

Significance
ClpB/Hsp100 chaperones protect cells from the devastating effects of protein inactivation and aggregation arising from extreme stress. This function is accomplished first by binding to the aggregates and then forcibly unraveling individual proteins by passing them through the central channel in the hexameric chaperones. Here, we investigate the role of the ClpB/Hsp100 N-terminal domain (NTD) in protein disaggregation. Our results demonstrate that ClpB recognizes exposed hydrophobic stretches in unfolded or aggregated client proteins via a substrate-binding groove in its NTD. We further show that the NTD has regulatory roles that include blocking the translocation channel in the absence of substrate and destabilizing client proteins upon binding, thus priming them for subsequent unfolding and disaggregation.
Keywords: Hsp104, ClpB, protein disaggregation, methyl-TROSY NMR, chaperones
Abstract
ClpB/Hsp100 is an ATP-dependent disaggregase that solubilizes and reactivates protein aggregates in cooperation with the DnaK/Hsp70 chaperone system. The ClpB–substrate interaction is mediated by conserved tyrosine residues located in flexible loops in nucleotide-binding domain-1 that extend into the ClpB central pore. In addition to the tyrosines, the ClpB N-terminal domain (NTD) was suggested to provide a second substrate-binding site; however, the manner in which the NTD recognizes and binds substrate proteins has remained elusive. Herein, we present an NMR spectroscopy study to structurally characterize the NTD–substrate interaction. We show that the NTD includes a substrate-binding groove that specifically recognizes exposed hydrophobic stretches in unfolded or aggregated client proteins. Using an optimized segmental labeling technique in combination with methyl-transverse relaxation optimized spectroscopy (TROSY) NMR, the interaction of client proteins with both the NTD and the pore-loop tyrosines in the 580-kDa ClpB hexamer has been characterized. Unlike contacts with the tyrosines, the NTD–substrate interaction is independent of the ClpB nucleotide state and protein conformational changes that result from ATP hydrolysis. The NTD interaction destabilizes client proteins, priming them for subsequent unfolding and translocation. Mutations in the NTD substrate-binding groove are shown to have a dramatic effect on protein translocation through the ClpB central pore, suggesting that, before their interaction with substrates, the NTDs block the translocation channel. Together, our findings provide both a detailed characterization of the NTD–substrate complex and insight into the functional regulatory role of the ClpB NTD in protein disaggregation.
The heat shock protein ClpB (Escherichia coli) or Hsp100 (eukaryotes) is the main protein disaggregase in bacteria, yeast, plants, and mitochondria of all eukaryotic cells, and it is essential for cell survival during severe stress (1–4). Recovery of functional proteins from aggregates by ClpB requires the synergistic interaction with a second molecular chaperone, DnaK (1). Through its cochaperone, DnaJ, DnaK initially binds to the aggregates, leading to the exposure of peptide segments that can be recognized by ClpB (5, 6). DnaK then recruits ClpB to the site of aggregation through direct physical interaction (7, 8), transferring the aggregate to ClpB. Using the energy derived from ATP hydrolysis, ClpB unravels the aggregate by threading single polypeptide chains, one at a time, through the central pore of its hexameric ring (9). Once released from the aggregate, the unfolded polypeptides can either refold spontaneously or fold with the help of additional cellular chaperones.
Like other Hsp100 proteins, ClpB forms a hexameric ring, with each protomer comprising an N-terminal domain (NTD) and two nucleotide binding domains (NBD1 and NBD2) separated by a unique regulatory coil–coil domain (10) essential for DnaK binding (7, 11) (Fig. 1 A and B). Both NBDs contain Walker A and Walker B motifs that are required for nucleotide binding and hydrolysis (12, 13), respectively, and a highly conserved tyrosine (Y243 in Thermus thermophilus ClpB) that plays a critical role in disaggregation. Each of the conserved tyrosines from a protomer is located in a so-called pore loop (14) (Fig. 1C) and extends into the axial channel to interact directly with positively charged and aromatic residues from the bound substrate (9, 15). Mutating these NBD-1 pore loop tyrosines leads to a partial reduction of the ClpB protein disaggregation activity (9, 15). When this mutation is combined with the deletion of the NTD, the resulting double mutant is completely inactive in substrate disaggregation (16), although each of these ClpB variants alone can reactivate protein aggregates (9, 15). The complete loss in activity only with the ClpB double mutant was suggested to result from overlapping substrate-binding functions for the NBD1 pore tyrosine residues and the ClpB NTD (16).
Fig. 1.

Structure and domain organization of the hexameric ClpB chaperone. Domain organization (A) and protomeric structure (B) of the ClpB chaperone [Protein Data Bank (PDB) ID code 1QVR (10)]. The ClpB protomer consists of an N-terminal domain (NTD; green), two nucleotide binding domains (NBD1, NBD2; dark and light blue, respectively), and a coil–coil domain insertion (CCD; yellow). (C) The monomers assemble into a hexamer consisting of three rings formed by NTDs (top ring; green), NBD1-CCD (blue-yellow), and NBD2 enclosing the central pore. The Inset shows a magnified view of the central pore loops of NBD1 with the conserved tyrosines (Y243; represented as red sticks) extending into the axial channel. This model of ClpB hexamers is based on cryo-electron microscopy structures of E. coli ClpB (EMD-2563) (52).
The ClpB NTD is a globular, 150-residue α-helical domain connected by an unstructured 17-residue linker to NBD1 (Fig. 1 A and B) (10). Its precise function remains unclear—it is not required for thermotolerance (17), yet it becomes important in vivo when Hsp70 activity is compromised (18, 19). Although it was reported that the NTD is not required for disaggregation of many small aggregates, it is involved in the reactivation of several strongly aggregated proteins (17, 18, 20). Here, we use NMR to structurally characterize its interaction with substrate proteins and to elucidate its functional role in protein disaggregation. Our results demonstrate that the NTD contains a substrate-binding groove that specifically recognizes hydrophobic residues exposed in unfolded or aggregated client proteins. Unlike the case for substrate binding involving the pore loops, the NTD–substrate interaction is independent both of the nucleotide state and conformational changes to ClpB that ATP hydrolysis promotes. Notably, we show that the NTD interaction destabilizes client proteins, priming them for subsequent unfolding and translocation. Finally, mutations in the NTD substrate-binding groove have a dramatic effect on protein translocation through the ClpB central pore, suggesting that NTDs block the translocation channel before interaction with substrates. Together, our findings provide molecular insight into the NTD–substrate complex as well as into the functional role of the NTD in both protein disaggregation and in regulating ClpB activity.
Results
Substrate Interactions with the ClpB NTD.
Previous studies have shown that ClpB interacts with unstructured proteins, thermally or chemically induced protein aggregates, and polypeptides enriched in positively charged and aromatic residues (15, 21, 22). Our understanding of substrate recognition by the ClpB chaperone and, in particular, the role played by its NTDs has, however, remained limited. To address this, we first studied the interactions between isolated NTDs and a diverse list of client proteins using solution NMR. A series of 1H–15N heteronuclear single-quantum coherence (HSQC) spectra were recorded of uniformly 15N-labeled NTD, both alone and in the presence of intrinsically disordered α-casein, soluble aggregates of firefly luciferase (FFL) or malate dehydrogenase (MDH), a series of globally folded proteins, or a 21-aa polypeptide (B1 peptide) shown to bind ClpB with high affinity (15, 22). Changes in NTD spectra were not detected upon addition of protein aggregates (Fig. S1 A and B), in agreement with previous observations that ClpB does not stably bind to aggregated proteins in the absence of the DnaK chaperone system (23, 24). Similarly, binding was not observed for globally folded proteins (GFP and DnaK) (Fig. S1 C and D). Large chemical shift perturbations (CSPs), however, were noted with the addition of α-casein, but not for the B1 peptide (Fig. 2A and Fig. S1 E and F), indicating that the NTD interacts preferentially with some unfolded substrates and not others.
Fig. S1.

Substrate interactions with NTD. Chemical shift perturbations (CSPs), plotted by residue number, of [1H, 15N]-labeled monomeric NTD upon binding to 3 molar equivalents of (A) chemically denatured firefly luciferase (FFL) aggregates, (B) heat-induced malate dehydrogenase (MDH) aggregates, (C) green fluorescent protein (GFP), (D) DnaK, (E) α-casein, (F) peptide B1, (G–K) reduced alkaline phosphatase (PhoA) fragments, (L) Src residues 1–85, (M) Sic1, (N) c-Jun, and (O) p13 (KLDSLIVFLREEA). CSPs were defined by the relation, , where ΔδH and ΔδN are proton and nitrogen chemical shift changes between apo and bound forms of the protein.
Fig. 2.

Interaction of client proteins with the isolated NTD of ClpB. (A) Overlay of 1H–15N HSQC spectra of ClpB NTD alone (in black) and in the presence of twofold excess of PhoA1-121 (red), PhoA119-243 (yellow), PhoA244-349 (purple), PhoA349-471 (green), α-casein (maroon), or p13 (cyan). Datasets were recorded at 11.7 T, 55 °C (500-MHz 1H frequency). (B, Top) ClpB NTD surface representation (PDB ID code 1QVR) (10) with NTD residues found by NMR to interact with the unstructured substrates from (A) colored orange. (B, Bottom) ClpB NTD structure colored by residue hydrophobicity (53) (white to red gradient) showing that the NTD substrate binding site is enriched in hydrophobic residues. (C) 1H–15N HSQC spectrum of an unstructured substrate protein, Sic1, alone (black) and upon interaction with ClpB NTD (red), 18 °C. Sic1 assignments (26) are indicated. (D) Percentage values of ClpB-client protein amino acids that show CSPs in the presence of NTD (shifts greater than 1 SD from the average). Values are normalized against the total number of each amino acid in the substrate sequences. From the data, NTD preferably binds to hydrophobic residues in its client proteins. Assignments of cross-peaks are as indicated in A and C.
Having established that certain clients are preferred, we further characterized substrate–NTD interactions by performing NMR titration experiments of 15N-labeled NTD with 10 different globally unfolded substrates. These varied in polypeptide chain length, net charge, amino acid composition, and in the amount of residual structure (Table S1), so as to include a wide range of diverse ClpB unfolded clients. Upon addition to the ClpB NTD, spectral changes were observed for all substrates (Fig. 2A and Fig. S1 E–N), with the exception of the B1 peptide (Fig. S1F), with calculated binding affinities in the range of 100–400 μM (Table S1). Interestingly, all shifting residues were in fast exchange on the NMR chemical shift timescale and the trajectories of the shifts were linear for all substrates, with the extent of perturbation correlated with the relative binding affinity (Fig. 2A, Fig. S2, and Table S1).
Table S1.
Binding affinities for a range of NTD substrates
| Protein | Molecular weight, kDa | PI | KD, μM | Protein type |
| α-Casein | ∼23 | 4.7 | 107 ± 23 | IDP |
| PhoA1-121 (P1) | 12.8 | 9.0 | 281 ± 73 | Unfolded |
| PhoA119-243 (P2) | 12.3 | 5.3 | 268 ± 62 | Unfolded |
| PhoA244-349 (P3) | 11.5 | 6.8 | 243 ± 42 | Unfolded |
| PhoA349-471 (P4) | 12.9 | 5.0 | 181 ± 56 | Unfolded |
| PhoA1-62 | 6.4 | 8.3 | 311 ± 57 | Unfolded |
| PhoA425-471 | 4.9 | 4.9 | 255 ± 35 | Unfolded |
| Sic1 | 9.6 | 12.2 | 342 ± 88 | IDP |
| c-Jun | 13.4 | 5.3 | 289 ± 58 | IDP |
| Src1-85 | 8.4 | 10.2 | 396 ± 74 | IDP |
| p13 | 1.3 | 4.3 | 320 ± 60 | Peptide |
Affinities quantified from fits of chemical shift changes in titration spectra of U-[1H,15N] NTD as a function of added substrate. All measurements were at 55 °C. IDP, intrinsically disordered protein.
Fig. S2.

