

Significance
Peptide neurotoxins that inhibit specific ion channels are valuable for research and clinical care but unknown for most targets. Here we consider KcsA, an orphan potassium channel with no known toxin. We build a phage-display library expressing natural toxins related to the sea anemone toxin ShK and 1.5 million novel combinatorial variants. Peptides that bind tightly to KcsA are isolated and two are described: Hui1 is novel and specific for KcsA, and HmK is natural and promiscuous. The 3D structure and action of Hui1 validate our strategy and reveal an unexpected basis for channel inhibition wherein an arginine side chain, too large to enter the conduction pathway, interacts with potassium ions traversing the pore from the other side of the membrane.
Keywords: Hui1 toxin, ShK toxin, HmK toxin, sea anemone, NMR
Abstract
Peptide neurotoxins are powerful tools for research, diagnosis, and treatment of disease. Limiting broader use, most receptors lack an identified toxin that binds with high affinity and specificity. This paper describes isolation of toxins for one such orphan target, KcsA, a potassium channel that has been fundamental to delineating the structural basis for ion channel function. A phage-display strategy is presented whereby ∼1.5 million novel and natural peptides are fabricated on the scaffold present in ShK, a sea anemone type I (SAK1) toxin stabilized by three disulfide bonds. We describe two toxins selected by sorting on purified KcsA, one novel (Hui1, 34 residues) and one natural (HmK, 35 residues). Hui1 is potent, blocking single KcsA channels in planar lipid bilayers half-maximally (Ki) at 1 nM. Hui1 is also specific, inhibiting KcsA-Shaker channels in Xenopus oocytes with a Ki of 0.5 nM whereas Shaker, Kv1.2, and Kv1.3 channels are blocked over 200-fold less well. HmK is potent but promiscuous, blocking KcsA-Shaker, Shaker, Kv1.2, and Kv1.3 channels with Ki of 1–4 nM. As anticipated, one Hui1 blocks the KcsA pore and two conserved toxin residues, Lys21 and Tyr22, are essential for high-affinity binding. Unexpectedly, potassium ions traversing the channel from the inside confer voltage sensitivity to the Hui1 off-rate via Arg23, indicating that Lys21 is not in the pore. The 3D structure of Hui1 reveals a SAK1 fold, rationalizes KcsA inhibition, and validates the scaffold-based approach for isolation of high-affinity toxins for orphan receptors.
Venomous animals produce neurotoxic peptides for defense and to capture prey. With potencies in the nanomolar range, the peptides act by modulating the function of target receptors. Toxins isolated from venoms have been used to identify and purify ion channels, to clarify their roles in physiology, to elucidate the structural basis for their function, and, recently, to diagnose and treat disease. Given their utility, it is frustrating that natural toxins cross-react with related receptors (or have no known target) and that most receptors lack a specific, high-affinity toxin. This state of affairs is easily understood; the small amounts of toxins isolated from natural sources makes target identification a challenge and their purpose in the wild does not favor target specificity. Here, we advance our approach to overcoming these problems, that is, creation of expression libraries of toxins allowing cloning based on target binding (1), by seeking a specific, high-affinity ligand for an orphan channel receptor.
Our strategy is to start with a known toxin and to design a phage-display library using the genetic database of its predicted homologs, in native and combinatorial fashion, so the encoded peptides share the same structural scaffold. As a proof of concept, we previously addressed a case of inadequate target discrimination by known natural toxins using a library of ∼11,200 peptides designed to share the fold in α-KTx scorpion toxins and a specific ligand for the human voltage-gated potassium channel Kv1.3 was isolated (1). Moka1, composed of domains from three scorpion species, blocks Kv1.3 with nanomolar affinity, allowing it to suppress T-cell-mediated immune responses, and is without unwanted side effects on gastrointestinal motility seen with natural toxins because it does not cross-inhibit Kv1.1 and Kv1.2. Supporting the design premise that encoded peptides are expressed, correctly folded, and accessible on the phage surface in a manner permissive of sorting based on target binding, the determined 3D structure of Moka1 revealed it to be constructed on an α-KTx scaffold. Here, we sought to extend our strategy by testing another scaffold and creation of a library sufficiently large to achieve isolation of peptides specific for a target with no known ligand.
KcsA is a prokaryotic channel with high potassium conductance and selectivity (2). The first potassium channel visualized at high resolution (3), KcsA has a single ion conduction pathway on the central axis of symmetry formed by four identical subunits, each with two transmembrane segments and a reentrant pore-forming loop (TM1-P-TM2). The 3D structure of KcsA confirmed explanations for selective ion permeation and conduction pathway gating deduced in the period before crystallization and its continued interrogation has been key to delineating the mechanistic bases for channel function (4–6). Although described 20 y ago (7), KcsA remains an orphan target so that studies with peptide toxins have required production of mutant channels with multiple mutations in the pore domain (8) or chimeras such as Kv1.3-KcsA, where the entire KcsA pore domain is replaced by the one in Kv1.3 (9).
To isolate toxins for KcsA, a peptide library was designed with ∼1,562,750 variants via combinatorial permutation of sequences related to the sea anemone type I (SAK1) toxin ShK. Phage sorting was performed on purified, wild-type KcsA channels. Peptides expressed on the enriched phage were synthesized and studied by surface plasmon resonance (SPR) to characterize their binding to purified KcsA and by voltage-clamp electrophysiology to assess channel blockade. Hui1, a novel and specific inhibitor of KcsA, HmK, a natural and promiscuous blocker, and Hui1 mutants were evaluated to identify toxin segments and residues responsible for specificity and affinity and to discern the mechanism of channel inhibition. The 3D structure of Hui1 determined by NMR, the 1:1 stoichiometry of KcsA inhibition via a pore-directed mechanism, and the role of two, canonical “dyad” residues (Lys21 and Tyr22) in high-affinity binding all met expectations for a SAK1-type toxin. In contrast, the influence of permeant trans ions (those traversing the channel after entering from the opposite side of the membrane) on dissociation of Hui1 from its external binding site indicated that Arg23, a residue with a side chain too bulky to fit snugly into the potassium conduction pore (10), was responsible for the voltage dependence of block rather than Lys21. This unexpected role for Hui1-Arg23 could reflect a new SAK1 binding orientation for the novel toxin; however, some models have located the ShK dyad Lys in the outer pore vestibule of Kv1.3 (11) rather than in the narrow portion of the conduction pathway (12). We posit that Hui1 binds and blocks like some, and perhaps most, natural SAK1 toxins.
Results
A SAK1 Scaffold Library with Over 1.5 Million Variants.
ShK, a SAK1 toxin from Stichodactyla helianthus, was chosen as the lead for design of the new phage library based on its ability to block many potassium channels and its physical stability. ShK has 35 residues including six Cys residues that form three disulfide bonds with a CysI–CysVI, CysII–CysIV, CysIII–CysV pattern (Fig. 1A) to yield the distinct SAK1 scaffold (13). Although different in tertiary structure, ShK blocks voltage-gated potassium channels similarly to scorpion α-KTx toxins, via binding and occlusion of the ion conduction pore (14). Of note, ShK-like domains in other animal proteins also modulate potassium channel function (15).
Fig. 1.

Library design and phage sorting. KcsA channels select phage-expressing Hui1 and HmK by binding. Phage preparation, library construction and sorting, and synthesis of KcsA and toxins are described in Materials and Methods. Single-letter codes for amino acids are standard. (A) SAK1 sequences (150) were aligned on conserved Cys residues (indicated by the gray highlighting) to define three domains that correspond to ShK residues Arg1-Gln16, Lys18-Phe27, and Arg29-Cys35, respectively. This yields 125, 94, and 133 unique A, B, and C domains, respectively (Table S1), which combine to produce ∼1.5 million ABC peptides. After five rounds of selection on KcsA, phage enrichment was observed compared with the NeutrAvidin (nAv) control (Fig. S1). (B) Hui1, a novel toxin isolated from the library, is composed of residues present in two parental toxins, AETX-K (blue) and HmK (green). Under each of the three domains in Hui1 is indicated its frequency of representation (in percent) among the 10 peptides enriched by phage selection on KcsA (Table S2).
The sequence of ShK was used as bait to search venom databases and 150 sequences with the same predicted arrangement of disulfide bonds were chosen (Table S1). To favor preservation of the scaffold architecture, the sequences were aligned on the conserved Cys residues and CysIII and CysIV were selected as boundaries for three combinatorial domains (Fig. 1A). The 150 sequences contain 125, 94, and 133 unique A, B, and C domains, respectively. Oligonucleotides were designed for unidirectional ligation to restrict permutations to genes encoding peptides with segments in the order A–B–C, yielding a calculated diversity of 1,562,600 de novo peptides as well as the 150 parental toxins. The peptides, ranging in length from 29 to 47 residues, were produced upstream and in-frame with the gene for phage coat protein pIII, as before (1).
Table S1.
