructured region independently of ubiquitination. Mol Cell Biol 2014; 34:4315–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim HH, Yang X, Kuwano Y, Gorospe M. Modification at HuR(S242) alters HuR localization and proliferative influence. Cell Cycle 2008; 7:3371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim HH, Abdelmohsen K, Lal A, et al. Nuclear HuR accumulation through phosphorylation by Cdk1. Genes Dev 2008; 22:1804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Doller A, Huwiler A, Muller R, Radeke HH, Pfeilschifter J, Eberhardt W. Protein kinase C alpha‐dependent phosphorylation of the mRNA‐stabilizing factor HuR: implications for posttranscriptional regulation of cyclooxygenase‐2. Mol Biol Cell 2007; 18:2137–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoon JH, Abdelmohsen K, Srikantan S, et al. Tyrosine phosphorylation of HuR by JAK3 triggers dissociation and degradation of HuR target mRNAs. Nucleic Acids Res 2014; 42:1196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU‐rich elements. Nucleic Acids Res 2011; 39(Database issue):D66–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kuwano Y, Kim HH, Abdelmohsen K, et al. MKP‐1 mRNA stabilization and translational control by RNA‐binding proteins HuR and NF90. Mol Cell Biol 2008; 28:4562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kuwano Y, Gorospe M. Protecting the stress response, guarding the MKP‐1 mRNA. Cell Cycle 2008; 7:2640–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sugiura R, Satoh R, Ishiwata S, Umeda N, Kita A. Role of RNA‐Binding Proteins in MAPK Signal Transduction Pathway. J Signal Transduct 2011; 2011:109746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emmons J, Townley‐Tilson WH, Deleault KM, et al. Identification of TTP mRNA targets in human dendritic cells reveals TTP as a critical regulator of dendritic cell maturation. RNA 2008; 14:888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bros M, Wiechmann N, Besche V, et al. The RNA binding protein tristetraprolin influences the activation state of murine dendritic cells. Mol Immunol 2010; 47:1161–70. [DOI] [PubMed] [Google Scholar]
- 53. Mukherjee N, Jacobs NC, Hafner M, et al. Global target mRNA specification and regulation by the RNA‐binding protein ZFP36. Genome Biol 2014; 15:R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raghavan A, Robison RL, McNabb J, Miller CR, Williams DA, Bohjanen PR. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J Biol Chem 2001; 276:47958–65. [DOI] [PubMed] [Google Scholar]
- 55. Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA‐binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest 2012; 122:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H. Involvement of KSRP in the post‐transcriptional regulation of human iNOS expression‐complex interplay of KSRP with TTP and HuR. Nucleic Acids Res 2005; 33:4813–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Turner M, Hodson D. Regulation of lymphocyte development and function by RNA‐binding proteins. Curr Opin Immunol 2012; 24:160–5. [DOI] [PubMed] [Google Scholar]
- 58. Zhang J, Modi Y, Yarovinsky T, et al. Macrophage beta2 integrin‐mediated, HuR‐dependent stabilization of angiogenic factor‐encoding mRNAs in inflammatory angiogenesis. Am J Pathol 2012; 180:1751–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Diaz‐Munoz MD, Bell SE, Fairfax K, et al. The RNA‐binding protein HuR is essential for the B cell antibody response. Nat Immunol 2015; 16:415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA‐binding protein HuR. J Immunol 2009; 182:6779–88. [DOI] [PubMed] [Google Scholar]
- 61. Katsanou V, Milatos S, Yiakouvaki A, et al. The RNA‐binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol 2009; 29:2762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghosh M, Aguila HL, Michaud J, et al. Essential role of the RNA‐binding protein HuR in progenitor cell survival in mice. J Clin Invest 2009; 119:3530–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Higashi AY, Ikawa T, Muramatsu M, et al. Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J Immunol 2009; 182:5633–40. [DOI] [PubMed] [Google Scholar]
