

Summary
Susceptibility to type 1 diabetes is associated strongly with human leucocyte antigen (HLA) genes, implicating T cells in disease pathogenesis. In humans, CD8 T cells predominantly infiltrate the islets, yet their activation and propagation probably requires CD4 T cell help. CD4 T cells can select from several differentiation fates following activation, and this choice has profound consequences for their subsequent cytokine production and migratory potential. In turn, these features dictate which other immune cell types T cells interact with and influence, thereby determining downstream effector functions. Obtaining an accurate picture of the type of CD4 T cell differentiation associated with a particular immune‐mediated disease therefore constitutes an important clue when planning intervention strategies. Early models of T cell differentiation focused on the dichotomy between T helper type 1 (Th1) and Th2 responses, with type 1 diabetes (T1D) being viewed mainly as a Th1‐mediated pathology. However, several additional fate choices have emerged in recent years, including Th17 cells and follicular helper T cells. Here we revisit the issue of T cell differentiation in autoimmune diabetes, highlighting new evidence from both mouse models and patient samples. We assess the strengths and the weaknesses of the Th1 paradigm, review the data on interleukin (IL)‐17 production in type 1 diabetes and discuss emerging evidence for the roles of IL‐21 and follicular helper T cells in this disease setting. A better understanding of the phenotype of CD4 T cells in T1D will undoubtedly inform biomarker development, improve patient stratification and potentially reveal new targets for therapeutic intervention.
Keywords: cytokine differentiation, diabetes, T cells
Introduction
Multiple lines of evidence support key roles for both CD4 and CD8 T cells in the immune response that drives type 1 diabetes (T1D). The primary human leucocyte antigen (HLA) associations with T1D are with the class II genes 1, the function of which is to activate CD4 T cells, and CD4 T cells are responsible for ‘licencing’ CD8 T cell activation 2, making an understanding of the CD4 T cell compartment particularly relevant. It has been shown recently that single nucleotide polymorphisms (SNPs) associated with T1D and other autoimmune diseases are enriched preferentially within CD4 T cell super‐enhancers 3, a subset of transcriptional enhancers important for cell identity 4. Intriguingly, super‐enhancer‐associated genes show a striking enrichment for cytokines, cytokine receptors and factors that regulate T cell differentiation 3, suggesting that control of T cell cytokine identity may be an important component of the genetic contribution to disease susceptibility. Understanding CD4 T cell differentiation may therefore hold the key to understanding T1D disease mechanisms and ultimately developing new therapeutic interventions.
The Th1 paradigm in T1D
Evidence in favour of the T helper type 1 (Th1) paradigm
Early models of CD4 T cell differentiation were based on a simple dichotomy between interferon (IFN)‐γ‐dominated Th1 responses and interleukin (IL)‐4‐dominated Th2 responses. Th1 cells can be induced by IL‐12 and are important for macrophage activation and clearance of intracellular pathogens, while Th2 cells provide defence against helminth infection, and are associated with allergic disorders (e.g. asthma, rhinitis, eczema) involving immunoglobulin (Ig)E, mast cells and eosinophils. Viewed through this lens, autoimmune diabetes appeared to fall firmly into the Th1 camp, with a seminal paper by Katz et al. demonstrating that T cells expressing a diabetogenic T cell receptor (TCR) elicited diabetes in neonatal NOD mice when differentiated to a Th1, but not a Th2, phenotype 5 (Fig. 1). Consistent with this idea, increasing levels of IFN‐γ were shown to correlate with progression to diabetes in non‐obese diabetic (NOD) mice 6, and IFN‐γ was shown to be required for diabetes in a virus‐induced model 7.
Figure 1.
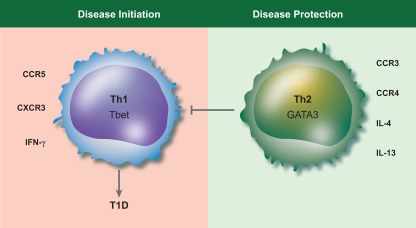
The original T helper type 1 (Th1)/Th2 paradigm in type 1 diabetes. Type 1 diabetes has been viewed traditionally as a Th1‐mediated pathology, with Th2 cells playing a protective role. The characteristic transcription factors and a selection of surface markers associated with Th1 cells or Th2 cells is shown.
A cornerstone of the Th1/Th2 dichotomy is the capacity of the products of one T cell subset to reciprocally inhibit the development of the other 8. In this respect, exogenous provision of IL‐4 was shown to inhibit diabetes in NOD mice 9, and transgenic expression of IL‐4 in the islets under the control of the insulin promoter completely prevented the development of diabetes 10. In addition, helminth infection, a strong driver of the Th2 response, was shown to protect from diabetes in animal models 11.
A number of studies have pointed to a direct role for the Th1 cell signature cytokine, IFN‐γ, in driving the disease process. Expression of IFN‐γ under the control of the human insulin promoter was shown to be sufficient to cause the development of diabetes in mice 12 and conversely blockade of IFN‐γ in NOD mice could prevent diabetes 13, 14. There are a number of ways in which IFN‐γ could be envisaged to contribute to the disease process including by up‐regulating expression of major histocompatibility complex (MHC) classes I and II, facilitating macrophage activation, and increasing leucocyte extravasation by inducing adhesion molecules and chemokines (reviewed in 15). Indeed IFN‐γ has been implicated in promoting the homing of diabetogenic T cells to the pancreatic islets in the NOD mouse 16. There is also a substantial literature directly implicating the IFN‐γ signalling pathway in beta cell death, the critical destructive event at the heart of autoimmune diabetes. IFN‐γ drives a persistent signal in pancreatic beta cells that can be inhibited by over‐expression of suppressor of cytokine signalling‐1 (SOCS1) 17, and islet expression of SOCS1 was found to be protective in the rat insulin promoter‐lymphocytic choriomeningitis virus (RIP‐LCMV) mouse model of diabetes 18. Both IFN‐γ–/– 16 and IFN‐γR–/– 19 islets are killed less effectively in vitro by CD8 T cells 16 and cytokines 19. It is particularly striking that beta cells lacking IFN‐γR show reduced sensitivity not just to IFN‐γ induced death, but also to TNF‐α‐ and IL‐1β‐induced death 19, highlighting the capacity of IFN‐γ to sensitize beta cells to multiple potential death triggers.
The balance between Th1 and Th2 responses has also been studied intensively in humans with T1D. Analysis of peripheral blood T cells from newly diagnosed adults (average age ∼29 years, average disease duration ∼5 weeks) provided support for an IFN‐γ‐dominated response to islet autoantigens, revealing that the balance between IFN‐γ and IL‐10 differed between patients and healthy controls. Individuals with T1D were more likely to have autoantigen‐specific T cells producing IFN‐γ alone, or to a lesser extent a mixed IFN‐γ and IL‐10 response, whereas non‐diabetic subjects showed a clear bias towards production of IL‐10 alone 20. Analogous results were obtained in a separate patient cohort with a similar demographic (average age 28·5 years, average diabetes duration 7 months): interestingly, first‐degree relatives also showed autoantigen‐specific responses that were characterized by more IFN‐γ and less IL‐10 than healthy controls, although the ratios were not as skewed as in T1D patients 21. A study assessing mRNA expression in whole blood revealed that levels of IFN‐γ mRNA were significantly higher in new‐onset T1D patients (average age ∼15 years, average diabetes duration 80 days) compared with an age‐matched at‐risk cohort 22. This could potentially reflect a heightening of the Th1 response during conversion to overt disease. Thus, a considerable body of evidence supported the concept that an IFN‐γ‐producing T cell could be responsible for the pathogenic process in T1D (Fig. 2a).
Figure 2.
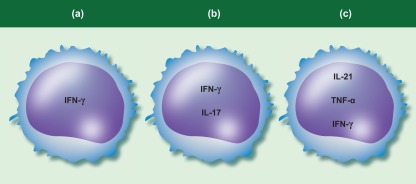
T cell cytokine production in type 1 diabetes (T1D). (a) Many studies have assessed interferon (IFN)‐γ in isolation as a measure of the T helper type 1 (Th1) response. (b) Some studies suggest T cells co‐expressing IFN‐ γ and interleukin (IL)‐17 may be expanded in people with type 1 diabetes [60,71]. (c) IL‐21‐producing T cells in the pancreas in mouse models of diabetes have been shown to co‐express tumour necrosis factor (TNF)‐α and IFN‐γ [106]. IL‐21‐producing T cells are elevated in T1D patients [67,106], and can co‐express TNF‐α and IFN‐γ [106].
Evidence against the Th1 paradigm
Although numerous studies support a Th1 bias in T1D, not all evidence is consistent with this conclusion. Some studies using the NOD mouse concluded that beta cell destruction was a Th2‐ rather than a Th1‐mediated event 23, while others concluded that both types of response were involved 24. At odds with data from short‐term Th2 clones 5, long‐term cultured Th2 clones derived from the same TCR transgenic animals have the capacity to induce diabetes, and could even enhance the ability of Th1 cells to cause disease 25. The effect of helminth products on the immune response was also shown to be more complex than anticipated originally, with effects on regulatory T cells and innate lymphoid cells 11, and it is now clear that helminth infection can protect from diabetes without necessarily invoking Th2 differentiation 26, 27.