Sequences of A, B, and C domains in the SAK1 scaffold library
| No. | Domain A | Link 1 | Domain B | Link 2 | Domain C |
| 1 | ACADSDATQCPLWAKAGE | C | AEDNMKYV | C | AETCGFC |
| 2 | ACADSSPSCDLFKNQGY | C | AKNPVWMKAN | C | AFTCRSC |
| 3 | ACEDIWDGCSHFLAP | C | ALSPTKMSKK | C | AKSCAFC |
| 4 | ACKDKSRYCRSWARSRY | C | ASPSHQQYLTAF | C | AKSCNSC |
| 5 | ACKDKVEHCATWSQNGQ | C | ASRPSYMMEK | C | AKSCRSC |
| 6 | ACKDNFAAATCKHVKENKN | C | DKPFYKSFMKLM | C | AKTCELC |
| 7 | ACKDYLPKSECTQFR | C | DKRREFMSVN | C | AKTCGFC |
| 8 | ACQNMYSFCGVLGKW | C | DNDWMKEN | C | AKTCGIC |
| 9 | ACSDRAHGHICESFKSF | C | DSKTREYMIDK | C | AKTCGKC |
| 10 | ACVDLKPECEAWAAQGE | C | DTPIMKRY | C | AKTCGRC |
| 11 | ACYDRYPHTCKQFDDY | C | DVRPTYMYKH | C | AKTCGYC |
| 12 | CIDKAQGCDATSSL | C | EDNKNFMLVY | C | AKTCKNC |
| 13 | DCFDKHHSCSDLLKY | C | EHRSMRDL | C | AKTCNLC |
| 14 | DCFNTQPELCDAWGRKGF | C | EIRKDFMPTE | C | ALSCGYC |
| 15 | DCHDNFAVCPTLGGMGY | C | ESASYQSIMAKK | C | ALTCGFC |
| 16 | DCIDLHERCPVWAGLGE | C | ESKYKAIMEKR | C | ARSCGSC |
| 17 | DCKDFSPACGTSLTSDL | C | ESNSYGIRTFLQTK | C | ARTCGLC |
| 18 | DCKDKGKDCESLADEGN | C | FNNPDKARMA | C | ASTCGFC |
| 19 | DCKDQMLSCPERALTGE | C | FNNPDKARNE | C | AVTCQLC |
| 20 | DCKDVADDCKAFGKLEKDDENS | C | FNYPDLMKIQ | C | AYTCDTC |
| 21 | DCRDEANNCAASINL | C | GEKFFDWMRKN | C | EATCGFC |
| 22 | DCRDRTNLCWRWIDR | C | GSQKYATN | C | EKTCGYC |
| 23 | DCSDNLSYCRYRRNL | C | HSESFFHDFMMSY | C | FNSCGRC |
| 24 | DCSDSHQHCSYWKSQGY | C | HTKAVYMLKS | C | GAACGLC |
| 25 | DCTDSQQMCLVWAKNGE | C | IKPGRNGRFIRYH | C | GKSCGFC |
| 26 | DCVDSFGQKMCEWLAQNNGY | C | IQTYVKSN | C | GNTCGLC |
| 27 | DCVDVDQQCSFWANSGF | C | IYDKDMMWKD | C | KASCKMC |
| 28 | ECAQPDIYGCVQWEQQGL | C | KAAHMAKWLKTN | C | KKACSVC |
| 29 | ECEDLIDSSICENKKSD | C | KAKPLPILSQ | C | KKKCKLC |
| 30 | ECIDSNAAICRILKSS | C | KAKPSPILSQ | C | KKSCARC |
| 31 | ECKDQYASADCKKAIGDDKWF | C | KANPNWMLGN | C | KKSCGTC |
| 32 | ECKDRYVNPQVCKSYAERGW | C | KDSGRNGVKLRAN | C | KKSCKQC |
| 33 | ECSDTNDECQLDMSP | C | KHSMKYRLSF | C | KKSCNLC |
| 34 | ECTDARGAACKDWADNRND | C | KRTPGYMLKY | C | KKSCNTC |
| 35 | ECVDKEAKCKSVLGTRADEM | C | KSHYDYMSKN | C | KKSCRVC |
| 36 | GCGDNQWYCNEWETKGL | C | LKKLNYAVGN | C | KKSCTDC |
| 37 | GCIDKAQGCDATSSL | C | LKNPVYMSSQ | C | KKTCGHC |
| 38 | GCKDNFSANTCKHVKANNN | C | LKYNSIMRES | C | KKTCGLC |
| 39 | GCQHVDKVPECQYYKSQGY | C | LNPTYQPVLRTR | C | KKTCGTC |
| 40 | GCVDVFPKCAEWKKNSNM | C | LRFPQFMSTE | C | KLSCELC |
| 41 | ICEDEIDCTGWERY | C | NADEWVQLN | C | KLYCNFC |
| 42 | ICNDKNTRCDEWAKKGE | C | NDSWLQIY | C | KRSCDFC |
| 43 | ICRDKLYSCKLWAKQGF | C | NEDWLKIN | C | KRSCGIC |
| 44 | KCADAHHDCRLIAVK | C | NFKKSFMEKN | C | KRSCGLC |
| 45 | KCFDQMTFCKKWARKGY | C | NHSKHKPYMVKN | C | KRSCKLC |
| 46 | KCKDHHPKCVKWAAKGE | C | NKNPKWMLEH | C | KRSCRIC |
| 47 | KCKDLAGERHCNGWKVKGD | C | NKNPNWMRPN | C | KSRCSFC |
| 48 | KCKDRSSFCRLTKHL | C | NQGSGYESYMETQ | C | MKMCGLC |
| 49 | KCLDDDPVYCKKYSKKY | C | NSGSWGIRSFMQKR | C | NKSCGLC |
| 50 | LCADKSKSEVCTTMAKF | C | NSNQYVKNN | C | PAMCGIC |
| 51 | LCEDKYPKCSSWASTGE | C | PKNAAWMKAN | C | PATCGYC |
| 52 | LCIDIYVSWCSDNEKILD | C | QDKHKLM | C | PFSCRFC |
| 53 | LCRDKFSKCGVMASSGL | C | QENWQEMVVQ | C | PHNCEVC |
| 54 | LCSDKSKPDICVNLKTL | C | QERDVAFT | C | PHSCRKC |
| 55 | LCYDRYVSSCEKLMNENEKYYKQI | C | QFHRRYMESV | C | PIKCGLC |
| 56 | MCVDIEVDQCNPDV | C | QKNKDWMSAN | C | PKKCGLC |
| 57 | NCGNQLPDCATRGNF | C | QKNPRYMLRN | C | PKKCKYC |
| 58 | NCHDSANNCGSLISY | C | QKNPTLMQKQ | C | PKMCNLC |
| 59 | NCPNNHSRCQEWADRGE | C | QRNTKWMFHY | C | PKMCNYC |
| 60 | NCQDKNVNMACWTVTQAQ | C | QSIPSYMKHY | C | PKSCGIC |
| 61 | NCQDTANDCANYVSV | C | QTAVEAMKLD | C | PKSCKIC |
| 62 | NCVDKESKEYCDNLVKGYA | C | RADYEKYLEE | C | PKSCRMC |
| 63 | NCVDKHPSCGYWRRTGE | C | RKRKYIMKKF | C | PKSCRTC |
| 64 | NCYDISKRCSKVKPL | C | RLYPTYMNKY | C | PKTCGAC |
| 65 | PCDNKDPDEECDHWAKLGK | C | RRNPAYMLAN | C | PKTCGKC |
| 66 | PCDTDPRCPFWGQYGY | C | RTNPGYMHKV | C | PKTCGRC |
| 67 | PCEDKHKSCVYWASIGV | C | RTNPKWMAKY | C | PKTCGVC |
| 68 | PCEDSHSNCAAWAKANE | C | RTSMKYKYTN | C | PKTCGYC |
| 69 | PCIDKDSRCPQWAQSGE | C | RTSMKYRLNL | C | PKTCKFC |
| 70 | PCINIHPKCIEWQKAGE | C | RTSQKYRAN | C | PKTCKIC |
| 71 | PCKDAITDCADYGRQA | C | RYGQRYYPYMKRY | C | PKTCNVC |
| 72 | PCKDEYSSCYFYANWYDY | C | SANFQWMHQN | C | PKTCQIC |
| 73 | PCQDTLENCSVYAAQSGY | C | SDSWTQQN | C | PLSCGFC |
| 74 | PCRDIYPHSCSQFFASYGGIK | C | SLGNLYLRTN | C | PLTCGFC |
| 75 | PCSDENQYCEKED | C | SNNNAWMSNN | C | PLTCGLC |
| 76 | PCTDQYPMCTAFAKNNY | C | SNNWVKGN | C | PLTCGYC |
| 77 | PCVDLNPNSCRQFKDVSGG | C | SNPKLAPFLRFG | C | PQTCGLC |
| 78 | PCVDQRLDCFTLLQNGH | C | SRNPRYMKVY | C | PQTCNVC |
| 79 | QCADRDPQGRCSQWRQF | C | STATYIDNH | C | PRSCDYC |
| 80 | QCEDRITVCWWTADR | C | SWHQYVREN | C | PRSCQVC |
| 81 | QCLDKGENCTGDPEQ | C | TAPSHQQYLTAF | C | PRTCGLC |
| 82 | QCVDRDPQGRCSQWRQF | C | TIDKDRMAKI | C | PRTCKFC |
| 83 | RCIDHDSLCPSWAQGGA | C | TMPIASEI | C | PRTCSLC |
| 84 | RSCIDTIPKSRCTAFQ | C | TNATYQAFMETK | C | PSTCNRC |
| 85 | RTCKDLIPVSECTDIR | C | TNAYFVNSLKIY | C | PVSCGIC |
| 86 | SCADRSNTNGINECPGWRSY | C | TNPEYSVHMNAY | C | PVSCKLC |
| 87 | SCEDINDREAEYCRQWEQAGY | C | TQQPPLAAEFS | C | PVTCGTC |
| 88 | SCEDSTFKGSSEFQCQKWAKAGE | C | TRNEYKTIMMRQ | C | PWTCRFC |
| 89 | SCKDSRSLCAQWARAGY | C | TRNVKFMLDK | C | PYNCDVC |
| 90 | SCMDKSGQCKSWAENGH | C | TSQLPLIAEFS | C | PYNCFVC |
| 91 | SCQDKINKCSQWFKQY | C | TSYKNYMMKN | C | PYSCGFC |
| 92 | SCQDSANDCANFVSV | C | VRLPDYMMQN | C | PYTCGFC |
| 93 | SCRDNDKRCPSWASS | C | VSEEKTMKLY | C | QAACEIC |
| 94 | SCRDQYRTCPGWGASGY | C | YTNPDWATEN | C | QKECGLC |
| 95 | SCRDYHPLCVEWAMKGR | C | C | QKSCDLC | |
| 96 | SCTDKYNTCEFSKPL | C | C | QKSCGFC | |
| 97 | SCTDTAGTDQCNYYKRF | C | C | QKSCNAC | |
| 98 | SCVDTSRSCSLTVSL | C | C | QKSCRTC | |
| 99 | TCADKTNAQGVNECPGLKSY | C | C | QKTCGLC | |
| 100 | TCENRPSRCDEWATRGE | C | C | QVSCELC | |
| 101 | TCFDHDECTQYSQVQ | C | C | RHSCGKC | |
| 102 | TCIDVYSGCRNFVSF | C | C | RISCNQC | |
| 103 | TCKDRFPKSQCSTYSTNGM | C | C | RKSCDFC | |
| 104 | TCQDRFPKSQCSTYSSNGL | C | C | RKSCGFC | |
| 105 | TCTDISPDCRAREAF | C | C | RKSCGHC | |
| 106 | VCADRNMYCPQYKQWGR | C | C | RKSCGTC | |
| 107 | VCDDAYNQCDSTI | C | C | RKSCKQC | |
| 108 | VCEDHPYCYKYSGID | C | C | RKSCKYC | |
| 109 | VCEDKLGFCPNYAPQ | C | C | RKTCGFC | |
| 110 | VCEDKLGLCPNYVRQ | C | C | RKTCGSC | |
| 111 | VCEDKYKDRCPGWASSGE | C | C | RKTCGTC | |
| 112 | VCEDSNRYCTKWSRAGY | C | C | RKTCMLC | |
| 113 | VCEDTDANCSEFVRQGF | C | C | RKTCNFC | |
| 114 | VCEDTSADCNRYSIEN | C | C | RKTCSHC | |
| 115 | VCFDENTYCHQWASAGH | C | C | RKTCSLC | |
| 116 | VCKDRTSWCRKWASYGY | C | C | RLACRVC | |
| 117 | VCKNNDREEKFGLCKIWKDLGF | C | C | RMSCGYC | |
| 118 | VCRDWFKETACRHAKSLGN | C | C | RQSCGLC | |
| 119 | VCSSADLSDCNDWAASGY | C | C | RQSCGQC | |
| 120 | VCVDSSTDCEGKRYL | C | C | RRSCGFC | |
| 121 | WCKDNERCYAYSADQ | C | C | RRTCGFC | |
| 122 | YCADKSSSCNLFSRQEKY | C | C | RSSCGKC | |
| 123 | YCDDKFPQCGVLHSA | C | C | RTSCKAC | |
| 124 | YCKDYDDSCKAFGKIAKKDKKS | C | C | SKSCGFC | |
| 125 | YCQDSHRSCSSWAGMGE | C | C | SKSCKQC | |
| 126 | C | SKSCLVC | |||
| 127 | C | SKTCGFC | |||
| 128 | C | SLSCGYC | |||
| 129 | C | SYTCNFC | |||
| 130 | C | TKSCKLC | |||
| 131 | C | TYSCGLC | |||
| 132 | C | WKSCSGC | |||
| 133 | C | WRSCSGC |
The public databases Pfam (pfam.xfam.org/) and UniProt (www.uniprot.org/) were evaluated for sequences homologous to ShK and bearing the same predicted pattern of Cys–Cys bonds, as described in Materials and Methods. This table lists the unique domains in the 150 parent sequences. Domains A (125), B (94), and C (133) segments were joined at the third and fourth Cys residues. Annotations: In bold are segments from ShK. In blue are segments from AETX-K. In green are segments from HmK. In red, yellow, and brown are C domains isolated after fifth phage panning on KcsA in Hui4-9. The segments in Hui1 are underlined.