- 64. Uhmann A, Dittmann K, Wienands J, Hahn H. Comment on ‘Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreER(T2)’. J Immunol 2009; 183:2891; author reply 2891–2. [DOI] [PubMed] [Google Scholar]
- 65. Nakamura H, Kawagishi H, Watanabe A, Sugimoto K, Maruyama M, Sugimoto M. Cooperative role of the RNA‐binding proteins Hzf and HuR in p53 activation. Mol Cell Biol 2011; 31:1997–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abdelmohsen K, Panda AC, Kang MJ, et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res 2014; 42:10099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Galban S, Martindale JL, Mazan‐Mamczarz K, et al. Influence of the RNA‐binding protein HuR in pVHL‐regulated p53 expression in renal carcinoma cells. Mol Cell Biol 2003; 23:7083–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mazan‐Mamczarz K, Galban S, Lopez de Silanes I, et al. RNA‐binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 2003; 100:8354–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhu H, Zhou HL, Hasman RA, Lou H. Hu proteins regulate polyadenylation by blocking sites containing U‐rich sequences. J Biol Chem 2007; 282:2203–10. [DOI] [PubMed] [Google Scholar]
- 70. Izquierdo JM. Hu antigen R (HuR) functions as an alternative pre‐mRNA splicing regulator of Fas apoptosis‐promoting receptor on exon definition. J Biol Chem 2008; 283:19077–84. [DOI] [PubMed] [Google Scholar]
- 71. Casolaro V, Fang X, Tancowny B, et al. Posttranscriptional regulation of IL‐13 in T cells: role of the RNA‐binding protein HuR. J Allergy Clin Immunol 2008; 121:853–9 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen J, Cascio J, Magee JD, et al. Posttranscriptional gene regulation of IL‐17 by the RNA‐binding protein HuR is required for initiation of experimental autoimmune encephalomyelitis. J Immunol 2013; 191:5441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Atasoy U, Curry SL, Lopez de Silanes I, et al. Regulation of eotaxin gene expression by TNF‐alpha and IL‐4 through mRNA stabilization: involvement of the RNA‐binding protein HuR. J Immunol 2003; 171:4369–78. [DOI] [PubMed] [Google Scholar]
- 74. Gubin MM, Techasintana P, Magee JD, et al. Conditional knockout of the RNA‐binding protein HuR in CD4(+) T cells reveals a gene dosage effect on cytokine production. Mol Med 2014; 20:93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yoon JH, S De, S Srikantan, et al. PAR‐CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun 2014; 5:5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sadri N, Lu JY, Badura ML, Schneider RJ. AUF1 is involved in splenic follicular B cell maintenance. BMC Immunol 2010; 11:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Veis DJ, Sorenson CM, Shutter JR, Korsmeyer SJ. Bcl‐2‐deficient mice demonstrate fulminant lymphoid apoptosis, polycystic kidneys, and hypopigmented hair. Cell 1993; 75:229–40. [DOI] [PubMed] [Google Scholar]
- 78. McDonnell TJ, Deane N, Platt FM, et al. bcl‐2‐immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 1989; 57:79–88. [DOI] [PubMed] [Google Scholar]
- 79. Diaz‐Munoz MD, Bell SE, Turner M. Deletion of AU‐rich elements within the Bcl2 3′UTR reduces protein expression and B cell survival in vivo. PLOS ONE 2015; 10:e0116899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jeltsch KM, Hu D, Brenner S, et al. Cleavage of roquin and regnase‐1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol 2014; 15:1079–89. [DOI] [PubMed] [Google Scholar]
- 81. Uehata T, Iwasaki H, Vandenbon A, et al. Malt1‐induced cleavage of regnase‐1 in CD4(+) helper T cells regulates immune activation. Cell 2013; 153:1036–49. [DOI] [PubMed] [Google Scholar]
