

Summary
Reliable risk assessment for biotherapeutics requires accurate evaluation of risk factors associated with immunogenicity. Immunogenicity risk assessment tools were developed and applied to investigate the immunogenicity of a fully human therapeutic monoclonal antibody, ATR‐107 [anti‐interleukin (IL)‐21 receptor] that elicited anti‐drug antibodies (ADA) in 76% of healthy subjects in a Phase 1 study. Because the ATR‐107 target is expressed on dendritic cells (DCs), the immunogenicity risk related to engagement with DC and antigen presentation pathways was studied. Despite the presence of IL‐21R on DCs, ATR‐107 did not bind to the DCs more extensively than the control therapeutic antibody (PF‐1) that had elicited low clinical ADA incidence. However, ATR‐107, but not the control therapeutic antibody, was translocated to the DC late endosomes, co‐localized with intracellular antigen‐D related (HLA‐DR) molecules and presented a dominant T cell epitope overlapping the complementarity determining region 2 (CDR2) of the light chain. ATR‐107 induced increased DC activation exemplified by up‐regulation of DC surface expression of CD86, CD274 (PD‐L1) and CD40, increased expansion of activated DC populations expressing CD86hi, CD40hi, CD83hi, programmed death ligand 1 (PD‐L1)hi, HLA‐DRhi or CCR7hi, as well as elevated secretion of tumour necrosis factor (TNF)‐α by DCs. DCs exposed to ATR‐107 stimulated an autologous T cell proliferative response in human donor cells, in concert with the detection of immunoglobulin (Ig)G‐type anti‐ATR‐107 antibody response in clinical samples. Collectively, the enhanced engagement of antigen presentation machinery by ATR‐107 was suggested. The approaches and findings described in this study may be relevant to identifying lower immunogenicity risk targets and therapeutic molecules.
Keywords: antigen processing and presentation, IL‐21 receptor, immunogenicity, T cell epitope, T cell proliferation
Introduction
Investigation of the immunogenicity of therapeutic antibody candidates has typically involved characterization of anti‐drug antibody (ADA) responses in clinical trials. This approach has provided limited information to investigate the underlying causes and to guide strategies for designing less immunogenic alternative molecules or therapeutic regimens. However, investigation capability has been enhanced in recent years by the introduction of new in‐silico, in‐vitro and human cellular immunogenicity risk assessment tools 1, 2, 3, 4. The studies presented here applied multiple new immunogenicity risk assessment tools and ADA characterization to investigate the underlying mechanisms of a strong immunogenic response induced by a human interleukin (IL)‐21R blocking therapeutic antibody, ATR‐107.
ATR‐107 is a fully human anti‐IL‐21 receptor (IL‐21R) monoclonal antibody (mAb) therapeutic candidate that was designed to block IL‐21 from binding and activating the receptor as a novel approach to treatment of lupus and other autoimmune diseases 5. As reported previously 6, ATR‐107 was immunogenic when administered to healthy human subjects, inducing anti‐ATR‐107 antibody development in 76% of subjects in a single ascending‐dose study. Of the patients who developed anti‐ATR‐107 antibodies, 74% developed low titre neutralizing antibodies and three subjects experienced hypersensitivity reactions.
Therapeutic mAbs and other biotherapeutics are thought to cause unwanted immunogenicity due to a combination of various product‐, patient‐ and treatment‐related factors 7, 8, 9. Among the most significant immunogenicity risk factors is the presence of sequences or structures in the biotherapeutic that differ from the human sequence and thus could form potential epitopes for recognition by T or B cell receptors. Although fully human mAbs have extensive sequence and structural homology to human immunoglobulin, the complementarity‐determining regions (CDRs) that form the antigen binding pocket are unique to each monoclonal antibody, and may be potential immunogenic epitopes 10. Other immunogenicity risk factors include immune status of the study subjects, the target or pharmacology of the biotherapeutic, and the route and frequency of administration.
ATR‐107 contains mutations in the Fc region that were designed to reduce effector function (resulting in no detectable antibody‐dependent cytotoxicity and complement‐binding activities) 5, which could also be potential immunogenic epitopes. ATR‐107 induced a high incidence of immunogenicity by both the subcutaneous (s.c.) and intravenous (i.v.) routes after a single dose, suggesting that factors other than route of administration must have contributed to the immunogenic response. The ATR‐107 target, IL‐21R, is known to be expressed on many lymphoid cells, including B cells, activated T cells, natural killer cells, monocytes and dendritic cells (DCs) 11, 12. ATR‐107 does not appear to activate the receptor or induce cytokine storm 13. The expected ATR‐107 mode of action is to block the effects of IL‐21 activation of its receptor, which include enhanced proliferation of lymphoid cells, B cell differentiation to memory cells and plasma cells, and development of T helper type 17 (Th17) cells 14, 15, 16, 17, 18. Anti‐inflammatory efficacy resulting from IL‐21R blockade has been observed in animal models 19. Thus, most of the effects of blocking IL‐21R might have been expected to reduce the risk of immunogenic response. However, targeting ATR‐107 to DCs, which are professional antigen‐presenting cells, might contribute to enhanced immunogenicity.
A novel biotherapeutic immunogenicity assessment model system that integrates DC binding, activation and intracellular trafficking tools was developed to investigate the hypothesis that ATR‐107 immunogenicity might involve the enhanced engagement of DC targeting machinery.
Materials and methods
Antibodies and reagents
Aqua live/dead cell tracker, CellLight® Late Endosomes‐GFP (BacMam 2·0), Hoechst 33342 were purchased from Life Technologies (Carlsbad, CA, USA). Recombinant human IL‐4 and granulocyte–macrophage colony‐stimulating factor (GM‐CSF) (carrier free) were purchased from R&D Systems (Minneapolis, MN, USA) and Cell Signaling (Danvers, MA, USA). Cell proliferation dye eFluor® 670 was purchased from eBioscience (San Diego, CA, USA). Anti‐CD4 and anti‐CD14 beads were purchased from Miltenyi Biotec (Bergisch Gladbach, Germany). The following fluorochrome‐labelled monoclonal antibodies were purchased from Biolegend (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA): IL‐21R (17A12), CD11c (3.9), CD14 (M5E2), CD40 (5C3), CD80 (2D10), CD86 (IT2·2), antigen‐D related (HLA‐DR) (L243), OX40L (11C3·1), CD83 (HB15E), inducible co‐stimulatory ligand (ICOSL) (2D3), programmed death ligand 1 (PDL‐1) (29E.2A3), CCR7 (G043H7) and CD4 (L200).
Cell isolation
Peripheral blood mononuclear cells (PBMCs), freshly purified from whole blood samples obtained from 11 healthy volunteers, were stained with anti‐CD14 or anti‐CD4 beads to isolate the CD14+ monocytes or CD4+ T cells, respectively. The CD14+ cells were cultured in RPMI‐1640 complete medium with the addition of IL‐4 (500 IU/ml) and GM‐CSF (1000 IU/ml) to derive to DCs.