ClpB NTD rapidly samples different binding sites from client proteins. Overlay of 1H–15N HSQC spectra of ClpB NTD alone (in black) and in the presence of twofold excess of PhoA1-121 (red), PhoA119-243 (yellow), PhoA244-349 (purple), PhoA349-471 (green), α-casein (maroon), or p13 (cyan) for selected NTD residues. Trajectories of the shifts are linear for all substrates (indicated by an arrow), with the exception of p13. The NTD–substrate complex is highly dynamic with the chemical shifts representing an average over a range of different environments. Shifts for p13 (cyan) do not lie on the same line as the rest of the substrates, as the peptide contains a single optimal NTD binding site and forms a stable (not averaged) complex.
The largest CSPs were observed for NTD residues M1–A17 (H1), G80–L91 (loop between H3–H4, H4), V106–V108 (loop between H4–H5), and L111 (H5) (Fig. S1 E–O). Mapping the CSPs onto the crystal structure of the ClpB NTD (10) revealed that these residues form a binding groove primarily enriched in hydrophobic amino acids (Fig. 2B). Thus, the ClpB NTD selectively interacts with globally unfolded substrate proteins via a newly identified hydrophobic groove.
ClpB NTD Specifically Binds to Hydrophobic Stretches in Client Proteins.
To better understand how the ClpB NTD “selects” its client proteins, we monitored the binding of a series of 15N-labeled globally unfolded substrates to unlabeled NTD by recording 1H–15N HSQC spectra (see example spectrum for Sic1, Fig. 2C). Those residues with 1H and 15N chemical shift changes in excess of 1 SD from the average change upon binding were classified as interacting with the ClpB NTD. A total of 235 out of 624 residues in the six substrates analyzed [four reduced fragments of alkaline phosphatase (25), Sic1 (26), and c-Jun] were selected and a primary sequence analysis of the ClpB binding sites revealed that they are highly enriched in hydrophobic amino acids (Fig. 2D), with potential NTD binding sites likely comprised of 3- to 6-aa-long stretches of residues. The fact that multiple binding sites are associated with a given client would explain the linear titration behavior observed for NTD residues with different substrates (see above, Fig. 2A and Fig. S2). Such linearity could occur if the NTD–substrate complex is highly dynamic, with the NTD rapidly sampling different binding sites on the substrate. In this manner, the chemical shift changes of NTD residues upon binding would represent averages over a range of different magnetic environments that would depend only very little on the nature of the substrate. A simple prediction, therefore, is that the CSPs of NTD residues in an NTD–substrate complex that is not averaged would not be on the same line as observed for the substrates considered. With this in mind, we designed a 13-mer peptide (referred to in what follows as p13) that, based on our NMR results, would contain a single optimal NTD binding site and would therefore be expected to form a stable complex. CSPs would thus reflect the unique 13-mer sequence rather than an average over a large number of hydrophobic sequences, as with the larger substrates. Notably, the p13 peptide (KLDSLIVFLREEA) interacted exclusively with the same residues of the NTD as the rest of the substrates (Fig. 2A and Fig. S1O), but the CSPs from the p13-NTD titration did not follow the same linear trajectory as for the other substrates (Fig. 2A and Fig. S2), thus confirming the dynamic nature of the substrate–NTD complexes studied in Fig. 2A.
The ClpB Chaperone Is Activated by NTD–Substrate Interactions.
Addition of substrate proteins has been widely reported to elevate ClpB ATPase activity (17), with some substrates such as α-casein stimulating ClpB in an NTD-dependent manner, while others inducing the same degree of activation for both ClpB and ClpBΔN (ClpB hexamers lacking the NTD). We thus wanted to establish whether substrates that bind to monomeric NTD also displayed an NTD-dependent activation of the hexameric ClpB chaperone. ATPase activity assays were measured for ClpBWT (WT ClpB) and ClpBΔN in the absence and presence of client proteins. Substrate proteins that showed CSPs upon addition of the isolated NTD in our NMR experiments also enhanced ATPase activity of ClpB by threefold to fivefold, similar to previously reported levels of activation by α-casein (17). These same substrates, however, were only able to activate ClpBΔN by 1.1- to 1.8-fold, suggesting that the direct interaction of the ClpB NTD with the substrate is essential for the full allosteric activation of the ClpB ATPase domains (Fig. 3A). Thus, the weak affinity interaction whereby unfolded client proteins bind to a specific grove in the NTDs of ClpB results in enhancement of ClpB activity.
Fig. 3.

Hexameric ClpB activation and segmental labeling. (A) Steady-state ATPase activity of ClpBWT and ClpBΔN measured alone and in the presence of PhoA1-121 (P1), PhoA119-243 (P2), PhoA244-349 (P3), PhoA350-471 (P4), α-casein (Cas), or 13-mer peptide (p13) substrates. The average values from three separate experiments are reported with their SD. Substrate activation of ClpB is shown to be highly dependent on the presence of the NTD. (B and C) Selected regions of 1H–13C HMQC methyl-TROSY spectra of the 580-kDa hexameric ClpB enzyme (B) uniformly [2H, 13CH3-Ile,Leu,Val]-labeled and (C) segmentally labeled at the NTD ([2H, 13CH3-Ile,Leu,Val]-ClpBNTD ligated to [2H,12C]-ClpBΔN). Datasets were recorded at 18.0 T (800-MHz 1H frequency), 55 °C. (D) Overlay of 1H–13C HMQC spectra of hexameric, segmentally labeled, ClpB (blue) and the isolated NTD (red). The high degree of overlap between the two spectra suggests that the structure of NTD does not change significantly upon ClpB hexamerization. Small changes between the two spectra, corresponding to residues in the linker region connecting NTD and NBD1 domains, are highlighted by an asterisk (*).
The newly identified NTD hydrophobic binding groove along with the previously known tyrosine loops comprise two distinct sites on ClpB for initial substrate engagement, with the tyrosine residues favoring binding to polypeptides enriched with positively charged and aromatic amino acids, such as the B1 peptide (15, 22), whereas the NTD preferentially recognizes and binds large aliphatic residues present in unfolded or aggregated substrates. To better understand the interplay between these two sites, we next studied the interaction of substrates with the hexameric ClpB chaperone using methyl-transverse relaxation optimized spectroscopy (TROSY) NMR.
NMR Characterization of a Segmentally Labeled 580-kDa Hexameric ClpB.
Methyl-TROSY NMR (27) in concert with labeling schemes whereby Ileδ1 and one of the two isopropyl methyl groups of Leu and Val are 13CH3-labeled in an otherwise highly deuterated background (referred to as ILV-protein) (28) have greatly increased the size limitations that previously were imposed on NMR studies of macromolecules (29–31). We have recently used such methodology to study the ClpB disaggregation machinery (7). However, due to the large size of each monomer of ClpB (97 kDa), NMR spectra of the ILV-labeled protein were very significantly overlapped, precluding detailed analyses of the full-length molecule (Fig. 3B). To overcome this limitation, in the current study, we implemented an intein-based (32) segmental isotopic labeling approach (Fig. S3), whereby only the NTD portion of ClpB is isotopically enriched with 13CH3-methyl groups, whereas the rest of the molecule is fully deuterated (NMR invisible). The optimized protocol (see Materials and Methods for details) enabled us to selectively methyl label only isolated NTDs [2H, 13CH3-ILVM] and then reassemble the full-length ClpB protein using the expressed protein ligation protocol (33, 34). Samples produced in this manner have allowed us to obtain very high-quality NMR spectra for the single NTD domain in the context of the full 580-kDa hexameric protein so that detailed studies can be undertaken (see below). This ligation method leaves a cysteine residue at the ligation point that could affect proper activity of the protein. Notably, however, both the T143C mutation and the ligation procedure had no effect on either the ATPase (Fig. S4A) or DnaK-dependent disaggregation activities of the enzyme (Fig. S4B).
Fig. S3.

Segmental labeling scheme for hexameric ClpB. To produce a hexameric ClpB protein, segmentally labeled at the NTD, NTD-intein fusion, and NBD1-CCD-NBD2 (ClpBΔN) ClpB regions were grown separately in M9 D2O minimal media supplemented with 14NH4Cl and [2H,12C]-glucose as the sole nitrogen and carbon sources, respectively. Methyl labeling precursors were added to the growth media of the NTD [Ile-δ1-[13CH3] and Val/Leu-[13CH3, 12CD3] (28)], but not to the ClpBΔN prep so that ClpBΔN will be “NMR invisible.” Both NTD-intein and ClpBΔN were purified as described in Materials and Methods. (Left Down) Cleavage of NTD from the intein fusion occurs via the following steps: (i) N → S acyl shift of cystein at the intein N terminus results in the formation of a reactive thioester. (ii) Thiol‐mediated cleavage: a nucleophilic attack on the thioester by a small thiol compound (MESNA) cleaves the precursor protein and generates a new thioester at the C terminus of the target protein. (Right Down) The ClpBΔN N-terminal tag is cleaved via TEV protease exposing an N-terminal cystein. The subsequent ligation reaction between ClpBΔN and ClpB NTD involves attack of the activated thioester in NTD by the α‐Cys residue of ClpBΔN. Next, spontaneous rearrangement of the thioester (via S → N acyl shift) results in the formation of a peptide bond between the two domains. The ligation procedure introduces a single cystein substitution at the ligation junction.
Fig. S4.