Sorting Yields Novel and Natural Toxins That Bind to Purified KcsA Channels.
Although KcsA was an orphan receptor without known ligands, we judged it feasible to isolate peptides that bound with high affinity from the SAK1 library because mutation of KcsA had previously allowed pore binding of α-KTx scorpion toxins, demonstrating its conserved structural relationship with eukaryotic K+ channels (8, 9). KcsA channels were synthesized as before (16) and used as the target for library screening after adherence in plastic wells. As before (1), toxin variants were encoded in-frame with phage particle coat protein pIII, thereby exposing the peptide on the phage surface. During each round of panning, phages were incubated in wells precoated with KcsA and those that associated poorly were separated from those that bound more avidly by washing. Stably bound phages were eluted from the wells with triethylamine, amplified, and subjected to another round of selective panning. After five cycles, DNA sequencing of 170 phagemids revealed that nine novel peptides and one native toxin (HmK) had been enriched on KcsA from the library of 1.5 million predicted variants (Table S2). Among the 10 peptides, two A domains, one B domain, and six C domains were represented. The A domains were from AETX-K and HmK. The B domain was from HmK and carried the conserved dyad residues (Lys and Tyr/Phe) that have proven important for high-affinity binding of the α-KTx (17) and ShK (18, 19) toxin families in potassium channel pores (Fig. 1B). Compared with their starting abundance in the library, the 10 peptides were enriched from 30,000-fold to 180,000-fold (Table 1 and Table S2). The novel peptide Hui1 and the native toxin HmK were enriched 140,000- and 90,000-fold, respectively, and represented 9% and 6% of the enriched phage. Hui1 phages were enriched ∼8- to 15-fold per cycle whereas total phage recovery increased ∼12-fold in wells with KcsA compared with a control protein over the five cycles (Fig. S1). Of note, the native toxin AETX-K that supplied the A and C domains in Hui1 to the library was present after four rounds at ∼1% and lost by the fifth selection cycle.
Table S2.
Binding of novel and parental toxins to KcsA assessed by SPR
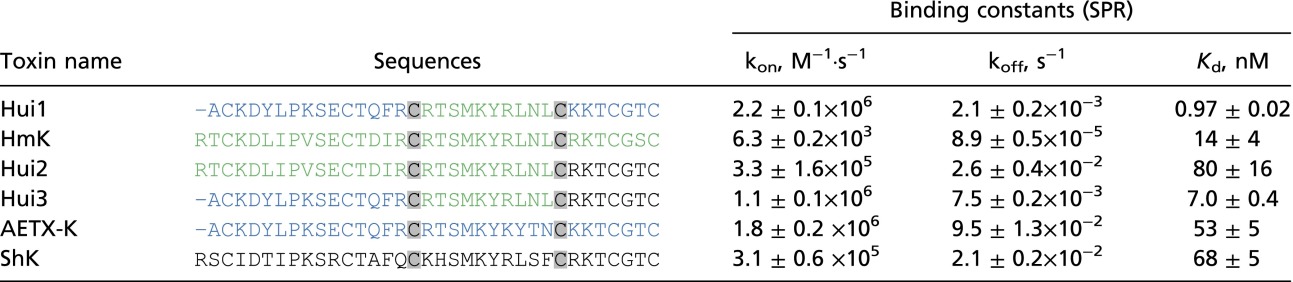 |
Six peptide toxins were synthesized and their interaction with purified KcsA channels studied by SPR as described in Materials and Methods. Each parameter is average ± SD for two or more measurements. In addition to Hui1-3 and HmK, five other novel toxins were isolated on KcsA in the fifth panning round (name and text colors as in Table S1, ACC#, % of isolates):
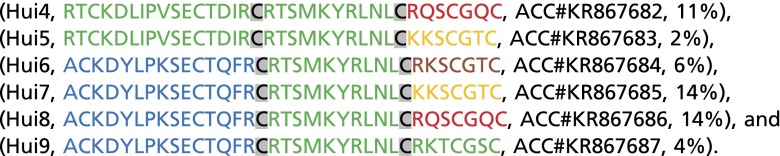
Table 1.
Novel and parental toxin blockade of KcsA and other K+ channels
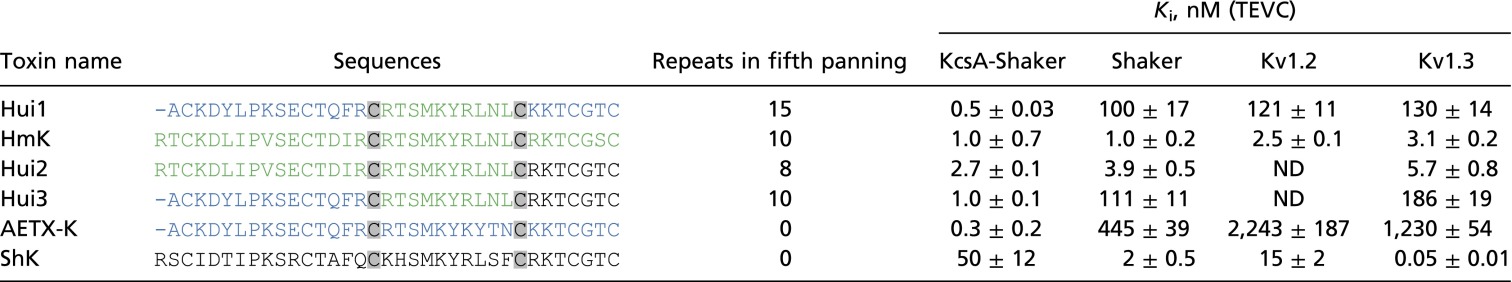 |
Six peptide toxins were synthesized and their inhibition of the indicated channels at equilibrium (Ki ± SEM) were determined by two-electrode voltage clamp (TEVC) as described in Fig. 3 with kinetic parameters assessed at 0 mV; ND, not determined; n = 5–24 oocytes. The on-rate and off-rate with KcsA-Shaker for Hui1 were kon = 1.1 × 106 ± 0.2 × 106/Ms and koff = 7.6 × 10−4 ± 0.4 × 10−4/s, whereas for ShK they were kon = 1.6 × 106 ± 0.2 × 106/Ms and koff = 5.8 × 10−2 ± 0.8 × 10−2/s; for HmK they were kon =1.3 × 106 ± 0.1 × 106/Ms and koff = 1.4 × 10−3 ± 0.3 × 10−3/s, and for AETX-K they were kon = 2.0 × 106 ± 0.4 × 106/Ms and koff = 4.7 × 10−4 ± 0.6 × 10−4/s. The kinetic parameters with Shaker for Hui1 were kon = 5.0 × 105 ± 0.3 × 105/Ms and koff = 3.0 ×10−2 ± 0.3 × 10−2/s, and for ShK they were kon = 1.3 × 107 ± 0.2 × 107/Ms and koff = 1.4 ×10−2 ± 0.3 × 10−2/s. The values with Kv1.3 for Hui1 were kon = 7.0 × 105 ± 1.2 × 105/Ms and koff = 4.8 × 10−2 ± 0.9 × 10−2/s, and for ShK they were kon = 2.7 × 107 ± 0.4 × 107/Ms and koff = 1.3 × 10−3 ± 0.1 × 10−3/s, respectively. Repeats are independent isolations of the same sequence in the fifth round of sorting on KcsA (170 phages studied). In total, phages enriched on KcsA in the fifth round included nine novel toxins (Hui1-9) and HmK (green text), a native toxin (Table S2); ShK was not isolated in any panning round, whereas AETX-K (blue text) was lost after the fourth sorting. Removing the N-terminal residue from ShK yields a toxin (ShK-ΔArg1) that inhibits KcsA-Shaker slightly more effectively (34 ± 6.0 nM).
Fig. S1.

Phage enrichment after five rounds of panning on KcsA. Phages eluted after the fifth round of panning were serially diluted and used to infect E. coli XL1-Blue for titering. Phage enrichment was observed with KcsA as the target compared with the control target protein, Neutravidin (nAv).
Three of the novel peptides expressed on the enriched phage (Hui1, Hui2, and Hui3) and three parental toxins that donated segments (AETX-K, HmK, and ShK) were synthesized and their interaction with purified KcsA channels studied using SPR spectroscopy. The six peptides showed equilibrium binding affinities that varied from 1 to 80 nM, whereas Moka1, the α-KTx blocker specific for Kv1.3 channels, showed no interaction with KcsA (Table S2). Hui1 had the highest affinity, ∼1 nM. The A and C segments of Hui1 (Fig. 1B) are derived from AETX-K, an Anemonia erythraea toxin that inhibits the binding of 125I-α-dendrotoxin to rat synaptosomal membrane (20) and the B segment from HmK is a Heteractis magnifica toxin known to inhibit Kv1.2 (21). In SPR studies, AETX-K and HmK bound to KcsA ∼50-fold and ∼14-fold less well than Hui1, respectively (Table S2).
Hui1 Blocks Single KcsA Channels.
KcsA channels were reconstituted into planar lipid bilayers and single-channel recording was used to assess the effects of Hui1. To overcome confounding inactivation, a point mutant (E71A) was introduced into KcsA that yields high open probability (Po) while retaining other native channel characteristics (16). Channels were reconstituted into liposomes using Escherichia coli lipids, fused into the bilayer, and studied under activating conditions (pHin 4.0, pHext 7.4 with symmetrical 100 mM KCl) by described methods (22). Without toxin, single KcsA-E71A channels had a Po of ∼0.98 at 100 mV (Fig. 2A). Adding 0.25 nM Hui1 to the external chamber induced long, nonconducting events. As the level of Hui1 increased, the channel Po decreased. Fitting the Po/toxin concentration relationship estimated the Ki of Hui1 for KcsA-E71A channels to be 0.93 ± 0.09 nM with a Hill coefficient of 0.99 ± 0.03, consistent with the expected 1:1 blocking mechanism of toxin via binding to the external face of the channel (Fig. 2B). In contrast, 50 nM Hui1 added into the internal chamber had no effect on the open probability of KcsA-E71A (0.98 ± 0.01, n = 3).
Fig. 2.