DC binding
At day 4 of DC culture, cells were stained with Alexa Fluor 647‐labelled ATR‐107, PF‐1 (control human therapeutic mAb) or allophycocyanin (APC)‐labelled mouse anti‐human IL‐21R mAb (BD Biosciences) for 20 min at 4°C. One group of cells were stained in the phosphate‐buffered saline (PBS) buffer with bovine serum albumin (BSA) and sodium azide (Pharmingen stain buffer; BD Biosciences); the other group of cells were pretreated with 50% human serum (Mediatech Inc., Manassas, VA, USA) or a Fc blocker (Miltenyi Biotec) for 10 min at room temperature, then stained with the Alexa Fluor 647‐labelled antibodies in the PBS buffer with 1% human serum. Cells from both groups were washed and subjected to flow cytometric analysis with BD LSR Fortessa.
DC intracellular trafficking to late endosomes
CD14+ cells were cultured with phenol‐free RPMI‐1640 complete medium with the presence of IL‐4 and GM‐CSF. At day 3 of culture, cells were transfected with CellLight Late Endosome‐GFP BacMam 2·0 (Life Technologies, Carlsbad, CA, USA) reagent at 3 µl per 10 000 cells following the manufacturer's procedure. After overnight culture, cells were stained with Hoechst 33342 (1 : 1000) and treated with Alexa Fluor 647‐labelled ATR‐107 or PF‐1 at 50 µg/ml for 30 min at 37°C. Cells were washed and replated into a 35‐mm glass‐bottomed dish (MatTek Corporation, Ashland, MA, USA). Live cell imaging was performed on a Perkin Elmer Ultraview LCI Spinning Disk Confocal microscope equipped with live‐cell chamber device (Solent Scientific, Fareham, UK) to maintain the 37°C condition during the experiment. Images were acquired with Perkin Elmer Volocity software in 10‐µm‐deep (0·2 µm sectioning) z‐stacked frames.
Immunofluorescence and intracellular HLA‐DR binding
At day 4 of DC culture, cells were treated with Alexa Fluor 647‐labelled ATR‐107 or PF‐1 at 50 µg/ml for 30 min at 37°C. The cells were washed and fixed with 4% paraformaldehyde (PFA) (pH 7·2 in PBS buffer) for 10 min at room temperature. After washing, cells were permeabilized with 0·1% Triton X‐100, then stained with fluorescein isothiocyanate (FITC)‐anti‐human HLA‐DR. Following the washing step, cells were harvested (Shandon Cytospin 4) and mounted onto slides with 4′,6‐diamidino‐2‐phenylindole (DAPI) mounting medium (Vector, Burlingame, CA, USA). Images were acquired on a Zeiss Axio imager fluorescence microscope and analysed using AxioVision 4·7·2 software (Zeiss International, Oberkochen, Germany.
Peptide presentation assay
Identification of ATR‐107 peptides presented by DCs following uptake and processing was performed by ProImmune (Oxford, UK) using the ProPresent® antigen presentation assay. Briefly, PBMCs from 10 HLA‐typed healthy donors were purified from whole blood by gradient density centrifugation. Immature monocyte‐derived DCs were generated in vitro and matured in the presence of test protein. DCs were harvested and washed prior to lysis in a detergent‐containing buffer solution. HLA molecules were recovered in a specific immunoaffinity step. Peptides were then eluted from the HLA complexes and processed for further analysis by sequencing mass spectrometry. Peptide samples were subsequently analysed by high‐resolution sequencing mass spectrometry (LC‐MS/MS). The resulting data were then compiled and analysed using sequence analysis software referencing the Human Uniprot_Complete_Proteome Database with the incorporated test item sequence.
In‐silico T cell epitope assessment
The sequences of ATR‐107 VH and VL were analysed by EpiMatrix 2 (Epivax, Providence, RI, USA). Each domain was parsed into overlapping 9‐mer peptides, with each peptide overlapping the last by eight amino acids. Each peptide was then scored for predicted binding to each of eight HLA Class II alleles (DRB1*0101, DRB1*0301, DRB1*0401, DRB1*0701, DRB1*0801, DRB1*1101, DRB1*1301 and DRB1*1501), which represent HLA supertypes covering up to 97% of human populations worldwide 20. Any peptide scoring above 1·64 on the EpiMatrix ‘Z’ scale (approximately the top 5% of the random peptide set) was classed as a ‘hit’ for binding to the major histocompatibility complex (MHC) molecule for which it was predicted. A previous study with a therapeutic protein demonstrated a correlation between an immunologically active peptide and the EpiMatrix prediction 4. As EpiMatrix predicts binding for alleles representative for the superfamilies of HLA types and not for each individual type, supplementary analysis of predicted peptide binding to HLA types present in the peptide presentation assay, but not included in EpiMatrix, was performed using the Immune Epitope Database (IEDB) analysis tools 21, 22, 23. Specific sequences were analysed using the ‘consensus’ analysis from IEDB with a top 10% ranked binder being considered a ‘hit’.
DC activation and cytokine measurement
At day 3 of DC culture, cells were treated with ATR‐107 or PF‐1 at 50 µg/ml. Cells untreated with test articles were used as control. Reference antigen keyhole limpet hemocyanin (KLH) was used as positive control to monitor assay performance. At day 5, DCs were washed to remove the excessive test articles, and then stained with antibody cocktails that include antibodies to CD11c, CD14, CD86, CD80, CD83, CD40, HLA‐DR, CD252 (OX40L), CD275 (ICOSL), CD274 (PD‐L1) and CD197 (CCR7). The expression of DC surface receptors and the percentages of activated/mature DCs were measured by flow cytometry (BD LSR Fortessa) and analysed using Cytobank (Fluidigm, San Francisco, CA, USA) and FlowJo (TreeStar Inc., Ashland, OR, USA). The cell supernatants were collected and analysed for secretion of cytokines interferon (IFN)‐γ, IL‐10, IL‐12p70, IL‐13, IL‐1β, IL‐2, IL‐4, IL‐6, IL‐8 and tumour necrosis factor (TNF)‐α using the V‐PLEX proinflammatory panel 1 (human) kit from Meso Scale Diagnostics, Rockville, MD, USA.
