

Abstract
The EglN2/PHD1 prolyl hydroxylase is an important oxygen sensor contributing to breast tumorigenesis. Emerging studies suggest that there is functional cross talk between oxygen sensing and mitochondrial function, both of which play an essential role for sustained tumor growth. However, the potential link between EglN2 and mitochondrial function remains largely undefined. Here, we show that EglN2 depletion decreases mitochondrial respiration in breast cancer under normoxia and hypoxia, which correlates with decreased mitochondrial DNA in a HIF1/2α‐independent manner. Integrative analyses of gene expression profile and genomewide binding of EglN2 under hypoxic conditions reveal nuclear respiratory factor 1 (NRF1) motif enrichment in EglN2‐activated genes, suggesting NRF1 as an EglN2 binding partner. Mechanistically, by forming an activator complex with PGC1α and NRF1 on chromatin, EglN2 promotes the transcription of ferridoxin reductase (FDXR) and maintains mitochondrial function. In addition, FDXR, as one of effectors for EglN2, contributes to breast tumorigenesis in vitro and in vivo. Our findings suggest that EglN2 regulates mitochondrial function in ERα‐positive breast cancer.
Keywords: EglN2, hypoxia, mitochondria, NRF1, tumorigenesis
Subject Categories: Cancer, Metabolism, Transcription
Introduction
The presence of hypoxic cells in the tumor microenvironment was proposed by Thomlinson and Gray more than 50 years ago (Thomlinson & Gray, 1955). These hypoxic cells confer radio‐ or chemotherapeutic resistance and therefore are hypothesized to be under selection for aggressive malignancy during the course of cancer development (Brown & Wilson, 2004). One central question is how hypoxic cancer cells sense their oxygen availability, adapt to the stressful environment, and proliferate out of control. The key proteins mediating oxygen sensing in these cells mainly involve proteins that are responsible for the hydroxylation of hypoxia‐inducible factor (HIF), namely the prolyl hydroxylases EglN1‐3. As a key EglN enzyme substrate, HIF1α is hydroxylated on prolines 402 and 564 under normoxic conditions. This promotes the binding of HIF1α to the von Hippel–Lindau (VHL) E3 ligase complex, leading to its subsequent ubiquitylation and proteasomal degradation (Kaelin & Ratcliffe, 2008). Under hypoxia, EglNs lose their ability to hydroxylate HIF1α and HIF2α. This leads to HIFα stabilization and dimerization with HIF1β (ARNT), thereby activating transcription of many key genes involved in cell proliferation, metabolism, and angiogenesis (Semenza, 2012). Therefore, EglNs couple oxygen availability to the transcription of many genes linked to hypoxic adaptation.
About a century ago, Otto Warburg proposed that unlike normal cells, cancer cells have the tendency to utilize glycolysis to produce ATP in the presence of oxygen, termed “aerobic glycolysis” (Warburg et al, 1924). The central idea of the Warburg theory is the transition from mitochondria‐mediated oxidative phosphorylation to a glycolytic metabolism in cancer cells (Warburg, 1925). However, tumor cells mostly maintain intact mitochondria, and increasing experimental evidence suggests that mitochondria still play important functions in cancer cells for sustained tumor growth (Nakashima et al, 1984; Guppy et al, 2002; Gatenby & Gillies, 2004; Zu & Guppy, 2004; Moreno‐Sanchez et al, 2007; Weinberg et al, 2010; Guo et al, 2011, 2013; Wallace, 2012; Strohecker et al, 2013). Chandel and colleagues recently showed that mitochondrial metabolism is essential for oncogenic Ras‐induced tumorigenicity (Weinberg et al, 2010). In support of this finding, recent studies showed that autophagy sustains mitochondrial metabolism that is important for oncogenic Ras‐ or Raf‐mediated tumorigenicity (Guo et al, 2011; Strohecker & White, 2014). In addition, glutamine‐mediated oxidative phosphorylation was reported to be a major ATP production resource under either normoxia or hypoxia (Fan et al, 2013). From the therapeutic perspective, recent studies implicated metformin, a mitochondrial complex I inhibitor, as an attractive therapeutic agent in cancer therapy (Owen et al, 2000; Zakikhani et al, 2006; Bost et al, 2012; Pollak, 2012; Sanchez‐Alvarez et al, 2013; Wheaton et al, 2014). Collectively, these studies suggest mitochondrial inhibitors to be promising treatment modalities for cancer, either alone or in combination with other therapies.
There are emerging studies clarifying the functional link between oxygen sensing, mitochondrial function, and metabolism (Papandreou et al, 2006; Simon, 2006; Zhang et al, 2007; Aragones et al, 2008). For example, HIF1α was shown to repress mitochondrial biogenesis or cellular respiration via regulating c‐Myc activity and pyruvate dehydrogenase kinase 1 (PDK1) expression (Kim et al, 2006; Papandreou et al, 2006; Zhang et al, 2007). However, the role of EglN family members, which serve as direct oxygen sensors, on mitochondrial function remains largely undefined in cancer. Previous studies from our group, as well as others, showed that EglN2 mRNA levels are induced by estrogen and are higher in ERα‐positive breast cancer compared to ERα‐negative breast cancer (Seth et al, 2002; Appelhoff et al, 2004; Zhang et al, 2009). Depletion of EglN2 decreases cell proliferation and breast tumorigenesis in an orthotopic breast cancer model by diminishing cyclin D1 transcription (Zhang et al, 2009). However, it remains largely unknown whether in breast cancer cells there is a functional link between EglN2 and mitochondria and if so, what is the underlying mechanism regulating the effect of EglN2 on mitochondrial function?
In this study, we show that EglN2 depletion causes diminished mitochondrial respiration in a HIF1/2α‐independent manner, resulting partly from decreased mtDNA content in breast cancer cells. Our integrative analyses of EglN2 ChIP‐Seq and gene expression profiling under hypoxic conditions unveil a unique NRF1 motif enrichment in EglN2‐upregulated genes. Mechanistically, by forming an activator complex with PGC1α and NRF1 on chromatin, EglN2 promotes FDXR gene expression and regulates mitochondrial function in a PGC1α‐ and NRF1‐dependent manner. FDXR, as an important downstream target of NRF1 and EglN2 signaling, at least partially mediates the effect of EglN2 on mtDNA content and contributes to tumorigenesis in ERα‐positive breast cancer.