Block of single KcsA channels by Hui1. Recordings of single KcsA-E71A channels at 100 mV with 100 mM symmetric KCl (external pH 7.4, internal pH 4.0) with Hui1 added at the levels indicated to the external chamber. (A) Representative recordings with no toxin and three concentrations of Hui1 are shown. The Po of KcsA-E71A decreased from 0.98 ± 0.01 to 0.15 ± 0.02 as Hui1 concentration increased from 0 to 5 nM. (B) Po as a function of added Hui1 (n = 3 for each concentration) fit to the Hill relationship, Po = (Po, max)/(1 + Ki/[Tx])h, where Po is the open probability, Po, max is the maximally obtained value for Po, Ki is the inhibition constant, [Tx] is the concentration of Hui1, and h is the Hill coefficient. The Ki of Hui1 for KcsA-E71A channels was estimated from the fit to be 0.93 ± 0.09 nM with h = 0.99 ± 0.03. Values are mean ± SEM. Some error bars are smaller than symbols.
Hui1 Blocks KcsA Specifically.
To facilitate investigation of the effects of voltage and ionic conditions on parameters of Hui1 blockade, we transplanted the KcsA pore into a Shaker voltage-gated K+ channel modified by N-terminal deletion to be noninactivating (KcsA-Shaker). As described by others (23), each subunit in this chimera carries the four transmembrane spans that form the Shaker channel voltage-sensing domain (S1–S4) but the native S5-P-S6 pore-forming segments are replaced by KcsA residues; fortunately, four of the hybrid subunits assemble in Xenopus oocytes to create functional channels, allowing ready assessment of currents using two-electrode voltage clamp. Hui1 was found to block KcsA-Shaker potently and reversibly (Fig. 3A and Table 1); fitting the dose–response relationship yielded a Ki for Hui1 of ∼0.5 nM with a Hill coefficient of 0.93 ± 0.05 (Fig. 3C), similar to the parameters determined on bona fide KcsA channels in bilayers. Hui1 was also found to be specific for KcsA-Shaker, showing 200-fold lower potency on Shaker and two other voltage-gated K+ channels, Kv1.2 and Kv1.3 (Table 1). Demonstrating that KcsA-Shaker in oocytes does not faithfully recapitulate the target used for phage sorting, the parent toxin AETX-K that bound 50-fold less well than Hui1 to pure KcsA (Table S2) blocked the chimera as potently as Hui1 and with greater specificity (Table 1).
Fig. 3.

Hui1 is a potent, specific, and voltage-dependent blocker of KcsA. KcsA-Shaker and other ion channels as indicated were expressed in oocytes and studied by two-electrode voltage clamp to assess equilibrium inhibition and kinetic blocking parameters using a holding voltage of −80 mV, 300-ms test pulses, and a 10-s interpulse interval (Tables 1 and 2), n = 6–12 cells for each condition. Values are mean ± SEM. Some error bars are smaller than symbols. (A) Representative current traces for KcsA-Shaker channels at steady state before (control), in the presence of 2 nM Hui1 (Hui1), and after toxin washout (wash) with steps of 10 mV from −80 mV to 80 mV. (B) The time course for block and unblock of KcsA-Shaker on acute application (bar) and washout of 2 nM Hui1. Peak currents recorded at 0 mV; every third point is shown. (C) Dose–response relationships for Hui1 inhibition of KcsA-Shaker (●) and Shaker (▲) studied as in B and fit to the Hill relationship, Fun = (1 + ([Tx]/Ki)h)−1, where Fun is the fraction of unblocked current at equilibrium, Ki is the dissociation constant, h is the Hill coefficient, and [Tx] is the concentration of Hui1. The Ki of Hui1 for KcsA-Shaker channels was estimated from the fit to be 0.50 ± 0.03 nM with h = 0.93 ± 0.05. (D) Effect of Hui1 concentration on blocking kinetics of KcsA-Shaker. The apparent first-order rate constants for association (♦, On, kon[Tx]) and dissociation (△, Off, koff) are plotted as a function of Hui1 concentration using the protocol in B. (E) Effect of voltage on Hui1 blocking kinetics of KcsA-Shaker channels. Each parameter was measured with test steps from −20 mV to 40 mV and normalized to its value at 0 mV; ♦, kon; Δ, koff; ●, Ki. On-rate and off-rate time constants were determined by single-exponential fits to the time course for block or unblock on acute application or washout of 5 nM Hui1. The inhibition constant Ki was calculated from the fraction of unblocked current at equilibrium and the rate constants (Materials and Methods). (F) Effect of voltage on blockade of KcsA-Shaker by Hui1 mutants. Ki for each toxin was determined from −20 mV to 40 mV based on the fraction of unblocked current at equilibrium (Fun) and plotted as a ratio to the value at 0 mV. The change in Ki with voltage is due to altered off-rate.
The A Domain Mediates Specific Block of KcsA.
Suggesting a role for the A domain in specific block of KcsA by Hui1, Hui3 carries the A domain in Hui1 (provided by AETX-K) and was similarly specific, whereas Hui2 has the A domain from HmK and was similarly promiscuous (Table 1). Further, ShK blocked KcsA-Shaker 100-fold less well than Hui1, and this was rationalized by four nonconservative differences between the two toxins in their A domains. Changing the sites in Hui1 individually to the residue in ShK produced mutant peptides that each inhibited KcsA-Shaker less effectively; thus, loss of potency for Hui1-Lys3Ile, Hui1-Glu10Arg, and Hui1-Arg15Gln explained ∼75% of the difference in inhibition between the two toxins (Table 2). One of these sites has been implicated in tight binding of ShK to Kv1.3 (24) and carries an opposite charge in Hui1 (ShK-Arg11 versus Hui1-Glu10). The fourth major difference between the two toxins in the A domain, an additional N-terminal residue in ShK, justified the remaining loss in affinity. Thus, eliminating Arg1 from ShK produced a somewhat more potent inhibitor (Table 1) whereas adding an Arg0 to Hui1 decreased blockade slightly (Table 2).
Table 2.
Inhibition of KcsA-Shaker by Hui1 variants
| Toxin | Ki, nM | Variant/Hui |
| Hui1 | 0.50 ± 0.03 | =1 |
| Hui1-Arg0 | 0.7 ± 0.1 | 1.4 |
| Hui1-Lys3Ile | 0.9 ± 0.1 | 1.8 |
| Hui1-Glu10Arg | 4.1 ± 0.4 | 8.2 |
| Hui1-Arg15Gln | 2.6 ± 0.5 | 5.2 |
| Hui1-Ser19Ala | 46 ± 3.5 | 92 |
| Hui1-Lys21Asn | 20 ± 2.0 | 40 |
| Hui1-Tyr22Ala | 4,260 ± 860 | 8,520 |
| Hui1-Arg23Ala | 0.25 ± 0.05 | 0.5 |
| Hui1-Arg23Lys | 0.45 ± 0.03 | 0.9 |
| Hui1-Lys28Ala | 0.40 ± 0.14 | 0.8 |
Equilibrium inhibition constants (Ki ± SEM) were determined by two-electrode voltage clamp as described in Fig. 3 (n = 5–12 oocytes). To test the influence of nonconservative differences between Hui1 and ShK on blockade, Hui1 toxins were produced with an added Arg0 or the changes Lys3Ile, Glu10Arg, or Arg15Gln. Hui1 residues Ser19, Lys21, Tyr22, and Arg23 were studied based on the importance of analogous residues in other SAK1 toxins.
The B domain of Hui1 carries the dyad residues that are critical for high-affinity binding of known SAK1 toxins to potassium channels (Fig. 1B). As expected, these residues, Lys21 and Tyr22, were important for Hui1 inhibition of KcsA (Table 2). Thus, a neutralizing mutation to produce Hui1-Lys21Asn decreased block 40-fold and altering the adjacent aromatic residue to create Hui1-Tyr22Ala decreased affinity over 8,000-fold. In addition, the conservative mutation of Hui1-Ser19 to Ala decreased inhibition ∼90-fold, a finding consistent with the impact of the homologous residue in ShK (Ser20) on block of Kv1.3 (25). Notwithstanding its role in affinity, the B region cannot be inferred to bestow specificity to Hui1 because HmK is homologous in this region and did not discriminate among the channels, inhibiting KcsA-Shaker, Shaker, Kv1.2, and Kv1.3 all with nanomolar affinity (Table 1).
We do not implicate the C domain of Hui1 in specific block of KcsA-Shaker for four reasons. First, the domain varied most widely, although modestly, among the phage isolated on KcsA (Table S2). Second, both Hui2 and Hui3 block KcsA-Shaker with high affinity despite carrying the C domain provided by ShK, a toxin that blocks the channel poorly (Table 1). Third, a nonconservative mutation in the domain to produce Hui1-Lys28Ala retained high affinity for KcsA-Shaker (Table 2). Finally, the NMR structure of Hui1 (discussed below) shows domain C residues on the opposite side of the toxin from those that most affect target binding.
Arg23, Not Lys21, Mediates the Voltage Dependence of Hui1 Block.
Hui1 block of KcsA meets expectations for a pore-directed toxin (26–28) whereby one molecule occludes the conduction pathway. Thus, the dose–response for Hui1 inhibition showed a Hill coefficient of ∼1 in studies of both single KcsA-E71A channels in planar bilayers and KcsA-Shaker channels in oocytes (Figs. 2B and 3C). Further, the Hui1 on-rate with KcsA-Shaker was dependent on toxin concentration in a linear manner and consistent with diffusion limitation (1.1 × 106 ± 0.2 × 106/Ms), whereas toxin off-rate was insensitive to added toxin (7.6 × 10−4 ± 0.4 × 10−4/s) (Fig. 3 B and D), as expected for a simple biomolecular interaction (28).
CTX blockade is sensitive to voltage because the positively charged ε-amino group of its dyad Lys enters the external portion of the conduction pathway and interacts with potassium ions traversing the pore from the intracellular (trans) compartment (10, 17, 28, 29). This mechanism has also been proposed for the ShK (12). Unexpectedly, neutralization of Hui Lys21 had no effect on the voltage dependence of toxin off-rate. Thus, the off-rate of Hui1 from KcsA-Shaker was sensitive to voltage with an effective valence (zδ) of ∼0.3 (Fig. 3E) and Hui1-Lys21Asn showed the same zδ as the wild-type toxin (Fig. 3F). In contrast, neutralization of Arg23 to produce Hui1-Arg23Ala eliminated the voltage dependence of toxin off-rate (Fig. 3F) even though the mutation had little effect on toxin affinity (Table 2). Supporting a role for a positively charged residue at position 23, the conservative mutation Hui1-Arg23Lys decreased but did not eliminate the voltage dependence of blockade (zδ ∼0.14), again with limited effect on affinity. The impact of Arg23 but not Lys21 on the voltage dependence of Hui1 was observed also when the mutant peptides were applied to a KcsA-Shaker channel variant that had a zδ for wild-type Hui1 of ∼0.6 (Fig. S2).
Fig. S2.

Effect of voltage on blockade of KcsA-Shaker-A54D channels by Hui1 and Hui1 mutants. Ki for each toxin was determined from −20 mV to 40 mV based on the fraction of unblocked current at equilibrium (Fun) and normalized to the value at 0 mV as in Fig. 3F. The change in Ki with voltage was due to changes in toxin off-rates (τoff). Some error bars are smaller than symbols.