The finding that NOD mice deficient in IFN‐γRα exhibited striking resistance to diabetes 28 appeared to provide strong support for the Th1 paradigm; however, protection was subsequently attributed to a closely linked gene on chromosome 10 that was carried over from the 129 background 29, 30. In fact, deficiency in IFN‐γ 31 or the β chain of its receptor 30 surprisingly leads to only a mild delay in diabetes development. Deficiency of IL‐4 failed to exacerbate disease in NOD mice 32, while injection of recombinant IFN‐γ did not accelerate diabetes 33 and, indeed, could even inhibit it 34. In certain experimental settings, the regulatory T cells protecting from diabetes actually required IFN‐γ 35. Perhaps most surprising was the revelation that the ability of Complete Freund's adjuvant (CFA) to protect NOD mice from diabetes, traditionally assumed to reflect IL‐4 or IL‐10 production, was largely dependent upon IFN‐γ 36. Collectively, these data questioned the traditional view that diabetes was caused by Th1 cells making IFN‐γ, and suggested that the situation might be rather more complex.
Data deriving from analysis of patient samples is also not clear‐cut. Insulin‐reactive T cells cloned from the pancreatic lymph nodes of individuals with long‐standing diabetes expressed IL‐13 but not IFN‐γ in response to stimulation 37. In individuals newly diagnosed with T1D, some reports suggested IFN‐γ production to be lower 38, 39, while others suggested an initial increase in IFN‐γ in the early weeks following diagnosis followed by a subsequent decrease 22, 40. In an analysis of serum cytokine levels in 44 newly diabetic children compared with 22 age‐matched controls, although Th1‐associated products such as regulated upon activation normal T cell expressed and secreted (RANTES) and macrophage inflammatory protein (MIP)‐1α were elevated, so too were factors considered indicative of a Th2 response such as IL‐4, IL‐5 and IL‐10 41. Similarly, the increase in IFN‐γ mRNA detected in whole blood from newly diabetic individuals compared with at‐risk individuals was mimicked by a similar increase in IL‐4 and IL‐10 22. A separate analysis concluded that T cells from people with T1D produced equivalent amounts of IFN‐γ and IL‐13 in response to autoantigens as T cells from control subjects 42. Regarding the prediabetic period, the Th1 response to autoantigen has been reported to be increased in one study 43 and decreased in another 44 in at‐risk individuals. One must be mindful when assaying peripheral blood that a lower response could potentially signify the migration of disease‐relevant T cells to the pancreas. Notwithstanding this consideration, it is clear that not all data from mouse models and patients provide consistent support for the dominance of a Th1 response in T1D.
The Th17 revision
Th17 cells in mouse models of autoimmune diabetes
The emergence of Th17 cells 45, 46 provided a major revision of the Th1/Th2 paradigm 47 and raised the possibility that tissue‐specific autoimmunity might be driven by IL‐17‐producing T cells rather than Th1 cells. However, the role of Th17 cells in diabetes remains far from clear. In mice, early work implicated IL‐17 in the pathogenic process 48, 49; however, it was shown subsequently that silencing IL‐17 expression did not protect NOD mice from diabetes 50. Furthermore, there were even suggestions that IL‐17 could protect from diabetes. Kriegel et al. 51 took advantage of the fact that colonization of the gastrointestinal tract with segmented filamentous bacteria (SFB) is known to cause Th17 induction 52, and asked whether SFB‐colonized NOD mice developed diabetes with different kinetics to their SFB‐negative counterparts. Strikingly, the presence of SFB appeared to delay diabetes, with only 16% of SFB+ mice developing diabetes by 30 weeks compared with 91% of SFB– animals. As expected, Th17 signature genes were up‐regulated strongly in SFB+ animals, whereas transcripts associated with Th1, Th2 and regulatory T cells (Treg) were unchanged 51. These data are clearly more consistent with a role for IL‐17, or other Th17 cell products, in delaying rather than promoting diabetes development. A similar conclusion emerged from studies in the diabetes‐prone Bio‐Breeding (BB) rat, in which oral transfer of a particular Lactobacillus strain promoted Th17 differentation and protected from diabetes 53. Furthermore, injection of Th17 polarized cells from CFA‐injected NOD mice delayed diabetes in NOD/severe combined immunodeficiency (SCID) recipients in a manner that depended, at least in part, on IL‐17 54.
The role of Th17 cells in diabetes has also been addressed using adoptive transfer of TCR transgenic T cells specific for pancreatic antigen. Highly purified Th17‐polarized BDC2·5 T cells were capable of inducing diabetes, but appeared to achieve this by differentiating further to a Th1 phenotype 55, 56. Indeed, the ensuing disease could be inhibited by antibodies to IFN‐γ but not IL‐17 55, 56. Conversely, two reports documented the ability of IFN‐γ–/– Th17 cells to transfer diabetes successfully 57, 58, arguing against a requirement for a Th1 transition. Interestingly, if T cells expressing a different pancreatic antigen‐specific TCR (BDC6·9) are used, far less Th17 to Th1 conversion is observed, yet diabetes is still induced 58. Thus, individual T cell clones may differ in the cytokines they use to elicit disease, perhaps depending upon the affinity of their TCR–antigen interactions.
Taken together, the murine studies to date suggest that although IL‐17 is up‐regulated in the early stages of diabetes development 58, 59, it does not necessarily follow that this cytokine, or indeed the Th17 subset, is necessary for disease.
Th17 cells in humans with type 1 diabetes
Several studies have indicated an increase in T cell IL‐17 production in humans with T1D, especially in the very early stages of disease. Children with new‐onset and long‐standing T1D (mean age 8·7 years) were shown to have more IL‐17‐positive T cells compared with age‐matched non‐diabetic controls 60. In a separate study, children within 6 months of T1D diagnosis (mean age 9·6 years) were shown to exhibit increased IL‐17 secretion from both CD4 and CD8 T cells 61. Both IL‐6 and IL‐1β can promote Th17 development 62, 63, so the demonstration that monocytes from T1D patients expressed elevated levels of mRNA for IL‐6 and IL‐1β provided a potential explanation for increased IL‐17 production 64. However, this has not been observed universally 60, and may depend upon the demographic of the cohort. A separate analysis of first‐degree relatives of T1D patients showed that monocytes from those who were autoantibody‐positive produced more IL‐1β and less IL‐6 in response to TLR ligation compared with those from seronegative individuals. IL‐1β plays multiple roles in autoimmune islet infiltration, and in addition to promoting IL‐17 production can also directly modify beta cell survival and function 65.
A key challenge associated with studying T cell differentiation in diabetes patients is the limitation of focusing only on peripheral blood samples. In an impressive attempt to circumvent this problem, Ferraro and colleagues studied T cells isolated from the pancreatic lymph nodes of T1D patients undergoing pancreas or pancreas/kidney transplant. These were compared with pancreatic lymph nodes from non‐diabetic donors. Careful analysis of T cell cytokine production and chemokine receptor profiles established that the pancreatic lymph nodes of type 1 diabetic subjects had a higher frequency of Th17 cells 66. More recently, an increase in the frequency of IL‐17+ cells was found in the peripheral blood of adult T1D patients when gating on CD45RA–CCR6+ population of CD4 T cells 67. Thus, several lines of evidence point to an increase in IL‐17 production in the T1D setting.
Possible role for Th1/17 cells in type 1 diabetes?
One area worthy of note in considering the contribution of Th17 cells to T1D is the role of cells with a propensity to make both IL‐17 and IFN‐γ (sometimes called Th1/17 cells). Cells co‐producing IL‐17 and IFN‐γ were identified originally in the gut of patients with Crohn's disease 68 and were shown subsequently to be present in the mouse colon in an adoptive transfer model of intestinal inflammation 69. Fate mapping experiments in mice established that in autoimmune settings, IL‐17‐positive T cells can initiate IFN‐γ production leading to the presence of a substantial number of T cells co‐expressing both cytokines 70. A closer analysis of the literature reveals hints that cells co‐producing IFN‐γ and IL‐17 may also be present in the T1D setting. By measuring IFN‐γ transcripts within sorted IL‐17‐producing cells, Reinert‐Hartwall et al. 71 found an increased propensity of IL‐17+ cells to make IFN‐γ in children with T1D (mean 8·3 years) compared with healthy controls. Interestingly, this phenomenon was even more striking in children who had not developed diabetes but exhibited advanced beta cell autoimmunity and impaired glucose tolerance (mean age 7·7 years) 71. Consistent with this theme, a separate study of IL‐17 production in the T1D setting found that T cells co‐producing IL‐17 and IFN‐γ were present in four of 11 of the diabetic children examined 60. In addition, when T cells from human pancreatic lymph nodes were examined, there was a suggestion that IFN‐γ as well as IL‐17 expression was up‐regulated in those deriving from T1D patients, although the results did not reach statistical significance 66. Finally, both IL‐17 and IFN‐γ were up‐regulated at the mRNA level within the pancreatic islets of an individual who died within 5 days of T1D diagnosis 72. Thus, the presence of T cells that co‐produce IL‐17 and IFN‐γ could potentially be a feature of T1D, and this area may warrant further investigation (Fig. 2b).