Results
EglN2 affects mitochondrial function in breast cancer
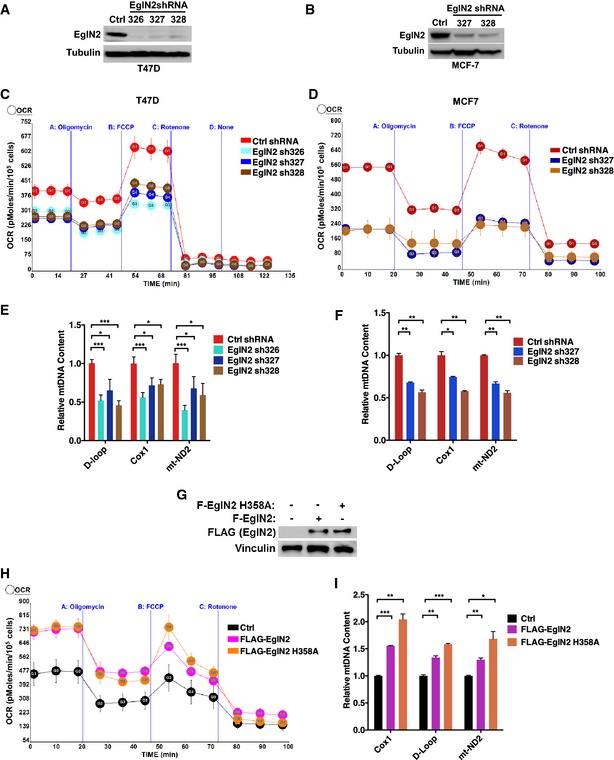
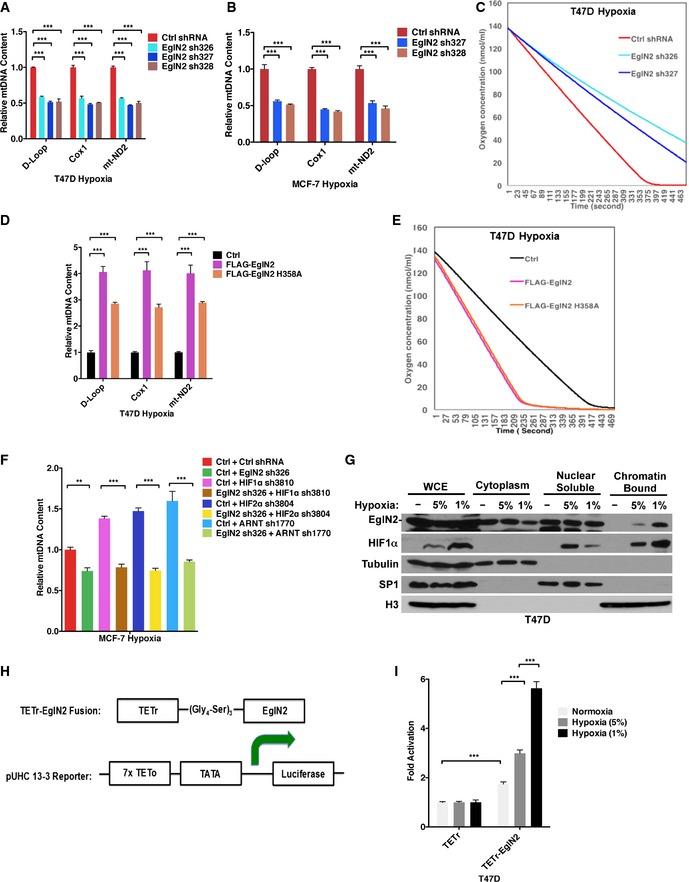
Our previous research demonstrated that EglN2 was critical to mediate ERα‐positive breast tumorigenesis (Zhang et al, 2009). Emerging literature suggests that in cancer cells, in addition to shifts in glycolytic activities, mitochondrial function also plays an important role for sustained tumor growth (Nakashima et al, 1984; Guppy et al, 2002; Gatenby & Gillies, 2004; Zu & Guppy, 2004; Moreno‐Sanchez et al, 2007; Weinberg et al, 2010; Guo et al, 2011, 2013; Strohecker et al, 2013). Furthermore, there exists the potential connection between oxygen sensing and mitochondrial function (Zhang et al, 2007; Aragones et al, 2008). However, it remains unknown whether there is a direct functional link between EglN2 and mitochondrial function in cancer. To address this, we firstly used three independent hairpins to deplete EglN2 expression in the ERα‐positive breast cancer cell line T47D as indicated by diminished EglN2 protein (Fig 1A). Next, we examined the effect of EglN2 knockdown on mitochondrial respiration, as measured by oxygen consumption rate (OCR) with XF‐24 extracellular flux analyzer. Breast cancer cells depleted of EglN2 displayed impaired mitochondrial respiration either under conditions of basal or maximal oxygen consumption induced by FCCP treatment (Fig 1C). To validate the on‐target effect of EglN2 shRNA, we also rescued EglN2 knockdown by infecting cells with an shRNA‐resistant EglN2 overexpression construct (Fig EV1A) and found that EglN2 overexpression rescued the phenotype of EglN2 shRNA on OCR in these cells (Fig EV1B).
Figure 1. EglN2 affects mitochondrial function in ERα‐positive breast cancer cells.
-
A, BImmunoblot from T47D (A) or MCF‐7 (B) cells infected with lentivirus encoding EglN2 shRNA (326, 327 or 328) or control shRNA (Ctrl).
-
C, DMeasurement of oxygen consumption rate (OCR) in T47D (C) or MCF‐7 (D) cells infected with lentivirus encoding EglN2 shRNAs or control shRNA (Ctrl shRNA) by using the Seahorse XF24 extracellular flux analyzer.
-
E, FqRT–PCR quantification of mtDNA from T47D (E) or MCF‐7 (F) cells infected with lentivirus encoding EglN2 shRNA (326, 327, or 328) or control shRNA.
-
G–IImmunoblot (G), measurement of OCR (H), and quantification of mtDNA (I) from T47D cells infected with lentivirus encoding FLAG‐EglN2 (F‐EglN2), FLAG‐EglN2 H358A, or control (Ctrl).
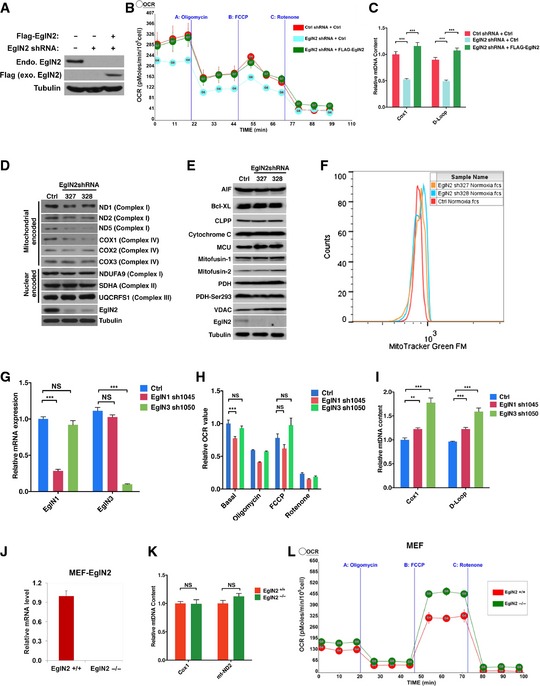
Figure EV1. EglN2 regulates mitochondrial function.
-
A–CImmunoblot of cell lysates (A), measurement of oxygen consumption rate (B), and qRT–PCR quantification of mtDNA (C) from T47D cells infected with lentivirus encoding either EglN2 shRNA or control (Ctrl) shRNA followed by another infection with lentivirus encoding either FLAG‐EglN2 or control (Ctrl).
-
D–FImmunoblot of cell lysates with indicated antibodies (D, E) and measurement of mitochondrial mass with MitoTracker Green staining followed by flow cytometry (F) from T47D cells infected with lentivirus encoding EglN2 shRNA (327 or 328) or control shRNA (Ctrl).
-
G–IqRT–PCR of mRNA (G), measurement of oxygen consumption rate (H), and qRT–PCR quantification of mtDNA (I) from T47D cells infected with lentivirus encoding either EglN1 shRNA (1045), EglN3 shRNA (1050), or control (Ctrl) shRNA.
-
J–LqRT–PCR of mRNA (J), measurement of oxygen consumption rate (K), and qRT–PCR quantification of mtDNA (L) from EglN2 KO or WT MEF cells.
Data information: Two‐tailed Student's t‐test was used to examine the P‐values from at least three replicate experiments. Error bars represent SEM. **P < 0.01, ***P < 0.005. NS denotes not significant.
To explore further how EglN2 contributes to mitochondrial function in these cells, we examined two critical determinants of mitochondrial respiratory function, mitochondrial DNA (mtDNA), and mitochondrial mass. To examine mtDNA content in these cells, we isolated genomic DNA followed by qRT–PCR for several different mtDNA markers, including D‐Loop, Cox1, and mt‐ND2. The amount of these mtDNA markers was normalized against nuclear DNA (nuDNA) to examine the relative abundance of mtDNA as described previously (Moiseeva et al, 2009). Depletion of EglN2 by several independent hairpins decreased mtDNA content in T47D cells (Fig 1E), the effect rescued by overexpression of shRNA‐resistant EglN2 (Fig EV1C). In addition, the effect EglN2 on mtDNA content was validated by the finding that EglN2 depletion led to decreased protein expression for a subset of mitochondrial‐encoded proteins, such as mitochondrial complex I subunits mt‐ND1, 2, and 5 (Fig EV1D), also some of mitochondrial complex IV subunits such as COX1 but not COX2 or 3. On the other hand, we did not detect distinctive difference in nuclear‐encoded mitochondrial proteins (NDUFA9, SDHA, and UQCRFS1) in some of these mitochondrial complexes (Fig EV1D), indicating a potential mitochondrial–nuclear protein imbalance upon EglN2 depletion. In addition to mtDNA content, we used two independent hairpins against EglN2 and examined their effect on mitochondrial mass in T47D cells. For this purpose, we examined protein levels of several mitochondrial‐located proteins, including AIF, Bcl‐XL, CLPP, cytochrome C, MCU, mitofusin‐1 and mitofusin‐2, PDH, and VDAC, and found the expression of these markers was not affected by EglN2 depletion (Fig EV1E), which was further supported by the lack of change for mitotracker green staining intensity upon EglN2 depletion (Fig EV1F). Therefore, our data suggest that EglN2 depletion decreases mtDNA content, but not mitochondrial mass, which is consistent with finding that mtDNA depletion disrupted mitochondrial function and breast tumorigenesis without affecting mitochondrial mass (Holmuhamedov et al, 2003; Yu et al, 2007).