Characterization of segmentally labeled hexameric ClpB. (A) ATPase activities of ClpBWT, ClpBT143C, and segmentally labeled (SL) ClpB measured in the absence (blue) or the presence (maroon) of 0.1 mg/mL α-casein. ATPase activities are expressed as turnover per monomer ClpB. (B) Disaggregation activity of the same ClpB variants in the presence of the DnaK, DnaJ, GrpE chaperone system. Amounts of reactivated firefly luciferase after 120-min incubation are expressed as a percentage of the activities before chemical inactivation. Average values from three separate experiments are shown along with SDs. The entry “cont” refers to control experiments performed in the absence of luciferase. Neither the T143C mutation nor the segmental labeling ligation procedure had any measurable effect on the ATPase (A) and DnaK-dependent disaggregation activities (B). (C) Translational diffusion coefficients were measured for a methyl-labeled ClpB sample, a segmentally methyl-labeled ClpB sample, and a monomeric NTD (16 kDa) using a variant of the SLED experiment that contains 13C editing (1). The slope of the ln(intensity) vs. squared gradient profile in each of the curves is proportional to the diffusion constant and inversely proportional to the cube root of the molecular weight, assuming that each of the diffusing particles is spherical. Diffusion coefficients for the two ClpB samples were identical to within error (1.28 ± 1.1 × 10−7 and 1.23 ± 0.6 × 10−7 cm2/s, respectively), indicating that the segmentally labeled sample also forms hexamers. The 16-kDa NTD diffused much more rapidly (5.7 ± 0.1 × 10−7 cm2/s), as expected. (D) Overlay of selected regions of 1H–13C HMQC correlation maps of segmentally labeled hexameric ATP-bound (green) and ADP-bound (blue) ClpB. No significant changes in spectra were observed between ATP- and ADP-bound states, with the exception of residues located in the linker connecting the NTD to NBD1.
To establish that our segmentally labeled NMR samples were indeed of the correct size, pulsed-field gradient diffusion experiments (35) (Fig. S4C) were recorded on a methyl-labeled ClpB sample, a segmentally methyl-labeled ClpB sample, and a monomeric NTD (16 kDa). The diffusion coefficients for the WT ClpB and the segmentally labeled sample were identical to within error (1.28 ± 0.11 × 10−7 and 1.23 ± 0.06 × 10−7 cm2/s, respectively, at 25 °C), indicating that the segmentally labeled sample also forms hexamers. By comparison, the 16-kDa NTD diffused much more rapidly (5.7 ± 0.1 × 10−7 cm2/s, 25 °C), as expected for a smaller protein.
The 1H–13C heteronuclear multiple-quantum coherence (HMQC) correlation map of segmentally labeled hexameric ClpB was of excellent quality (Fig. 3C), and all 39 ILVM residues were resolved and could be assigned (Materials and Methods). Assignments were facilitated by the close similarity of chemical shifts in spectra of the hexamer and the monomer, with the exception of residues located in the linker connecting the NTD to NBD1 in the hexamer (Fig. 3D). Additionally, no significant changes in spectra of the NTDs were observed between ATP- and ADP-bound ClpB (Fig. S4D), despite the fact that the hexamer undergoes large conformational changes upon nucleotide hydrolysis (10). Preliminary spectra establish, therefore, that the structure of the NTD does not change significantly upon ClpB hexamerization or nucleotide hydrolysis. Moreover, the strong correlation between the slow components of the methyl 1H relaxation rates, R2S (36), measured for ATP- and ADP-bound ClpB (Fig. 4A), strongly suggests that the rapid side-chain dynamics of the NTD are likewise independent of nucleotide state.
Fig. 4.

NMR characterization of hexameric ClpB. (A) Linear correlation plot of the slow component of methyl 1H transverse relaxation rates (R2s) (36) measured on ATP- and ADP-bound ClpB samples (55 °C) that are segmentally methyl labeled in the NTD domain. The high degree of correlation between the two nucleotide-bound states establishes that the fast timescale dynamics of the NTD residues do not change upon ClpB nucleotide hydrolysis. (B) Build-up curves of experimental intensity ratios (|Iforb/Iallow|) from methyl 1H triple-quantum experiments plotted against relaxation delay for selected NTD residues. Iforb is the intensity of the triple-quantum methyl 1H signal that can only be excited from differential transverse relaxation between 1H transitions, whereas Iallow is the intensity of the corresponding single quantum signal (37). Plotted are curves for hexameric ClpB in ATP-bound (green) and ADP-bound (blue) states (55 °C), as well as profiles for the isolated NTD, 25 °C (red). Fits to Eq. S1 (SI Materials and Methods) are represented by solid lines.
To characterize the overall dynamics of the NTD within the hexamer, we next estimated the rotational correlation time of the domain from the buildup of “forbidden” methyl 1H triple-quantum (3Q) coherences (37) (Fig. 4B). Interestingly, the measured correlation times for both ADP- and ATP-bound states, (87 ± 26 ns and 82 ± 17 ns, respectively, at 55 °C, where the error denotes the distribution of residue specific correlation times) was more than two times smaller than the expected value for a 580-kDa protein (∼180 ns) and an order of magnitude larger than that of the isolated NTD (8.1 ± 0.1 ns at 55 °C). Therefore, the NTD rotates more rapidly in solution than the rest of the hexameric ClpB protein and appears to be only partially docked to NBD1 in both ATP- and ADP-bound states (Fig. 4B, green and blue, respectively). The measured correlation times, consistent with only partial restricted motion, are in keeping with previous studies establishing that the NTD does not maintain direct physical contact with the rest of ClpB (10), no doubt because of the intervening unstructured 17-residue linker.
Client Interactions with the Two Substrate-Engagement Sites in Hexameric ClpB.
As described above, previous studies have shown that ClpB can engage substrates via its NTDs and tyrosine pores that line the axial channel into which substrates are directed. The high-quality 1H–13C correlation spectra that can be recorded on ClpB samples with segmentally labeled NTD provides an avenue for characterizing and quantifying the interactions of substrates with the ClpB hexamer. Initially, an NMR titration of unlabeled α-casein with segmentally labeled ClpB in the ATP-bound state (NTD-[2H, 13CH3-ILVM], ΔN-[2H, 12C], where ΔN refers to the NBD1-CCD-NBD2 domains of a protomer) was carried out and compared with the corresponding results from a similar titration involving the monomeric NTD (2H, 13CH3-ILVM). To simplify the analysis, the conserved tyrosine (Y243) of the tyrosine loops in ClpB was mutated so that the interactions of substrates with ClpB would be limited only to the NTD. In addition, samples were prepared with a ClpBΔN E271A/E668A variant that does not hydrolyze ATP (Materials and Methods). Fig. 5 A and B show the measured CSPs of monomeric NTD and of the hexameric ClpB Y243A mutant (ClpBY243A), respectively, upon addition of α-casein. Both the isolated NTD and the hexameric ClpBY243A show changes to the same methyl residues in the NTD hydrophobic binding groove upon addition of α-casein, and these changes are similar in both magnitude and direction. As might be expected, the titration data of the monomeric NTD is consistent with a simple 1:1 binding model and a KD value of 98 ± 23 μM is obtained at 55 °C (Fig. 5C). By contrast, the titration data for the hexamer could not be well fit to a model in which each of the six NTDs of the complex binds an α-casein molecule or where two NTDs are required for binding a single α-casein (Fig. S5; n = 6, 3). Fits were of similarly good quality when three or six NTDs were assumed to bind a single α-casein (Fig. S5; n = 2, 1, corresponding to either two or one substrate molecule per NTD), in agreement with a previously reported study where multiple protomers of ClpB where shown to interact with one substrate (22), with extracted KD values ranging from 13 ± 3 μM (three NTDs per casein) to 29 ± 4 μM (six NTDs per casein; Fig. 5D), 55 °C. Binding of one α-casein molecule to multiple NTDs simultaneously is consistent with both the size of α-casein and the large number of potential NTD binding sites in the molecule (see above). Notably, comparable affinities were obtained for binding to the monomer and hexamer from a similar titration using the p13 peptide that contains a single, well-defined NTD binding site, assuming a 1:1 binding model (one peptide per NTD) in both cases (Table S2). This result supports the notion that the microscopic affinity of substrate for a given NTD does not increase in the hexamer, but rather that larger substrates can simultaneously bind several NTDs in ClpB, thereby increasing the overall affinity via an avidity effect.
Fig. 5.

Substrate binding to hexameric ClpB. (A and B) Chemical shift perturbations induced by α-casein binding to (A) [2H, 13CH3-ILVM]-labeled isolated NTD and (B) segmentally [2H, 13CH3-ILVM]-ClpBNTD,[2H, 12C]-ClpBΔN labeled ClpBY243A. CSPs are defined by the relation , where ΔδH and ΔδC are methyl 1H and 13C chemical-shift changes between apo and bound forms of the protein and α (β) is 1 SD of the methyl 1H (13C) chemical shifts deposited in the Biological Magnetic Resonance Data Bank [α is 0.29 (I), 0.28 (L), 0.27 (V), and 0.41 (M), whereas β is 1.65 (I), 1.6 (L), 1.4 (V), and 1.54 (M)]. (C and D) Titration curves for NTD (C) and segmentally labeled ClpBY243A (D), 55 °C. Either 1H or 13C chemical-shift changes, Δδ (in parts per million), are plotted as a function of α-casein concentration, from which KD values for the NTD–casein (C) and ClpBY243A–casein (D) interactions were obtained as described in the text. (E) CSPs for segmentally ILVM-labeled ClpBWT at the endpoint of the titration with α-casein, 55 °C. Residues in the NTD-NBD1 linker showing measurable CSPs for ClpBWT, but not for ClpBY243A, are colored in green. Inset shows cartoon representation highlighting the NTD and NBD1 domains of ClpB, with stick representations of NTD substrate-binding methyl residues (blue) and linker methyl residues (green). (F and G) Titration of α-casein into (F) ATP-bound and (G) ADP-bound hexameric ClpB with segmentally methyl-labeled NTD. Δδ as a function of α-casein concentration for residues L132, L135, and V141 (reporting on binding to the tyrosine loops) are shown in green. The corresponding curves for binding of α-casein to the NTDs of the hexamer are shown in blue. All fits (solid lines) in F and G correspond to the case of three α-casein molecules bound per hexamer that produced the best fits (Fig. S6). Calculated dissociation constants for the α-casein interaction with hexameric ClpB are reported in Table S2.
Fig. S5.