- 82. Baou M, Jewell A, Murphy JJ. TIS11 family proteins and their roles in posttranscriptional gene regulation. J Biomed Biotechnol 2009; Article ID 634520, 11 pages, doi:10.1155/2009/634520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Blackshear PJ, Phillips RS, Ghosh S, Ramos SB, Richfield EK, Lai WS. Zfp36l3, a rodent X chromosome gene encoding a placenta‐specific member of the Tristetraprolin family of CCCH tandem zinc finger proteins. Biol Reprod 2005; 73:297–307. [DOI] [PubMed] [Google Scholar]
- 84. Lai WS, Stumpo DJ, Kennington EA, et al. Life without TTP: apparent absence of an important anti‐inflammatory protein in birds. Am J Physiol Regul Integr Comp Physiol 2013; 305:R689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schubert CM, Lin R, de Vries CJ, Plasterk RH, Priess JR. MEX‐5 and MEX‐6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol Cell 2000; 5:671–82. [DOI] [PubMed] [Google Scholar]
- 86. Varnum BC, Ma QF, Chi TH, Fletcher B, Herschman HR. The TIS11 primary response gene is a member of a gene family that encodes proteins with a highly conserved sequence containing an unusual Cys‐His repeat. Mol Cell Biol 1991; 11:1754–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. Phosphorylation of tristetraprolin, a potential zinc finger transcription factor, by mitogen stimulation in intact cells and by mitogen‐activated protein kinase in vitro. J Biol Chem 1995; 270:13341–7. [DOI] [PubMed] [Google Scholar]
- 88. Lai WS, Thompson MJ, Taylor GA, Liu Y, Blackshear PJ. Promoter analysis of Zfp‐36, the mitogen‐inducible gene encoding the zinc finger protein tristetraprolin. J Biol Chem 1995; 270:25266–72. [DOI] [PubMed] [Google Scholar]
- 89. Busse M, Schwarzburger M, Berger F, Hacker C, Munz B. Strong induction of the Tis11B gene in myogenic differentiation. Eur J Cell Biol 2008; 87:31–8. [DOI] [PubMed] [Google Scholar]
- 90. Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mitogen‐activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol Cell Biol 2001; 21:6461–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor‐alpha production by tristetraprolin. Science 1998; 281:1001–5. [DOI] [PubMed] [Google Scholar]
- 92. Gomperts M, Corps AN, Pascall JC, Brown KD. Mitogen‐induced expression of the primary response gene cMG1 in a rat intestinal epithelial cell‐line (RIE‐1). FEBS Lett 1992; 306:1–4. [DOI] [PubMed] [Google Scholar]
- 93. Corps AN, Brown, KD. Insulin and insulin‐like growth factor I stimulate expression of the primary response gene cMG1/TIS11b by a wortmannin‐sensitive pathway in RIE‐1 cells. FEBS Lett 1995; 368:160–4. [DOI] [PubMed] [Google Scholar]
- 94. DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor‐inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem 1990; 265:19185–91. [PubMed] [Google Scholar]
- 95. Taylor GA, Carballo E, Lee DM, et al. A pathogenic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996; 4:445–54. [DOI] [PubMed] [Google Scholar]
- 96. Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU‐rich elements: implications for joint and gut‐associated immunopathologies. Immunity 1999; 10:387–98. [DOI] [PubMed] [Google Scholar]
- 97. Carballo E, Gilkeson GS, Blackshear PJ. Bone marrow transplantation reproduces the tristetraprolin‐deficiency syndrome in recombination activating gene‐2 (–/–) mice. Evidence that monocyte/macrophage progenitors may be responsible for TNFalpha overproduction. J Clin Invest 1997; 100:986–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Qiu LQ, Stumpo DJ, Blackshear PJ. Myeloid‐specific tristetraprolin deficiency in mice results in extreme lipopolysaccharide sensitivity in an otherwise minimal phenotype. J Immunol 2012; 188:5150–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kratochvill F, Machacek C, Vogl C, et al. Tristetraprolin‐driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol Syst Biol 2011; 7:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hudson BP, Martinez‐Yamout MA, Dyson HJ, Wright PE. Recognition of the mRNA AU‐rich element by the zinc finger domain of TIS11d. Nat Struct Mol Biol 2004; 11:257–64. [DOI] [PubMed] [Google Scholar]