In a parallel set of experiments, we depleted EglN2 by two independent hairpins in another ERα‐positive breast cancer cell line MCF‐7 (Fig 1B). Consistently, EglN2 knockdown diminished mitochondrial respiration under basal or maximal oxygen consumption condition in MCF‐7 cells (Fig 1D). Depletion of EglN2 also decreased mtDNA content in these cells, similarly as the phenotype observed in T47D cells (Fig 1F). In a complementary set of experiments, overexpression of EglN2 in T47D cells increased mitochondrial respiration (Fig 1G and H), corresponding to increased mtDNA content in these cells (Fig 1I). It is important to note that the catalytic dead EglN2 H358A mutant, as described previously (Epstein et al, 2001; Zhang et al, 2009), increased mitochondrial respiration and mtDNA content, indicating the effect of EglN2 on mtDNA is independent of its enzymatic activity (Fig 1H and I).
To examine whether the effect of OCR is EglN2 specific, we also depleted EglN1 or EglN3 expression by respective shRNAs and found that OCR was not significantly affected by EglN1 or EglN3 under maximal oxygen consumption condition (Fig EV1G and H). Under basal condition, EglN3 depletion did not affect OCR but EglN1 depletion modestly diminished OCR (Fig EV1H). Interestingly, depletion of EglN1 or 3 led to modest upregulation of mtDNA, opposite to the phenotype observed in EglN2‐depleted cells (Fig EV1I). It is worth noting that the effect of EglN2 on OCR and mtDNA is specific for breast cancer cells tested, but not for murine embryonic fibroblast (MEFs) as EglN2 knockout MEFs displayed similar OCR and mtDNA content as the littermate wild‐type control (Fig EV1J–L). Accumulatively, our data suggest that EglN2 positively regulates mitochondrial function in ERα‐positive breast cancer cells.
EglN2 regulates mitochondrial function in a HIF1/2α‐independent manner
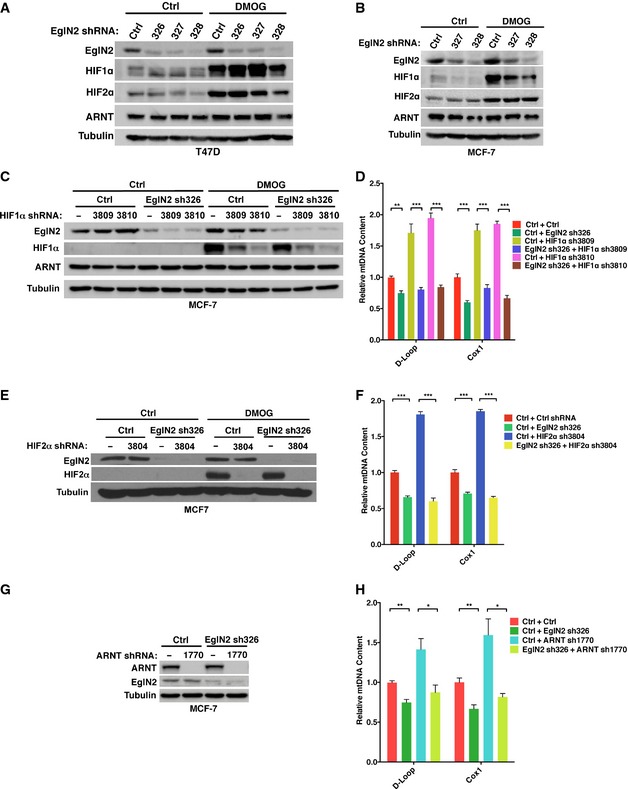
Previous research showed that HIF1α repressed mitochondrial biogenesis or cellular respiration via regulating c‐Myc activity and pyruvate dehydrogenase kinase 1 (PDK1) expression (Kim et al, 2006; Papandreou et al, 2006; Zhang et al, 2007). Although all of EglN family members can hydroxylate HIFα in vitro, EglN1 remains the dominant HIF prolyl hydroxylase regulating HIFα based on data from cell culture and genetically engineered mice (Berra et al, 2003; To & Huang, 2005; Takeda et al, 2006; Minamishima et al, 2008). To examine whether EglN2 knockdown might decrease mitochondrial function by inducing HIFα stabilization, we firstly determined HIF1α and HIF2α protein levels upon EglN2 depletion in T47D and MCF‐7 cells. In both cell lines, EglN2 depletion by multiple independent shRNAs did not lead to HIF1α or HIF2α upregulation, which is consistent with previously published data obtained with EglN2 siRNAs for MCF‐7 cells (Fig 2A and B) (Appelhoff et al, 2004). This finding suggests that EglN2 regulates mitochondrial function in a HIF1/2α‐independent manner. To further confirm this finding, we generated MCF‐7 cell lines depleted of HIF1α expression with two independent hairpins (Fig 2C). Consistent with the previous publication, HIF1α depletion led to increased mtDNA content as well as increased OCR (Figs 2D and EV2A) (Zhang et al, 2007). However, EglN2 depletion still led to decreased OCR as well as mtDNA content in these cells depleted of HIF1α, strengthening the notion that the effect of EglN2 on mitochondrial function is independent of HIF1α (Figs 2D and EV2A). In addition to HIF1α, we also depleted HIF2α in these cells (Fig 2E) and found similarly that EglN2 knockdown decreased mtDNA content and OCR independent of HIF2α (Figs 2F and EV2B).
Figure 2. EglN2 regulates mitochondrial function in a HIF1/2α‐independent manner.
-
A, BImmunoblot from T47D (A) or MCF‐7 (B) cells infected with lentivirus encoding EglN2 shRNA (326, 327, or 328) or control shRNA (Ctrl).
-
C–HImmunoblot of cell lysates (C, E, G) and qRT–PCR quantification of mtDNA (D, F, H) from MCF‐7 cells infected with lentivirus encoding either HIF1α shRNAs (3809, 3810) (C), HIF2α shRNA (3804) (E), ARNT shRNA (1770) (G), or control (−) followed by another infection with lentivirus encoding either EglN2 shRNA (326) or control (Ctrl) shRNA.
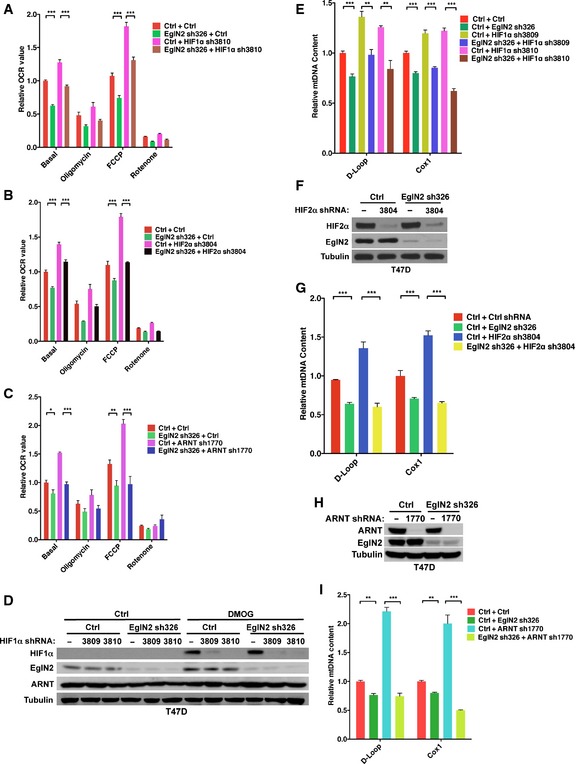
Figure EV2. EglN2 regulates mitochondrial function in a HIF1/2α‐independent manner.
-
A–CMeasurement of oxygen consumption from MCF7 cells infected with lentivirus encoding either HIF1 shRNAs (3809, 3810) (A), HIF2 shRNA (3804) (B), ARNT shRNA (1770) (C), or control (Ctrl) shRNA followed by another infection with lentivirus encoding either EglN2 shRNA or control (Ctrl) shRNA.