IL‐21 production in type 1 diabetes
IL‐21 is required for autoimmune diabetes in mice
IL‐21 is familiar to the diabetes community as a candidate gene at the diabetes susceptibility locus Idd3 73, 74, 75, and levels of IL‐21 mRNA have been shown to increase during diabetes development in mice 76, 77, 78 (Table 1). IL‐21 is a member of the common‐γ chain receptor family of cytokines that includes IL‐2, IL‐4, IL‐7, IL‐9 and IL‐15. Its receptor is a heterodimer, comprising the common γ‐chain and an IL‐21Rα subunit, which is broadly expressed on a wide range of haematopoietic cell types. It came as something of a surprise when two groups reported that IL‐21 signalling was required critically for diabetes in NOD mice 78, 79. In one report, fewer than 10% of IL‐21R–/– NOD had developed diabetes by 35 weeks 79, while in the other none of the IL‐21R–/– NOD animals were diabetic even at 60 weeks of age, a time‐point at which > 90% of IL‐21R‐sufficient animals had developed disease 78.
Table 1.
Interleukin (IL)‐21 production at the site of the autoimmune attack in mouse models of type 1 diabetes. Table shows data relating to IL‐21 expression in mouse models of diabetes.
| Publication | Findings | Mouse model |
|---|---|---|
| Clough et al. 77 | Increased IL‐21 mRNA in pancreatic lymph nodes of diabetic compared with non‐diabetic mice | DO11 × rip‐OVA |
| Sutherland et al. 78 | Increased IL‐21 mRNA were in the pancreas of diabetic compared with non‐diabetic mice | NOD |
| McGuire et al. 84 | Enrichment of IL‐21‐producing T cells within the pancreas compared with peripheral lymphoid organs. Fewer IL‐21+ T cells were observed in the pancreas of NOD mice bearing the B6 Idd3 region | NOD |
| Kenefeck et al. 106 | Enrichment of IL‐21‐producing T cells within the pancreas compared with peripheral lymphoid organs. The IL‐21‐producing T cells co‐express TNF‐α and IFN‐γ but not IL‐17 | DO11 × rip‐OVA |
NOD = non‐obese diabetic; OVA = ovalbumin; IFN = interferon; TNF = tumour necrosis factor.
The timing of these reports coincided with the discovery that IL‐21 can enhance Th17 differentiation and can itself be produced by Th17 cells to exert feedback in an autocrine fashion 80, 81, 82. This prompted the question of whether a defect in Th17 differentiation might underlie the lack of diabetes in IL‐21R–/– mice. Support for such a notion came from the observation that decreased numbers of IL‐17‐producing T cells were detected in IL‐21R –/– mice in one study 79. However, in the other study 78, IL‐17‐producing T cells were slightly increased, and the amount of IL‐17 following in‐vitro restimulation was actually slightly higher in IL‐21R–/– mice, arguing against a reduction in IL‐17 production being responsible for disease protection. Therefore, the role of IL‐21 in the development of diabetes appeared to be more than just an effect on Th17 differentiation.
Possible roles for IL‐21 in autoimmune diabetes
If IL‐21 did not exert its pro‐diabetogenic effects by IL‐17 up‐regulation, how else could its ability to promote disease be explained? The answer to this critical question is not yet elucidated fully. It seems unlikely that IL‐21 acts directly on pancreatic beta cells, as they appear to lack expression of the IL‐21 receptor 78. However, the presence of IL‐21 local to beta cells is sufficient to trigger the cascade necessary for diabetes induction, even in non‐autoimmune prone C57BL/6 mice 78. Accordingly, expression of IL‐21 under the human insulin promoter elicited spontaneous diabetes in ∼80% of mice, with substantial islet infiltration by CD4 T cells and macrophages as well as dendritic cells (DC) and B cells 78. Forced expression of IL‐21 was therefore sufficient to trigger islet infiltration – but is IL‐21 required for spontaneous islet infiltration occurring in the absence of transgenic over‐expression? The answer appears to be yes, as mice deficient in IL‐21 signalling were virtually devoid of inflammatory infiltration in the islets 78, 79. Furthermore, short‐term blockade of the IL‐21 pathway appeared to reverse established insulitis in NOD mice, resulting in a significantly reduced number of lymphocytes with the islet lesion 83.
Which cell is the key target for the pro‐diabetogenic effects of IL‐21? IL‐21 is an extraordinarily pleiotropic cytokine with the capacity to act on a broad array of cell types, including CD4 and CD8 T cells, natural killer (NK) cells, B cells, macrophages and DC (Fig. 3). In this respect, McGuire et al. 84 found that when diabetes was induced in NOD/SCID mice by adoptive transfer of CD4 and CD8 T cells, it was the CD8 T cells that required IL‐21R in order for diabetes to develop. Loss of IL‐21 sensitivity in the CD4 compartment led to only a partial reduction in diabetes incidence. In contrast, a similar experiment by Van Belle et al. 85 concluded that CD4 T cells were the obligate targets of IL‐21, with IL‐21 responsiveness in CD8 T cells having only a partial affect. While, at first sight, these findings are hard to reconcile, it is probably reasonable to conclude that IL‐21 can act on both CD4 and CD8 T cells to promote diabetes, with the relative importance of each axis being dependent upon the precise experimental context.
Figure 3.
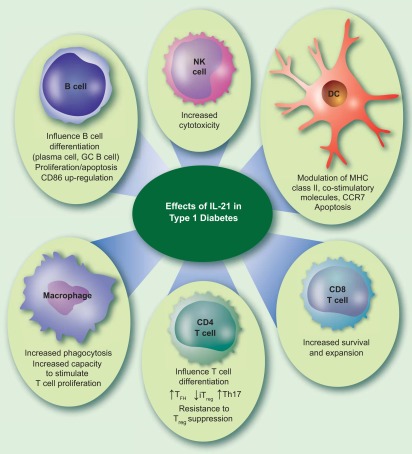
Potential effects of interleukin (IL)‐21 on immune cells in type 1 diabetes. IL‐21 is a highly pleiotropic cytokine and could potentially act on several different cell types in the context of type 1 diabetes development (see text for references).
The effects of IL‐21 on the CD4 compartment include the promotion of cell survival, as illustrated by the increased sensitivity of IL‐21R–/– T cells to activation‐induced cell death 85. In addition, IL‐21 may act on macrophages, increasing their capacity to stimulate CD4 T cell proliferation 86. A further manner in which IL‐21 may contribute to autoimmunity is by imparting resistance to Treg suppression. Several investigators have shown that IL‐21 is able to counteract the suppressive function of Treg 77, 87 and that this requires the IL‐21 to act on conventional CD4 T cells 85, 88, rather than the Treg themselves. Interestingly, resistance to Treg suppression has been reported in both mice 77 and humans with T1D 89, 90, although many other factors in addition to IL‐21 are likely to contribute to this effect 91.
IL‐21 is also known to play key roles in orchestrating T cell : B cell interactions, and this may be relevant in the light of the possible contribution of B cells to diabetes pathogenesis 92, 93, 94. IL‐21 can promote the formation of antibody‐producing plasma cells 95, 96, and can also instruct germinal centre B cell development by up‐regulating expression of the transcription factor B cell lymphoma 6 protein (Bcl6) 97, 98. It was shown recently that IL‐21 can up‐regulate B cell CD86 expression 99 which, in turn, can influence the homeostasis of follicular helper T cells (Tfh) 100 (see later) and facilitate further B cell stimulation. IL‐21 could therefore conceivably contribute to diabetes development by acting on the B cell compartment. Interestingly, recent data suggest that an alteration at the IL‐2/IL‐21 locus that confers increased risk for T1D is associated with decreased production of IL‐10 in memory B cells 101. Thus, there are likely to be additional IL‐21‐dependent control points relevant to diabetes pathogenesis that are not yet elucidated fully.
In an interesting development, it has also been shown that IL‐21 can act on DCs to influence their maturation and migration. In a virus‐induced diabetes model, pancreatic DCs required IL‐21R signals to acquire CCR7 and MHC class II and migrate to the draining lymph node 85. Indeed, disease resistance associated with IL‐21R deficiency could be overcome by the adoptive transfer of IL‐21R‐sufficient DC 85. This contrasts with the capacity of IL‐21 to inhibit DC maturation in vitro 102, and suggests that its effects may be highly context‐dependent. In this regard, it has emerged that IL‐21 is a potent inducer of DC apoptosis, an effect that is reversed in the presence of granulocyte–macrophage colony‐stimulating factor (GM‐CSF) 103. This is reminiscent of the situation in B cells, where IL‐21 can induce either activation or apoptosis depending on the context 104, 105, and suggests that the capacity of IL‐21 to influence DC biology may be far greater that appreciated previously.