- 101. Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin‐related zinc finger proteins to AU‐rich elements and destabilization of mRNA. J Biol Chem 2000; 275:17827–37. [DOI] [PubMed] [Google Scholar]
- 102. Blackshear PJ, Lai WS, Kennington EA, et al. Characteristics of the interaction of a synthetic human tristetraprolin tandem zinc finger peptide with AU‐rich element‐containing RNA substrates. J Biol Chem 2003; 278:19947–55. [DOI] [PubMed] [Google Scholar]
- 103. Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT. RNA binding properties of the AU‐rich element‐binding recombinant Nup475/TIS11/tristetraprolin protein. J Biol Chem 2002; 277:48558–64. [DOI] [PubMed] [Google Scholar]
- 104. Lai WS, Kennington EA, Blackshear PJ. Tristetraprolin and its family members can promote the cell‐free deadenylation of AU‐rich element‐containing mRNAs by poly(A) ribonuclease. Mol Cell Biol 2003; 23:3798–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lykke‐Andersen J, Wagner E. Recruitment and activation of mRNA decay enzymes by two ARE‐mediated decay activation domains in the proteins TTP and BRF‐1. Genes Dev 2005; 19:351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fenger‐Gron M, Fillman C, Norrild B, Lykke‐Andersen J. Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol Cell 2005; 20:905–15. [DOI] [PubMed] [Google Scholar]
- 107. Sandler H, Kreth J, Timmers HT, Stoecklin G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res 2011; 39:4373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vindry C, Lauwers A, Hutin D, et al. dTIS11 Protein‐dependent polysomal deadenylation is the key step in AU‐rich element‐mediated mRNA decay in Drosophila cells. J Biol Chem 2012; 287:35527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bell SE, Sanchez MJ, Spasic‐Boskovic O, et al. The RNA binding protein Zfp36l1 is required for normal vascularisation and post‐transcriptionally regulates VEGF expression. Dev Dyn 2006; 235:3144–55. [DOI] [PubMed] [Google Scholar]
- 110. Stumpo DJ, Byrd NA, Phillips RS, et al. Chorioallantoic fusion defects and embryonic lethality resulting from disruption of Zfp36L1, a gene encoding a CCCH tandem zinc finger protein of the Tristetraprolin family. Mol Cell Biol 2004; 24:6445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Espel E. The role of the AU‐rich elements of mRNAs in controlling translation. Semin Cell Dev Biol 2005; 16:59–67. [DOI] [PubMed] [Google Scholar]
- 112. Qi MY, Wang ZZ, Zhang Z, et al. AU‐rich‐element‐dependent translation repression requires the cooperation of tristetraprolin and RCK/P54. Mol Cell Biol 2012; 32:913–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Franks TM, Lykke‐Andersen J. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU‐rich elements. Genes Dev 2007; 21:719–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stumpo DJ, Broxmeyer HE, Ward T, et al. Targeted disruption of Zfp36l2, encoding a CCCH tandem zinc finger RNA‐binding protein, results in defective hematopoiesis. Blood 2009; 114:2401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hodson DJ, Janas ML, Galloway A, et al. Deletion of the RNA‐binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol 2010; 11:717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Frasca D, Landin AM, Alvarez JP, Blackshear PJ, Riley RL, Blomberg BB. Tristetraprolin, a negative regulator of mRNA stability, is increased in old B cells and is involved in the degradation of E47 mRNA. J Immunol 2007; 179:918–27. [DOI] [PubMed] [Google Scholar]
- 117. Khabar KS. The AU‐rich transcriptome: more than interferons and cytokines, and its role in disease. J Interferon Cytokine Res 2005; 25:1–10. [DOI] [PubMed] [Google Scholar]