Multiple NTDs simultaneously bind to a client protein. Titration curves reporting changes in 1H chemical shifts (Δδ, in parts per million) of methyl probes of ClpBY243A as a function of α-casein concentration. The isotherms are fit to models (solid lines) assuming (A) each of the six NTDs in the ClpB hexamer binds one α-casein molecule, (B) two NTDs are required for binding to a single α-casein, (C) three NTDs interact with a single α-casein, or (D) all six NTDs bind a single α-casein molecule. A schematic of each binding possibility is shown, with n the number of casein molecules required to saturate a ClpB hexamer. Dissociation constants (KD) were obtained by fitting experimental data to the appropriate equation as described in Materials and Methods. (E and F) RMSD plots showing the deviation of the calculated and experimental chemical shifts (in parts per million) based on fits of titration data for ATP-ClpBY243A (E) and ADP-ClpBY243A (F). Notably, good fits were obtained for n = 1 and n = 2, where six or three NTDs, respectively, are assumed to bind to a single α-casein.
Table S2.
Binding affinities of α-casein or 13-mer peptide to ClpB variants and NTD
| ClpB variant | Substrate | Nucleotide | N* | KD, μM |
| ClpBY243A | α-Casein | ATP | 6 | 29 ± 4 |
| ClpBY243A | α-Casein | ADP | 6 | 43 ± 6 |
| ClpBWT | α-Casein | ATP | 2 | 46 ± 5 (NTD) |
| (<0.09)† (Y243) | ||||
| ClpBWT | α-Casein | ADP | 2 | 85 ± 12 (NTD) |
| 4 ± 0.4 (Y243) | ||||
| ClpBY243A | p13 | ATP | 1 | 380 ± 80 |
| NTD | α-Casein | — | 1 | 98 ± 23 |
| NTD | p13 | — | 1 | 390 ± 50 |
Affinities derived from fits of chemical shift changes in 1H–13C HMQC spectra of 13CH3-ILV–labeled ClpB variants as a function of added substrate. All measurements were performed at 55 °C.
Number of ClpB NTDs involved in the interaction with a single substrate molecule. N is related to n, the number of casein or p13 ligands that bind to ClpB (Figs. S5 and S6) according to 6/N = n.
Y243 corresponds to binding to the tyrosine pores. Measurement of nanomolar KD values is very difficult by NMR. An upper value of 90 nM is reported here, which encompasses 85% of the values obtained from a bootstrap analysis of the titration data (Fig. S7).
To establish whether substrate interactions with the ClpB NTD are affected by the nucleotide state of the hexamer, we performed a second set of experiments by titrating α-casein into ADP-bound ClpBY243A. As with the ATP-ClpBY243A titration experiments, fits were only satisfactory for models where either one or two α-casein molecules bind to the hexamer, with a fitted KD value of 43 ± 6 μM for a 1:1 α-casein to hexamer ratio, 55 °C, that is similar to the value obtained with the ATP-bound hexamer (Fig. S5 and Table S2). Thus, the interactions of substrates with ClpB NTD are not nucleotide dependent.
Having shown that ClpB NTDs engage substrates in a similar manner as the isolated NTD (Fig. 5 A and B), we next studied substrate interactions with ClpBWT, which contains, in addition to the newly characterized NTD binding site, conserved tyrosine pores that play a role in substrate binding (15, 16). Upon titration of α-casein into segmentally labeled ClpBWT, a CSP pattern very similar to that for ClpBY243A was observed, with the exception of additional large CSPs for methyl groups from L132, L135, and V141 (compare Fig. 5 B and E, green), located on the linker connecting the NTD to NBD1 and the preceding helix, in close proximity to the tyrosine pores. The location of L132, L135, and V141 and the absence of the associated CSPs in the case of the ClpBY243A mutant, suggests that these residues serve as reporters of substrate interactions with the tyrosine pores. It thus becomes possible to separate substrate binding to the NTDs from binding to the tyrosine pore loops and to obtain independent thermodynamic parameters in the context of the WT hexamer. This is achieved by fitting the titration data to a model in which binding to at least one NTD precedes interactions with the tyrosine loops in the pore (see below), as illustrated in Fig. S6 and described in Materials and Methods. Fig. 5F shows the titration isotherms corresponding to binding to the tyrosine pores (green) and NTDs (blue) of ClpBWT in the ATP state. Clear differences are apparent in casein binding affinities to NTD (KD = 46 ± 5 μM) and the tyrosine pores (KD < 90 nM, 85% confidence limit), although a precise value for binding to the tyrosine pores is difficult to obtain because of the very high affinity (Table S2 and Fig. S7). Notably, NTD isotherms do not fit well to a model involving only a single casein per hexamer, but rather one in which three substrates interact (i.e., one casein binds two NTDs). The fact that more than a single casein is required to saturate binding may reflect the fact that this particular substrate is not sufficiently long to bind all six NTDs and the pore loops simultaneously, thereby allowing additional molecules to engage the hexamer. α-Casein titrations were then repeated for ADP-bound, segmentally labeled ClpBWT, and KD values of 4 ± 0.4 μM (Fig. 5G, green) and 85 ± 12 μM (Fig. 5G, blue) were measured for binding to the tyrosine loop pores and NTDs, respectively. Once again, the binding data were best fit to a model where three casein substrates bind to the hexamer (Fig. S6). The affinities of substrates for the NTDs in both ADP- and ATP-bound ClpBWT are comparable (Fig. S7), as expected from studies on ClpBY243A described above. Moreover, the substantially lower substrate affinity for the tyrosine pores in the ADP state (Fig. S7) is consistent with previous studies showing that the affinity of the pore loops for client proteins is significantly reduced upon nucleotide hydrolysis (14).
Fig. S6.

Interaction of WT hexameric ATP- and ADP-bound ClpB with α-casein. (A) Binding schemes representing different scenarios for the interaction of α-casein with hexameric ClpB, with n the number of ligand molecules (α-casein) bound to one ClpB hexamer. Each scheme assumes that a molecule of α-casein binds to a different number of NTDs, with n = 1 denoting one α-casein per six NTDs (i.e., one binding site per ClpB hexamer), n = 2 corresponding to two binding sites (each binding site comprises three NTDs), and so forth. This is illustrated schematically where the purple line denotes a ligand, each of the green circles is an NTD, and the tyrosine pore is depicted in yellow. In each of the schemes, P and L are protein (ClpB) and ligand (α-casein), respectively, PLk corresponds to the case where k α-casein molecules are bound to ClpB (one ligand per 6/n NTDs corresponding to the n NTD binding sites in each of the models above), PHLk-1 corresponds to ClpB with k bound ligands, 1 that is bound to 6/n NTDs and the tyrosine pore site simultaneously, whereas the remaining k − 1 ligands are bound exclusively to NTDs. Thus, in the case of n = 3, each ligand binding event involves interaction of α-casein with a pair of NTDs so that PL1 is ClpB bound to one ligand (involving two NTDs) that does not engage the tyrosine pore site and PH denotes the case when the ligand is bound to both NTD and the pore. Note that only a single ligand can engage the tyrosine pore site and that this engagement can occur only after binding to the first ligand binding site in the ClpB ring (i.e., to at least 6/n NTDs in each of the models above), although it does not have to. The basis for the fact that binding to NTDs must precede binding to the tyrosine pore site is shown in Fig. 6E, where if binding to the NTD is knocked out there is no engagement with the pore and subsequent translocation. Note that each NTD binding event, as depicted by the horizontal traces above (P + L…PLn), is assumed to be independent of all others and to occur with the same microscopic association constant, 1/KD. The appropriate multiplicative factors associated with each KD value can be derived by counting the number of ways that a ligand can bind to each of the states. The ratio KR is defined as [PL1]/[PH] and the affinity that ligand has for the pore is independent of whether the pore binding event occurs immediately after the first substrate binds NTDs or after subsequent ligand binding events (the multiplicative factors for each KR take into account the number of ways that k bound substrates can engage a single pore, for example). The product KD⋅KR can be thought of as the overall equilibrium dissociation constant of a ligand molecule that is engaged both by the NTDs and the tyrosine pore. (B–E) Titration curves reporting changes in chemical shifts of methyl probes of ClpBWT-ADP as a function of α-casein concentration fit to models (solid lines) indicated in A, assuming (B) all six NTDs bind a single α-casein molecule, n = 1, (C) three NTDs interact with a single α-casein, n = 2, (D) two NTDs are required for binding a single α-casein, n = 3, or (E) each of the six NTDs in the ClpB hexamer binds one α-casein molecule, n = 6. (F and G) Δδ RMSD plots showing the deviation of the fitted chemical shifts from the experimental values (in parts per million) for ATP-ClpBWT (F) and ADP-ClpBWT (G) titrations with α-casein.
Fig. S7.

The affinity of the tyrosine pore for substrate is nucleotide dependent. Histograms showing the distribution of fitted KR (A), KD (B), and KD⋅KR (C) values for the binding of α-casein to ATP-ClpB (green) and ADP-ClpB (blue). Distributions were obtained using a bootstrap procedure (51) involving 200 trials.
In summary, the conserved tyrosine pores, known to be required for protein translocation through the ClpB central channel (9, 15), interact in a high-affinity manner with α-casein in the ATP-bound state. The significant affinity decrease upon ATP hydrolysis is thought to provide the driving force for substrate translocation (15). The NTD, in contrast, is bound to α-casein with an affinity that is not dependent on the nucleotide state of the ClpB chaperone. Finally, it is worth emphasizing the unique strength of the NMR approach, whereby it is possible to distinguish the two substrate engagement sites within ClpB, as well as to estimate the affinity of substrate for each. This is possible despite the large difference in binding affinities, which would have masked the weaker interaction using most other techniques.
Binding of Client Proteins to ClpB NTD Stabilizes Their Unfolded Conformation.
We have shown that the ClpB–NTD interactions with client proteins are dynamic, involving hydrophobic residues on both partners. It is of interest to examine how the conformations of substrates are affected by binding to NTDs, which are the initial substrate engagement sites before protein unfolding and translocation by ClpB. To address how the thermodynamics of protein folding change upon NTD binding and hence what conformations might be stabilized, we measured temperature titration profiles of the Pin1WW domain, which contains a potential NTD binding site (V26-F29), in the absence and presence of the isolated NTD. The Pin1WW domain folds on the microsecond timescale (38) and its folding transition can be monitored by NMR, with peak positions given by population-weighted averages of chemical shifts of corresponding probes in the unfolded and folded states (Fig. 6A). 1H–13C HMQC spectra were acquired over a temperature range of 15–80 °C (Fig. 6 A and B, without and with NTD, respectively), from which the melting profiles in Fig. 6C were generated. The free Pin1WW domain folds in solution by means of a two-state mechanism, with Ile, Leu, Val, and Met probes reporting a melting temperature (TM) of 60.1 ± 0.4 °C (Fig. 6C, blue curves). Upon addition of NTD, the stability of Pin1WW was reduced (compare red vs. blue melting profiles in Fig. 6C), with the TM value decreasing by 10 °C to 50.7 ± 0.8 °C. This result suggests that a substrate’s unfolded state is stabilized by engagement with the NTDs of ClpB, potentially priming it for subsequent translocation.
Fig. 6.