IL‐21 in human T1D
It has been found recently that CD4 T cells from T1D patients produce higher levels of IL‐21 in response to in‐vitro restimulation than T cells from non‐diabetic individuals 67, 106. Work by Kenefeck et al. 106 focused on adult patients, with a mean age of 37, while analysis by Ferreira and colleagues 67 also included children (median age 14, range 6–42 years). Importantly, both studies normalized their analysis to the memory cell population to avoid variation in the frequency of memory T cells affecting their results. Interestingly, the IL‐21‐producing T cells in T1D patients co‐expressed high levels of IFN‐γ and TNF‐α 106 (Fig. 2c). T cells co‐expressing IL‐21, IFN‐γ and TNF‐α were also detected infiltrating the pancreas in a TCR transgenic mouse model of diabetes 106.
In a separate study, mRNA levels of IL‐21 were found to be higher in total CD4 T cells from T1D patients 107, although Kenefeck et al. found no significant difference in IL‐21 mRNA levels, even when homing in on the memory T cell population 106. It is possible that IL‐21 production is more pronounced at the earlier disease stages, as the former study 107 examined individuals within 2 years of onset while most participants in the latter study 106 were long‐standing diabetes patients. In all three studies, the increase in IL‐21 was observed in bulk CD4 T cells and the authors did not attempt to identify autoantigen‐specific cells. The changes in IL‐17 expression in T1D discussed earlier 60, 66, 71 were also detected in polyclonal T cell populations. Conceivably, the factors that influence the propensity of T cells to produce a particular cytokine may act on all CD4 T cells, rather than solely those responding to islet antigens; however, this issue warrants further investigation.
Taken together, analysis of mouse models has clearly demonstrated that the loss of IL‐21 signalling inhibits diabetes development while, conversely, its local production is sufficient to initiate immune‐mediated islet destruction. The over‐production of IL‐21 in T1D patients highlights this cytokine as worthy of further investigation in respect of disease pathogenesis in humans.
Follicular helper T cells in T1D
Expansion of Tfh in mice and humans with autoimmune diabetes
Given the link between IL‐21 production and diabetes pathogenesis, a key question becomes what is the identity of the IL‐21‐producing cell? IL‐21 is the signature cytokine for Tfh, the subset that specializes in providing help for B cell antibody production 108, 109. In this regard, a recent unbiased microarray analysis of T cells responding to islet antigen in the pancreatic lymph node of mice revealed a striking signature of Tfh differentiation 106. The top 20 most significantly up‐regulated genes in T cells responding to islet antigen included four archetypal Tfh genes (CXCR5, PD‐1, IL‐21, Bcl6), and flow cytometry analysis demonstrated that T cells with a Tfh phenotype were over‐represented in the pancreatic lymph nodes 106. Tfh are so‐named due to their capacity to enter the B cell follicles of secondary lymphoid tissues, where they initiate the formation of germinal centres. These are specialized structures where B cells mutate their immunoglobulin molecules, so that those with higher affinity for antigen can be selected to enter the long‐lived plasma cell and memory B cell pools. The capacity of B cells to solicit help from Tfh within the germinal centre is a key factor in the selection procedure. Importantly, germinal centres could be demonstrated in the pancreatic lymph nodes (LN) of diabetic mice by confocal microscopy 106, consistent with the presence of a functional Tfh population.
The finding that diabetes was associated with Tfh differentiation in mice prompted an analysis of Tfh cells in the peripheral blood of humans with T1D. Within the memory pool, the % CXCR5+ and CXCR5+inducible T cell co‐stimulator (ICOS)+ T cells was found to be significantly higher in individuals with T1D 106. Consistent with this, an increase in peripheral blood T cells with a Tfh phenotype, has also been reported in two independent cohorts of T1D patients, one of which comprised exclusively new onset patients (within 2 years’ diagnosis, mean age 23) 107, while the other included individuals with disease duration ranging from 2 to 20 years (median age 32) 67. Thus, in both mouse models and in humans, an expansion of cells with a Tfh phenotype appears to be a feature of autoimmune diabetes (Table 2). Regarding the issue of whether Tfh might represent the source of IL‐21 in T1D, both Kenefeck et al. and Ferreira et al. demonstrated a highly significant correlation between the frequency of Tfh and the frequency of IL‐21+ T cells 67, 106, providing strong support for such a notion. However, as IL‐21 can also be produced by other T cells, including Th17 cells 81, 82, and immunosuppressive Tr1 cells 110, the contribution of non‐Tfh populations cannot be excluded.
Table 2.
Link between follicular helper T cells (Tfh) differentiation and autoimmune diabetes. Table collating some of the evidence that suggests Tfh differentiation may be a feature of autoimmune diabetes in mice and humans.
| Mouse | Human |
|---|---|
| Increase in Tfh genes in microarray of T cells responding to islet antigen in the pancreatic LN 106 | Increased production of IL‐21 after ex‐vivo stimulation of memory T cells from T1D patients compared with matched controls 67, 106 |
| Increased Tfh differentiation due to roquin mutation exacerbates diabetes 137 | Increased IL‐21 mRNA in memory T cells from new onset T1D patients compared with matched controls 107 |
| CXCR5‐enriched T cells preferentially induce diabetes upon adoptive transfer 106 | Increased numbers of cells with a Tfh phenotype in blood of T1D patients compared with controls 67, 106, 107. Correlation between frequency of Tfh and IL‐21+ T cells 67, 106 |
IL = interleukin; T1D = type 1 diabetes; LN = lymph node.
The precise relationship between CXCR5+ T cells in the blood and bona fide Tfh cells remains controversial 111. There is now good evidence that Tfh can become circulating memory cells 112, 113, 114, 115, but in so doing they down‐regulate many of their characteristic Tfh markers, although these can be regained upon antigen re‐encounter 116. Interestingly, one study showed that Tfh that lose their phenotype after antigen deprivation retain intermediate levels of CXCR5 117, suggesting that this marker may offer the best chance to track such cells. The presence of blood‐borne Tfh in SAP‐deficient mice and humans suggests that they arise prior to intimate T cell : B cell interactions within the Germinal Center 118, and while it is technically possible for bona fide Tfh to exit the GC to enter the circulation, this appears to be a rare event 119. Thus, the CXCR5+ cells in the circulation of T1D patients may derive from pre‐Tfh cells that have bifurcated from those entering the GC, choosing instead to commit to a memory pathway. The generation of blood‐borne Tfh‐phenotype cells prior to the development of GC 118 is consistent with recent data highlighting that the homeostasis of this population in LN and blood can be strikingly different 120. Despite the many ongoing controversies, it is clear that the blood‐borne CXCR5+ fraction, while heterogeneous, contains circulating Tfh memory cells that can traffic to B cell follicles of secondary lymphoid tissues and contribute to GC reactions 120.
Tfh and autoantibody status
The presence of autoantibodies is a key predictor of diabetes development in at‐risk individuals, with the number of antibodies and the timing of their appearance being particularly telling 121, 122. The key role of Tfh in class‐switching and affinity maturation is consistent with a role for these cells in autoantibody production. Intriguingly, one study focusing on newly diagnosed patients identified a small difference in the percentage of Tfh cells between individuals that were either positive or negative for certain autoantibodies. Tfh numbers appeared to be independent of glutamate decarboxylase (GAD) autoantibody status, but were higher in those deemed positive for ZnT8 or IA‐2 autoantibodies compared with autoantibody‐negative individuals 107. The relationship between circulating cells with a Tfh phenotype and the emergence of autoantibodies will be important to elucidate fully in future studies. Given the role of Tfh in honing the quality of the B cell response, it is noteworthy that autoantibodies to islet antigens frequently exhibit affinity maturation 123 and, indeed, the presence of high‐affinity autoantibodies may help to identify those individuals most likely to progress from at‐risk status to overt diabetes 124, 125, 126. It is possible that measuring peripheral blood Tfh, as well as autoantibodies, could be useful in at‐risk individuals and might provide additional power to predict progression to overt disease.
IL‐2 signalling impairs Tfh differentiation
One notable aspect of Tfh biology is that IL‐2 signalling is known to impair Tfh differentiation. Accordingly, signals generated by the IL‐2 receptor during early T cell activation can influence the balance between Tfh differentiation and other effector T cell fates 127, 128 via signal transducer and activator of transcription (STAT)‐5‐dependent skewing of the Bcl6/PR domain zinc finger protein 1 (BLIMP1) ratio 129. Elegant experiments have used MHC class II tetramers to home in on antigen‐specific T cells following influenza infection in mice and have shown that IL‐2 administration selectively decreases the number of Tfh cells (CXCR5+PD1+), but not other effector T cells (CXCR5‐PD1‐) 130. Conversely, it has been shown that under conditions of IL‐2 deprivation, Th1 cells can acquire a Tfh phenotype 131. Thus, the availability of IL‐2 in the local environment has significant consequences for the development and homeostasis of the Tfh population. These findings may be relevant to the observed increase in Tfh in type 1 diabetes, as multiple defects in the IL‐2 signalling pathway have been associated with this disease setting 132, 133, 134, 135, 136. Interestingly Kenefeck et al. found an inverse relationship between the ability of T cells from type 1 diabetes patients to respond to IL‐2 and propensity to acquire a Tfh phenotype in vitro 106. It is therefore possible that suboptimal IL‐2 signalling could contribute to increased Tfh differentiation in T1D.