-
D–IImmunoblot of cell lysates (D, F and H) and qRT–PCR quantification of mtDNA (E, G and I) from T47D cells infected with lentivirus encoding either HIF1α shRNAs (3809, 3810) (D), HIF2α shRNA (3804) (F), ARNT shRNA (1770) (H), or control (−) followed by another infection with lentivirus encoding either EglN2 shRNA (326) or control (Ctrl) shRNA.
Data information: Two‐tailed Student's t‐test was used to examine the P‐values from at least three replicate experiments. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.005.
As a complementary approach, we also depleted the expression of HIFα binding partner ARNT, which is essential for HIFα‐mediated transcriptional regulation machinery (Fig 2G). Similar to our findings obtained from HIF1α or HIF2α knockdown cell lines, ARNT depletion in MCF‐7 cells also led to increased mtDNA content and OCR (Figs 2H and EV2C). Consistently, EglN2 depletion was still able to decrease mtDNA content and OCR in the cells depleted of ARNT (Figs 2H and EV2C). To rule out the cell type‐specific effect of EglN2 on mitochondrial function in a HIFα‐independent manner, we also depleted either HIF1α, HIF2α, or ARNT in T47D cells (Fig EV2D, F, and H). In accordance with the data we obtained from MCF‐7 cells, EglN2 depletion in these HIF knockdown cells still led to decreased mtDNA content (Fig EV2E, G and I). In summary, our data suggest that EglN2 regulates mitochondrial function in a HIF1/2α‐independent manner.
EglN2 binds to chromatin and regulates mitochondrial function under hypoxia
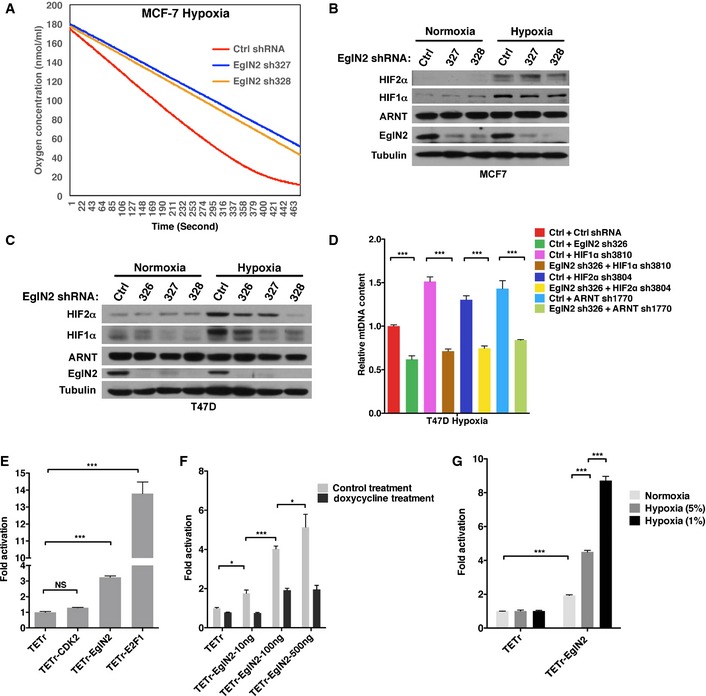
Considering that most solid tumors are characterized by the existence of hypoxic regions compared to normal tissues, we also examined the effect of EglN2 on mtDNA contents under hypoxia. Similar to the effects observed under normoxia, depletion of EglN2 by several independent hairpins decreased mtDNA content in both T47D and MCF‐7 cells under hypoxia (Fig 3A and B). Of note, depletion of EglN2 showed modest but more consistent downregulation of mtDNA content under hypoxia than normoxia (comparing Fig 3A to 1C, Fig 3B to 1F). Consistent with the mtDNA phenotype, cells depleted of EglN2 by two independent hairpins in both T47D and MCF‐7 showed decreased oxygen consumption rate under hypoxia measured by an oxytherm electrode unit (Figs 3C and EV3A). Conversely, EglN2 WT or catalytic dead (H358A) overexpression in T47D cells increased mtDNA content under hypoxia, with higher mtDNA induction compared to normoxia (compare Fig 3D to 1I, average fourfold versus 1.5‐fold). These EglN2‐overexpressed cells also displayed increased oxygen consumption rate compared to control under hypoxia (Fig 3E). To determine whether EglN2 regulates mitochondrial function via HIF under hypoxia, we also examined HIF1α, HIF2α, or ARNT protein levels upon EglN2 depletion in T47D or MCF‐7 cells (Fig EV3B and C). In MCF‐7 cells, EglN2 depletion did not affect any of these protein levels (Fig EV3B). In T47D cells, EglN2 depletion led to downregulation of HIF1α and HIF2α while not affecting ARNT protein levels (Fig EV3C), further arguing against the involvement of HIFα in EglN2‐depletion‐induced OCR or mtDNA downregulation since HIFα downregulation would increase OCR or mtDNA content as described previously as well as in our experiment system (Zhang et al, 2007). To further confirm this, we also depleted HIF1α, HIF2α, or ARNT expression by using validated shRNA used previously followed by examination of mtDNA content by EglN2 depletion under hypoxia. Consistently, in both MCF‐7 and T47D cells, our results showed that EglN2 depletion led to decreased mtDNA content in a HIF1/2α‐independent manner under hypoxia (Figs 3F and EV3D).
Figure 3. EglN2 binds to chromatin and regulates mitochondrial function under hypoxia.
-
A, BqRT–PCR quantification of mtDNA from T47D (A) or MCF‐7 (B) cells infected with lentivirus encoding EglN2 shRNA (326, 327, or 328) or control shRNA and treated with hypoxia (5% O2).
-
CMeasurement of oxygen consumption by an oxytherm electrode unit as a function of time for T47D cells infected with lentivirus encoding EglN2 shRNA (326 or 327), or control shRNA and treated with hypoxia (5% O2).
-
DqRT–PCR quantification of mtDNA from T47D cells infected with lentivirus encoding FLAG‐EglN2, FLAG‐EglN2 H358A, or control (Ctrl) and treated with hypoxia (5% O2).
-
EMeasurement of oxygen consumption by an oxytherm electrode unit as a function of time for T47D cells infected with lentivirus encoding FLAG‐EglN2, FLAG‐EglN2 H358A, or control (Ctrl) (E) treated with hypoxia (5% O2).
-
FqRT–PCR quantification of mtDNA from MCF‐7 cells infected with lentivirus encoding HIF1α shRNA (3810), HIF2α shRNA (3804), ARNT shRNA (1770), or control (−) followed by another infection with lentivirus encoding either EglN2 shRNA (326) or control (Ctrl) shRNA and treated with hypoxia (5% O2).
-
GImmunoblot of fractionated cell lysates (WCE, cytoplasmic, nuclear soluble, and chromatin bound) from T47D (1 × 107) cells treated with hypoxia (5%, 1%) or control (−).
-
HSchematics of TETr‐EglN2 and luciferase reporter plasmids.
-
IDetermination of luciferase activity in T47D cells transfected with plasmids encoding the indicated TETr‐fusion proteins along with the pUHC 13‐3 reporter and a CMV‐Renilla plasmid followed by the indicated treatment.
Figure EV3. EglN2 binds to chromatin and regulates mitochondrial function under hypoxia.
-
AMeasurement of oxygen consumption by an oxytherm electrode unit as a function of time for MCF7 cells infected with lentivirus encoding EglN2 shRNA (327 or 328) or control shRNA treated with hypoxia (5% O2).
-
B, CImmunoblot from MCF7 (B) or T47D (C) cells infected with lentivirus encoding EglN2 shRNA (326, 327, or 328) or control shRNA (Ctrl) with or without hypoxia (5% O2) treatment.
-
DqRT–PCR quantification of mtDNA from MCF7 cells infected with lentivirus encoding HIF1α shRNA (3810), HIF2α shRNA (3804), ARNT shRNA (1770), or control (−) followed by another infection with lentivirus encoding either EglN2 shRNA (326) or control (Ctrl) shRNA with hypoxia (5% O2) treatment.
-
E–GDetermination of luciferase activity in 293FT cells with plasmids encoding the indicated TETr‐fusion proteins.
Data information: Two‐tailed Student's t‐test was used to examine the P‐values from at least three replicate experiments. Error bars represent SEM. *P < 0.05, ***P < 0.005. NS denotes not significant.