To confirm that the voltage dependence of Hui1 block was due to trans ion enhanced toxin dissociation, the kinetics of Hui1 inhibition of KcsA-Shaker were studied with various levels of added monovalent cations inside the oocytes (Fig. 4), a convenient method that is crude compared with ionic reconstitution without cellular constituents, but effective (30, 31). As expected, increasing the concentration of intracellular potassium from ∼90 mM to 190 mM did not modify Hui1 on-rate but did speed toxin off-rate in a manner linearly dependent on the amount of microinjected potassium. In contrast, similar increases in the level of intracellular sodium, lithium and N-methyl glucamine (NMG), three impermeant cations, had no effect on either the on-rate of Hui1 or dissociation of the toxin from its extracellular site. Consistent with interaction of Hui Arg23 and ions traversing the pore, Hui-Arg23Ala was insensitive to increased levels of either intracellular potassium or NMG, whereas Hui-Arg23Lys was sensitive but less so than wild-type Hui1.
Fig. 4.

Hui1-Arg23 is responsible for voltage-dependent block. KcsA-Shaker was expressed in oocytes and studied by two-electrode voltage clamp to assess equilibrium inhibition and kinetic blocking parameters by the protocol in Fig. 3 at 0 mV after microinjection of 40 nL of 0.25 M to 1 M of the indicated salt (Tables 1 and 2). The change in Ki with voltage was due to changes in toxin dissociation rates. The dissociation rate of Hui1 was increased by microinjected KCl, but not by NMGCl, NaCl, or LiCl. The dissociation rate of Hui-Arg23Ala was insensitive to microinjected KCl or NMGCl, whereas the dissociation rate of Hui-Arg23Lys was less sensitive to microinjected KCl than wild-type Hui1. Oocyte solute space has been estimated to be ∼400 nL and intracellular potassium concentration to be ∼90 mM; therefore, a 20 nmol KCl microinjection should raise intracellular potassium by ∼50 mM to 140 mM, leading to an 11-mV shift in EK and a 50% increase in outward current (30, 31), close to the observed values here of 10 ± 2 mV and a 45 ± 9% (n = 4–6 oocytes). The new Ki were stable during the period of recording, that is, from 30 to 120 min after microinjection. Values are mean ± SEM.
Hui1 Employs a SAK1 Scaffold.
The 3D solution structure of Hui1 was determined by well-established NMR methods (32) to assess the structural basis for toxin function. A total of 324 distance and dihedral constraints resulted in an Hui1 solution structure with an rmsd of 0.46 (1.02) Å for the polypeptide backbone (all heavy atoms) (Fig. 5A, Fig. S3, and Tables S3 and S4). Superimposition of Hui1 and ShK showed that Hui1 retained a SAK1 scaffold despite containing three segments from two SAK1 parents. Structural similarity between Hui1 and ShK was strongest in the central helix-kink-helix motif (Hui residues 13–25) spanning domains A and B, showing a backbone rmsd value of 0.76 Å between the two toxins whereas the value overall was 1.41 Å (Fig. 5B). The C domain showed the greatest structural difference between the two toxins, forming a short C-terminal helix (Hui1 residues 28–31) that had a more open conformation and different orientation for the Cys17-Cys32 bond in Hui1, but had a limited impact on function, presumably because of its distance from residues mediating binding and specificity.
Fig. 5.

Structure of Hui1 by NMR in comparison with the ShK toxin. The Hui1 structure was determined from 324 distance and dihedral constraints derived from homo- and heteronuclear 2D-NMR spectra (Materials and Methods, Fig. S3, and Table S3). (A) Hui1 structure (PDB ID code 2N6B). The 25 lowest-energy structures obtained from distance-geometry/simulated annealing determination are shown. (B) Superposition of Hui1 (orange) and crystal structure of ShK (4LFQ, residues 2–35, gray) based on alignment of the central helix-kink-helix motif (Hui1 residues 13–25) shows Hui1 retains an ShK-like scaffold. Side chains of Hui1 residues 21–23 are shown. Comparing Hui1 to an NMR structure of ShK (Fig. S4) suggests lower structural similarity between the two toxins. (C) Comparison of Hui1 and ShK side chains showing four residues contributing to the channel-binding surface. Domains A (blue), B (green), and C (blue-purple), as identified in Table 1, are indicated by translucent color rendering; domain C is on the backside and visible in D. (D) Comparison of Hui1 and ShK side chains in the A domain that contribute to altered affinity of the two toxins for KcsA-Shaker (Table 2) with surface coloring as in C. The orientation is related to C by the indicated 60° turn around the horizontal axis. (E) Electrostatic surface rendering of Hui1 colored according to interpolated charge (red/blue = negative/positive) oriented as in C using the Discovery Studio 3.0 suite (Accelrys). Side chains of the four binding-site and three nonconserved amino acid differences between Hui1 and ShK are shown. (F) Electrostatic surface rendering of ShK (residues 2–35, 4LFQ) with coloring code, orientation, and side chains homologous to Hui1 in E.
Fig. S3.

NMR determination of the Hui1 structure with method details. Homonuclear fingerprint spectra of Hui1. (Top) HN{F2}/Hali{F1} overlaid regions of the 2D TOCSY (red) and NOESY (blue) spectra of Hui1 acquired in H2O-based buffer. Line connecting spin systems and assignments are shown. (Bottom) Haro{F2}/Haro{F1} region of the TOCSY spectrum with assignments of the three aromatic spin systems.
Table S3.
1H, 13C resonance assignments for Hui1
| Residue | HN | Hα | Cα | Hβ | Cβ | Others |
| Ala1 | ND | ND | ND | ND | ||
| Cys2 | ND | 4.75 | 55 | 2.93, 3.16 | ||
| Lys3 | 8.1 | 4.52 | 54.8 | 1.61, 1.76 | 35.3 | CγH2 0.93; CδH2 1.29; CεH2 2.84 |
| Asp4 | 8.59 | 4.96 | 54.2 | 2.69, 3.17 | ||
| Tyr5 | 9.04 | 4.67 | 58.2 | 2.92, 3.42 | 39.1 | C2,6H 7.23; C3,5H 6.87 |
| Leu6 | 7.06 | 4.85 | 51.8 | 1.49, 1.76 | 43.5 | CγH 1.66; CδH3 1.02, 1.05 |
| Pro7 | — | 4.48 | 62.3 | 1.98, 2.06 | Cγ H2 2.48; CδH2 3.49, 3.82 | |
| Lys8 | 8.46 | 4.06 | 62.7 | 1.82, 1.90 | CγH2 1.52; CδH2 1.72; CεH2 3.03 | |
| Ser9 | 8.14 | 4.03 | 59.3 | 3.86, 3.94 | 62.1 | |
| Glu10 | 7.82 | 4.39 | 58.1 | 2.08, 2.33 | CγH2 2.23, 2.44 | |
| Cys11 | 7.62 | 5.12 | 52.0 | 2.87 | 37.5 | |
| Thr12 | 6.91 | 4.49 | 59.0 | 4.65 | 70.9 | CγH3 1.19 |
| Gln13 | 8.96 | 3.83 | 59.0 | 2.04, 2.12 | CγH2 2.47; NεH2 6.74, 7.63 | |
| Phe14 | 8.44 | 4.15 | 61.3 | 2.77, 3.27 | 39.2 | C2,6H 7.10; C3,5H 7.31 |
| Arg15 | 7.61 | 3.59 | 59.6 | 1.77, 1.92 | Cγ H2 1.61,1.20; CδH2 1.30, 2.63 | |
| Cys16 | 8.25 | 4.20 | 56.5 | 3.05, 3.19 | 35.9 | |
| Arg17 | 7.73 | 4.13 | 58.0 | 1.59, 1.82 | Cγ H2 1.72; CδH2 3.18 | |
| Thr18 | 7.40 | 4.31 | 61.8 | 4.02 | 71.2 | CγH3 0.79 |
| Ser19 | 8.01 | 5.06 | 56.1 | 3.91, 4.07 | 63.2 | |
| Met20 | 9.24 | 4.15 | 58.7 | 2.07, 2.17 | 32.3 | CγH2 2.60, 2.73 |
| Lys21 | 8.32 | 4.11 | 59.2 | 1.60, 1.84 | CγH2 1.12; CδH2 1.27; CεH2 2.77, 2.84 | |
| Tyr22 | 7.65 | 4.04 | 60.2 | 2.67, 3.00 | C2,6H 7.37; C3,5H 6.82 | |
| Arg23 | 8.21 | 4.07 | 60.0 | 1.78, 2.43 | Cγ H2 1.53, 1.62; CδH2 3.24, 3.36; NεH 7.32 | |
| Leu24 | 8.37 | 4.41 | 55.8 | 1.81, 1.54 | 42.7 | CγH 1.68; CδH3 0.91, 0.95 |
| Asn25 | 7.14 | 5.10 | 55.1 | 2.95 | ||
| Leu26 | 7.97 | 4.40 | 55.1 | 1.58 | 43.5 | CγH 1.46; CδH3 0.37, 0.76 |
| Cys27 | 8.58 | 5.46 | 53.6 | 3.00, 3.15 | ||
| Lys28 | 8.31 | 3.82 | 60.2 | 1.52, 1.64 | CγH2 1.24, 1.34; CδH2 1.80; CεH2 3.10 | |
| Lys29 | 7.33 | 4.15 | 58.7 | 1.73, 1.76 | CγH2 1.31; CδH2 1.62; CεH2 2.98 | |
| Thr30 | 10.56 | 3.8 | 68.4 | 4.00 | 67.8 | CγH3 1.14 |
| Cys31 | 8.82 | 4.84 | 52.7 | 2.91, 3.31 | 35.5 | |
| Gly32 | 7.83 | 4.1 | 46.7 | — | — | |
| Thr33 | 8.84 | 4.32 | 62.6 | 4.48 | 68.1 | CγH3 1.10 |
| Cys34 | 7.71 | 4.28 | 56.2 | 3.10, 3.28 |
Chemical shifts for Hui1 1H, 13C nuclei in 20 mM phosphate buffer, pH 6.6, 10 mM NaCl, and 0.02% NaN3, determined at 16.4 T and 298 K. ND, not determined.
Table S4.
Structural statistics for Hui1
| NMR distance restraints | |
| Total interresidue restraints | 258 |
| Sequential (|i − j| = 1) | 122 |
| Medium range (1 < |i − j| < 5) | 80 |
| Long range (|i − j| ≥ 5) | 56 |
| NOE violations, Å | |
| Maximum single violation | 0.5 |
| Rmsd of NOE violations | 0.057 ± 0.001 |
| Dihedral angle restraints | |
| Total dihedral restraints | 66 |
| Experimental (φ) | 8 |
| TALOS derived (φ,ψ) | 58 |
| Dihedral angle violations, ° | |
| Maximum single violation | 5 |
| Rmsd of dihedral violation | 1.00 ± 0.09 |
| Deviation from idealized geometry | |
| Bond lengths, Å | 0.004 ± 0.000 |
| Bond angles, ° | 0.600 ± 0.008 |
| Improper angles, ° | 0.520 ± 0.021 |
| Mean rmsd values, Å | |
| Backbone | 0.46 |
| Heavy atoms | 1.02 |
| Backbone (residues 14–26) | 0.12 |
| Heavy atoms (residues 14–26) | 0.76 |
Fig. S4.