Could Tfh be responsible for driving disease?
Whether Tfh cells are directly responsible for autoimmune pathology is hard to assess in patients; however, data from mouse models provides support for such a notion. In a TCR transgenic diabetes model, a mutation in Roquin leading to dysregulated Tfh generation dramatically accelerated diabetes development 137. In a second TCR transgenic model, based on a different pancreatic antigen and different transgenic T cells, enriching for T cells with a Tfh phenotype increased their capacity to cause diabetes upon adoptive transfer 106. Anecdotal evidence also links the Tfh response with diabetes in NOD mice: the spontaneous formation of germinal centres in NOD spleen has been documented 138, and germinal centres have even been detected within the lymphoid mass infiltrating the pancreas itself 139, 140.
Although IL‐21 is the characteristic cytokine associated with Tfh cells, they are also known to be capable of producing other cytokines, including IFN‐γ, which could explain the association of this cytokine with T1D 112, 141. Indeed, human Tfh isolated from lymph nodes of chronically HIV‐infected subjects were shown to be capable of substantial co‐production of cytokines, including IL‐21, TNF‐α and IFN‐γ 142. Several studies have implicated persistent antigen presentation in Tfh differentiation 117, 143, a notion that might fit with the inability of self‐antigens to be cleared in autoimmune settings. Interestingly, Tfh differentiation is subject to regulation by a number of pathways that are linked genetically to autoimmunity, including the CD28/cytotoxic T lymphocyte antigen (CTLA)‐4 axis 144, 145, 146, 147, 148, 149, 150, IL‐2 127, 128, 129, 130 and the lymphoid‐specific tyrosine phosphatase (LYP) encoded by protein tyrosine phosphatase, non‐receptor type 22 (PTPN22) 151. The link between these loci and disease initiation is complex, but modulation of Tfh differentiation adds an additional consideration to the other known roles of the candidate genes in these locations.
Concluding comments
The complexities of CD4 T cell differentiation are still emerging, but we have clearly moved beyond the era of a simple Th1/Th2 dichotomy. Recent data suggest that early T cell differentiation is likely to be even more diverse than was appreciated previously 152, with a broad selection of functional phenotypes being generated, the most useful of which are then expanded selectively. Moreover, intermediate differentiation states exist, including a Tfh‐like transitional stage shared by Tfh and Th1 cells 153, and phenotypical conversions are possible, such as the switch from Th1 to Tfh under conditions of limiting IL‐2 131. These developments argue for a more nuanced view of T cell differentiation in type 1 diabetes that does not focus solely on Th1 cells, but also encompasses the possible involvement of IL‐21‐producing T cells such as Tfh, as well as T cells co‐producing IFN‐γ and IL‐17.
The demonstration of an augmented Tfh population in type 1 diabetes 67, 106, 107 is in line with a growing appreciation that Tfh differentiation is a feature of several autoimmune diseases (154 and reviewed in 155). Increasingly refined genetic analysis suggests that T1D may be more similar to other autoantibody‐positive diseases, such as juvenile idiopathic arthritis and rheumatoid arthritis, than to conditions lacking characteristic autoantibodies, such as ulcerative colitis and Crohn's disease 156. This is clearly consistent with the potential involvement of Tfh cells in the underlying immunological processes.
Tfh cell numbers in the peripheral blood are being linked increasingly with protective anti‐viral immunity 157, 158, 159. This is interesting, given the long‐standing debate regarding the putative contribution of viral infection to diabetes initiation 160, 161. One could envisage that evolutionary selection for characteristics that confer an advantage in infectious settings might have also influenced susceptibility to autoimmunity in parallel.
One potential benefit of broadening our perception of T cell differentiation in diabetes beyond the simple Th1 paradigm is the prospect of identifying new disease biomarkers (Fig. 4). Exploration of CXCR5, or other markers of circulating Tfh‐like cells, may present new opportunities for assessing diabetes risk, tracking disease progression or gauging response to therapeutic interventions. We envisage that an increasingly refined understanding of the CD4 T cell population in type 1 diabetes will help us to monitor the autoimmune response and ultimately deploy effective immunomodulatory strategies in this disease setting.
Figure 4.
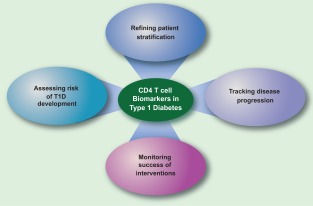
Potential utility of biomarkers arising from a better understanding of the T cell phenotype in type 1 diabetes (T1D). Surface markers and/or secreted products from CD4 T cells can potentially be used to gauge the risk of T1D development, in conjunction with established risk indicators. They may also be used to refine patient stratification, perhaps selecting groups that might be predicted to benefit from a particular immune intervention. Longitudinal studies may reveal whether particular markers can be used to stage the disease process. Phenotypical markers may also be of utility in assessing the efficacy of therapeutic interventions, perhaps in combination with tetramer technology.
Disclosure
L.S.K.W. declares no conflicts of interest. M.v.H. declares a commercial interest in developing IL‐21 blockade reagents at NovoNordisk.
Acknowledgements
L.S.K.W. is funded by an MRC Senior Fellowship and received a Diabetes UK studentship to study IL‐21 production in T1D and a JDRF project grant to study follicular helper T cells in T1D. M.v.H. is Vice President and head of the NovoNordisk R&D Center in Seattle and professor at the La Jolla Institute. We are grateful to Linda Wicker for helpful comments on the manuscript.
References
- 1. Todd JA. Etiology of type 1 diabetes. Immunity 2010; 32:457–67. [DOI] [PubMed] [Google Scholar]
- 2. Bevan MJ. Helping the CD8(+) T‐cell response. Nat Rev Immunol 2004; 4:595–602. [DOI] [PubMed] [Google Scholar]
- 3. Vahedi G, Kanno Y, Furumoto Y et al Super‐enhancers delineate disease‐associated regulatory nodes in T cells. Nature 2015; 520: 558–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hnisz D, Abraham BJ, Lee TI et al Super‐enhancers in the control of cell identity and disease. Cell 2013; 155:934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Katz JD, Benoist C, Mathis D. T helper cell subsets in insulin‐dependent diabetes. Science 1995; 268:1185–8. [DOI] [PubMed] [Google Scholar]
- 6. Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Therapeutic intervention by immunostimulation? Diabetes 1994; 43:613–21. [DOI] [PubMed] [Google Scholar]
- 7. von Herrath MG, Oldstone MB. Interferon‐gamma is essential for destruction of beta cells and development of insulin‐dependent diabetes mellitus. J Exp Med 1997; 185:531–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 1989; 7:145–73. [DOI] [PubMed] [Google Scholar]
- 9. Rapoport MJ, Jaramillo A, Zipris D et al Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. J Exp Med 1993; 178:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mueller R, Krahl T, Sarvetnick N. Pancreatic expression of interleukin‐4 abrogates insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. J Exp Med 1996; 184:1093–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zaccone P, Cooke A. Helminth mediated modulation of type 1 diabetes (T1D). Int J Parasitol 2013; 43:311–8. [DOI] [PubMed] [Google Scholar]
- 12. Sarvetnick N, Liggitt D, Pitts SL, Hansen SE, Stewart TA. Insulin‐dependent diabetes mellitus induced in transgenic mice by ectopic expression of class II MHC and interferon‐gamma. Cell 1988; 52:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Debray‐Sachs M, Carnaud C, Boitard C et al Prevention of diabetes in NOD mice treated with antibody to murine IFN gamma. J Autoimmun 1991; 4:237–48. [DOI] [PubMed] [Google Scholar]
- 14. Campbell IL, Kay TW, Oxbrow L, Harrison LC. Essential role for interferon‐gamma and interleukin‐6 in autoimmune insulin‐dependent diabetes in NOD/Wehi mice. J Clin Invest 1991; 87:739–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 2004; 75:163–89. [DOI] [PubMed] [Google Scholar]
- 16. Savinov AY, Wong FS, Chervonsky AV. IFN‐gamma affects homing of diabetogenic T cells. J Immunol 2001; 167:6637–43. [DOI] [PubMed] [Google Scholar]
- 17. Chong MM, Thomas HE, Kay TW. Gamma‐interferon signaling in pancreatic beta‐cells is persistent but can be terminated by overexpression of suppressor of cytokine signaling‐1. Diabetes 2001; 50:2744–51. [DOI] [PubMed] [Google Scholar]
- 18. Barral AM, Thomas HE, Ling EM et al SOCS‐1 protects from virally‐induced CD8 T cell mediated type 1 diabetes. J Autoimmun 2006; 27:166–73. [DOI] [PubMed] [Google Scholar]
- 19. Yi Z, Li L, Garland A et al IFN‐gamma receptor deficiency prevents diabetes induction by diabetogenic CD4+, but not CD8+, T cells. Eur J Immunol 2012; 42:2010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arif S, Tree TI, Astill TP et al Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest 2004; 113:451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petrich de Marquesini LG, Fu J, Connor KJ et al IFN‐gamma and IL‐10 islet‐antigen‐specific T cell responses in autoantibody‐negative first‐degree relatives of patients with type 1 diabetes. Diabetologia 2010; 53:1451–60. [DOI] [PubMed] [Google Scholar]