ADA isotype characterization
Characterization of ADA isotype was performed by immunodepleting serum IgM or IgG immunoglobulins from samples confirmed positive for ATR‐107 ADA prior to analysis of residual anti‐ATR‐107 activity. Anti‐human IgG or anti‐human IgM agarose uncoated sepharose beads were centrifuged at 14 000 × g for 5 min to remove storage buffer and then washed three times with wash buffer prior to reconstitution in assay buffer (1% BSA in PBS‐Tris). The reconstituted beads were centrifuged for 10 min at 14 000 × g and supernatant was removed from the bead pellet. Confirmed ADA‐positive samples or negative control (pooled human serum) were diluted 1 : 25 in assay buffer and 250 µl of each diluted sample was added to the pelleted beads. After mixing, the bead–serum sample mixtures were rotated end‐over‐end for approximately 1–1·5 h at room temperature. The mixtures were centrifuged for 10 min at 14 000 × g and the supernatants were removed carefully to new tubes and centrifuged for an additional time to pellet any residual bead. The supernatant samples, along with positive and negative controls, were analysed in the electrochemiluminescent bridging anti‐ATR‐107 ADA assay described previously 6. Samples were co‐incubated with biotinylated ATR‐107 and ruthenylated ATR‐107. Complexes formed by anti‐ATR‐107 ADA bridging these reagents were captured and measured using a Meso Scale Discovery electrochemiluminescence assay platform. The percentage increase in signal generated after preincubation with anti‐IgM‐ or anti‐IgG‐coated beads was calculated based on the signal generated after preincubation with the uncoated beads.
CD4+ T cell proliferation
DCs derived from purified CD14+ cells were cultured in the presence of IL‐4 and GM‐CSF. At day 3 of culture, cells were loaded with ATR‐107, PF‐1 or tuberculin‐purified protein derivative (PPD) at 50 μg/ml. Untreated cells were used as control. After an additional 2‐day culture, cells were washed and co‐cultured with the purified autologous CD4+ T cells (3 × 105 cells/well, labelled with cell proliferation dye eFluor 670) at a ratio of 1 : 10 (DC : CD4+ T cells) in 96‐well round‐bottomed plates. After 7 days of DC–CD4+ T cell co‐culture, detection of CD4+ T cell proliferation was performed by co‐staining with anti‐human CD4 antibody. Cells were acquired on a BD LSR Fortessa and analysed using FlowJo software (TreeStar Inc.).
Statistical analysis
Statistical analysis was performed using two‐tailed and paired Student t‐tests. Statistical significance was set at P < 0·05.
Results
DC binding, internalization and trafficking to late endosomes
T cell antigen‐specific responses are initiated by the interaction of T cell receptors with peptide–MHC complexes presented on the surfaces of DCs or other antigen‐presenting cells. The IL‐21R target is found on DCs, and therefore we investigated whether ATR‐107 engagement with DCs is distinguishable from that of a low‐immunogenicity mAb, PF‐1. PF‐1 is also an IgG1 mAb bearing the same Fc mutations as ATR‐107, but elicited 3% ADA after administration of a single dose in healthy human subjects and has no known target expression on DCs.
In these studies, DCs were derived from CD14+ monocytes that were isolated from healthy subject whole blood samples and cultured in the presence of IL‐4 and GM‐CSF. On day 4, cells were tested for binding to ATR‐107 and PF‐1, respectively, by flow cytometry. Both ATR‐107 and PF‐1 bound to the DCs with similar binding fluorescence intensity (Fig. 1a). The expression of IL‐21R on human DCs was confirmed by staining with a commercial mouse anti‐human IL‐21R; although the binding appeared to be modest, it was in alignment with the previously published expression level of IL‐21R on human monocytes and GM‐CSF‐derived macrophages 11. Application of Fc blockers did not reduce the binding signal to a large extent, indicating that the DC surface FcγR played a role, but was not the major mediator of the ATR‐107 and PF‐1 binding to DCs (Fig. 1b).
Figure 1.
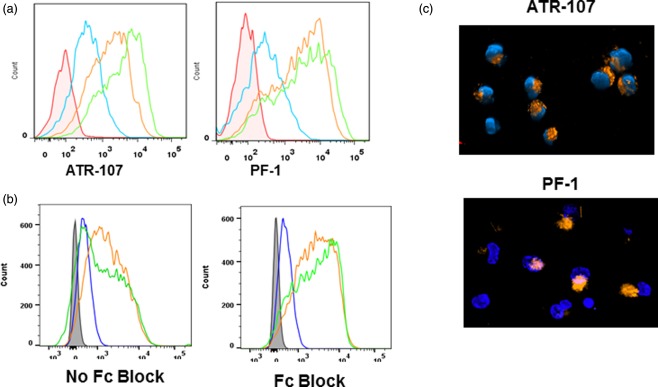
Analysis of dendritic cell (DC) binding and internalization. (a) Titration of human IL‐21R blocking therapeutic antibody (ATR‐107) and control human therapeutic antibody (PF‐1) binding to DCs in the absence of Fc blockers. Red = untreated cells; blue = 1 µg/ml; orange = 10 µg/ml; green = 50 µg/ml. (b) Comparison of ATR‐107 (20 µg/ml) and PF‐1 (20 µg/ml) binding to the DCs in the presence and absence of Fc blockers. Tinted grey = untreated cells; blue = commercial mouse anti‐human interleukin (IL)‐21R; green = PF‐1; orange = ATR‐107. (c) DC internalization of ATR‐107 and PF‐1. Orange = Alexa Fluor 647‐labelled ATR‐107 or PF‐1; blue = nucleus (Hoechst 33342 stain).
Upon antigen engagement, DCs endocytose protein antigens avidly through a variety of mechanisms, including non‐specific macropinocytosis and specific receptor‐mediated endocytosis. We next assessed the DC internalization and intracellular trafficking of ATR‐107 and PF‐1 to the late endosomal compartments. Similar to ATR‐107, PF‐1 was internalized by DC quickly, despite the lack of a targeting receptor on the DC surface. The internalization was observed within 2 min of the therapeutic antibody addition (data not shown). At 30 min, internalization of both mAbs was observed more readily, from which ATR‐107 exhibited a more polarized distribution pattern than PF‐1 (Fig. 1c). By applying live cell three‐dimensional confocal imaging, ATR‐107, but not PF‐1, was found to be present in DC late endosomes (Fig. 2a), indicating its translocation into the compartment in which binding to HLA molecules can occur. Further analysis by immunofluorescence microscopy confirmed that ATR‐107 was co‐localized with the intracellular HLA‐DR molecules (Fig. 2b). Except for showing the lack of translocation to late endosomes and binding to intracellular HLA‐DR, the fate of PF‐1 intracellular trafficking was not evaluated. Collectively, the data suggested that ATR‐107 engaged the DC antigen processing and presentation machinery more efficiently than PF‐1.
Figure 2.