Overlay of Hui1 and the ShK NMR structures. Alignment of Hui1 and an NMR-based structure of ShK (PDB 1ROO). Structural similarity of Hui1 (orange) and this NMR-based structure of ShK (gray) is lower than for the alignment with a crystal structure of ShK shown in Fig. 5B. The N-terminal Arg1 residue in ShK (indicated by arrow) is not present in Hui1.
Our results have linked the A domain with specificity and the B domain with high-affinity binding (Tables 1 and 2). Comparing the structures of Hui1 and ShK in these domains (Fig. 5C), we observed that the side chains of Hui1 residues that contribute strongly to binding (Ser19, Lys21, and Tyr22; Table 2), as well as Arg23 that is responsible for the voltage dependence of blockade (Fig. 4), cluster in the B domain and superimpose with the analogous residues of ShK. In contrast, the three nonconservative differences between Hui1 (Lys3, Glu10, and Arg15) and ShK (Ile4, Arg11, and Gln16) as well as ShK-Arg1, absent from Hui1, all lie in the A domain. Structural similarity is lower in the A domain, consistent with a role in determining selectivity, and yet comparable side-chain orientations are exhibited in the two toxins (Fig. 5D). We also evaluated the electrostatic surface potential of Hui1 and ShK, observing similar patterns for the B and C domains and notable differences in the A domain (Fig. 5 E and F). Although the two toxins differ by only a single charge overall (in keeping with their similar on-rates; Table S2), the surface of the A domain is positive in ShK and mixed in Hui1, primarily due to Hui1-Glu10. In light of the strong structural resemblance of the two proteins, electrostatic differences seem to play an important role in determining binding selectivity mediated by the A domain.
Discussion
Peptide Toxin Phage Display for Orphan Receptors.
Peptide toxins isolated from venoms have been useful to identify ion channel subtypes in tissues and subcellular locales, to reveal the roles channels play in physiology, and to delineate mechanism, for example, to correlate structure, including conformational changes, and function (27, 33, 34). Recently, peptide toxins have also gained use in medical practice both as diagnostic tools and as therapeutics (35, 36), notably to treat intractable pain through ion channel blockade (37). The study of channels without known toxins has sometimes been made possible by pore transplantation (9) or multisite mutations that allow toxin blockade (38), but neither strategy is useful to examine native channels. Although scorpions, spiders, snakes, sea anemones, and cone snail venoms all contain peptides that target ion channels (39–41), the small amounts of toxins in venom make purification and target identification a challenge.
The emergence of genomic and proteomic databases for venoms suggested to us the potential for development of a high-throughput screen to select among toxins expressed on phage for desired attributes. Thus, despite the enormous number of natural peptide toxins extrapolated from genetic studies (>12 million), a limited number of resilient scaffolds that tolerate wide residue diversity are used (42). Further supporting the utility of phage display, peptide toxins are small (∼20–100 residues) and stabilized by two to five disulfide bridges. To originally test the strategy of combining structure-based peptide design and the power of phage display, we considered the case of Kv1.3, a channel with known natural peptide toxin ligands that were inadequately specific. Construction and sorting of a library with ∼104 native and de novo peptides sharing a scorpion α-KTx scaffold allowed isolation of Moka1, a specific Kv1.3 blocker (1).
In this paper, we advance the strategy, creating a library of over 106 peptides on a scaffold from sea anemone and isolate Hui1, a de novo toxin specific for KcsA, an orphan target without previously known ligands. The NMR structure of Hui1 shows the SAK1 scaffold used to design the peptides in the library has been recapitulated. The structural basis for the affinity and specificity of Hui1 were explored through comparison of its function as a KcsA channel inhibitor, including studies of the basis for voltage-dependent blockade, with Hui1 mutants and the related toxins, Hui2, Hui3, HmK, AETX-K, and ShK.
Voltage-Sensitive Block by Pore-Directed Toxins.
Whereas Hui1 blocks the KcsA pore in a simple bimolecular manner, Arg23 on the toxin interaction surface, rather than a canonical Lys, was found to confer sensitivity to voltage. This was unexpected because CTX blocks classical potassium channels such that Lys27 enters the pore to impede passage of potassium ions (26–28). This endows CTX off-rate with dependence on membrane voltage due to electrostatic repulsion between permeating potassium ions in the conduction pathway and the basic Lys residue. Neutralization of CTX Lys27 abolishes the dependence of toxin off-rate on voltage (17, 28, 29), whereas neutralizing two Arg residues on the CTX interaction surface does not (17). This mechanism was recently visualized in a crystal structure of CTX in complex with a voltage-gated potassium channel—the long alkyl side chain and the hydrogens of ε-amino group of Lys27 were noted to be uniquely suited for close contact with the carbonyl oxygens in the first (S1) potassium ion site in the pore filter, unlike the terminal guanidinium group of Arg, and was observed to alter distribution of potassium ions in the conduction pathway (10).
Although some have proposed that ShK Lys22 binds in the Kv1.3 selectivity filter (12), this remains a matter of controversy. Molecular dynamic simulations have modeled ShK Lys22 in the external vestibule of Kv1.3 away from the narrow filter region (11), as we posit here for Hui1 Lys21 with KcsA. Suggesting a pore location, weak energetic coupling of ShK Lys22 and a Tyr in the selectivity filter of Kv1.3 has been observed using mutant cycle analysis (14); however, weak coupling is also consistent with interaction through an intervening residue (10). Consistent with localization of ShK Lys22 in the outer vestibule rather than the filter of a Ca2+-activated potassium channel, mutant cycle analysis showed coupling of potassium in the external bath solution and the toxin residue (25). Whereas neutralization of CTX Lys27 (17, 28, 29) and Hui1 Arg23 (Fig. 4) produces toxins that dissociate in a voltage-insensitive manner and no longer show “knock-off” by trans ions, we are not aware of similar studies with ShK Lys22 in support of its proximity to the Kv1.3 selectivity filter. ShK bearing a charge-reversing unnatural amino acid at position 22 was modeled with the residue in the outer vestibule of Kv1.3 (18). These disparate observations are consistent with the notion that SAK1 toxins do not locate Lys in the selectivity filter or that they bind with multiple orientations. Of note, a role for Hui1 Arg23 is consistent with inhibition of Kv1.3 by an ShK peptidomimetic bearing the three-residue B domain epitope Lys-Tyr-Arg (24).
What can we surmise about Hui1 residues Lys21-Tyr22 -Arg23 relative to the KcsA pore before structural determination of this bimolecular complex? Because neutralization of Hui1 Arg23, but not of Lys21, abolishes voltage dependence of KcsA-Shaker blockade, we infer that Arg23 is closer to the selectivity filter than Lys21. Given that the S1 potassium binding site in KcsA is similar in size and geometry to that in the voltage-gated potassium channel studied by Banerjee et al. (10), Hui1-Arg23 is too large to fit snugly into the site, suggesting the residue is in the vestibule. Supporting a less intimate association of Hui1-Arg23 and the narrow portion of the conduction pathway, the conservative mutation CTX-Lys27Arg destabilized binding to a Kv channel ∼1,000-fold (43), whereas Hui1-Arg23Ala had only a 0.5-fold effect on affinity (Table 2). Consistent with a weaker interaction with ions in permeation pathway, the voltage dependence of Hui1 block of KcsA was about half that of CTX with a Kv channel (28) and the smaller position 23 residue in Hui1-Arg23Lys yielded a toxin with decreased sensitivity to trans ions, as if the charged side-chain was even more distant from ions traversing the pore. Also consonant with a vestibule location for Hui-Arg23 at some distance from the pore, the voltage dependence of Hui dissociation was doubled by a mutation in the KcsA vestibule and neutralization of Arg23 (but not of Lys21) again ablated the effect of voltage (Fig. S2). Because the mutations Hui-Lys21Asn and Hui-Tyr22Ala decreased affinity so significantly, ∼40-fold and 8,000-fold, respectively (Table 2), we hypothesize these dyad residues interact intimately with residues in the KcsA vestibule.
Structural Context for the Interaction of Hui1 and KcsA.
The electrophysiology data establish that the Hui1 B domain is mandatory for tight binding to KcsA (explaining its uniform representation among the phage enriched from the library) whereas the more variable A domain and channel specificity are intimately related (Table 1). That four sequence differences between Hui1 and ShK alter the charge of the site (Arg0, Lys3/Ile4, Glu10/Arg11, and Arg15/Gln16) and that each contributes to the ∼100-fold difference in their affinity for KcsA (∼10, 10, 50, and 30%, respectively) makes clear that channel recognition is a function of several nondominating influences and implicates electrostatic differences in the A domain as important for toxin specificity. However, the common reflection of electrostatics is on the kinetics of complex formation, and on-rate of the two toxins for KcsA-Shaker is similar, whereas the Hui1 off-rate is ∼75-fold slower, indicative of more stable binding (Table 1). More consistent with expectations, the higher affinity of ShK for Shaker and Kv1.3 was due in large measure to enhanced on-rates that were ∼26-fold and ∼39-fold faster than Hui1. Naturally, verification of molecular explanations for channel specificity, the promiscuity of HmK, and trans ion-enhanced toxin dissociation mediated by Arg rather than Lys remain speculative until the relevant toxin–channel cocomplex structures are elucidated.
Effect of Target Milieu on Toxin Binding.
It is thought-provoking, and perhaps serendipitous, that Hui1 demonstrates such similar affinity for KcsA (∼1 nM) studying binding to purified protein in detergent by SPR (Table S2), blockade of single KcsA-E71A channels in planar lipid bilayers (Fig. 2), and inhibition of KcsA-Shaker channels expressed in oocytes (Table 1) because the three assays, ionic conditions, and target constructs vary and pore-directed peptide toxins often show differences in affinity for the same target in different environments. Thus, AgTX2, a scorpion α-KTx family toxin, inhibits the toxin-sensitive KcsA mutant KcsA3M-Shaker expressed in oocytes with subnanomolar affinity (23) but binds to purified KcsA3M three orders of magnitude less well (8). Indeed, the effects of environment on target conformation and target–toxin interaction may explain the unexpected behavior of AETX-K, the toxin that donated its A and C domains to Hui1. AETX-K binds to pure KcsA with an affinity 50-fold lower than Hui1 as judged by SPR (Fig. S1), an observation that rationalizes its presence at low levels after four rounds of phage panning but its loss in the fifth cycle. In contrast, AETX-K inhibits with high affinity and even greater specificity than Hui1 when KcsA is linked to the S1–S4 domains of Shaker and expressed in the plasma membrane of oocytes (Table 1). The higher affinity of Hui1 for pure KcsA suggests it is preferable for biophysical studies, our purpose in seeking a toxin here, whereas AETX-K may prove useful to differentiate KcsA-Shaker from mammalian channels in cell membranes. Although these contextual effects have not interfered with use of purified targets to select toxins active on channels in biological membranes, here or in the past (1), the findings highlight the important role of target milieu on phage selection and toxin activity.