- 22. Han D, Leyva CA, Matheson D et al Immune profiling by multiple gene expression analysis in patients at‐risk and with type 1 diabetes. Clin Immunol 2011; 139:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Anderson JT, Cornelius JG, Jarpe AJ, et al Insulin‐dependent diabetes in the NOD mouse model. II. Beta cell destruction in autoimmune diabetes is a TH2 and not a TH1 mediated event. Autoimmunity 1993; 15:113–22. [DOI] [PubMed] [Google Scholar]
- 24. Azar ST, Tamim H, Beyhum HN, Habbal MZ, Almawi WY. Type I (insulin‐dependent) diabetes is a Th1‐ and Th2‐mediated autoimmune disease. Clin Diagn Lab Immunol 1999; 6:306–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Poulin M, Haskins K. Induction of diabetes in nonobese diabetic mice by Th2 T cell clones from a TCR transgenic mouse. J Immunol 2000; 164:3072–8. [DOI] [PubMed] [Google Scholar]
- 26. Mishra PK, Patel N, Wu W, Bleich D, Gause WC. Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL‐10 in the absence of a Th2‐type response. Mucosal Immunol 2013; 6:297–308. [DOI] [PubMed] [Google Scholar]
- 27. Hubner MP, Shi Y, Torrero MN et al Helminth protection against autoimmune diabetes in nonobese diabetic mice is independent of a type 2 immune shift and requires TGF‐beta. J Immunol 2012; 188:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang B, Andre I, Gonzalez A et al Interferon‐gamma impacts at multiple points during the progression of autoimmune diabetes. Proc Natl Acad Sci USA 1997; 94:13844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanagawa O, Xu G, Tevaarwerk A, Vaupel BA. Protection of nonobese diabetic mice from diabetes by gene(s) closely linked to IFN‐gamma receptor loci. J Immunol 2000; 164:3919–23. [DOI] [PubMed] [Google Scholar]
- 30. Serreze DV, Post CM, Chapman HD, Johnson EA, Lu B, Rothman PB. Interferon‐gamma receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes 2000; 49:2007–11. [DOI] [PubMed] [Google Scholar]
- 31. Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of gamma‐interferon delays but does not prevent diabetes in NOD mice. Diabetes 1996; 45:812–7. [DOI] [PubMed] [Google Scholar]
- 32. Wang B, Gonzalez A, Hoglund P, Katz JD, Benoist C, Mathis D. Interleukin‐4 deficiency does not exacerbate disease in NOD mice. Diabetes 1998; 47:1207–11. [DOI] [PubMed] [Google Scholar]
- 33. Satoh J, Seino H, Abo T et al Recombinant human tumor necrosis factor alpha suppresses autoimmune diabetes in nonobese diabetic mice. J Clin Invest 1989; 84:1345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sobel DO, Han J, Williams J, Yoon JW, Jun HS, Ahvazi B. Gamma interferon paradoxically inhibits the development of diabetes in the NOD mouse. J Autoimmun 2002; 19:129–37. [DOI] [PubMed] [Google Scholar]
- 35. Fousteri G, Dave A, Bot A, Juntti T, Omid S, von Herrath M. Subcutaneous insulin B:9‐23/IFA immunisation induces Tregs that control late‐stage prediabetes in NOD mice through IL‐10 and IFNgamma. Diabetologia 2010; 53:1958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez‐Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol 2001; 166:1352–9. [DOI] [PubMed] [Google Scholar]
- 37. Kent SC, Chen Y, Bregoli L et al Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 2005; 435:224–8. [DOI] [PubMed] [Google Scholar]
- 38. Halminen M, Simell O, Knip M, Ilonen J. Cytokine expression in unstimulated PBMC of children with type 1 diabetes and subjects positive for diabetes‐associated autoantibodies. Scand J Immunol 2001; 53:510–3. [DOI] [PubMed] [Google Scholar]
- 39. Kukreja A, Cost G, Marker J et al Multiple immuno‐regulatory defects in type‐1 diabetes. J Clin Invest 2002; 109:131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karlsson Faresjo MG, Ernerudh J, Ludvigsson J. Cytokine profile in children during the first 3 months after the diagnosis of type 1 diabetes. Scand J Immunol 2004; 59:517–26. [DOI] [PubMed] [Google Scholar]
- 41. Chatzigeorgiou A, Harokopos V, Mylona‐Karagianni C, Tsouvalas E, Aidinis V, Kamper EF. The pattern of inflammatory/anti‐inflammatory cytokines and chemokines in type 1 diabetic patients over time. Ann Med 2010; 42:426–38. [DOI] [PubMed] [Google Scholar]
- 42. Strom A, Menart B, Simon MC et al Cellular interferon‐gamma and interleukin‐13 immune reactivity in type 1, type 2 and latent autoimmune diabetes: action LADA 6. Cytokine 2012; 58:148–51. [DOI] [PubMed] [Google Scholar]
- 43. Karlsson MG, Lawesson SS, Ludvigsson J. Th1‐like dominance in high‐risk first‐degree relatives of type I diabetic patients. Diabetologia 2000; 43:742–9. [DOI] [PubMed] [Google Scholar]
- 44. Karlsson Faresjo MG, Ludvigsson J. Diminished Th1‐like response to autoantigens in children with a high risk of developing type 1 diabetes. Scand J Immunol 2005; 61:173–9. [DOI] [PubMed] [Google Scholar]
- 45. Park H, Li Z, Yang XO et al A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 2005; 6:1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harrington LE, Hatton RD, Mangan PR et al Interleukin 17‐producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 2005; 6:1123–32. [DOI] [PubMed] [Google Scholar]
- 47. Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell‐mediated tissue damage. Nat Med 2007; 13:139–45. [DOI] [PubMed] [Google Scholar]
- 48. Jain R, Tartar DM, Gregg RK et al Innocuous IFNgamma induced by adjuvant‐free antigen restores normoglycemia in NOD mice through inhibition of IL‐17 production. J Exp Med 2008; 205:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Emamaullee JA, Davis J, Merani S et al Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 2009; 58:1302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Joseph J, Bittner S, Kaiser FM, Wiendl H, Kissler S. IL‐17 silencing does not protect nonobese diabetic mice from autoimmune diabetes. J Immunol 2012; 188:216–21. [DOI] [PubMed] [Google Scholar]
- 51. Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA 2011; 108:11548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ivanov, II , Atarashi K, Manel N et al Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009; 139:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lau K, Benitez P, Ardissone A et al . Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2‐mediated Th17 bias. J Immunol 2011; 186:3538–46. [DOI] [PubMed] [Google Scholar]
- 54. Nikoopour E, Schwartz JA, Huszarik K et al Th17 polarized cells from nonobese diabetic mice following mycobacterial adjuvant immunotherapy delay type 1 diabetes. J Immunol 2010; 184:4779–88. [DOI] [PubMed] [Google Scholar]
- 55. Bending D, De la Pena H, Veldhoen M et al Highly purified Th17 cells from BDC2.5NOD mice convert into Th1‐like cells in NOD/SCID recipient mice. J Clin Invest 2009; 119:565–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Martin‐Orozco N, Chung Y, Chang SH, Wang YH, Dong C. Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol 2009; 39:216–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu SM, Lee DH, Sullivan JM et al Differential IL‐21 signaling in APCs leads to disparate Th17 differentiation in diabetes‐susceptible NOD and diabetes‐resistant NOD.Idd3 mice. J Clin Invest 2011; 121:4303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li CR, Mueller EE, Bradley LM. Islet antigen‐specific Th17 cells can induce TNF‐alpha‐dependent autoimmune diabetes. J Immunol 2014; 192:1425–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vukkadapu SS, Belli JM, Ishii K et al Dynamic interaction between T cell‐mediated beta‐cell damage and beta‐cell repair in the run up to autoimmune diabetes of the NOD mouse. Physiol Genomics 2005; 21:201–11. [DOI] [PubMed] [Google Scholar]
- 60. Honkanen J, Nieminen JK, Gao R et al IL‐17 immunity in human type 1 diabetes. J Immunol 2010; 185:1959–67. [DOI] [PubMed] [Google Scholar]
- 61. Marwaha AK, Crome SQ, Panagiotopoulos C et al Cutting edge: increased IL‐17‐secreting T cells in children with new‐onset type 1 diabetes. J Immunol 2010; 185:3814–8. [DOI] [PubMed] [Google Scholar]
- 62. Acosta‐Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor‐beta are essential for the differentiation of interleukin 17‐producing human T helper cells. Nat Immunol 2007; 8:942–9. [DOI] [PubMed] [Google Scholar]
- 63. Wilson NJ, Boniface K, Chan JR et al Development, cytokine profile and function of human interleukin 17‐producing helper T cells. Nat Immunol 2007; 8:950–7. [DOI] [PubMed] [Google Scholar]