In order to determine the mechanism by which EglN2 contributes to mitochondrial function, we treated T47D cells with various hypoxic conditions (5 and 1%) followed by cell fractionations to examine EglN2 protein levels in different cellular compartments. Interestingly, not only was EglN2 localized in the cytoplasm and nucleus as previously described (Metzen et al, 2003; Steinhoff et al, 2009; Fujita et al, 2012), but also EglN2 displayed increased chromatin‐bound levels upon exposure to hypoxia (Fig 3G), raising the possibility that EglN2 interacts with the chromatin and affects transcriptional machinery. In order to test this possibility, we generated mammalian expression plasmids encoding fusion proteins consisting of the TET repressor DNA‐binding domain (TETr) fused to EglN2 (or CDK2 and E2F1 serving as negative and positive control, respectively) with a flexible linker containing Gly4‐Ser repeats as described previously (Fig 3H) (Kim & Kaelin, 2001). Next, cells were transiently transfected with plasmids encoding various TETr‐fusion proteins and a luciferase reporter containing seven TETo‐binding sites to examine the effect of fusion proteins on transcription (Fig 3H). Consistent with previous literature, E2F1 dramatically induced transcription while CDK2 fusion protein did not (Fig EV3E) (Kim & Kaelin, 2001). EglN2 modestly induced transcription in a dose‐dependent manner (Fig EV3F). To examine whether the effect of TETr‐EglN2 on transcription was direct, we treated these cells with doxycycline, which blocks the binding of TETr to TETo. Doxycycline treatment abrogated the effect of EglN2 on transcriptional activation (Fig EV3F). In addition, we treated cells with various hypoxic conditions (5% and 1%) and found that EglN2 induced more robust transcriptional activation under hypoxia compared to normoxia in T47D or 293T cells, corresponding to increased EglN2 chromatin binding upon hypoxic treatment (Figs 3G and I, and EV3G). Accumulatively, our results suggest that EglN2 activates transcription, with more profound effect under hypoxia.
EglN2 binds to NRF1 and PGC1α complex on chromatin
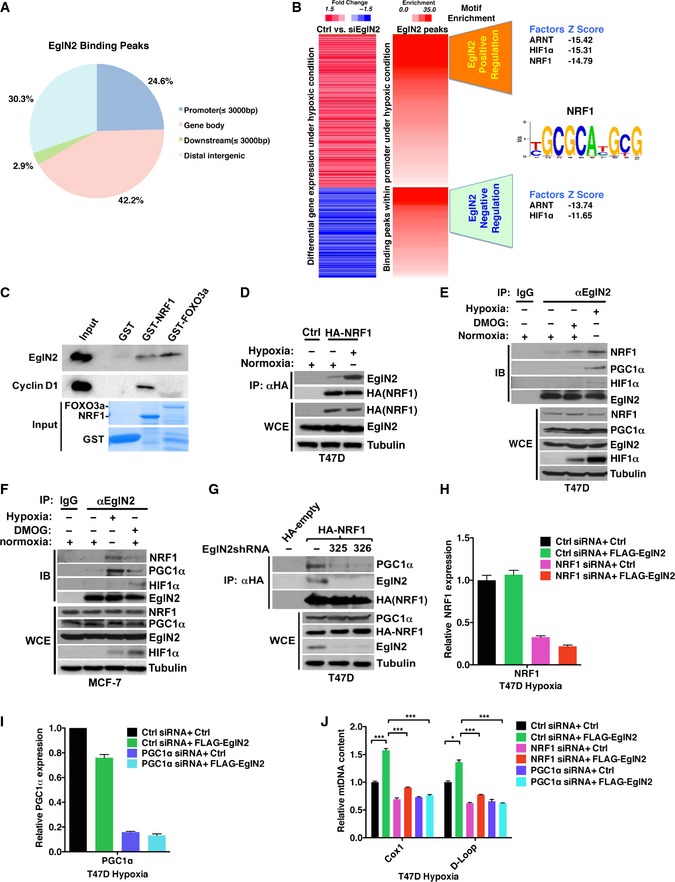
Hypoxia is a vital determinant that affects gene expression during tumor growth and progression. There is a close link between pathologically low oxygen levels and therapy‐resistant tumors (Shannon et al, 2003; Brown & Wilson, 2004). In addition, EglN2 shows a more profound effect on mtDNA content as well as more robust transcriptional activation under hypoxia compared to normoxia. Therefore, we wondered whether the effect of EglN2 on mitochondria has any connection with transcriptional activation of EglN2 under hypoxia in breast cancer. Since EglN2 binds to chromatin robustly under hypoxic conditions, next we aimed to map the EglN2 binding sites on a genomewide scale. For this purpose, we conducted HA‐EglN2 ChIP‐Seq in the T47D cells that overexpressed HA‐EglN2 and were cultured under hypoxic condition. T47D parental cells treated with the same condition followed by HA ChIP‐Seq served as the control to filter non‐specific binding. Consistent with our hypothesis, EglN2 displayed robust chromatin binding with 32,382 binding peaks under false discovery rate (FDR) of 0.05, with significant occurrence of binding sites observed at gene‐proximal promoters by CEAS analysis (Shin et al, 2009) (Fig 4A and Table EV1).
Figure 4. EglN2 interacts with NRF1 and PGC1α.
-
AGenomewide distribution of EglN2 binding peaks under hypoxia (1% O2) with concurrent DMOG (1 mM) treatment for 16 h.
-
BMotif enrichment of integrated analyses of gene expression profiling (Ctrl vs EglN2) and EglN2 binding peaks from T47D cells treated with DMOG (1 mM) and hypoxia (1% O2) for 16 h.
-
CImmunoblot (IB) analysis showing the binding between in vitro translated EglN2 or cyclin D1 and GST, GST‐NRF1, or GST‐FOXO3a.
-
DImmunoblot (IB) assays of whole‐cell extract (WCE) and immunoprecipitation (IP) of T47D (1 × 107) cells (expressing Ctrl or HA‐NRF1) treated with either control (normoxia) or hypoxia (5% O2).
-
E, FImmunoblot (IB) assays of whole‐cell extract (WCE) and immunoprecipitation (IP) of T47D (E) or MCF‐7 (F) (5 × 107) cells treated with either control (normoxia), DMOG (1 mM), or hypoxia (5% O2).
-
GImmunoblot (IB) assays of whole‐cell extracts (WCE) and immunoprecipitation (IP) of T47D (5 × 107) cells infected with lentivirus encoding either EglN2 shRNAs (325 and 326) or control (−) followed by transfection with either HA‐NRF1 or HA‐empty control.
-
H–JqRT–PCR of mRNA (H and I) or mtDNA (J) from T47D cells infected with lentivirus encoding either FLAG‐EglN2 or control (Ctrl) followed by transfecting with the NRF1 pool siRNA (H), PGC1α siRNA #4 (I), or control siRNA (Ctrl) and hypoxia (5% O2) treatment.
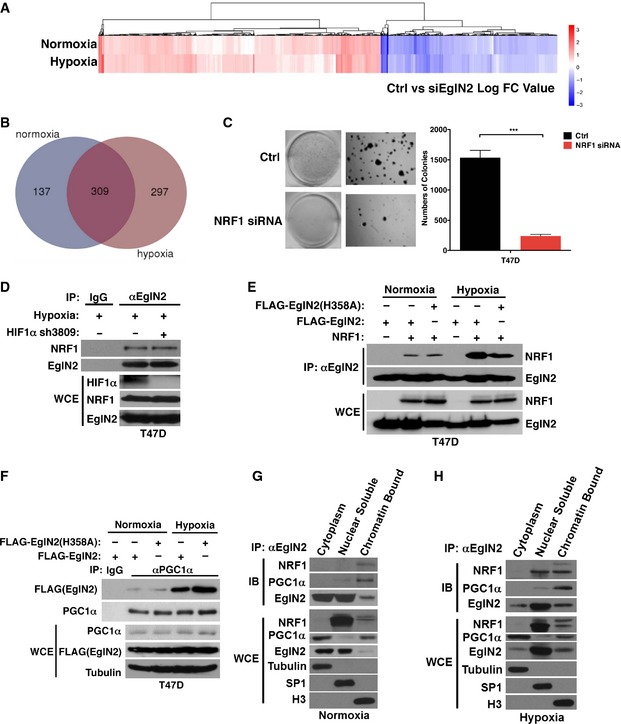
To further examine the potential effect of EglN2 on downstream target genes, we performed transcriptome microarray analysis using an EglN2 siRNA or control siRNA followed by hypoxic treatment. A total of 919 genes were differentially expressed upon EglN2 depletion (Limma algorithm, P‐value cutoff of e‐05) (Smyth, 2004), including 606 positively regulated and 313 negatively regulated genes (Table EV2). For comparison, we also performed microarray analysis under normoxia (Table EV3) and found that there are more EglN2 positively regulated under hypoxia compared to normoxia (Fig EV4A and B), consistent with more robust effect of EglN2 on transcriptional activation under hypoxia (Figs 3I and EV3G). As described above, we observed significant enrichment of EglN2 binding peaks on gene‐proximal promoters (Fig 4A). To identify unique transcription factors that regulate EglN2 target gene either positively or negatively, we selected the strongest 1,000 EglN2 peaks within promoters of EglN2 positively and negatively regulated genes (P‐value cutoff of 0.05) and performed motif enrichment analysis by using the method described previously (He et al, 2010; Liu et al, 2011). We identified nuclear respiratory factor 1 (NRF1) motif uniquely enriched in promoters of EglN2 positively regulated genes (Fig 4B), suggesting that NRF1 and EglN2 might cooperate to activate transcription. In addition, depletion of NRF1 decreased anchorage‐independent growth, an important indicator for tumor growth (Fig EV4C), which is reminiscent of the phenotype we observed previously with EglN2 depletion in ERα‐positive breast cancer cells (Zhang et al, 2009). It is worth noting that the consensus recognition motif for HIF1α and ARNT was enriched in the promoters of both EglN2‐activated and EglN2‐repressed genes (Fig 4B). Since our results showed that the effects of EglN2 on mitochondrial function under normoxia or hypoxia were largely independent of HIF1/2α signaling, we focus our efforts on elucidating the role of NRF1 on mitochondrial signaling affected by EglN2.