Hui1 is specific for KcsA, an example of achieving target specificity by driving selection for high affinity, as seen with Moka1 (1). Conversely, HmK is promiscuous, demonstrating that sorting for high-affinity binding does not demand high specificity (Table 1). The small α-KTx library that yielded Moka1 did not show enrichment on KcsA (1); given greater library diversity, we suspect that an α-KTx–based toxin could also be designed to block KcsA. The isolation of Moka1 on an α-KTx scaffold (1), and Hui1 on SAK1 toxin scaffold, supports the notion that other scaffolds will also be amenable to sorting novel and natural peptides, allowing analogous work on other channels and different types of membrane receptors important to human health and disease.
Materials and Methods
Library Construction.
The sequence of ShK was used as template for BLAST searching of the UniProt and Pfam database (44, 45). The combinatorial library was built based on 150 SAK1 sequences (Table S1). Three domains were defined by the third and fourth of the six conserved Cys residues that form disulfide bridges, allowing for insertions and deletions (Fig. 1) and the 150 SAK1 sequences provided 125, 94, and 133 unique sequences for domains A, B, and C, respectively (Table S1). Nucleotides duplexes encoding the third and fourth Cys were ligated unidirectionally to produce toxin genes with only an ABC pattern. Complementary nucleotide pairs for each unique domain were synthesized, phosphorylated, and annealed individually. To achieve and monitor domain incorporation, 94 separate reactions were performed to ligate the ABC inserts into pAS62 in-frame with phage particle coat protein pIII, as before (1). Each reaction contained one B domain, 125 A domain duplexes, and 133 C domain duplexes with equimolar amounts of A, B, and C. Ligation mixtures were transformed into SS320 Electrocompetent Cells (Lucigen). Sequencing of 376 plaques supported unbiased insert utilization.
Library Screening.
Three wells in a NUNC-Immuno MaxiSorp 96-well plate (Thermo Fisher Scientific) were coated overnight at 4 °C with 1 μg of KcsA in 50 μL of 100 mM NaHCO3 per well, 1 mM n-dodecyl-β-d-maltopyranoside (DDM, pH 9.0). Another three wells were coated in the same buffer with NeutrAvidin (Life Technologies), a deglycosylated form of avidin, as the negative control. Wells were washed twice with TBS (25 mM Tris⋅HCl, 140 mM NaCl, 3 mM KCl, and 1 mM DDM, pH 7.4) containing 0.1% Tween 20 and 0.5% BSA (TBSTB) and then blocked with TBS containing 2% BSA (TBSB) before washing twice more with TBSTB. For each well, an equal number of library phage particles, from 108 to 1010, were added in 50 μL TBSB and incubated on a rocking shaker for 1 h. Poorly adherent phage particles were removed by washing three to five times with 200 μL TBSTB. Bound phage particles were eluted with 100 μL of 0.1 M triethylamine by incubation for 10 min on a rocking shaker. The pH of the eluate was adjusted to between 7.0 and 8.5 with 1 M Tris⋅HCl (pH 8.0) and used to infect E. coli XL1-Blue for phage amplification. Phage particles were quantified by titering before and after selective library sorting and genotyped by DNA sequencing.
KcsA and Toxin Production.
KcsA and KcsA-E71A were produced with a C-terminal hexahistidine-tag, as before (5), for phage panning, SPR, and reconstitution into planar lipid bilayers. Briefly, KcsA-E71A mutation was produced using a Quickchange Site-Directed Mutagenesis kit (Agilent Technologies) and confirmed by DNA sequencing. KcsA proteins were expressed in E. coli BL21 (DE3) cells and extracted with 100 mM KCl, 50 mM DM, and 20 mM Tris⋅HCl, pH 7.5, and then purified by cobalt-affinity and size-exclusion chromatography. Hui1 (KR608403), Hui2 (KR608404), Hui3 (KR608405), HmK (O16846), AETX-K (Q0EAE5), ShK (P29187), and mutant Hui1 peptides were produced with CSBio. Folding reactions were quenched by acidification and products purified by reverse-phase HPLC, as before (1). Toxin concentrations were determined using a spectrophotometer. Toxin peptides were synthesized in solid state, folded, and purified by HPLC. Fractions with the desired protein were identified by analytical LC-MS, judged to be more than 95% pure, and lyophilized before storage at −80 °C.
SPR Spectroscopy.
Binding kinetics were determined using a BIAcore-3000 with immobilization of 600–1,000 response units of KcsA on a Ni-NTA chip via the C-terminal hexahistidine tag (GE Healthcare). KcsA protein was purified as previously described (5) and stored at 4 °C in 150 mM KCl, 50 mM Tris-Cl, 1 mM DDM, pH 7.5, before use. Peptide toxins were studied by applying 60 μL of five serial dilutions between 0.25–10 nM in running buffer (10 mM Hepes, 150 mM NaCl, 3 mM KCl, 50 µM EDTA, and 0.005% Tween 20, pH 7.4). Toxin binding was corrected by subtraction of responses on a blank flow cell. Association and dissociation kinetics were studied with a flow rate of 25 μL⋅min−1. Data were fitted with a 1:1 Langmuir model to estimate kon and koff using BIAevaluation 3.1 software; Kd values were calculated from the ratio of koff and kon. The binding parameters for Hui1 were validated using a ProteOn XPR36 Protein Interaction Array System (BioRad).
Single KcsA Channel Recording.
Liposomes were formed with KcsA-E71A from a solution of 35 mM CHAPS, 20 mg/mL E. coli polar lipids (Avanti Polar Lipids), and 1 μg KcsA/mL in reconstitution buffer (200 mM KCl and 10 mM Mops, pH 7.0) by dialysis against reconstitution buffer at room temperature, and the resulting suspension of liposomes frozen at −80 °C in 50-μL aliquots. For recording, a liposome aliquot was diluted with 50 μL of 1.5 M KCl, refrozen, thawed, and then sonicated for 10 s in a cylindrical bath sonicator. Single channels were recorded at room temperature in a horizontal planar bilayer system with two aqueous chambers separated by an 80-μm-thick partition hand-crafted from overhead transparency film, as described (22). Briefly, bilayers were formed of POPE/POPG (7.5:2.5 mg/mL in n-decane) on a 50-μm hole. The “internal” chamber contained 100 mM KCl adjusted to pH 4.0 with succinic acid and the “external” chamber contained the 100 mM KCl, 10 mM Mops adjusted to pH 7.0 with KOH. The pH gradient ensures that channels operate here with the cytoplasmic side of the protein facing the internal solution. Current was recorded with an Axopatch-200B amplifier (Molecular Devices) with signals sampled at 10 kHz after low-pass filtering (eight-pole Bessel, 0.75- to 1-kHz corner frequency). Single-channel recordings were obtained by squirting 0.5 μL of a liposome suspension onto the internal side of the bilayer and waiting 2–5 min. On appearance of a channel (at 100 mV), chambers were perfused with fresh solutions. After recording for 3 min to confirm proper channel characteristics, 0.25–5 nM Hui1 toxin was perfused across the external solution. Recordings were idealized using Clampfit and filtered at 300 Hz before analysis.
Two-Electrode Voltage Clamp.
Xenopus laevis oocytes were injected with cRNA encoding KcsA-Shaker (23), KcsA-E71A-Shaker, Shaker (NM_167596, the variant used here is the noninactivating variant Shaker-IR Δ6–46), human Kv1.2 (NM_004974), and human Kv1.3 (NM_002232) carried in a laboratory vector with the 5′ and 3′ portions of the Xenopus laevis β-globin gene, a cytomegalovirus promoter for expression in mammalian cells, and a T7 promoter for in vitro transcription. Recording solution was (in millimolar): 10 KCl, 90 NMDG, 1 MgCl2, 0.3 CaCl2, 10 Hepes, pH 7.5, with 0.1% BSA (Fraction V). Recordings were performed with constant gravity flow of solution at 2 mL/min yielding chamber exchange in ∼5 s. Currents were recorded 1–3 d after cRNA injection using an Oocyte clamp amplifier OC-725C (Warner Instruments) and electrodes filled with 3 M KCl with resistance of 0.3–1 MΩ. Data were filtered at 1 kHz and digitized at 20 kHz using pClamp software and assessed with Clampfit v9.0 and Origin 6.0. Equilibrium inhibition for Hui1 was determined by fitting dose–response curves whereas it was calculated for other toxins from the fraction of unblocked current achieved by one to three toxin concentrations that inhibited 20–80% of the current.
kon and koff were estimated from fits of the kinetics of toxin wash-in and wash-out calculated. Thus, according to a bimolecular scheme with a single toxin-bound state and rapid changes in toxin concentration compared with the speed of block and unblock, one can assess steady-state and kinetic parameters of blockade, as before (28, 38). Thus, the unblocked fractional current at equilibrium (Fun) with a toxin concentration [Tx] relates to the dose dependence of inhibition according to Fun = (1 + [Tx]/Ki)−1; Fun is related to the second-order association rate constant kon (M−1⋅s−1) and first-order dissociation rate constant koff (s−1) according to Fun = koff/(kon[Tx] + koff); and the equilibrium inhibition constant for half-maximal blockade is Ki = koff/kon. Time constants determined by single exponential fits of the inhibition of current on toxin wash-in and its restoration on wash-out are related to these parameters by τon = (kon[Tx] + koff)−1 and τoff = (koff)−1. Because τoff is a direct measure of the dissociation rate constant it provides an internal validation of the determined kinetic parameters.
NMR Spectroscopy and Structure Determination.
Lyophilized Hui1 was dissolved to a final concentration of 0.6–0.7 mM in phosphate buffer (20 mM, pH 6.6) containing 10 mM NaCl and 0.02% NaN3 in 7% or 99% 2H2O. NMR spectra were collected on a Bruker DRX700 spectrometer equipped with cryogenic triple-resonance TCI probe with z axis pulsed field gradients. All experiments were conducted at 16.4 T and 298 K. Resonance assignments and structural restraints (distances and dihedral angles) were obtained from 2D homonuclear and heteronuclear spectra (Fig. S3) and secondary chemical shift analysis using TALOS (46). Detailed experimental conditions are provided in Supporting Information. Structure calculation was performed using the CNS (Crystallography and NMR System) software (47) using distance-geometry and simulating annealing protocols. Calculations were performed iteratively, introducing short-range and later long-range distance constraints, followed by dihedral constraints, to eliminate erroneous assignments. Structure quality of the final ensemble was validated by the PROCHECK_NMR program (48). Statistics of the final structures are in Table S4.
Detailed Experimental Methods for Structure Determination
Acquisition of NMR Data.