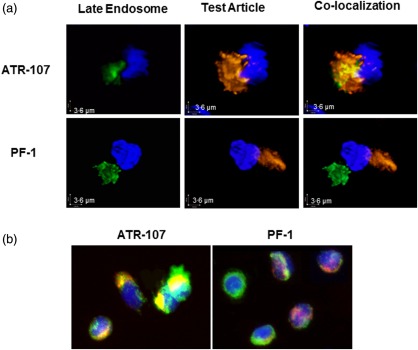
Analysis of intracellular trafficking to the late endosomal compartments in dendritic cells (DCs). (a) Live cell three‐dimensional confocal imaging of translocation to DC late endosomes. Orange = Alexa 647‐labelled ATR‐107 or control human therapeutic antibody (PF‐1); green = CellLight® Late Endosomes‐GFP (BacMam 2·0); blue = nucleus (Hoechst 33342 stain). (b) Immunofluorescence imaging of intracellular human leucocyte antigen‐D related (HLA‐DR) binding. Red = Alexa Fluor 647‐labelled ATR‐107 or PF‐1; green = HLA‐DR‐fluorescein isothiocyanate (FITC); blue = nucleus [4′,6‐diamidino‐2‐phenylindole (DAPI)].
Potential T cell epitope identification
To determine which mAb sequences are likely to be processed and presented, DCs from 10 HLA‐typed healthy donors were pulsed with ATR‐107 and PF‐1 and subjected to processing and presentation. HLA‐DRB1 molecules were immunoprecipitated and bound peptides were eluted and analysed by mass spectrometry. Peptides specifically derived from ATR‐107 were detected in six of 10 of the donor samples. Only peptides derived from the light chain of ATR‐107 were detected. The most commonly presented peptides were from the light chain and contained some or all the CDR2 region (Fig. 3a).
Figure 3.
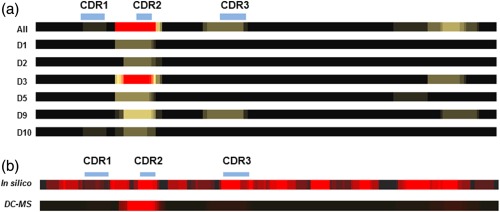
Correlation between in‐silico prediction of potential T cell epitopes from human IL‐21R blocking therapeutic antibody (ATR‐107) VL and presentation of peptides determined by dendritic cells (DCs). (a) Individual donor (designated Dn) heat‐maps of sequences processed and presented by DCs identify subtle variations in presentation, although the CDR2 is always represented by the most peptides. Donors with no ATR‐107 VL peptides presented were omitted. (b) Heat‐map of sequences predicted to be bound by eight HLA supertypes (9mer) compared to a heat‐map of peptides processed and presented by DCs in a DC‐MS assay. The in‐silico heat map was constructed using predictions for each overlapping 9‐mer with the eight human leucocyte antigen (HLA) supertypes analysed. Each potential HLA binding 9mer was stacked, so that core binding residues that interact with multiple HLA types appear brightest. The DC‐MS heat‐map was generated from peptides identified in the assay, with nested sets being represented by bright ‘cores’ with less intense flanking regions.
In‐silico assessment of T cell epitopes was performed using EpiMatrix (Epivax), with potential epitopes identified as being predicted to be top 5% binders to at least four of eight HLA supertypes. Further classification of probable T cell epitopes as being of foreign origin was made on the basis of whether each 9‐mer identified differed by at least one residue from the germline sequence of this antibody. This analysis identified two potential overlapping epitopes in the heavy chain CDR2 and a single potential epitope in the light chain CDR2. No additional peptides were identified using the same criteria applied to analysis using the IEDB. Additional peptides from all the CDRs were predicted to bind to between one and three HLA types.
In‐silico analysis is usually considered to be over‐predictive, and therefore a more limited repertoire of peptides may actually be processed and presented by APCs. A comparison with potential epitopes determined by in‐silico predictions with the repertoire of peptides actually processed and presented in the DC–MS assay is shown in Fig. 3b. Each donor presented one or more peptides corresponding to the CDR2 of the light chain, with few individual donors presenting peptides derived from CDR1 and CDR3. Sporadic presentation of peptides from the constant domain of the light chain was also detected.
In‐silico assessment for PF‐1 indicated only a single predicted HLA binding peptide in the non‐germline region, which coincided with the light chain CDR2. This peptide was not detected in the DC–MS assay (data not shown), suggesting either that processing was incomplete or that other peptides from the molecule were presented more favourably.
DC activation and phenotypical changes
DCs present MHC–peptide complexes at their cell surface upon maturation and up‐regulate cell surface receptors that act as co‐receptors to modulate activation of T cells 24. To determine the effect of ATR‐107 treatment on DC activation, DCs derived from 11 healthy donor samples were pulsed with ATR‐107 or PF‐1 at day 3 of culture in the presence of IL‐4 and GM‐CSF. The expression of DC surface receptors CD86, CD80, CD83, CD11c, CD40, HLA‐DR, CD252 (OX40L), CD275 (ICOSL), CD274 (PD‐L1) and CD197 (CCR7) was analysed at day 5. Modulation of the expression levels of the DC surface receptors was illustrated by changes in median fluorescence intensity (MFI). As shown in Fig. 4, ATR‐107, but not PF‐1, up‐regulated significantly the expression of CD86, CD40 and CD274 (PD‐L1). Expression of other tested DC receptors was not modulated significantly by ATR‐107. In comparison with unstimulated cell controls, 73% (eight of 11) of the healthy donor samples responded to ATR‐107 and exhibited a greater than 100% increase of the CD86 expression, whereas 36% (four of 11), 82% (nine of 11) and 64% (seven of 11) of the donor samples responded to ATR‐107 and exhibited a greater than 50% increase of HLA‐DR, CD86 and PD‐L1, respectively (Table 1). Upon PF‐1 treatment, a greater than 50% increase of the tested DC receptors was not observed, except for CD80, which was up‐regulated by 51% above the unstimulated cells in one of the 11 tested donor samples.
Figure 4.
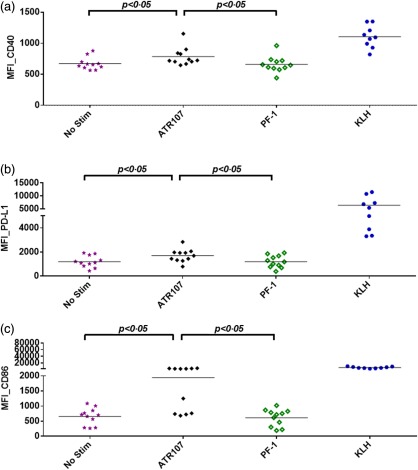
Flow cytometric analysis of dendritic cell (DC) surface receptor expression levels. (a) Median fluorescence intensity (MFI)_CD40; (b) MFI_programmed death ligand 1 (PD‐L1); (c) MFI_CD86. No Stim: non‐test article‐treated DC control.
Table 1.