- 64. Bradshaw EM, Raddassi K, Elyaman W et al Monocytes from patients with type 1 diabetes spontaneously secrete proinflammatory cytokines inducing Th17 cells. J Immunol 2009; 183:4432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wong FS, Wen L. Therapy: immunotherapy for T1DM – targeting innate immunity. Nat Rev Endocrinol 2013; 9:384–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ferraro A, Socci C, Stabilini A et al Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes 2011; 60:2903–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ferreira RC, Simons HZ, Thompson WS et al IL‐21 production by CD4 effector T cells and frequency of circulating follicular helper T cells are increased in type 1 diabetes patients. Diabetologia 2015; 58:781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Annunziato F, Cosmi L, Santarlasci V et al Phenotypic and functional features of human Th17 cells. J Exp Med 2007; 204:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ahern PP, Schiering C, Buonocore S et al Interleukin‐23 drives intestinal inflammation through direct activity on T cells. Immunity 2010; 33:279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hirota K, Duarte JH, Veldhoen M et al Fate mapping of IL‐17‐producing T cells in inflammatory responses. Nat Immunol 2011; 12:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reinert‐Hartwall L, Honkanen J, Salo HM et al Th1/Th17 plasticity is a marker of advanced beta cell autoimmunity and impaired glucose tolerance in humans. J Immunol 2015; 194:68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Arif S, Moore F, Marks K et al Peripheral and islet interleukin‐17 pathway activation characterizes human autoimmune diabetes and promotes cytokine‐mediated beta‐cell death. Diabetes 2011; 60:2112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhernakova A, Alizadeh BZ, Bevova M et al Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am J Hum Genet 2007; 81:1284–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Todd JA, Walker NM, Cooper JD et al Robust associations of four new chromosome regions from genome‐wide analyses of type 1 diabetes. Nat Genet 2007; 39:857–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McGuire HM, Vogelzang A, Hill N, Flodstrom‐Tullberg M, Sprent J, King C. Loss of parity between IL‐2 and IL‐21 in the NOD Idd3 locus. Proc Natl Acad Sci USA 2009; 106:19438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell 2004; 117:265–77. [DOI] [PubMed] [Google Scholar]
- 77. Clough LE, Wang CJ, Schmidt EM et al Release from regulatory T cell‐mediated suppression during the onset of tissue‐specific autoimmunity is associated with elevated IL‐21. J Immunol 2008; 180:5393–401. [DOI] [PubMed] [Google Scholar]
- 78. Sutherland AP, Van Belle T, Wurster AL et al Interleukin‐21 is required for the development of type 1 diabetes in NOD mice. Diabetes 2009; 58:1144–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL‐21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci USA 2008; 105:14028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Korn T, Bettelli E, Gao W et al IL‐21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007; 448:484–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wei L, Laurence A, Elias KM, O'Shea JJ. IL‐21 is produced by Th17 cells and drives IL‐17 production in a STAT3‐dependent manner. J Biol Chem 2007; 282:34605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nurieva R, Yang XO, Martinez G et al Essential autocrine regulation by IL‐21 in the generation of inflammatory T cells. Nature 2007; 448:480–3. [DOI] [PubMed] [Google Scholar]
- 83. McGuire HM, Walters S, Vogelzang A et al Interleukin‐21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes 2011; 60:867–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McGuire HM, Vogelzang A, Ma CS et al A subset of interleukin‐21+ chemokine receptor CCR9+ T helper cells target accessory organs of the digestive system in autoimmunity. Immunity 2011; 34:602–15. [DOI] [PubMed] [Google Scholar]
- 85. Van Belle TL, Nierkens S, Arens R, von Herrath MG. Interleukin‐21 receptor‐mediated signals control autoreactive T cell infiltration in pancreatic islets. Immunity 2012; 36:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ruckert R, Bulfone‐Paus S, Brandt K. Interleukin‐21 stimulates antigen uptake, protease activity, survival and induction of CD4+ T cell proliferation by murine macrophages. Clin Exp Immunol 2008; 151:487–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Peluso I, Fantini MC, Fina D et al IL‐21 counteracts the regulatory T cell‐mediated suppression of human CD4+ T lymphocytes. J Immunol 2007; 178:732–9. [DOI] [PubMed] [Google Scholar]
- 88. Attridge K, Wang CJ, Wardzinski L et al IL‐21 inhibits T cell IL‐2 production and impairs Treg homeostasis. Blood 2012; 119:4656–64. [DOI] [PubMed] [Google Scholar]
- 89. Lawson JM, Tremble J, Dayan C et al Increased resistance to CD4+CD25hi regulatory T cell‐mediated suppression in patients with type 1 diabetes. Clin Exp Immunol 2008; 154:353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner J. The effector T cells of diabetic subjects are resistant to regulation via CD4+FOXP3+ regulatory T cells. J Immunol 2008; 181:7350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Walker LS. Regulatory T cells overturned: the effectors fight back. Immunology 2009; 126:466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pescovitz MD, Greenbaum CJ, Krause‐Steinrauf H et al Rituximab, B‐lymphocyte depletion, and preservation of beta‐cell function. N Engl J Med 2009; 361:2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wong FS, Wen L, Tang M et al Investigation of the role of B‐cells in type 1 diabetes in the NOD mouse. Diabetes 2004; 53:2581–7. [DOI] [PubMed] [Google Scholar]
- 94. Smith MJ, Packard TA, O'Neill SK et al Loss of anergic B cells in pre‐diabetic and new onset T1D patients. Diabetes 2014; 64:1703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ozaki K, Spolski R, Ettinger R et al Regulation of B cell differentiation and plasma cell generation by IL‐21, a novel inducer of Blimp‐1 and Bcl‐6. J Immunol 2004; 173:5361–71. [DOI] [PubMed] [Google Scholar]
- 96. Ettinger R, Sims GP, Fairhurst AM et al IL‐21 induces differentiation of human naive and memory B cells into antibody‐secreting plasma cells. J Immunol 2005; 175:7867–79. [DOI] [PubMed] [Google Scholar]
- 97. Zotos D, Coquet JM, Zhang Y et al IL‐21 regulates germinal center B cell differentiation and proliferation through a B cell‐intrinsic mechanism. J Exp Med 2010; 207:365–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Linterman MA, Beaton L, Yu D et al IL‐21 acts directly on B cells to regulate Bcl‐6 expression and germinal center responses. J Exp Med 2010; 207:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Attridge K, Kenefeck R, Wardzinski L et al IL‐21 promotes CD4 T cell responses by phosphatidylinositol 3‐kinase‐dependent upregulation of CD86 on B cells. J Immunol 2014; 192:2195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Salek‐Ardakani S, Choi YS, Rafii‐El‐Idrissi Benhnia M et al B cell‐specific expression of B7‐2 is required for follicular Th cell function in response to vaccinia virus. J Immunol 2011; 186:5294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Thompson WS, Pekalski ML, Simons HZ et al Multi‐parametric flow cytometric and genetic investigation of the peripheral B cell compartment in human type 1 diabetes. Clin Exp Immunol 2014; 177:571–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brandt K, Bulfone‐Paus S, Foster DC, Ruckert R. Interleukin‐21 inhibits dendritic cell activation and maturation. Blood 2003; 102:4090–8. [DOI] [PubMed] [Google Scholar]
- 103. Wan CK, Oh J, Li P et al The cytokines IL‐21 and GM‐CSF have opposing regulatory roles in the apoptosis of conventional dendritic cells. Immunity 2013; 38:514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mehta DS, Wurster AL, Whitters MJ, Young DA, Collins M, Grusby MJ. IL‐21 induces the apoptosis of resting and activated primary B cells. J Immunol 2003; 170:4111–8. [DOI] [PubMed] [Google Scholar]
- 105. Jin H, Carrio R, Yu A, Malek TR. Distinct activation signals determine whether IL‐21 induces B cell costimulation, growth arrest, or Bim‐dependent apoptosis. J Immunol 2004; 173:657–65. [DOI] [PubMed] [Google Scholar]
- 106. Kenefeck R, Wang CJ, Kapadi T et al Follicular helper T cell signature in type 1 diabetes. J Clin Invest 2015; 125:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xu X, Shi Y, Cai Y et al Inhibition of increased circulating Tfh cell by anti‐CD20 monoclonal antibody in patients with type 1 diabetes. PLOS ONE 2013; 8:e79858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005; 5:853–65. [DOI] [PubMed] [Google Scholar]
- 109. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621–63. [DOI] [PubMed] [Google Scholar]
- 110. Pot C, Jin H, Awasthi A et al Cutting edge: IL‐27 induces the transcription factor c‐Maf, cytokine IL‐21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL‐10‐producing Tr1 cells. J Immunol 2009; 183:797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Schmitt N, Bentebibel SE, Ueno H. Phenotype and functions of memory Tfh cells in human blood. Trends Immunol 2014; 35:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Luthje K, Kallies A, Shimohakamada Y et al The development and fate of follicular helper T cells defined by an IL‐21 reporter mouse. Nat Immunol 2012; 13:491–8. [DOI] [PubMed] [Google Scholar]
- 113. Liu X, Yan X, Zhong B et al Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med 2012; 209:1841–52, S1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Weber JP, Fuhrmann F, Hutloff A. T‐follicular helper cells survive as long‐term memory cells. Eur J Immunol 2012; 42:1981–8. [DOI] [PubMed] [Google Scholar]