Figure EV4. EglN2 binds to NRF1 and PGC1α complex on chromatin.
-
A, BAnalysis and comparison of gene expression profiling (siCtrl vs siEglN2) between normoxia and hypoxia (1% O2) in T47D.
-
CRepresentative images (left) and quantification (right) of anchorage‐independent growth assay from T47D cells transfected with NRF1 siRNA or control siRNA (Ctrl). Two‐tail Student's t‐test was used to examine the P‐values from at least three replicate experiments. Error bars represent SEM. ***P < 0.005.
-
DImmunoblot (IB) assays of whole‐cell extracts (WCE) and immunoprecipitation (IP) of T47D (5 × 107) cells infected with lentivirus encoding either HIF1 shRNA or control (−) with hypoxia (5% O2) treatment.
-
E, FImmunoblot (IB) assays of whole‐cell extracts (WCE) and immunoprecipitation (IP) of T47D (1 × 107) cells infected with lentivirus encoding either FLAG‐EglN2, FLAG‐EglN2 H358A, or control (−) followed by transfection with either NRF1 or empty control with or without hypoxia (5% O2) treatment.
-
G, HCell fractionation using subcellular protein fractionation kit followed by immunoblot and immunoprecipitation (IP) of T47D (5 × 107) cells under normoxia (G) and hypoxia (5% O2) (H).
NRF1 was first identified as an activator for cytochrome C gene involved in the mitochondrial respiratory chain (Evans & Scarpulla, 1989). As a key transcription factor of nucleus‐encoded genes associated with mitochondrial function, NRF1 affects various genes (such as TFAM, TFB1M, and TFB2M) required for mitochondrial respiratory functions by employing PGC1α as the essential transcriptional co‐activator (Kelly & Scarpulla, 2004; Scarpulla, 2006; Zhang et al, 2007). Abnormally active mitochondrial function was known to predict poor clinical outcome in human breast cancer patients (Sotgia et al, 2012). Our results above showed that EglN2 regulated mitochondrial function, which motivated us to examine the potential functional link between EglN2 and NRF1 in this process. We first asked whether EglN2 and NRF1 directly interact in vitro by performing glutathione S‐transferase (GST) pull‐down assays with purified GST or GST‐NRF1 and in vitro translated EglN2. As positive controls, we interrogated the previously described interactions between NRF1 and cyclin D1 as well as EglN2 and FOXO3a (Wang et al, 2006; Zheng et al, 2014). We also observed the binding between NRF1 and EglN2 (Fig 4C). Next, to examine the interaction between EglN2 and NRF1 in cells, we treated T47D breast cancer cells expressing HA‐NRF1 under the control of a weak heterologous promoter with normoxia or hypoxia followed by co‐immunoprecipitation. Whereas the interaction between NRF1 and EglN2 was weak under normoxia, it was significantly enhanced under hypoxia (Fig 4D). Consistently, we observed enhanced binding between endogenous EglN2 and NRF1 in T47D or MCF‐7 cells under hypoxia (Fig 4E and F). Despite the robust upregulation of HIF1α protein levels under hypoxia, we only detected minimal binding between EglN2 and HIF1α (Fig 4E and F). This was further supported by using another hypoxia mimetic, DMOG, which only modestly induced endogenous EglN2 binding with HIF1α or NRF1 in some setting (Fig 4E and F). The observation that the interaction between NRF1 and EglN2 was not affected upon HIF1α depletion in these cells under hypoxia (Fig EV4D) is consistent with HIF1α‐independent mitochondrial regulation by EglN2. To further examine whether NRF1 and EglN2 binding is dependent on EglN2 enzymatic activity, we also examined the binding of EglN2 H358A catalytic mutant with NRF1 and found that the mutant bound to NRF1 similarly as the wild type, arguing that this binding is independent of EglN2 enzymatic activity (Fig EV4E).
Our results suggest a potential link between hypoxia‐induced EglN2‐NRF1 association and mitochondrial function in ERα‐positive breast cancer. Since PGC1α is the essential transcriptional co‐activator and binding partner of NRF1 that orchestrates mitochondrial function (Scarpulla, 2006), we postulated that hypoxia might similarly induce EglN2 and PGC1α binding. To test this, we examined the interaction between EglN2 and PGC1α and observed enhanced binding under hypoxia compared to normoxia (Fig 4E and F). Similar to the binding between EglN2 and NRF1, the interaction of EglN2 and PGC1α is independent of EglN2 enzymatic activity (Fig EV4F). To further examine which cellular compartment where EglN2 binds with NRF1 and PGC1α complex, we performed cell fractionation followed by endogenous EglN2 immunoprecipitation under either normoxia or hypoxia. Whereas EglN2 bound with NRF1 and PGC1α weakly in chromatin‐bound fractions under normoxia, these interactions were significantly enriched under hypoxia condition (Fig EV4G and H). Based on these findings, we hypothesized that EglN2 might mediate the interaction between PGC1α and NRF1, therefore affecting target gene expression involved in modulating mitochondrial function (Murphy, 2009). Indeed, EglN2 depletion dramatically decreased the association between PGC1α and NRF1 (Fig 4G), suggesting that EglN2 plays an important role mediating the PGC1α/NRF1 interaction. We further examined mtDNA content in cells overexpressing EglN2 followed by depletion of either NRF1 or PGC1α siRNA (Fig 4H and I). Under hypoxic condition, the increase in mtDNA content upon EglN2 overexpression was abrogated by either NRF1 or PGC1α knockdown (Fig 4J), suggesting that the effect of EglN2 on mitochondrial function is NRF1‐ and PGC1α‐dependent. It is notable that comparing to single FLAG‐EglN2‐infected cells, this cell line with another round of transfection with ctrl siRNA displayed less robust increase in mtDNA content (comparing Fig 3D with Fig 4J), which could possibly be due to the longer term culture of latter cells that develop some adaptive response.
EglN2 regulates FDXR in breast cancer
To further identify the downstream effectors of EglN2 and NRF1/PGC1α complex that might mediate the effect of EglN2 on mitochondrial function under hypoxia, we carried out NRF1 ChIP‐Seq under hypoxic condition in these cells and determined how EglN2 and NRF1 interact on a genomewide scale. NRF1 ChIP‐Seq analysis demonstrated its binding sites predominantly located in the proximal promoter regions (Fig 5A and Table EV4) and largely overlapped with EglN2 binding sites (Fig 5B and Table EV5), reinforcing the functional interaction between EglN2 and NRF1.
Figure 5. EglN2 and NRF1 regulate FDXR in ERα‐positive breast cancer cells.