Donor response frequency based on expression of dendritic cell (DC) surface receptors.
| MFI ≥ 50% increasea | MFI ≥ 100% increasea | |||||
|---|---|---|---|---|---|---|
| %Responders | KLH | ATR‐107 | PF‐1 | KLH | ATR‐107 | PF‐1 |
| HLA‐DR | 78 | 36 | 0 | 56 | 9 | 0 |
| CD80 | 67 | 9 | 9 | 44 | 9 | 0 |
| OX40L | 0 | 0 | 0 | 0 | 0 | 0 |
| CD86 | 100 | 82 | 0 | 100 | 73 | 0 |
| CD83 | 44 | 9 | 0 | 11 | 0 | 0 |
| CD40 | 56 | 0 | 0 | 0 | 0 | 0 |
| ICOSL | 0 | 0 | 0 | 0 | 0 | 0 |
| PD‐L1 | 100 | 64 | 0 | 100 | 0 | 0 |
| CCR7 | 22 | 0 | 0 | 11 | 0 | 0 |
Increases over baseline (cells untreated with test articles). ICOSL = inducible co‐stimulatory ligand; HLA‐DR = human leucocyte antigen‐D related; PD‐L1=programmed death ligand 1; KLH = keyhole limpet hemocyanin; MFI = median fluorescence intensity; ATR‐107 = human IL‐21R blocking therapeutic antibody.
Expansion of the activated DC populations that highly express DC co‐receptors is a reflection of enhanced DC activation and function. The effect of the ATR‐107 or PF‐1 treatment was analysed by the increased percentages of the activated DC populations over those not treated with test molecules (baseline), and illustrated in Fig. 5a. Both test molecule‐treated and untreated cells were cultured in the presence of IL‐4 and GM‐CSF. Upon ATR‐107 treatment, the percentages of the CD11chiCD86hi, CD11chiCD40hi, CD11chiCD83hi, CD11chiHLA‐DRhi, CD11chiPD‐L1hi and CD11chiCCR7hi cell populations were augmented significantly over the baseline (Fig. 5b–g). With the exception of the CD11chiCCR7hi cells, the increased cell percentages described above were shown to be significantly different from those resulting from the PF‐1 treatment. The donor response frequency was calculated further based on the thresholds of 50% or 100% increase over the unstimulated cells. With the ATR‐107 treatment, enhanced expansion of the activated DC populations was detected in most of the tested donor samples whereas, with the PF‐1 treatment, expansion of the activated DC populations was observed sparsely (Table 2).
Figure 5.
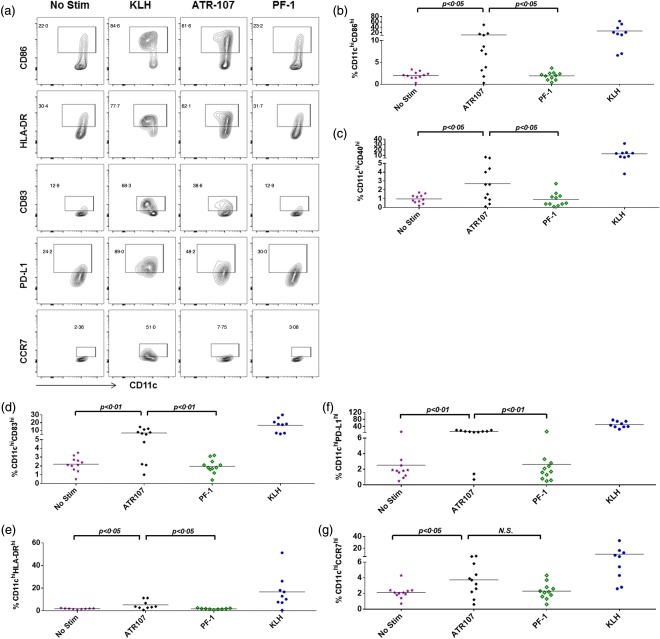
Flow cytometric analysis of percentages of activated dendritic cell (DC) populations. (a) Exampled illustrations of flow cytometric gating for test molecule‐activated DC populations. Gates for test article‐activated DC populations were drawn by comparing with the keyhole limpet hemocyanin (KLH)‐treated cells (positive control) and the test molecule untreated cells (baseline). (b) % CD11chiCD86hi; (c) % CD11chiCD40hi; (d) %CD11chiCD83hi; (e) %CD11chiHLA‐DRhi; (f) % CD11chiPD‐L1hi; (g) % CD11chiCCR7hi. No Stim = non‐test article‐treated DC control; n.s. = no significant difference.
Table 2.
Donor response frequency based upon %activated dendritic cell (DC) populations.
| %Cells ≥ 50% increasea | %Cells ≥ 100% increasea | |||||
|---|---|---|---|---|---|---|
| %Responders | KLH | ATR‐107 | PF‐1 | KLH | ATR‐107 | PF‐1 |
| %HLA‐DRhi CD11chi | 89 | 64 | 36 | 78 | 64 | 18 |
| %CD80hi CD11chi | 89 | 55 | 9 | 78 | 36 | 0 |
| %OX40Lhi CD11chi | 100 | 45 | 9 | 89 | 18 | 0 |
| %CD86hi CD11chi | 100 | 73 | 9 | 100 | 73 | 0 |
| %CD83hi CD11chi | 100 | 73 | 9 | 89 | 64 | 9 |
| %CD40hi CD11chi | 100 | 64 | 18 | 100 | 45 | 9 |
| %ICOSLhi CD11chi | 11 | 18 | 18 | 0 | 0 | 18 |
| %PD‐L1hi CD11chi | 100 | 82 | 0 | 100 | 73 | 0 |
| %CCR7hi CD11chi | 78 | 45 | 9 | 67 | 27 | 0 |
Increases over baseline (cells untreated with test articles). KLH = keyhole limpet hemocyanin; ATR107 = human IL‐21R blocking therapeutic antibody; PF‐1 = control human therapeutic antibody; HLA‐DR = human leucocyte antigen‐D related; ICOSL= inducible co‐stimulatory ligand; PD‐L1 = programmed death ligand 1.
Collectively, the data suggested that ATR‐107 induced more potent DC activation and maturation than PF‐1.
DC cytokine secretion
Part of the DC activation process is cytokine secretion, which has important effects on initiation and sustainability of the innate and adaptive immune response 25, 26, 27, 28. Supernatants of the DCs treated with ATR‐107 or PF‐1 were measured for secretion of the proinflammatory cytokines IFN‐γ, IL‐10, IL‐12p70, IL‐13, IL‐1β, IL‐2, IL‐4, IL‐6, IL‐8 and TNF‐α using the Meso Scale Diagnostics human proinflammatory cytokine kit. As shown in Fig. 6, ATR‐107 treatment augmented the secretion of TNF‐α and IL‐13 by DCs. Other cytokines were either not detected or did not show a significant difference from those secreted by the unstimulated cells or cells treated with PF‐1 (data not shown).
Figure 6.