- 115. Choi YS, Yang JA, Yusuf I et al Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol 2013; 190:4014–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hale JS, Youngblood B, Latner DR et al Distinct memory CD4+ T cells with commitment to T follicular helper‐ and T helper 1‐cell lineages are generated after acute viral infection. Immunity 2013; 38:805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Baumjohann D, Preite S, Reboldi A et al Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 2013; 38:596–605. [DOI] [PubMed] [Google Scholar]
- 118. He J, Tsai LM, Leong YA et al Circulating precursor CCR7(lo)PD‐1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013; 39:770–81. [DOI] [PubMed] [Google Scholar]
- 119. Shulman Z, Gitlin AD, Targ S et al T follicular helper cell dynamics in germinal centers. Science 2013; 341:673–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory‐like properties. J Clin Invest 2014; 124:5191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ziegler AG, Rewers M, Simell O et al Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013; 309:2473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Steck AK, Vehik K, Bonifacio E et al . Predictors of progression from the appearance of islet autoantibodies to early childhood diabetes: the environmental determinants of diabetes in the young (TEDDY). Diabetes Care 2015; 38:808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Skarstrand H, Krupinska E, Haataja TJ, Vaziri‐Sani F, Lagerstedt JO, Lernmark A. Zinc transporter 8 (ZnT8) autoantibody epitope specificity and affinity examined with recombinant ZnT8 variant proteins in specific ZnT8R and ZnT8W autoantibody‐positive type 1 diabetes patients. Clin Exp Immunol 2015; 179:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Achenbach P, Koczwara K, Knopff A, Naserke H, Ziegler AG, Bonifacio E. Mature high‐affinity immune responses to (pro)insulin anticipate the autoimmune cascade that leads to type 1 diabetes. J Clin Invest 2004; 114:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Curnock RM, Reed CR, Rokni S, Broadhurst JW, Bingley PJ, Williams AJ. Insulin autoantibody affinity measurement using a single concentration of unlabelled insulin competitor discriminates risk in relatives of patients with type 1 diabetes. Clin Exp Immunol 2012; 167:67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bender C, Schlosser M, Christen U, Ziegler AG, Achenbach P. GAD autoantibody affinity in schoolchildren from the general population. Diabetologia 2014; 57:1911–8. [DOI] [PubMed] [Google Scholar]
- 127. Choi YS, Kageyama R, Eto D et al ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011; 34:932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ, Jenkins MK. Opposing signals from the Bcl6 transcription factor and the interleukin‐2 receptor generate T helper 1 central and effector memory cells. Immunity 2011; 35:583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med 2012; 209:243–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ballesteros‐Tato A, Leon B, Graf BA et al Interleukin‐2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity 2012; 36:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Oestreich KJ, Mohn SE, Weinmann AS. Molecular mechanisms that control the expression and activity of Bcl‐6 in TH1 cells to regulate flexibility with a TFH‐like gene profile. Nat Immunol 2012; 13:405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lowe CE, Cooper JD, Brusko T et al Large‐scale genetic fine mapping and genotype‐phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet 2007; 39:1074–82. [DOI] [PubMed] [Google Scholar]
- 133. Dendrou CA, Plagnol V, Fung E et al Cell‐specific protein phenotypes for the autoimmune locus IL2RA using a genotype‐selectable human bioresource. Nat Genet 2009; 41:1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Long SA, Cerosaletti K, Bollyky PL et al Defects in IL‐2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T‐cells of type 1 diabetic subjects. Diabetes 2010; 59:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Garg G, Tyler JR, Yang JH et al Type 1 diabetes‐associated IL2RA variation lowers IL‐2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol 2012; 188:4644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin‐2 in type 1 diabetes. Diabetes 2012; 61:14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Silva DG, Daley SR, Hogan J et al Anti‐islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet‐reactive CD4 T cells. Diabetes 2011; 60:2102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Luzina IG, Atamas SP, Storrer CE et al Spontaneous formation of germinal centers in autoimmune mice. J Leukoc Biol 2001; 70:578–84. [PubMed] [Google Scholar]
- 139. Kendall PL, Yu G, Woodward EJ, Thomas JW. Tertiary lymphoid structures in the pancreas promote selection of B lymphocytes in autoimmune diabetes. J Immunol 2007; 178:5643–51. [DOI] [PubMed] [Google Scholar]
- 140. Astorri E, Bombardieri M, Gabba S, Peakman M, Pozzilli P, Pitzalis C. Evolution of ectopic lymphoid neogenesis and in situ autoantibody production in autoimmune nonobese diabetic mice: cellular and molecular characterization of tertiary lymphoid structures in pancreatic islets. J Immunol 2010; 185:3359–68. [DOI] [PubMed] [Google Scholar]
- 141. Reinhardt RL, Liang HE, Locksley RM. Cytokine‐secreting follicular T cells shape the antibody repertoire. Nat Immunol 2009; 10:385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Perreau M, Savoye AL, De Crignis E et al Follicular helper T cells serve as the major CD4 T cell compartment for HIV‐1 infection, replication, and production. J Exp Med 2013; 210:143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Deenick EK, Chan A, Ma CS et al Follicular helper T cell differentiation requires continuous antigen presentation that is independent of unique B cell signaling. Immunity 2010; 33:241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen‐specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA‐4. Immunity 2014; 41:1013–25. [DOI] [PubMed] [Google Scholar]
- 145. Sage PT, Paterson AM, Lovitch SB, Sharpe AH. The coinhibitory receptor CTLA‐4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014; 41:1026–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wang CJ, Heuts F, Ovcinnikovs V et al CTLA‐4 controls follicular helper T‐cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci USA 2015; 112:524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Linterman MA, Denton AE, Divekar DP et al CD28 expression is required after T cell priming for helper T cell responses and protective immunity to infection. Elife 2014; 3. doi:10.7554/eLife.03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Linterman MA, Vinuesa CG. Signals that influence T follicular helper cell differentiation and function. Semin Immunopathol 2010; 32:183–96. [DOI] [PubMed] [Google Scholar]
- 149. Walker LS, Wiggett HE, Gaspal FM et al Established T cell‐driven germinal center B cell proliferation is independent of CD28 signaling but is tightly regulated through CTLA‐4. J Immunol 2003; 170:91–8. [DOI] [PubMed] [Google Scholar]
- 150. Walker LS, Gulbranson‐Judge A, Flynn S et al Compromised OX40 function in CD28‐deficient mice is linked with failure to develop CXC chemokine receptor 5‐positive CD4 cells and germinal centers. J Exp Med 1999; 190:1115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Maine CJ, Marquardt K, Cheung J, Sherman LA. PTPN22 controls the germinal center by influencing the numbers and activity of T follicular helper cells. J Immunol 2014; 192:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Becattini S, Latorre D, Mele F et al T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 2015; 347:400–6. [DOI] [PubMed] [Google Scholar]
- 153. Nakayamada S, Kanno Y, Takahashi H et al Early Th1 cell differentiation is marked by a Tfh cell‐like transition. Immunity 2011; 35:919–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Linterman MA, Rigby RJ, Wong RK et al Follicular helper T cells are required for systemic autoimmunity. J Exp Med 2009; 206:561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tangye SG, Ma CS, Brink R, Deenick EK. The good, the bad and the ugly ‐ TFH cells in human health and disease. Nat Rev Immunol 2013; 13:412–26. [DOI] [PubMed] [Google Scholar]
- 156. Onengut‐Gumuscu S, Chen WM, Burren O et al Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet 2015; 47:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Locci M, Havenar‐Daughton C, Landais E et al Human circulating PD‐(+)1CXCR3(–)CXCR5(+) memory Tfh cells are highly functional and correlate with broadly neutralizing HIV antibody responses. Immunity 2013; 39:758–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Cubas RA, Mudd JC, Savoye AL et al Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med 2013; 19:494–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Bentebibel SE, Lopez S, Obermoser G et al Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med 2013; 5:176ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Morgan NG, Richardson SJ. Enteroviruses as causative agents in type 1 diabetes: loose ends or lost cause? Trends Endocrinol Metab 2014; 25:611–9. [DOI] [PubMed] [Google Scholar]
- 161. Schneider DA, von Herrath MG. Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia 2014; 57:2009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]