The ClpB NTD blocks the translocation channel in the substrate-unbound state and stabilizes the unfolded form of bound client proteins. (A and B) Selected regions from 1H–13C HMQC correlation maps showing the temperature dependence of cross-peak positions from methyl groups of L7 of the PinWW domain (A) alone and (B) in the presence of the isolated NTD domain. (C) Representative melting curves for L7δ1 and M15ε of free (blue) and NTD-bound (red) Pin1WW fitted to standard equations for two-state unfolding (54) (solid lines; SI Materials and Methods). The TM value of the substrate is reduced by ∼10 °C upon NTD binding (60.1 ± 0.4 °C, free vs. 50.7 ± 0.8 °C, bound). The NTD remains stable over the course of the entire temperature titration (TM = 105.4 ± 0.02 °C). (D) Steady-state ATPase activity of ClpB variants measured in the absence (red) and presence of α-casein (green), p13 (yellow), or peptide B1 (blue). Notably, binding of α-casein to ClpBΔN showed partial activation of ATP hydrolysis, in contrast to ClpBNTD-4A for which enhanced activity was not observed. (E) Threading activities of ClpB variants. α-Casein was incubated with ClpP and either BAP, BAPY243A, BAPΔN, BAPNTD-4A, or BAPΔN/Y243A, and hydrolysis of α-casein measured at the indicated time points (0, 15, 40, 60, 90, or 120 min). Significantly slower hydrolysis rates were observed for the NTD mutated ClpB variant (BAPNTD-4A) compared with variants of ClpB lacking the NTD (BAPΔN). Reactivation of α-glucosidase (F) or firefly luciferase (G) aggregates monitored in the presence of indicated ClpB mutants and the DnaK/DnaJ/GrpE chaperone system. Reactivation after 120 min is shown as a fraction of WT ClpB disaggregation, and SEs of three independent assays are displayed.
The Hydrophobic Groove in the NTD Can Play a Critical Role in Substrate-Dependent Activation of ClpB.
Fig. 3A shows that the NTD plays an important role in engagement of substrates by ClpB, leading to enhanced ATPase activity relative ClpBΔN. To better understand the functional role of the NTD interaction with client proteins, in particular focusing on the hydrophobic binding groove, we generated a ClpB variant (ClpBNTD-4A) containing four mutations in the NTD (W6A, L14A, L91A, L111A), which disrupt the hydrophobic substrate-binding site. As expected, the isolated NTD-4A showed no CSPs upon the addition of p13 peptide (Fig. S8A), confirming that these mutations do indeed abolish NTD–substrate binding. Biochemical experiments performed on full-length ClpBNTD-4A showed that it is hexameric with WT level ATPase activity and activation by the DnaK chaperone (Fig. S8B). We then compared the substrate-dependent enhancement of ATPase activity for ClpBWT, ClpBNTD-4A, ClpBY243A, ClpBΔN, and ClpBΔNY243A. Three substrates were chosen for this experiment: (i) α-casein, which can interact with both the NTD and NBD1 tyrosine pores; (ii) p13 peptide, which binds specifically to the NTD; and (iii) B1 peptide (15), which interacts with the tyrosine pores with high affinity (22). Notably, the ATPase activity of ClpBNTD-4A was not enhanced upon addition of p13 or α-casein (Fig. 6D). The ClpBWT ATPase activity was enhanced by fourfold to sixfold upon addition of any one of the three substrates, whereas ClpBΔN/Y243A, which does not contain either of the two initial engagement sites, showed no activation (Fig. 6D). Mutating the conserved tyrosine pores (ClpBY243A) had no effect on activation by the p13 peptide, which interacts only with the NTD, but did, however, abolish B1-dependent activation, as the tyrosine pores act as the B1 binding site (Fig. 6D). Both the ClpBNTD-4A and ClpBΔN variants were, as expected, still activated fivefold by the B1 peptide, but not by p13 (Fig. 6D). Interestingly, mutating the substrate-binding site in the NTD had a somewhat different effect on α-casein–dependent activation than was the case when this domain was deleted entirely. Although ClpBNTD-4A showed essentially no enhancement in ATP hydrolysis by α-casein, ClpBΔN showed partial activation (Fig. 6D), suggesting that the NTD blocks the entrance to the ClpB central channel before substrate engagement (see below) and hence in the case of ClpBNTD-4A, where client binding is not possible, significantly limits α-casein engagement by the tyrosine loops.
Fig. S8.