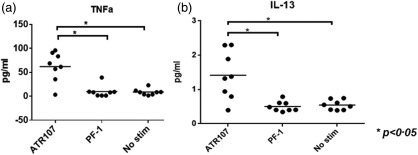
Analysis of cytokine secretion by dendritic cells (DCs) after treatment with human IL‐21R blocking therapeutic antibody (ATR‐107) or control human therapeutic antibody (PF‐1). (a) Tumour necrosis factor (TNF)‐α; (b) interleukin (IL)‐13. No Stim = non‐test article‐treated DC control. *P < 0·05.
Anti‐ATR‐107 ADA isotype evaluation
The capability of inducing specific T cell activation and proliferation is a functional hallmark of mature DCs. However, DC antigen presentation and expression of co‐stimulatory signals may not always lead to an enhanced T cell response; specific T cells expressing receptors that can recognize the presented MHC class II and antigen peptide complex at the DC surface must also be available. Unfortunately, cellular samples were not available from the clinical study to investigate functional T cell responses. Identification of the ADA isotype may help to elucidate the mechanism of ADA induction; in particular, it may indicate whether the response was likely to have involved a T cell response. To determine the involvement of a T cell response in the induction of the ADA response to ATR‐107, an immunodepletion technique using anti‐immunoglobulin beads was used to isotype the clinical study samples. In this study, 15 ADA‐positive samples (from 12 subjects), representing a range of ADA titres, were incubated with anti‐human IgG‐coated beads or uncoated beads. Negative controls were also incubated with beads. Following the incubation period and removal of the beads, the resulting supernatants were analysed in the ATR‐107 ADA assay, which used an electrochemiluminescence platform. The results (Table 3) showed that samples had lower signal [response unit (RU) values] after incubation with the anti‐IgG‐coated beads relative to the signals generated after incubation with uncoated beads, indicating the presence of IgG‐specific ADA in these samples. In some samples with lower signals, negative inhibition was observed, potentially reflecting interference by residual beads in the sample. Samples were also incubated with anti‐IgM beads; however, no marked level of inhibition was observed (data not shown), indicating that little detectable IgM was present at the time of sampling.
Table 3.
Evaluation of anti‐ATR‐107 antibody isotypes.
| ADA end‐point log10 titre result | Uncoated beads (RU) | Anti‐human IgG‐beads (RU) | %Inhibition of signal (IgG) |
|---|---|---|---|
| 4·11 | 11 669 | 472 | 96·0 |
| 4·08 | 11 647 | 668 | 94·3 |
| 4·04 | 7976 | 684 | 91·4 |
| 3·83 | 4118 | 290 | 93·0 |
| 3·52 | 2829 | 253 | 91·1 |
| 3·51 | 1779 | 338 | 81·0 |
| 3·11 | 1005 | 327 | 67·5 |
| 2·88 | 717 | 426 | 40·6 |
| 2·7 | 269 | 524 | −95·2 |
| 2·55 | 213 | 389 | −82·8 |
| 2·29 | 146 | 346 | −137·5 |
| 1·91 | 153 | 435 | −184·3 |
| 1·86 | 134 | 706 | −426·5 |
| 1·83 | 132 | 251 | −90·5 |
| 1·76 | 112 | 218 | −94·6 |
| Negative control | 92 | 203 | −120·7 |
| Negative control | 79 | 421 | −435·7 |
ADA = anti‐drug antibodies; Ig = immunoglobulin; RU = response unit.
These results indicate that ATR‐107 induced an IgG response in multiple healthy subjects, and suggest that ATR‐107 was likely to have induced T cell responses.
CD4+ T cell proliferation
To evaluate the priming of the T cell response to ATR‐107 antigen presentation by DCs ex vivo, DCs treated with ATR‐107 or PF‐1 were co‐cultured with autologous CD4+ T cells that were isolated from eight healthy donors. As shown in Fig. 7, ATR‐107 stimulated increased CD4+ T cell proliferation over the unstimulated cells or PF‐1 treatment. The donor response frequency was 50% (four of eight) based on a threshold of 50% increase over the unstimulated cells and 37.5% (three of eight) based on a threshold of 100% increase over the unstimulated cells. None of the donors exhibited a greater than 50% increase of the CD4+ T cell proliferative response to PF‐1.
Figure 7.
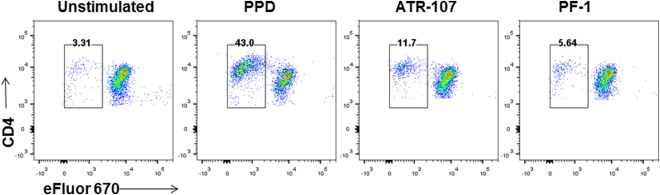
Representative data showing flow cytometric analysis of CD4+ T cell proliferative response to dendritic cells (DCs) treated with human IL‐21R blocking therapeutic antibody (ATR‐107). Cell division dye (eFluor 670) was used to label autologous CD4+ T cells prior to incubation with DCs that were treated with ATR‐107 or control human therapeutic antibody (PF‐1). Cells in plots were gated from live CD4+ T cells. PPD = tuberculin‐purified protein derivative.
Discussion
The risk of inducing unwanted immune responses is a major concern for all biotherapeutic development programmes due to the potential impact of such responses on the pharmacokinetic, pharmacodynamic, efficacy and safety profiles of the therapeutic; thus, there is a need for reliable immunogenicity risk prediction based upon better understanding of the immunogenicity risk factors. Fully human mAbs are generally expected to have a low risk of inducing immune responses in humans; however, the fully human mAb ATR‐107 induced ADA responses in 76% of healthy subjects after a single dose. Because ATR‐107 was designed to block the IL‐21R on DCs and other immune cells, we investigated whether its interaction with DCs would result in increased DC antigen processing and presentation machinery as a potential underlying mechanism for its strong immunogenic response.
In spite of the presence of IL‐21R on DCs, in these studies ATR‐107 did not bind to DCs more extensively than PF‐1, the control therapeutic mAb that induced a low incidence of clinical ADA. It is known that DC uptake can be mediated by DC surface receptors, as well as through the heightened levels of non‐specific pinocytosis or phagocytosis that is characteristic of antigen presentation cells. The non‐specific pinocytosis or phagocytosis complicates the evaluation of the DC surface receptor expression and binding events. Human antigen‐presenting cells were reported previously to express functional IL‐21R at a modest but significant level. Treatment with IL‐21 enhances phagocytosis in IL‐21R‐expressing monocytes and GM‐CSF‐derived macrophages 11. We did not specifically evaluate the effect of ATR‐107 on DC pinocytosis or phagocytosis; however, our staining results with the commercial mouse anti‐human IL‐21R mAb demonstrated low‐level expression of IL‐21R on DCs in this in vitro culture system. We postulate that the non‐specific pinocytosis and phagocytosis may have contributed collectively to the indistinguishable DC binding between ATR‐107 and PF‐1. This postulation was supported by the lack of significant binding signal reduction when excessive amounts of the unlabelled ATR‐107 were used to compete with the fluorochrome‐labelled ATR‐107 for binding to the DCs (data not shown). The role of FcR‐mediated endocytosis appeared to be limited, as would be expected by removing effector functions through mutations introduced to the Fc regions of ATR‐107 and PF‐1 and by observation of binding to DCs in the presence of Fc blockers. Nevertheless, ATR‐107 differed from PF‐1 in DC intracellular trafficking to the late endosomal compartment. Translocation to late endosomes and binding to intracellular HLA‐DR molecules are indicators of antigen processing and presentation. Indeed, the DC mass spectrometric analysis revealed a dominant peptide overlapping the CDR2 region of the ATR‐107 light chain and a few other peptides in the CDR1 and CDR3 that are presented less frequently by the DCs. In contrast, none of the non‐human germline sequences was presented for PF‐1.