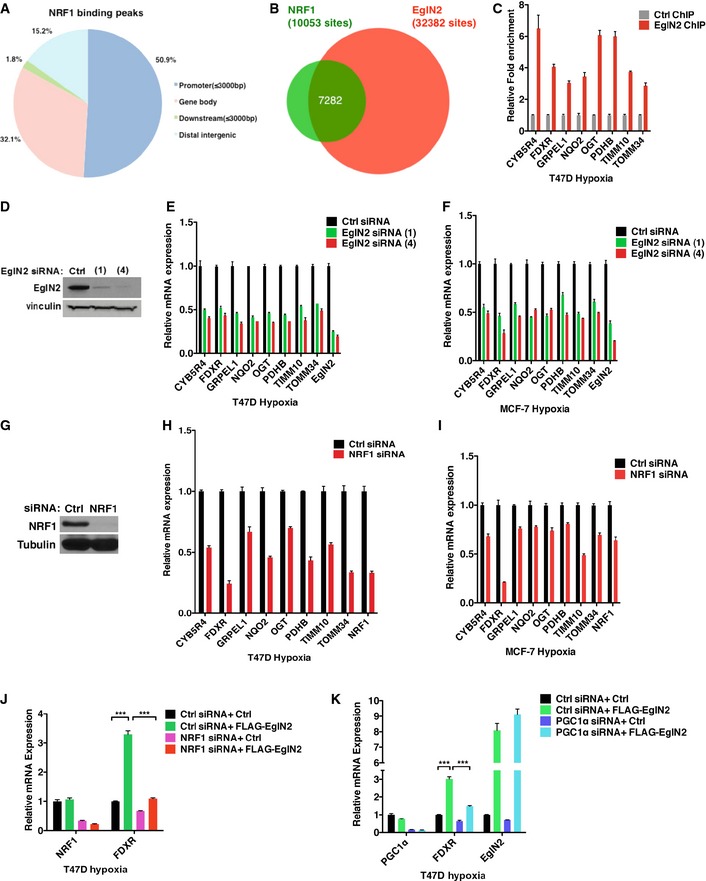
-
AGenomewide distribution of NRF1 binding peaks under hypoxia (1% O2) with concurrent DMOG (1 mM) treatment for 16 h.
-
BVenn diagram showing the overlap between the EglN2 and NRF1 cistromes in T47D cells.
-
CqRT–PCR of genomic DNA retrieved from either control IgG (Ctrl) or EglN2 antibody ChIP under normoxia or hypoxia.
-
D–FImmunoblots of lysates (D), qRT–PCR of mRNA from T47D cells (E) or MCF‐7 (F) transfected with EglN2 siRNA (1) and (4) or control (Ctrl) followed by hypoxia treatment (5% O2).
-
G–IImmunoblots of lysates (G), qRT–PCR of mRNA from T47D (H) or MCF‐7 (I) transfected with the NRF1 pool siRNA followed by hypoxia treatment (5% O2).
-
J, KqRT–PCR of mRNA from T47D cells infected with lentivirus encoding either FLAG‐EglN2 or control (Ctrl) and, after selection, transfected with NRF1 siRNA (J), PGC1α siRNA (K), or control siRNA (Ctrl) followed by hypoxia (5% O2) treatment for 16 h.
To identify the essential genes responsible for the effect of EglN2 and NRF1 on mitochondrial function under hypoxia, we selected eight mitochondrial‐related pathway genes that were bound by both EglN2 and NRF1 on their promoters (Appendix Fig S1A–H) and showed reduced expression upon EglN2 knockdown (Table EV2). Consistent with the ChIP‐Seq data, ChIP‐PCR confirmed the binding of endogenous EglN2 on their promoters (Fig 5C) under hypoxia. In addition, EglN2 depletion decreased their expression in T47D cells under hypoxia (Fig 5D and E). Similar findings were found for MCF‐7 cell under hypoxia (Fig 5F). In accordance with the data that EglN2 bound with NRF1 weakly under normoxia and regulates mitochondrial function, EglN2 depletion also decreased these gene expressions under normoxia (Fig EV5A). Among these genes, FDXR was the one showing the most robustly decreased expression upon NRF1 depletion under normoxia or hypoxia in T47D cells (Figs 5G and H, and EV5B), and also the most downregulated one upon NRF1 knockdown in MCF‐7 cells under hypoxia (Fig 5I). Therefore, we hypothesized that FDXR may be one of essential downstream effectors in EglN2/NRF1‐regulated mitochondrial pathway. To this end, we set to examine the role of FDXR in EglN2‐ and NRF1‐regulated mitochondrial phenotype in breast cancer cells. Consistent with stronger binding of EglN2 on FDXR gene promoter under hypoxia compared to normoxia, hypoxia treatment modestly upregulated FDXR mRNA expression in T47D cells (Fig EV5C). In addition, overexpression of EglN2 increased FDXR mRNA expression and this effect was ameliorated upon NRF1 depletion (Fig 5J). Similarly, depletion of PGC1α by siRNAs attenuated both basal and EglN2‐induced FDXR expression under hypoxia (Figs 5K and EV5D). Collectively, these results suggest that NRF1 and PGC1α are required for the regulation of FDXR by EglN2 and that an EglN2, NRF1, and PGC1α complex is required for FDXR expression in ERα‐positive cancer.
Figure EV5. EglN2/NRF1/PGC1α regulates FDXR in breast cancer.
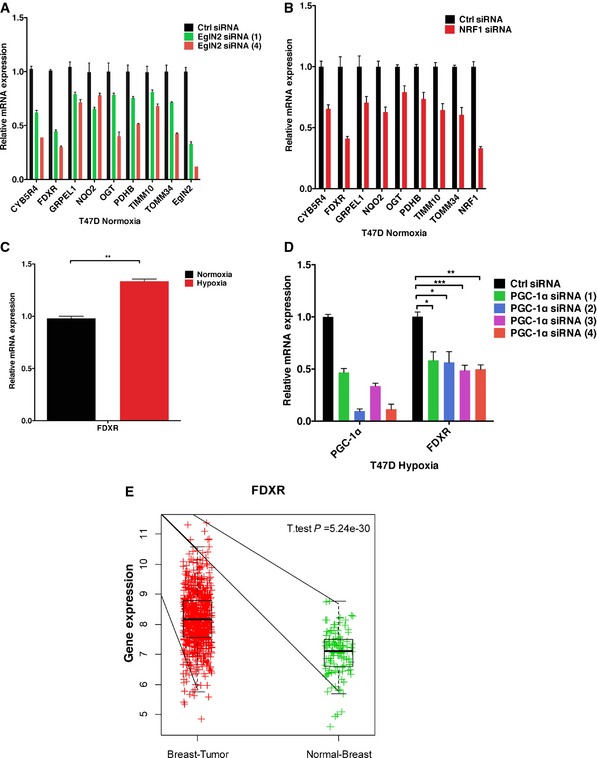
-
A, BqRT–PCR of mRNA from T47D transfected with either EglN2 siRNA (1, 4) (A) or NRF1 pool siRNA (B) under normoxia.
-
CqRT–PCR of mRNA from T47D with or without hypoxia (5% O2) treatment.
-
DqRT–PCR of mRNA from T47D cells transfected with either PGC‐1α siRNAs (1, 2, 3, and 4) or control (Ctrl) followed by hypoxia (5% O2) treatment for 16 h.
-
EFDXR regulates ERα‐positive breast tumorigenesis. Unpaired two‐sample t‐test comparing expression of FDXR in the TCGA dataset between the indicated patients.
Data information: Two‐tailed Student's t‐test was used to examine the P‐values from at least three replicate experiments. Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.005.
FDXR contributes to ERα‐positive breast tumorigenesis
FDXR is an important mitochondrial flavoprotein that initiates electron transport from NADPH to its substrates, which contributes to p53‐mediated apoptosis through the generation of oxidative stress in mitochondria in colon cancer (Hwang et al, 2001). However, its role in mitochondrial regulation and tumorigenesis in other cancers is relatively unexplored. By serving as a key downstream target of EglN2 and NRF1, FDXR may be essential for their ability to regulate mtDNA content. To test this, first we depleted FDXR expression by two independent shRNAs (FDXR sh434 or sh435, Fig 6A), and indeed observed decreased mtDNA content compared with control under hypoxia (Fig 6B). In accordance with FDXR acting downstream of EglN2 on mitochondrial signaling, FDXR overexpression rescued the mtDNA defect induced by EglN2 loss (Fig 6C and D). By examining oxygen consumption rate using the XF‐24 extracellular flux analyzer, we observed decreased basal oxygen consumption upon EglN2 depletion, an effect rescued by FDXR overexpression (Fig 6E). Next, we examined the anchorage‐independent growth in these cells, an important marker for tumor phenotype. Consistent with our previously published data with orthotopic tumor growth, EglN2 depletion decreased anchorage‐independent growth (Fig 6F) (Zhang et al, 2009). Overexpression of FDXR can at least partially rescue EglN2 loss‐induced soft agar growth defect (Fig 6F). These results suggest that FDXR, as a downstream target of EglN2 and NRF1, at least partially mediates the effect of EglN2 on mitochondrial function and anchorage‐independent growth.