NMR measurements were conducted with Bruker DRX700 spectrometer equipped with cryogenic triple-resonance TCI probe, that is equipped with z axis pulsed field gradients. The experiments were conducted at 16.4 T and 298 K. Backbone chemical shift assignments, of the Hui1 toxin, were obtained from homonuclear 2D total correlation spectroscopy (TOCSY) and 2D NOESY spectra (Fig. S3) acquired over 4,096 data points with spectral widths of 12 ppm. Chemical shift assignments of 13C and 15N amides were obtained using 2D 1H, 13C (natural abundance) heteronuclear multiple quantum coherence (HMQC), and 2D 1H 15N heteronuclear single quantum coherence (HSQC) experiments. HMQC spectra were acquired in QF fashion with 1,200 complex points, with acquisition times of 53 ms in the 13C dimension with 4,096 complex points and with an acquisition time of 208 ms in the observed dimension. HSQC spectra were acquired in an echo–antiecho fashion with 280 complex points with acquisition times of 61.6 ms in the 15N dimension, and 2,048 complex points and acquisition times of 91 ms in the observed dimension. At all spectra, water suppression was achieved using selective pulses or low-power irradiation of the water signal during the relaxation delay. The Hα-HN 3J-coupling constants were obtained from a one-dimensional experiment with 16,384 data points and 256 scans.
Spectra Analysis.
TOCSY and NOESY spectra were assigned as described earlier (30). Distance constraints were derived from the NOESY spectra by comparison with a reference intensity (e.g., 2.5 Å for the Hδ-Hε in the aromatic ring of Tyr and Phe residues) and using the well-known relation that peak intensity is proportional to the inverse sixth power of the distance. Backbone dihedral angles constraints were obtained from J-coupling measurements and TALOS analysis of the chemical shifts of HN, Hα, 15Namidic (obtained from the HSQC spectrum), and 13Cα (obtained from the HMQC spectrum). In addition, eight χ1 angle constraints were estimated by comparing the resonance intensities in the NOESY spectra of Hα/ Hβ1 and Hα/Hβ2 belonging to the same residue.
Structure Determination.
The Hui1 structure was calculated using the CNS software. A molecular topology file and all constraints (distance and dihedral) served as input for the calculation, using distance-geometry and simulating annealing protocols to produce structure sufficing experimental constraints. To eliminate erroneous constraints, the calculations were performed iteratively, with gradual introduction of constraints. The growing number of constraints with each cycle of calculations increases the precision of the accepted NMR structure ensembles. Quality and structure statistics of the final ensemble of structure models were validated by the PROCHECK_NMR program.
Acknowledgments
The authors thank Crina Nimigean, David Posson, and Dorothy Kim for guidance and material support for bilayer studies (and Chris Miller for allowing us to first contaminate his apparatus). This work was supported by NIH Grant R01HL105959 (to S.A.N.G.) and United States–Israel Binational Science Foundation Grant BSF2013185 (to J.H.C. and S.A.N.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: NMR, atomic coordinates, chemical shifts, and restraints have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 2N6B).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1514728112/-/DCSupplemental.
References
- 1.Takacs Z, et al. A designer ligand specific for Kv1.3 channels from a scorpion neurotoxin-based library. Proc Natl Acad Sci USA. 2009;106(52):22211–22216. doi: 10.1073/pnas.0910123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LeMasurier M, Heginbotham L, Miller C. KcsA: It’s a potassium channel. J Gen Physiol. 2001;118(3):303–314. doi: 10.1085/jgp.118.3.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doyle DA, et al. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414(6859):43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 5.Cuello LG, et al. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature. 2010;466(7303):272–275. doi: 10.1038/nature09136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng WW, McCoy JG, Thompson AN, Nichols CG, Nimigean CM. Mechanism for selectivity-inactivation coupling in KcsA potassium channels. Proc Natl Acad Sci USA. 2011;108(13):5272–5277. doi: 10.1073/pnas.1014186108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schrempf H, et al. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans. EMBO J. 1995;14(21):5170–5178. doi: 10.1002/j.1460-2075.1995.tb00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacKinnon R, Cohen SL, Kuo A, Lee A, Chait BT. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280(5360):106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- 9.Legros C, et al. Generating a high affinity scorpion toxin receptor in KcsA-Kv1.3 chimeric potassium channels. J Biol Chem. 2000;275(22):16918–16924. doi: 10.1074/jbc.275.22.16918. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A, Lee A, Campbell E, Mackinnon R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K(+) channel. eLife. 2013;2:e00594. doi: 10.7554/eLife.00594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin L, Wu Y. Molecular mechanism of the sea anemone toxin ShK recognizing the Kv1.3 channel explored by docking and molecular dynamic simulations. J Chem Inf Model. 2007;47(5):1967–1972. doi: 10.1021/ci700178w. [DOI] [PubMed] [Google Scholar]
- 12.Beeton C, et al. The D-diastereomer of ShK toxin selectively blocks voltage-gated K+ channels and inhibits T lymphocyte proliferation. J Biol Chem. 2008;283(2):988–997. doi: 10.1074/jbc.M706008200. [DOI] [PubMed] [Google Scholar]
- 13.Tudor JE, Pallaghy PK, Pennington MW, Norton RS. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat Struct Biol. 1996;3(4):317–320. doi: 10.1038/nsb0496-317. [DOI] [PubMed] [Google Scholar]
- 14.Kalman K, et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273(49):32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- 15.Rangaraju S, et al. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J Biol Chem. 2010;285(12):9124–9136. doi: 10.1074/jbc.M109.071266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordero-Morales JF, et al. Molecular driving forces determining potassium channel slow inactivation. Nat Struct Mol Biol. 2007;14(11):1062–1069. doi: 10.1038/nsmb1309. [DOI] [PubMed] [Google Scholar]
- 17.Park CS, Miller C. Mapping function to structure in a channel-blocking peptide: Electrostatic mutants of charybdotoxin. Biochemistry. 1992;31(34):7749–7755. doi: 10.1021/bi00149a002. [DOI] [PubMed] [Google Scholar]
- 18.Lanigan MD, et al. Mutating a critical lysine in ShK toxin alters its binding configuration in the pore-vestibule region of the voltage-gated potassium channel, Kv1.3. Biochemistry. 2002;41(40):11963–11971. doi: 10.1021/bi026400b. [DOI] [PubMed] [Google Scholar]
- 19.Dauplais M, et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J Biol Chem. 1997;272(7):4302–4309. doi: 10.1074/jbc.272.7.4302. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa Y, et al. Isolation and cDNA cloning of a potassium channel peptide toxin from the sea anemone Anemonia erythraea. Toxicon. 2006;48(5):536–542. doi: 10.1016/j.toxicon.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Gendeh GS, et al. A new potassium channel toxin from the sea anemone Heteractis magnifica: Isolation, cDNA cloning, and functional expression. Biochemistry. 1997;36(38):11461–11471. doi: 10.1021/bi970253d. [DOI] [PubMed] [Google Scholar]
- 22.Piasta KN, Theobald DL, Miller C. Potassium-selective block of barium permeation through single KcsA channels. J Gen Physiol. 2011;138(4):421–436. doi: 10.1085/jgp.201110684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu Z, Klem AM, Ramu Y. Ion conduction pore is conserved among potassium channels. Nature. 2001;413(6858):809–813. doi: 10.1038/35101535. [DOI] [PubMed] [Google Scholar]
- 24.Harvey AJ, Gable RW, Baell JB. A three-residue, continuous binding epitope peptidomimetic of ShK toxin as a Kv1.3 inhibitor. Bioorg Med Chem Lett. 2005;15(13):3193–3196. doi: 10.1016/j.bmcl.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Rauer H, Pennington M, Cahalan M, Chandy KG. Structural conservation of the pores of calcium-activated and voltage-gated potassium channels determined by a sea anemone toxin. J Biol Chem. 1999;274(31):21885–21892. doi: 10.1074/jbc.274.31.21885. [DOI] [PubMed] [Google Scholar]
- 26.Anderson CS, MacKinnon R, Smith C, Miller C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J Gen Physiol. 1988;91(3):317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacKinnon R, Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J Gen Physiol. 1988;91(3):335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein SA, Miller C. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys J. 1993;65(4):1613–1619. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park CS, Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 1992;9(2):307–313. doi: 10.1016/0896-6273(92)90169-e. [DOI] [PubMed] [Google Scholar]
- 30.Wang K-W, Tai K-K, Goldstein SAN. MinK residues line a potassium channel pore. Neuron. 1996;16(3):571–577. doi: 10.1016/s0896-6273(00)80076-8. [DOI] [PubMed] [Google Scholar]
- 31.Tai KK, Goldstein SAN. The conduction pore of a cardiac potassium channel. Nature. 1998;391(6667):605–608. doi: 10.1038/35416. [DOI] [PubMed] [Google Scholar]
- 32.Wuthrich K. NMR of Proteins and Nucleic Acids. Wiley; Hoboken, NJ: 1986. [Google Scholar]
- 33.Miller C. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 1995;15(1):5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 34.Posson DJ, Ge P, Miller C, Bezanilla F, Selvin PR. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 2005;436(7052):848–851. doi: 10.1038/nature03819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beraud E, Chandy KG. Therapeutic potential of peptide toxins that target ion channels. Inflamm Allergy Drug Targets. 2011;10(5):322–342. doi: 10.2174/187152811797200696. [DOI] [PubMed] [Google Scholar]
- 36.Vetter I, Lewis RJ. Therapeutic potential of cone snail venom peptides (conopeptides) Curr Top Med Chem. 2012;12(14):1546–1552. doi: 10.2174/156802612802652457. [DOI] [PubMed] [Google Scholar]
- 37.Prommer E. Ziconotide: A new option for refractory pain. Drugs Today (Barc) 2006;42(6):369–378. doi: 10.1358/dot.2006.42.6.973534. [DOI] [PubMed] [Google Scholar]
- 38.Chen H, Kim LA, Rajan S, Xu S, Goldstein SAN. Charybdotoxin binding in the I(Ks) pore demonstrates two MinK subunits in each channel complex. Neuron. 2003;40(1):15–23. doi: 10.1016/s0896-6273(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 39.Cao Z, Di Z, Wu Y, Li W. Overview of scorpion species from China and their toxins. Toxins (Basel) 2014;6(3):796–815. doi: 10.3390/toxins6030796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang S. Proteome and peptidome profiling of spider venoms. Expert Rev Proteomics. 2008;5(5):731–746. doi: 10.1586/14789450.5.5.731. [DOI] [PubMed] [Google Scholar]
- 41.Diochot S, Lazdunski M. Sea anemone toxins affecting potassium channels. Prog Mol Subcell Biol. 2009;46:99–122. doi: 10.1007/978-3-540-87895-7_4. [DOI] [PubMed] [Google Scholar]
- 42.Mouhat S, Jouirou B, Mosbah A, De Waard M, Sabatier JM. Diversity of folds in animal toxins acting on ion channels. Biochem J. 2004;378(Pt 3):717–726. doi: 10.1042/BJ20031860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldstein SA, Pheasant DJ, Miller C. The charybdotoxin receptor of a Shaker K+ channel: Peptide and channel residues mediating molecular recognition. Neuron. 1994;12(6):1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- 44.Jungo F, Bougueleret L, Xenarios I, Poux S. The UniProtKB/Swiss-Prot Tox-Prot program: A central hub of integrated venom protein data. Toxicon. 2012;60(4):551–557. doi: 10.1016/j.toxicon.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen Y, Delaglio F, Cornilescu G, Bax A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR. 2009;44(4):213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystal. D Biol. Crystal. 1998;54(5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 48.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]