ATR‐107 induced increased DC activation exemplified by the significant up‐regulation of DC surface receptor (CD86, PD‐L1, CD40) expression levels, increased expansion of activated DC populations, as well as elevated secretion of TNF‐α. ATR‐107 treatment up‐regulated the secretion of IL‐13 by DCs significantly; although the secretion level is probably too low to justify its functional impact, increased IL‐13 secretion is known to favour the development of antibody response by B cells. The CD4+ T cell proliferative response to ATR‐107‐treated DCs was found to be relatively variable; 50% of the tested donors’ samples responded based on a threshold of 50% increase over the baseline and 37.5% of the donors responded based on a threshold of 100% increase over the baseline. The heterogeneity of human individuals, especially in HLA haplotypes and the T cell receptor specificity to recognize the DC‐presented peptide–HLA complexes, might have accounted for the variability. In addition, priming and sustainability of the CD4+ T cell proliferative response requires the presence of proper co‐stimulatory signals and cytokine environments that are probably variable among human individuals 24, 29, 30. Although ATR‐107 robustly up‐regulated the expression level of CD86 on DCs, and to a lesser extent up‐regulated CD40 and PD‐L1, it is not possible to know whether the level of up‐regulation will be sufficient to stimulate T cells effectively in vivo. However, extensive expansion of the activated DCs by ATR‐107 would increase the possibility for DC–T interaction and priming of the T cell response.
Given the lack of information on the HLA haplotypes and functional T cell responses of individuals in the ATR‐107 clinical study, it is not possible to make a comparison between clinical ADA response, HLA haplotype and the non‐clinical assessments in vitro. Future inclusion of HLA typing of clinical study subjects and allowance for blood sample collection in study protocols, if deemed necessary to characterize the cellular immune response, would facilitate clinical validation of the in‐vitro immunogenicity risk assessment tools and improve understanding of the immunogenicity risk of genetic status associated with HLA haplotypes.
In light of the reported presence of IL‐21R expression on DCs 31 and other immune cells, the test results described herein may reflect the interplay of the pharmacological effects of the IL‐21R blocking and the induced immunogenic response by ATR‐107. It is possible that other mechanisms may also play a role in the ATR‐107 immunogenicity. IL‐21 is involved in development of B regulatory cells and inhibition of DC maturation 31, 32, 33, thus the immunogenic potential of ATR‐107 could be enhanced further through blocking these effects. However, both of these mechanisms might be expected to affect the immunogenicity of a therapeutic that blocks either IL‐21 or the IL‐21 receptor. Evidence to date suggests that IL‐21 cytokine antagonists may be less immunogenic than ATR‐107 (data not shown).
DCs take up protein antigens and transport them to endosomal compartments where the proteins are unfolded by low pH conditions and reductases, and are then cleaved proteolytically to produce peptide fragments. Some of the peptides produced via this DC processing machinery may bind with high affinity to Class II MHCs that are co‐located in the late endosomal compartment 34, 35, 36. The peptide–MHC complex is then transported to the cell surface, where it may stimulate CD4+ T helper cells bearing receptors specific for the peptide–MHC complex. Functions of activated T helper cells are required for supporting B cell functions, including isotype switching, mutation of binding sites to produce higher affinity antibody and differentiation to plasma and memory cells. The hypothesis that targeting ATR‐107 to DCs may have affected the antigen presentation pathway, leading ultimately to enhanced B cell production of ADA, was supported by work in the field of vaccine sciences 37. DCs are considered a natural adjuvant, and vaccines are being designed by attaching immunogenic peptides to monoclonal antibodies specific for various antigen‐presenting cell receptors to achieve more effective immune protection against microbes and cancer 38. To date, few investigators have evaluated the effects of biotherapeutics on DC activation and intracellular trafficking to endosomal compartments.
DCs perform multiple functions that contribute to inflammation, autoimmunity and responses to infectious disease and cancer; thus, there is significant interest in developing DC‐targeting therapeutics for either reducing or enhancing immune responses 39, 40. However, the findings described here suggest that therapeutic mAbs that efficiently engage antigen presentation machinery may increase the risk of unwanted immunogenicity via their potential for enhancing antigen presentation.
Immunogenicity prediction tools such as in‐silico analysis of T cell epitopes, in‐vitro PBMC and DC triggered T cell assays are becoming used more widely to assess the immunogenicity risk of therapeutic protein candidates. Each predictive tool explores a limited aspect of the immune response, indicating that using a single method provides an incomplete assessment of the overall immunogenicity risk of a product. The approaches described here, which evaluated the risk for DC antigen presentation and T cell proliferation by integrating multiple established tools (in‐silico, DC‐T, DC‐mass spectrometry) with newly developed tools to assess DC binding, intracellular trafficking and DC activation provided results consistent with the high clinical immunogenicity observed for ATR‐107. The methods applied and findings described here are relevant to identifying lower immunogenicity risk targets and therapeutic molecules.
Disclosure
The authors have no disclosures.
Acknowledgements
The authors would like to thank Jun Zhang for conducting the DC activation experiments and Yang Cong for fluorochrome conjugation of test articles.