- 118. Lukong KE, Chang KW, Khandjian EW, Richard S. RNA‐binding proteins in human genetic disease. Trends Genet 2008; 24:416–25. [DOI] [PubMed] [Google Scholar]
- 119. Gabut M, Chaudhry S, Blencowe BJ. SnapShot: the splicing regulatory machinery. Cell 2008; 133:192–19e1. [DOI] [PubMed] [Google Scholar]
- 120. Leskov I, Pallasch CP, Drake A, et al. Rapid generation of human B‐cell lymphomas via combined expression of Myc and Bcl2 and their use as a preclinical model for biological therapies. Oncogene 2013; 32:1066–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Aukema SM, Siebert R, Schuuring E, et al. Double‐hit B‐cell lymphomas. Blood 2011; 117:2319–31. [DOI] [PubMed] [Google Scholar]
- 122. Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic BCL‐2 is required for maintenance of a model leukemia. Cancer Cell 2004; 6:241–9. [DOI] [PubMed] [Google Scholar]
- 123. Nabors LB, Gillespie GY, Harkins L, King PH. HuR, a RNA stability factor, is expressed in malignant brain tumors and binds to adenine‐ and uridine‐rich elements within the 3′ untranslated regions of cytokine and angiogenic factor mRNAs. Cancer Res 2001; 61:2154–61. [PubMed] [Google Scholar]
- 124. Dixon DA, Tolley ND, King PH, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase‐2 expression in colon cancer cells. J Clin Invest 2001; 108:1657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Raymond V, Atwater JA, Verma IM. Removal of an mRNA destabilizing element correlates with the increased oncogenicity of proto‐oncogene fos. Oncogene Res 1989; 5:1–12. [PubMed] [Google Scholar]
- 126. Rimokh R, Berger F, Bastard C, et al. Rearrangement of CCND1 (BCL1/PRAD1) 3′ untranslated region in mantle‐cell lymphomas and t(11q13)‐associated leukemias. Blood 1994; 83:3689–96. [PubMed] [Google Scholar]
- 127. Zucconi BE, Wilson GM. Modulation of neoplastic gene regulatory pathways by the RNA‐binding factor AUF1. Front Biosci (Landmark Ed) 2011; 16:2307–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Hahn CN, Venugopal P, Scott HS, Hiwase DK. Splice factor mutations and alternative splicing as drivers of hematopoietic malignancy. Immunol Rev 2015; 263:257–78. [DOI] [PubMed] [Google Scholar]
- 129. Stoecklin G, Gross B, Ming XF, Moroni C. A novel mechanism of tumor suppression by destabilizing AU‐rich growth factor mRNA. Oncogene 2003; 22:3554–61. [DOI] [PubMed] [Google Scholar]
- 130. Al‐Souhibani N, Al‐Ahmadi W, Hesketh JE, Blackshear PJ, Khabar KS. The RNA‐binding zinc‐finger protein tristetraprolin regulates AU‐rich mRNAs involved in breast cancer‐related processes. Oncogene 2010; 29: 4205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Selmi T, Martello A, Vignudelli T, et al. ZFP36 expression impairs glioblastoma cell lines viability and invasiveness by targeting multiple signal transduction pathways. Cell Cycle 2012; 11:1977–87. [DOI] [PubMed] [Google Scholar]
- 132. Shimada H, Ichikawa H, Nakamura S, et al. Analysis of genes under the downstream control of the t(8;21) fusion protein AML1‐MTG8: overexpression of the TIS11b (ERF‐1, cMG1) gene induces myeloid cell proliferation in response to G–CSF. Blood 2000; 96:655–63. [PubMed] [Google Scholar]
- 133. Abba MC, Sun H, Hawkins KA, et al. Breast cancer molecular signatures as determined by SAGE: correlation with lymph node status. Mol Cancer Res 2007; 5:881–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Carrick DM, Blackshear PJ. Comparative expression of tristetraprolin (TTP) family member transcripts in normal human tissues and cancer cell lines. Arch Biochem Biophys 2007; 462:278–85. [DOI] [PubMed] [Google Scholar]
- 135. Crick F. Central dogma of molecular biology. Nature 1970; 227:561–3. [DOI] [PubMed] [Google Scholar]
- 136. Anderson P. Post‐transcriptional regulons coordinate the initiation and resolution of inflammation. Nat Rev Immunol 2010; 10:24–35. [DOI] [PubMed] [Google Scholar]