Hydrophobic residues in the NTD are required for interaction with substrates. (A) Superposition of selected regions of 1H–15N HSQC spectra of the uniformly 15N-labeled NTD (Left) or NTD-4A (W6A, L14A, L91A, L111A) (Right) with (red) or without (black) threefold excess p13. There are no changes to the spectrum of NTD-4A upon addition of p13. (B) ATPase activities of ClpBWT (WT), ClpBNTD-4A (NTD-4A), and ClpBΔN (ΔN) measured in the absence (blue) or the presence (maroon) of DnaK. Average values from three separate experiments are shown along with SDs. ClpBNTD-4A shows WT level ATPase activity and fourfold activation by the DnaK chaperone.
NTD Blocks the Entrance to the ClpB Central Channel Before Substrate Engagement.
As a follow-up on the observed differences in ATPase activities of ClpBNTD-4A and ClpBΔN, described above, we directly tested for potential defects in substrate translocation by introducing the NTD mutations described above into the BAP ClpP system. BAP is a ClpB variant that physically interacts and cooperates with a second chaperone, ClpP, in the degradation of substrates (9). BAP transfers model peptides and α-casein into the proteolytic chamber of the associated ClpP for degradation, so that the translocation activity of ClpB variants can be established by monitoring the efficacy of substrate degradation over time. NTD-4A, Y234A, and ΔN mutations were introduced into the BAP background and substrate translocation efficiencies compared as shown in Fig. 6E. Although α-casein was efficiently degraded in a control assay containing BAPWT/ClpP, close to 50% of the starting substrate remained upon mutating the tyrosine pores (BAPY234A), and ∼30% with the deletion of the NTD (BAPΔN) (Fig. 6E). Consistent with the earlier ATP hydrolysis experiments, introduction of NTD-4A into BAP led to a near-complete reduction of α-casein hydrolysis. The appreciable difference in α-casein degradation rates between BAP chaperones with a mutated NTD (BAPNTD-4A, little α-casein hydrolysis) and those lacking the NTD (BAPΔN; Fig. 6E) provides further evidence that the role of the NTD is to block the channel entrance in the absence of substrate engagement.
As a next step, we tested the impact of the NTD–substrate interaction on ClpB disaggregation, a reaction that requires additional chaperones (1). Reactivation rates of aggregated α-glucosidase and firefly luciferase were measured for ClpB, ClpBΔN, and ClpBNTD-4A in the presence of the DnaK chaperone system. WT ClpB, in collaboration with DnaK/DnaJ, was able to reactivate ∼70% of aggregated FFL and ∼45% of α-glucosidase. Deletion of the NTD reduced the disaggregation activity by ∼40% for α-glucosidase (Fig. 6F) and by ∼25% for FFL (Fig. 6G), indicating that the two aggregates have somewhat different dependencies on the NTD for disaggregation. Introduction of the 4A mutation into the ClpB NTD, however, had a much more significant effect, with reduced reactivation rates for both substrates of ∼70% compared with WT ClpB (Fig. 6 F and G). This suggests that, even in the presence of the DnaK chaperone system, the NTD at least partially blocks the channel before interaction with substrates.
SI Materials and Methods
Translational Diffusion.
Translational diffusion coefficients were measured by recording a series of 1D 13C-edited spectra, 25 °C, using a pulse scheme that is similar to an 15N-edited experiment published previously with 15N and 13C pulses interchanged (35). A diffusion delay of 200 ms was used in conjunction with encode/decode gradient strengths ranging from 5 to 54 G/cm. The resulting 1H signal was integrated over the methyl 1H frequency range to obtain intensities as a function of encoding/decoding gradient strength.
T1, T2, and Heteronuclear 1H–15N NOE Measurements.
Measurements of backbone 15N R1 and R2 relaxation rates and steady-state heteronuclear 1H–15N NOEs (HNOEs) were carried out on a 1 mM uniformly 15N-enriched NTD sample at 25 °C, 14.1 T, using TROSY-based pulse schemes (2). The 15N R1 data were acquired using relaxation delays ranging from 10 to 1,150 ms while 15N R1ρ rates were quantified using relaxation delays from 2 to 80 ms. R2 values were calculated from R1 and R1ρ rates according to the equation R1ρ = R1cos2θ + R2sin2θ, where θ = arctan(ωSL/Δω), Δω is the resonance offset from the spin-lock carrier, and ωSL is the spin-lock field strength (1.84 kHz). Values of relaxation rates were obtained by nonlinear least-squares fitting of the experimental data to a monoexponential decay function, Aexp(−RiT), Ri = {R1,R1ρ}. Heteronuclear NOE values were obtained by recording spectra in the presence (7-s relaxation delay) and absence (13-s relaxation delay) of a 1H saturation period of 6 s. Values of R1, R2, and NOE were fitted using the model-free approach with ModelFree4 software (55) assuming isotropic overall motion, to extract a single correlation time (τC) and residue-specific values of order parameters squared (S2) and effective internal correlation times (τe).
Measurement of Methyl 1H R2s Rates.
Residue-specific relaxation rates of slowly relaxing methyl 1H single-quantum transitions were measured using the pulse scheme of Tugarinov and Kay (36). A set of 2D 13C, 1H datasets were recorded where a relaxation delay, T, was varied from 1–20 ms for segmentally labeled hexameric ClpB (NTD-[2H, 13CH3-ILVM], ClpBΔN-[2H, 12C]) and from 1 to 100 ms for the monomeric NTD. R2s rates were obtained from exponential fits of peak intensity, I, as a function of relaxation delay, using the relation I = Ioexp(−R2sT).
Measurement of S2axisτc Values.
Methyl group S2axisτc values were measured on samples of segmentally labeled hexameric ClpB (NTD-[2H, 13CH3-ILVM], ClpBΔN-[2H, 12C]) and on monomeric [2H, 13CH3-ILVM]-labeled NTD. Data analysis was as described in ref. 37 using an approach that quantifies the time dependencies of sums (Iallow) and differences (Iforb) of magnetization derived from methyl 1H single- and triple-quantum transitions, respectively. For hexameric ClpB, Iallow and Iforb were measured at 55 °C with relaxation time (T) values of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 12, 14, and 16 ms, whereas for the monomeric NTD values were obtained at 25 °C using delays of T = 2, 4, 7, 10, 15, 20, 25, 30, 35, 40, 45, and 50 ms. The profiles Iforb/Iallow were fitted to the following:
| [S1] |
where
| [S2] |
In Eq. S1, η is as given in Eq. S2, where P2(x) = (3x2 − 1)/2, θaxis,HH is the angle (90°) between the methyl threefold axis and the vector that connects a pair of methyl 1H nuclei, τC is the assumed isotropic tumbling time of the protein, S2axis is the square of an order parameter quantifying the amplitude of motion of the methyl threefold symmetry axis, is Planck’s constant divided by 2π, γH is the gyromagnetic ratio of a proton spin, and rHH is the distance between pairs of methyl protons (1.813 Å). The parameter δ in Eq. S1 takes into account the 1H density around the methyl group in question. Values of S2axisτC measured for the NTD were used to obtain S2axis values on a per-residue basis assuming a value for τC from 15N spin relaxation experiments discussed above. The resultant order parameters were subsequently extrapolated to 55 °C by assuming temperature-dependent changes in S2axis values as described in ref. 56. These extrapolated order parameters were then used to obtain per-residue values of τC for methyl probes attached to the NTD of segmentally labeled ClpB for which S2axisτc values had been measured. To compare correlation times for the attached NTD and the “rigid” regions of the assumed isotropically tumbling hexameric ClpB complex a value of τC = 180 ns, 55 °C, based on the estimated value of τC = 120 ns for the Thermoplasma acidophilum α7α7 complex, 50 °C (31), was assumed by taking into account the viscosity dependence of D2O as a function of temperature (57) as well as the molecular-weight differences between the two particles.
NMR Thermal Melt.
Two-dimensional 1H–13C HMQC spectra were collected on a 0.4 mM sample of the Pin1 WW domain, alone and in the presence of 1 mM ClpB NTD, between the temperatures of 15 and 80 °C at intervals of 5 °C. The positions of many peaks shift considerably over the temperature range and can be used to monitor the unfolding of the protein (58), as it is known that Pin1 WW folds via a two-state mechanism (38). A thermal denaturation midpoint temperature, Tm, was determined by fitting changes in chemical shifts on a per-residue basis according to the following equation:
| [S3] |
In Eq. S3, the chemical shift of a methyl probe in the folded state changes linearly with temperature, T, as bF + mF*T, whereas the corresponding shift in the unfolded state is temperature independent, bU; ΔH is enthalpy change from unfolding at Tm; and ΔCp is the change in heat capacity upon unfolding.
Differential Scanning Calorimetry.
Differential scanning calorimetry (DSC) experiments were carried out with a VP-DSC calorimeter from MicroCal at a scan rate of 1.0 K/min. DSC data were analyzed with a two-state unfolding model (59), using theoretical heat capacity baselines for the unfolded state (60) and optimizing a linear heat capacity baseline for the folded state. The fits had five adjustable parameters: the denaturation midpoint, the enthalpy of denaturation at the midpoint, two parameters to describe the linear folded-state baseline, and a vertical offset correction for the unfolded baseline (61). The analyses were done in triplicate and yielded a denaturation temperature midpoint of 105.4 ± 0.1 °C and ΔHND = 12.92 ± 0.03 kcal⋅mol−1.
ClpB ATPase Activity Determination.
The ATPase activities of WT or mutant ClpB (10 μM) were measured spectrophotometrically (62) with an ATP-regeneration system that contained 2.5 mM phosphoenolpyruvate, 0.2 mM NADH, 50 μg/mL pyruvate kinase, 50 μg/mL lactate dehydrogenase, and 3 mM ATP at 55 °C (63). When indicated, α-casein (0.1 mg/mL), p13 (50 μM), peptide B1 (50 μM), or an ATP hydrolysis inactive T199A mutant of DnaK (25 μM), was added to the reaction. The changes in absorbance at 340 nm were monitored in a V-650 spectrophotometer. All ATPase assays were performed in triplicate.
Degradation Reactions.
All degradation reactions were carried out using 1 μM BAP variants, 1.5 μM ClpP, and 2 μM α-casein supplemented by an ATP regenerating system (63). α-Casein degradation was followed by gel electrophoresis and Coomassie blue staining. The amounts of remaining substrate at the indicated time points (0, 5, 10, 15, 30, 60, and 120 s) were quantified using ImageJ software.
Disaggregation Reactions.
Recombinant firefly luciferase (0.2 μM) was incubated for 30 min at 30 °C in denaturation buffer (25 mM Hepes-KOH, pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 6 M urea). To form irreversible aggregates, denatured luciferase was diluted 125-fold into renaturation buffer at 4 °C (25 mM Hepes, pH 7.5, 50 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.1 mg/mL BSA, 1 mM ATP, 20 mM creatine phosphate, 6 μg of creatine kinase). The refolding reaction was initiated through the addition of 5 μM WT ClpB or mutants of ClpB, 3 μM DnaK, 0.8 μM DnaJ, and 0.4 μM GrpE to 2 nM luciferase aggregates. Luminescence was measured following 2 h of disaggregation and subsequent addition of 50 μM luciferase reagent (Promega) to 2 μL of the refolding reaction.
α-Glucosidase from Bacillus stearothermophilus (Sigma) (0.1 μM) was denatured for 10 min at 75 °C in reaction buffer (25 mM Hepes-KOH, pH 7.5, 50 mM KCl, 10 mM MgCl2). Chaperones were then added (3.5 μM WT ClpB or mutants of ClpB, 3.0 μM DnaK, 0.8 μM DnaJ, 0.2 μM GrpE) before refolding at 37 °C. Following 2 h of disaggregation, the recovered α-glucosidase activity was measured as described previously (64).
Discussion
This study provides important insights into both the ClpB NTD–substrate interaction and the function of the NTD in the aggregate reactivation reaction. Using NMR spectroscopy, we have identified a substrate-binding site within the ClpB NTD that specifically interacts with stretches of hydrophobic residues in client proteins. In this context, it is noteworthy that protein aggregates, unlike degradation targets (39), are not tagged by any known mechanism, but do contain specific surface motifs such as hydrophobic stretches that do not typically occur on the surface of folded proteins. It is, therefore, interesting to speculate that the specificity of this binding site for clusters of hydrophobic amino acids may well be an important factor in how ClpB distinguishes between aggregates and properly folded proteins.