References
- 1. Wullner D, Zhou L, Bramhall E et al Considerations for optimization and validation of an in vitro PBMC derived T cell assay for immunogenicity prediction of biotherapeutics. Clin Immunol 2010; 137:5–14. [DOI] [PubMed] [Google Scholar]
- 2. Schafer JR, Jesdale BM, George JA, Kouttab NM, De Groot AS. Prediction of well‐conserved HIV‐1 ligands using a matrix‐based algorithm, EpiMatrix. Vaccine 1998; 16:1880–4. [DOI] [PubMed] [Google Scholar]
- 3. Rombach‐Riegraf V, Karle AC, Wolf B et al Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T‐cell responses in vitro . PLOS ONE 2014; 9:e86322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koren E, De Groot AS, Jawa V et al Clinical validation of the ‘in silico’ prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol 2007; 124:26–32. [DOI] [PubMed] [Google Scholar]
- 5. Vugmeyster Y, Guay H, Szklut P et al In vitro potency, pharmacokinetic profiles, and pharmacological activity of optimized anti‐IL‐21R antibodies in a mouse model of lupus. MAbs 2010; 2:335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hua F, Comer GM, Stockert L et al Anti‐IL21 receptor monoclonal antibody (ATR‐107): safety, pharmacokinetics, and pharmacodynamic evaluation in healthy volunteers: a phase I, first‐in‐human study. J Clin Pharmacol 2014; 54:14–22. [DOI] [PubMed] [Google Scholar]
- 7. Rosenberg AS, Worobec AS. A risk‐based approach to immunogenicity concerns of therapeutic protein products. Part 1. Considering consequences of the immune response to a protein. BioPharm Int 2004; 17:22–6. [Google Scholar]
- 8. Rosenberg AS, Worobec AS. A risk‐based approach to immunogenicity concerns of therapeutic protein products. Part 2. Considering host‐specific and product‐specific factors impacting immunogenicity. BioPharm Int 2004; 17:34–42. [Google Scholar]
- 9. Rosenberg AS, Worobec AS. A risk‐based approach to immunogenicity concerns of therapeutic protein products, Part 3. Effects of manufacturing changes in immunogenicity and the utility of animal immunogenicity studies. BioPharm Int 2005; 18:32–6. [Google Scholar]
- 10. Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs 2010; 2:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vallieres F, Girard D. IL‐21 enhances phagocytosis in mononuclear phagocyte cells: identification of spleen tyrosine kinase as a novel molecular target of IL‐21. J Immunol 2013; 190:2904–12. [DOI] [PubMed] [Google Scholar]
- 12. Spolski R, Leonard WJ. Interleukin‐21: a double‐edged sword with therapeutic potential. Nat Rev Drug Discov 2014; 13:379–95. [DOI] [PubMed] [Google Scholar]
- 13. Guo Y, Hill AA, Ramsey RC et al Assessing agonistic potential of a candidate therapeutic anti‐IL21R antibody. J Transl Med 2010; 8:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spolski R, Leonard WJ. Interleukin‐21: basic biology and implications for cancer and autoimmunity. Annu Rev Immunol 2008; 26:57–79. [DOI] [PubMed] [Google Scholar]
- 15. Rankin AL, MacLeod H, Keegan S et al IL‐21 receptor is critical for the development of memory B cell responses. J Immunol 2011; 186:667–74. [DOI] [PubMed] [Google Scholar]
- 16. Leonard WJ, Zeng R, Spolski R. Interleukin 21: a cytokine/cytokine receptor system that has come of age. J Leukoc Biol 2008; 84:348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kasaian MT, Whitters MJ, Carter LL et al IL‐21 limits NK cell responses and promotes antigen‐specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity 2002; 16:559–69. [DOI] [PubMed] [Google Scholar]
- 18. Desjardins M, Mazer BD. B cell memory and primary immune deficiencies: interleukin‐21 related defects. Curr Opin Allergy Clin Immunol 2013; 13:639–45. [DOI] [PubMed] [Google Scholar]
- 19. Young DA, Hegen M, Ma HL et al Blockade of the interleukin‐21/interleukin‐21 receptor pathway ameliorates disease in animal models of rheumatoid arthritis. Arthritis Rheum 2007; 56:1152–63. [DOI] [PubMed] [Google Scholar]
- 20. Southwood S, Sidney J, Kondo A et al Several common HLA‐DR types share largely overlapping peptide binding repertoires. J Immunol 1998; 160:3363–73. [PubMed] [Google Scholar]
- 21. Nielsen M, Lundegaard C, Lund O. Prediction of MHC class II binding affinity using SMM‐align, a novel stabilization matrix alignment method. BMC Bioinformatics 2007; 8:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bui HH, Sidney J, Peters B et al Automated generation and evaluation of specific MHC binding predictive tools: ARB matrix applications. Immunogenetics 2005; 57:304–14. [DOI] [PubMed] [Google Scholar]
- 23. Sturniolo T, Bono E, Ding J et al Generation of tissue‐specific and promiscuous HLA ligand databases using DNA microarrays and virtual HLA class II matrices. Nat Biotechnol 1999; 17:555–61. [DOI] [PubMed] [Google Scholar]
- 24. Bakdash G, Sittig SP, van Dijk T, Figdor CG, de Vries IJ. The nature of activatory and tolerogenic dendritic cell‐derived signal II. Front Immunol 2013; 4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev 2008; 19:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Saint‐Vis B, Fugier‐Vivier I, Massacrier C et al The cytokine profile expressed by human dendritic cells is dependent on cell subtype and mode of activation. J Immunol 1998; 160:1666–76. [PubMed] [Google Scholar]
- 27. Nagorsen D, Marincola FM, Panelli MC. Cytokine and chemokine expression profiles of maturing dendritic cells using multiprotein platform arrays. Cytokine 2004; 25:31–5. [DOI] [PubMed] [Google Scholar]
- 28. Wesa A, Galy A. Increased production of pro‐inflammatory cytokines and enhanced T cell responses after activation of human dendritic cells with IL‐1 and CD40 ligand. BMC Immunol 2002; 3:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Coates PT, Colvin BL, Hackstein H, Thomson AW. Manipulation of dendritic cells as an approach to improved outcomes in transplantation. Exp Rev Mol Med 2002; 4:1–21. [DOI] [PubMed] [Google Scholar]
- 30. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brandt K, Bulfone‐Paus S, Foster DC, Ruckert R. Interleukin‐21 inhibits dendritic cell activation and maturation. Blood 2003; 102:4090–8. [DOI] [PubMed] [Google Scholar]
- 32. Yoshizaki A, Miyagaki T, DiLillo DJ et al Regulatory B cells control T‐cell autoimmunity through IL‐21‐dependent cognate interactions. Nature 2012; 491:264–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang X, Yang J, Chu Y et al T follicular helper cells mediate expansion of regulatory B cells via IL‐21 in lupus‐prone MRL/lpr mice. PLOS ONE 2013; 8:e62855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol 2008; 8:607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Niel G, Wubbolts R, Stoorvogel W. Endosomal sorting of MHC class II determines antigen presentation by dendritic cells. Curr Opin Cell Biol 2008; 20:437–44. [DOI] [PubMed] [Google Scholar]
- 36. Roche PA, Furuta K. The ins and outs of MHC class II‐mediated antigen processing and presentation. Nat Rev Immunol 2015; 15:203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steinman RM. Dendritic cells in vivo: a key target for a new vaccine science. Immunity 2008; 29:319–24. [DOI] [PubMed] [Google Scholar]
- 38. Chappell CP, Giltiay NV, Dresch C, Clark EA. Controlling immune responses by targeting antigens to dendritic cell subsets and B cells. Int Immunol 2014; 26:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic‐cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol 2007; 7:790–802. [DOI] [PubMed] [Google Scholar]
- 40. Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol 2004; 4:24–34. [DOI] [PubMed] [Google Scholar]