It has long been known that the rate of hydrolysis of nucleotides by ClpB can be enhanced through interactions with substrates (17). Here, we show that this enhancement is initially due to client protein binding to the NTD and later with the tyrosine pores. This sequential ordering of binding is suggested by ATPase activity assays (Fig. 6D), where α-casein is not able to stimulate ATP hydrolysis in ClpBNTD-4A because it cannot bind to the mutated NTD, although the tyrosine pores are intact in this construct and hence available for binding in a WT manner. Further evidence for sequential binding can be found in the ClpP-coupled α-casein degradation assays and DnaK-dependent protein disaggregation. Here too, mutations in the NTD substrate-binding groove have a dramatic effect on ClpB chaperone activation, whereas deletion of the entire NTD only modestly decreased the efficacy of coupled substrate translocation/degradation or disaggregation. This can be seen both in assays with the BAP/ClpP system (Fig. 6E) and in DnaK-dependent protein disaggregation reactions (Fig. 6 F and G). Thus, our results strongly suggest that for efficient ClpB function substrate contact with the NTDs precedes binding to the tyrosine pores.
Our work has also established that substrate interactions with the ClpB NTD hydrophobic groove stabilize the unfolded state of the client protein. Thus, the NTD may not only identify substrates, but also prime them for subsequent unfolding and translocation by the ClpB tyrosine pores. We also show that the NTD–substrate interaction is nucleotide independent so that the initial substrate engagement platform remains unaffected by the conformational changes in the ClpB machine during the unfolding and translocation process. This may be important for preventing premature substrate release.
Taken together, the NMR and biochemical results presented here imply that the ClpB NTD plays an important regulatory role through its interaction with substrates. Before establishing contact with clients, the NTD blocks the translocation channel in a way that significantly hinders substrate proteins from entering. This regulatory effect may ensure that partially unfolded or intrinsically disordered regions of properly folded, functional proteins are not mistakenly identified by the tyrosine loops and unfolded by ClpB. Once substrates bind to the hydrophobic groove of the NTD, the inhibitory effect is released, ClpB ATPase activity is stimulated, and interaction with the NTD stabilizes the unfolded state of the client protein. Thus, although the ClpB NTD may be dispensable for thermotolerance, it nevertheless plays an important role both in substrate recognition and in ClpB regulation throughout the disaggregation reaction.
Materials and Methods
Construct Preparation for Segmental Labeling of ClpB.
The DNA fragment encoding Thermus thermophilus ClpBΔN (amino acids 143–854) was generated by PCR using Thermus thermophilus ClpB (7) as a template and subsequently cloned into a modified pET28b vector, with the thrombin cleavage site replaced by a six-histidine tag followed by a tobacco etch virus (TEV) protease cleavage site. The NTD domain of ClpB (amino acids 1–142) was prepared by a ligation-free cloning method (40, 41) with residues 1–142 of ClpB PCR amplified with appropriate primers from a full-length ClpB vector and inserted into a pTWIN1 vector (NEB) immediately in front of the Mxe GyrA intein (an entire chitin binding domain, Ssp DnaB intein, and the MCS cassette was replaced by residues 1–142 of ClpB). An uncleavable hexahistidine tag was added at the N terminus of the construct by ligation of a duplex oligonucleotide containing five his codons (flanked by NdeI overhangs on both sides) into a NdeI-cleaved NTD plasmid. Mutations to all constructs, including those used in intein studies, were introduced using the QuikChange (Stratagene) approach and verified by DNA sequencing.
Protein Purification.
ClpB WT and mutants (7), NTD (amino acids 1–141) (7), ClpBΔN (amino acids 143–854) (7) and associated mutants, DnaK (7), DnaJ (42), GrpE (43), firefly luciferase (44), GFP (45), PhoA fragments (25), Sic1 (26), BAP (7), and ClpP (46) were prepared as described.
Purification of Segmentally Methyl-Labeled Hexameric ClpB Samples.
Plasmids encoding the NTD-intein or a ClpBΔN E271A/E668A/T143C variant that does not hydrolyze ATP (referred to as WT in the text) were transformed into E. coli BL21-CodonPlus (DE3) cells. Cells were grown at 37 °C in M9 D2O media supplemented with 14NH4Cl and [2H,12C]-glucose as the sole nitrogen and carbon sources, respectively. In the case of the NTD, methyl labeling of the Ile-δ1-[13CH3], Leu/Val-[13CH3,12CD3] variety (referred to as ILV-protein) was achieved following the procedure of Tugarinov et al. (28). Cells were grown to OD600 ∼ 0.8 and expression was induced by addition of 1 mM isopropyl β-d-1-thiogalactopyranoside and allowed to proceed overnight at 25 °C. Following expression, bacteria were harvested and proteins were purified on Ni-NTA resin (GE Healthcare). The NTD was cleaved from the intein fusion by incubating the protein overnight in a solution of 50 mM Hepes, pH 8.0, 300 mM NaCl, and 200 mM sodium mercaptoethane sulfonate (MESNA). The cleaved protein was further purified on a Ni-NTA column to remove the intein tag. All buffers used in this purification were supplemented with 20 mM MESNA to slow down the hydrolysis of the C-terminal α-thioester.
The ClpBΔN N-terminal 6His-tag was cleaved by TEV protease, followed by further purification on a Ni-NTA column. Cleaved products (NTD with a C-terminal α-thioester and ClpBΔN with a free N-terminal Cys) were partially unfolded in 2 M GnHCl and concentrated to the millimolar range. Ligation reactions were set up by mixing the products (NTD with a C-terminal α-thioester and ClpBΔN with a free N-terminal Cys) at a 3:1 ratio in 20 mM Hepes, pH 8.0, 300 mM NaCl, 2 M GnHCl, 2 mM tris(2-carboxyethyl)phosphine (TCEP), and 1 mM EDTA for 3–5 d. Ligation efficiency was 50–70%.
To separate the ligation products from the mixture, we introduced an uncleavable six-histidine tag at the N terminus of the NTD construct. The ligated product was separated from ClpBΔN lacking the six-histidine tag by purification over a Ni-NTA column under denaturing conditions (6 M GuHCl). Ligated ClpB was then refolded on the Ni-NTA column by gradually reducing the concentration of denaturing agent in the wash buffer (from 6 to 0 M GuHCl). The unligated NTD fragment was removed by further purification of the mixture over a HiPrep DEAE FF 16/10 column. All purifications were done under reducing conditions by supplementing the buffers with 2 mM TCEP.
Pure ligation product was assembled into hexameric ClpB by incubating the monomers for 1 h in 50 mM Hepes, pH 8.0, 20 mM KCl, 10 mM MgCl2, 2 mM TCEP, and 2 mM ATP. The hexameric protein was then purified on a HiLoad 16/60 Superdex 200-pg gel filtration column (GE Healthcare) and equilibrated with 50 mM Hepes, 20 mM KCl, pH 8.0, 2 mM ATP/ADP, and 0.03% NaN3. Purity and composition of the ligation product were confirmed by SDS/PAGE and electrospray ionization–MS.
NMR Spectroscopy.
All NMR experiments were carried on Varian INOVA spectrometers of 11.7 T (500 MHz), 14.1 T (600 MHz), or 18.8 T (800 MHz). The 600-MHz spectrometer was equipped with a cryogenically cooled probe. NMRPipe (47) and Sparky (48) were used to process and visualize NMR spectra, respectively.
NMR Chemical Shift Perturbations.
The interaction of the isolated ClpB NTD with client proteins was monitored by 2D 1H–15N TROSY HSQC experiments (49), 11.7 T, 55 °C. U-[1H,15N] NTD samples (200 μM concentration) were titrated with unlabeled, twofold excess (400 μM) α-casein, PhoA1-122, PhoA119-243, PhoA244-349, PhoA349-471, PhoA425-471, Sic1, κ-casein, c-Jun, UD, GFP, DnaK, heat-denatured MDH, chemically denatured FFL, peptide B1, and 13-mer peptide (p13). Additional binding studies were performed by recording spectra of U-[2H,15N] PhoA1-122, PhoA119-243, PhoA234-349, PhoA349-471, PhoA425-471, Sic1, or c-Jun samples each at a concentration of 300 μM in the presence of unlabeled NTD (600 μM), 18.8 T, 18 °C.
CSPs were calculated from the following relation:
| [1] |
with Δδ the corresponding amide 1H (ΔδH) or 15N (ΔδN) chemical shift change between free and bound NTD forms. CSPs between free NTD and NTD–substrate mixtures that were greater than 1 SD from the mean were considered significant.
To monitor the interaction of hexameric ClpB with substrates, NMR titrations were carried out using 2D 1H–13C HMQC experiments, 18.8 T, 55 °C. Unlabeled α-casein or p13 (600 μM) was added to segmentally labeled ClpBWT and ClpBY243A [NTD-2H, 13CH3-ILVM, ΔN-2H, 12C] samples (200 μM monomer concentration). CSPs were calculated from the following relation:
| [2] |
where ΔδH(C) is the shift change between methyl group 1H (13C) nuclei in apo and fully saturated forms of the protein, α (β) is 1 SD of the methyl 1H (13C) chemical shifts [separate values of α (β) are used for different methyl groups], as tabulated in the Biological Magnetic Resonance Data Bank (www.bmrb.wisc.edu). CSPs greater than 1 SD from the mean were considered significant.
NMR Titrations.
To estimate dissociation constants for the interaction of the isolated ClpB NTD with a number of unstructured proteins, U-[1H,15N] NTD samples (200 μM) were titrated with increasing amounts of unlabeled α-casein, PhoA1-122, PhoA119-243, PhoA234-349, PhoA349-471, PhoA425-471, Sic1, c-Jun, Src11-85, and p13 (net concentrations of 20, 40, 60, 80, 100, 150, 200, 250, 300, 400, 500, 600, and 800 μM) and the positions of cross-peaks monitored by recording 2D 1H–15N TROSY-HSQC spectra, 11.7 T, 55 °C. Additional titrations were carried out with [2H,13CH3-ILVM]-samples of NTD (200 μM) and α-casein or p13 (10, 20, 30, 40, 50, 75, 100, 150, 200, 300, and 400 μM) using 2D 1H–13C HMQC experiments.
For all of the above titrations Kd values were calculated by a nonlinear least-squares analysis using the following equation:
| [3] |
where [P]T and [L]T are the total protein (NMR labeled) and ligand (unlabeled) concentrations at each aliquot, Δδ′ is the difference in peak position after each aliquot, and Δδ′MAX is the change in shifts between apo and fully bound states of the protein. Binding isotherms were quantified separately for 1H, 15N, or 13C chemical shifts.
An NMR titration of ClpBY243A with p13 was carried out by recording a series of 2D 1H–13C-HMQC experiments where to a segmentally labeled [NTD-2H, 13CH3-ILVM, ΔN-2H, 12C] sample (200 μM) was added increasing amounts of unlabeled p13 (20, 40, 60, 80, 100, 150, 200, 250, 300, 400, 500, and 600 μM), 18.8 T, 55 °C. NMR titrations of ClpBWT or ClpBY243A with α-casein were performed at 55 °C, 18.8 T in a similar manner. Segmentally labeled ClpBWT (200 μM) or ClpBY243A (300 μM) [NTD-2H, 13CH3-ILVM, ΔN-2H, 12C] samples were titrated with increasing amounts of unlabeled α-casein (10, 20, 40, 60, 80, 100, 150, 200, 300, and 550 μM).
The CSPs from the titrations of ClpBY243A, for which there are only NTD binding sites, were fitted with the model of Eq. 4, which assumes that each microscopic binding event is energetically equivalent (multiplicative factors of KD take into account the number of ways that binding events can occur):
| [4] |
It was assumed that one molecule of p13 binds to each NTD site on ClpB (i.e., 6 sites per ClpB) so that n = 6. This is reasonable because p13 is a small peptide (13 residues). By contrast, titration profiles for α-casein were fit for different values of n and RMSDs between experimental and calculated chemical shifts used to establish the best fit (best value of n). The isotherms were fitted to
| [5] |
where CSPMAX is the chemical shift change at the completion of the titration and [PT] is the total protein concentration.
In the case of ClpBWT, where α-casein can bind to both NTD and tyrosine loop sites, isotherms were fitted with the model in Eq. 6:
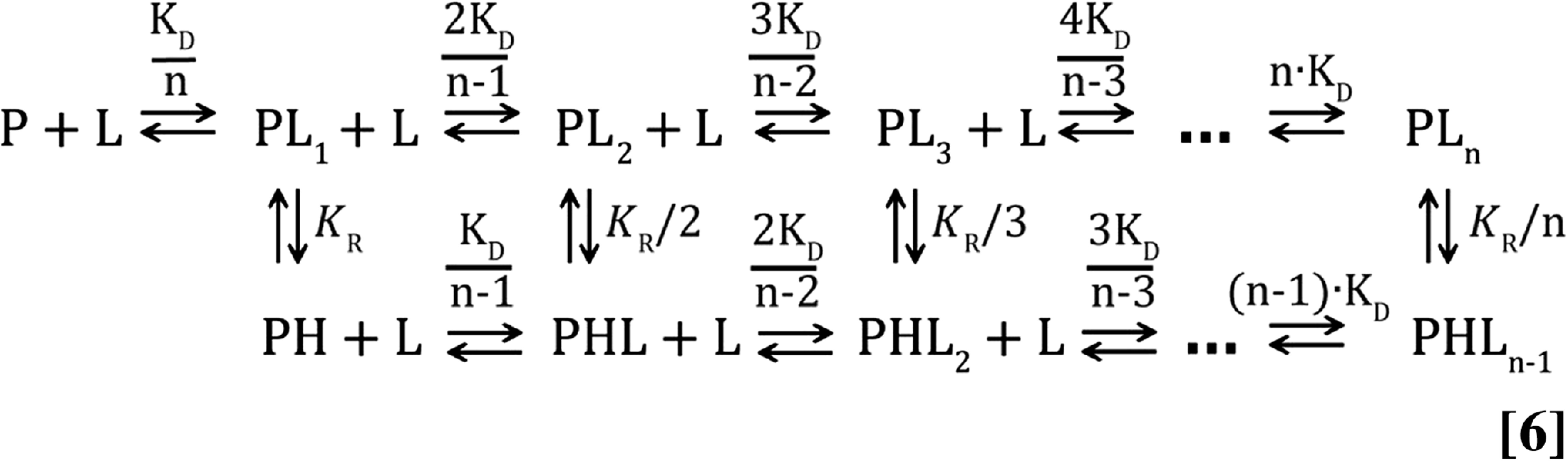 |
Details of this model are given in the legend to Fig. S6. As with the simpler model of Eq. 4, each binding event is assumed to have the same intrinsic affinity (i.e., same microscopic dissociation constant), whereas the multiplicative factors associated with each K take into account the multiplicity of ways that a ligand can bind to a particular state (50). Each isotherm was fitted assuming different values of n in the range from 1 to 6 (i.e., between one and six ligand binding sites on the ClpB hexamer). During the course of the fits, the concentrations of each state in Eq. 6 were obtained by numerically solving the resultant equations relating concentration to dissociation constants, including mass-conservation for all components of the reaction (ClpBWT and α-casein). Concentrations were converted to CSPs for low (NTD, CSPL) and high (tyrosine pore, CSPH) affinity binding sites, using the following equations:
| [7.1] |
| [7.2] |
where PHL0 = PH. All isotherms were fit together to extract global parameters KD and KR and residue-specific CSPMAX values. Distributions of fitted parameters were obtained by running 200 bootstrap simulations (51).
Fits of all titration data and bootstrap analyses were performed using in-house written programs (Python 2.7), exploiting the optimization function leastsq from the SciPy 9.0 library.
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research and the Natural Sciences and Engineering Research Council of Canada (to L.E.K.). WT ClpB and PhoA constructs were generously provided by F. Tsai (Baylor College of Medicine) and B. Kalodimos (Rutgers University), respectively. L.E.K. holds a Canada Research Chair in Biochemistry.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1512783112/-/DCSupplemental.
References
- 1.Glover JR, Lindquist S. Hsp104, Hsp70, and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell. 1998;94(1):73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez Y, Lindquist SL. HSP104 required for induced thermotolerance. Science. 1990;248(4959):1112–1115. doi: 10.1126/science.2188365. [DOI] [PubMed] [Google Scholar]
- 3.Doyle SM, Wickner S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem Sci. 2009;34(1):40–48. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 4.Hodson S, Marshall JJ, Burston SG. Mapping the road to recovery: The ClpB/Hsp104 molecular chaperone. J Struct Biol. 2012;179(2):161–171. doi: 10.1016/j.jsb.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 5.Zietkiewicz S, Lewandowska A, Stocki P, Liberek K. Hsp70 chaperone machine remodels protein aggregates at the initial step of Hsp70-Hsp100-dependent disaggregation. J Biol Chem. 2006;281(11):7022–7029. doi: 10.1074/jbc.M507893200. [DOI] [PubMed] [Google Scholar]
- 6.Mogk A, Kummer E, Bukau B. 2015. Cooperation of Hsp70 and Hsp100 chaperone machines in protein disaggregation. Front Mol Biosci 2:22.
- 7.Rosenzweig R, Moradi S, Zarrine-Afsar A, Glover JR, Kay LE. Unraveling the mechanism of protein disaggregation through a ClpB-DnaK interaction. Science. 2013;339(6123):1080–1083. doi: 10.1126/science.1233066. [DOI] [PubMed] [Google Scholar]
- 8.Doyle SM, et al. Interplay between E. coli DnaK, ClpB and GrpE during protein disaggregation. J Mol Biol. 2015;427(2):312–327. doi: 10.1016/j.jmb.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weibezahn J, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119(5):653–665. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 10.Lee S, et al. The structure of ClpB: A molecular chaperone that rescues proteins from an aggregated state. Cell. 2003;115(2):229–240. doi: 10.1016/s0092-8674(03)00807-9. [DOI] [PubMed] [Google Scholar]
- 11.Seyffer F, et al. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat Struct Mol Biol. 2012;19(12):1347–1355. doi: 10.1038/nsmb.2442. [DOI] [PubMed] [Google Scholar]
- 12.Wendler P, Ciniawsky S, Kock M, Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 2012;1823(1):2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 13.Hanson PI, Whiteheart SW. AAA+ proteins: Have engine, will work. Nat Rev Mol Cell Biol. 2005;6(7):519–529. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 14.Lee S, Choi JM, Tsai FT. Visualizing the ATPase cycle in a protein disaggregating machine: Structural basis for substrate binding by ClpB. Mol Cell. 2007;25(2):261–271. doi: 10.1016/j.molcel.2007.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlieker C, et al. Substrate recognition by the AAA+ chaperone ClpB. Nat Struct Mol Biol. 2004;11(7):607–615. doi: 10.1038/nsmb787. [DOI] [PubMed] [Google Scholar]
- 16.Doyle SM, Hoskins JR, Wickner S. DnaK chaperone-dependent disaggregation by caseinolytic peptidase B (ClpB) mutants reveals functional overlap in the N-terminal domain and nucleotide-binding domain-1 pore tyrosine. J Biol Chem. 2012;287(34):28470–28479. doi: 10.1074/jbc.M112.383091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beinker P, Schlee S, Groemping Y, Seidel R, Reinstein J. The N terminus of ClpB from Thermus thermophilus is not essential for the chaperone activity. J Biol Chem. 2002;277(49):47160–47166. doi: 10.1074/jbc.M207853200. [DOI] [PubMed] [Google Scholar]
- 18.Barnett ME, Nagy M, Kedzierska S, Zolkiewski M. The amino-terminal domain of ClpB supports binding to strongly aggregated proteins. J Biol Chem. 2005;280(41):34940–34945. doi: 10.1074/jbc.M505653200. [DOI] [PubMed] [Google Scholar]
- 19.Chow IT, Barnett ME, Zolkiewski M, Baneyx F. The N-terminal domain of Escherichia coli ClpB enhances chaperone function. FEBS Lett. 2005;579(20):4242–4248. doi: 10.1016/j.febslet.2005.06.055. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka N, Tani Y, Hattori H, Tada T, Kunugi S. Interaction of the N-terminal domain of Escherichia coli heat-shock protein ClpB and protein aggregates during chaperone activity. Protein Sci. 2004;13(12):3214–3221. doi: 10.1110/ps.04780704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle SM, Hoskins JR, Wickner S. Collaboration between the ClpB AAA+ remodeling protein and the DnaK chaperone system. Proc Natl Acad Sci USA. 2007;104(27):11138–11144. doi: 10.1073/pnas.0703980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li T, Lin J, Lucius AL. Examination of polypeptide substrate specificity for Escherichia coli ClpB. Proteins. 2015;83(1):117–134. doi: 10.1002/prot.24710. [DOI] [PubMed] [Google Scholar]
- 23.Acebrón SP, Martín I, del Castillo U, Moro F, Muga A. DnaK-mediated association of ClpB to protein aggregates. A bichaperone network at the aggregate surface. FEBS Lett. 2009;583(18):2991–2996. doi: 10.1016/j.febslet.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 24.Winkler J, Tyedmers J, Bukau B, Mogk A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol. 2012;198(3):387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saio T, Guan X, Rossi P, Economou A, Kalodimos CG. Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science. 2014;344(6184):1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mittag T, et al. Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc Natl Acad Sci USA. 2008;105(46):17772–17777. doi: 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H–13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125(34):10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- 28.Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat Protoc. 2006;1(2):749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 29.Rosenzweig R, Kay LE. Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu Rev Biochem. 2014;83:291–315. doi: 10.1146/annurev-biochem-060713-035829. [DOI] [PubMed] [Google Scholar]
- 30.Gelis I, et al. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131(4):756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445(7128):618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 32.Shah NH, Muir TW. Inteins: Nature’s gift to protein chemists. Chem Sci (Camb) 2014;5(1):446–461. doi: 10.1039/C3SC52951G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: A general method for protein engineering. Proc Natl Acad Sci USA. 1998;95(12):6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Severinov K, Muir TW. Expressed protein ligation, a novel method for studying protein-protein interactions in transcription. J Biol Chem. 1998;273(26):16205–16209. doi: 10.1074/jbc.273.26.16205. [DOI] [PubMed] [Google Scholar]
- 35.Choy WY, et al. Distribution of molecular size within an unfolded state ensemble using small-angle X-ray scattering and pulse field gradient NMR techniques. J Mol Biol. 2002;316(1):101–112. doi: 10.1006/jmbi.2001.5328. [DOI] [PubMed] [Google Scholar]
- 36.Tugarinov V, Kay LE. Relaxation rates of degenerate 1H transitions in methyl groups of proteins as reporters of side-chain dynamics. J Am Chem Soc. 2006;128(22):7299–7308. doi: 10.1021/ja060817d. [DOI] [PubMed] [Google Scholar]
- 37.Sun H, Kay LE, Tugarinov V. An optimized relaxation-based coherence transfer NMR experiment for the measurement of side-chain order in methyl-protonated, highly deuterated proteins. J Phys Chem B. 2011;115(49):14878–14884. doi: 10.1021/jp209049k. [DOI] [PubMed] [Google Scholar]
- 38.Kubelka J, Hofrichter J, Eaton WA. The protein folding “speed limit.”. Curr Opin Struct Biol. 2004;14(1):76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Kästle M, Grune T. Interactions of the proteasomal system with chaperones: Protein triage and protein quality control. Prog Mol Biol Transl Sci. 2012;109:113–160. doi: 10.1016/B978-0-12-397863-9.00004-3. [DOI] [PubMed] [Google Scholar]
- 40.Chen GJ, Qiu N, Karrer C, Caspers P, Page MG. 2000. Restriction site-free insertion of PCR products directionally into vectors. BioTechniques 28(3):498–500, 504–505. [DOI] [PubMed]
- 41.van den Ent F, Löwe J. RF cloning: A restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods. 2006;67(1):67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 42.Klostermeier D, Seidel R, Reinstein J. The functional cycle and regulation of the Thermus thermophilus DnaK chaperone system. J Mol Biol. 1999;287(3):511–525. doi: 10.1006/jmbi.1999.2636. [DOI] [PubMed] [Google Scholar]
- 43.Klostermeier D, Seidel R, Reinstein J. Functional properties of the molecular chaperone DnaK from Thermus thermophilus. J Mol Biol. 1998;279(4):841–853. doi: 10.1006/jmbi.1998.1816. [DOI] [PubMed] [Google Scholar]
- 44.Lum R, Niggemann M, Glover JR. Peptide and protein binding in the axial channel of Hsp104. Insights into the mechanism of protein unfolding. J Biol Chem. 2008;283(44):30139–30150. doi: 10.1074/jbc.M804849200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guterman A, Glickman MH. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J Biol Chem. 2004;279(3):1729–1738. doi: 10.1074/jbc.M307050200. [DOI] [PubMed] [Google Scholar]
- 46.Religa TL, Ruschak AM, Rosenzweig R, Kay LE. Site-directed methyl group labeling as an NMR probe of structure and dynamics in supramolecular protein systems: Applications to the proteasome and to the ClpP protease. J Am Chem Soc. 2011;133(23):9063–9068. doi: 10.1021/ja202259a. [DOI] [PubMed] [Google Scholar]
- 47.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 48. Goddard TD, Kneller DG (1999) SPARKY 3 (University of California, San Francisco)
- 49.Pervushin K, Riek R, Wider G, Wüthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci USA. 1997;94(23):12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cantor CR, Schimmel PR (1971) Biophysical Chemistry (Freeman, San Francisco)
- 51.Press WH, Teukolsky SA, Vetterling WT, Flannery BP. 1988. Numerical Recipes in C (Cambridge Univ Press, Cambridge, UK)
- 52.Carroni M, et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. eLife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eisenberg D, Schwarz E, Komaromy M, Wall R. Analysis of membrane and surface protein sequences with the hydrophobic moment plot. J Mol Biol. 1984;179(1):125–142. doi: 10.1016/0022-2836(84)90309-7. [DOI] [PubMed] [Google Scholar]
- 54.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. Freeman; New York: 1999. [Google Scholar]
- 55.Mandel AM, Akke M, Palmer AG., 3rd Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J Mol Biol. 1995;246(1):144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 56.Song XJ, Flynn PF, Sharp KA, Wand AJ. Temperature dependence of fast dynamics in proteins. Biophys J. 2007;92(6):L43–L45. doi: 10.1529/biophysj.106.102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cho CH, Urquidi J, Singh S, Robinson GW. Thermal offset viscosities of liquid H2O, D2O, and T2O. J Phys Chem B. 1999;103(11):1991–1994. [Google Scholar]
- 58.Ruschak AM, Religa TL, Breuer S, Witt S, Kay LE. The proteasome antechamber maintains substrates in an unfolded state. Nature. 2010;467(7317):868–871. doi: 10.1038/nature09444. [DOI] [PubMed] [Google Scholar]
- 59.Freire E. Thermal denaturation methods in the study of protein folding. Methods Enzymol. 1995;259:144–168. doi: 10.1016/0076-6879(95)59042-0. [DOI] [PubMed] [Google Scholar]
- 60.Häckel M, Hinz HJ, Hedwig GR. A new set of peptide-based group heat capacities for use in protein stability calculations. J Mol Biol. 1999;291(1):197–213. doi: 10.1006/jmbi.1999.2952. [DOI] [PubMed] [Google Scholar]
- 61.Privalov G, Kavina V, Freire E, Privalov PL. Precise scanning calorimeter for studying thermal properties of biological macromolecules in dilute solution. Anal Biochem. 1995;232(1):79–85. doi: 10.1006/abio.1995.9957. [DOI] [PubMed] [Google Scholar]
- 62.Nørby JG. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- 63.Stiggall DL, Galante YM, Hatefi Y. 1979. Preparation and properties of complex V. Methods Enzymol 55:308–315, 819–821.
- 64.Biter AB, Lee S, Sung N, Tsai FT. Structural basis for intersubunit signaling in a protein disaggregating machine. Proc Natl Acad Sci USA. 2012;109(31):12515–12520. doi: 10.1073/pnas.1207040109. [DOI] [PMC free article] [PubMed] [Google Scholar]