Figure 6. FDXR regulates mitochondrial function and ERα‐positive breast tumorigenesis.
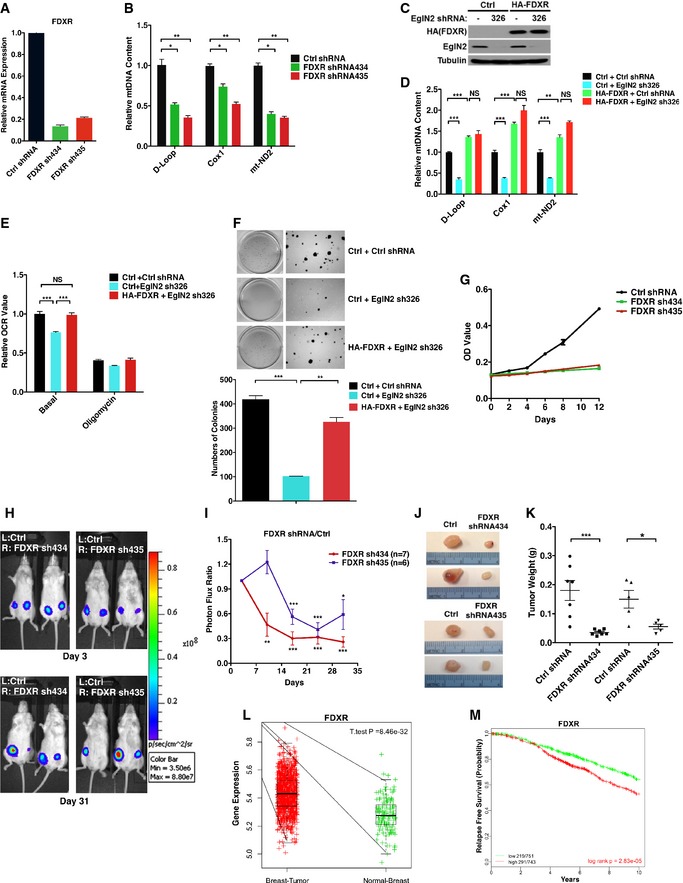
-
A, BqRT–PCR of mRNA (A) and mitochondrial DNA (B) from T47D cells infected with lentivirus encoding either FDXR shRNA (434, 435) or control (Ctrl) shRNA followed by hypoxia treatment (5% O2).
-
C–FImmunoblot of cell lysates (C), qRT–PCR of mitochondrial DNA (D), measurement of oxygen consumption rate (E), and anchorage‐independent growth assay (F) from T47D cells infected with lentivirus encoding either HA‐FDXR or control (Ctrl) followed by another infection with lentivirus encoding either EglN2 shRNA (326) or control (−) shRNA.
-
GCell proliferation assay for T47D cells infected with lentivirus encoding either FDXR shRNA (434, 435) or control (Ctrl) shRNA as a function of time.
-
HRepresentative bioluminescence imaging from Day 3 and Day 31 post‐implantation of cancer cells injected orthotopically into the mammary fat pads as indicated.
-
IQuantitation of imaging studies carried out in (H). See Materials and Methods for normalization description.
-
JRepresentative tumor gross appearance at the necropsy.
-
KTumor weight plots at the necropsy.
-
LUnpaired two‐sample t‐test comparing expression of FDXR in METABRIC datasets between the indicated patient samples.
-
MKaplan–Meier OS curves for ERα‐positive patients in METABRIC cohorts (Curtis et al, 2012). Patients were rank‐ordered and divided into two equal groups (low in green and high in red), using the FDXR gene expression level.
As a downstream effector of EglN2, we further examined the function of FDXR in ERα‐positive breast tumorigenesis. To this end, we generated T47D cells expressing firefly luciferase followed by infection with lentivirus encoding either FDXR shRNAs (FDXR sh434 or sh435, Fig 6A) or control. Firstly, cells depleted of FDXR by two different shRNAs displayed a proliferation defect compared to control (Fig 6G). Next, T47D FDXR knockdown (sh434 or sh435) and control cells were orthotopically injected bilaterally into the mammary fat pads of mice supplemented with estrogen pellets to promote the growth of breast cancer cells followed by noninvasive bioluminescent imaging. The initial imaging was taken at day 3 post‐implantation of tumor cells to verify equal injection on bilateral mammary glands, and further imaging was performed on a weekly basis over 4 weeks. Over time, there was a progressive decline in tumor bioluminescent signals from FDXR shRNAs compared to control shRNA (Fig 6H and I). Consistent with the bioluminescent signal, the tumors formed from FDXR shRNA cells were significantly smaller than those formed from control shRNA cells (Figures 6J and K), indicating that FDXR depletion inhibits breast tumor growth in vivo.
To further examine the functional importance of FDXR in breast cancer patients, we compared FDXR expression between ERα‐positive breast cancer versus normal breast samples in METABRIC and TCGA breast cancer cohorts (Cancer Genome Atlas, 2012; Curtis et al, 2012). In both datasets, FDXR expression was higher in cancer cohorts than in normal cohorts (Figs 6L and EV5E). Finally, higher expression of FDXR is associated with worse clinical prognosis in ERα‐positive breast cancer patients (Fig 6M), suggesting FDXR to be a potential prognostic marker in ERα‐positive cancer.
Discussion
In this study, we demonstrate that EglN2 contributes to maintenance of mitochondrial function in ERα‐positive breast cancer under hypoxia. By integrative analyses of transcriptome profiles and genomewide binding of EglN2 under hypoxic condition, we discovered that EglN2 promotes transcriptional activation by interacting with NRF1. Mechanistically, by forming an activator complex with PGC1α and NRF1, EglN2 promotes FDXR gene expression and regulates mitochondrial function in a PGC1α‐ and NRF1‐dependent manner. FDXR is a common downstream target of this trimeric protein complex that at least partially mediates the effect of EglN2 on mitochondrial function. FDXR depletion decreases breast cancer cell proliferation and xenograft growth in vivo. Increased FDXR expression also predicts worse prognosis in ERα‐positive breast cancer.
EglN2 has previously been shown to be involved in reprogramming metabolism pathways. Elimination of EglN2 decreases the oxygen consumption in murine skeletal muscle by reprogramming glucose metabolism from oxidative to anaerobic ATP production through inducing a PPARα pathway (Aragones et al, 2008). Under normal conditions, this will impair the skeletal muscle performance since EglN2 knockout mice have lower oxygen consumption. However, under ischemia injury condition, these mice displayed reduced oxidative stress with less reaction oxygen species (ROS) production. Therefore, these mice are more resistant to hypoxic tolerance. Our study in breast cancer cells demonstrates depletion of EglN2 decreases oxygen consumption rate, yet by a different mechanism involving the PGC1α and NRF1 complex.
Mechanistically, we propose that EglN2 regulates FDXR and modulates mitochondrial functions in breast cancer cells with more profound effects under the hypoxic condition. Our integrative analyses of EglN2 and NRF1 ChIP‐Seq data, as well as EglN2‐mediated differential mRNA expression, identify genes potentially involved in mitochondrial functions. Among these genes, FDXR is the most robustly regulated genes by both EglN2 and NRF1 under normoxia or hypoxia. It is important to note that there still exists the possibility that other gene may be involved in regulating the mitochondrial function affected by EglN2/NRF1 under normoxia or hypoxia, which was also reflected by the partial rescue of anchorage‐independent growth by FDXR overexpression in EglN2‐depleted cells. Nonetheless, our findings establish FDXR as an important downstream regulator for EglN2 and NRF1 that modulates mitochondrial function in ERα‐positive breast cancer.
EglN2 enzymatic activity depends on oxygen, and it is lower under hypoxic than normoxic conditions. Under hypoxic conditions, EglN2 depletion decreases mitochondrial function while its overexpression increases mitochondrial function, suggesting EglN2's role in mitochondrial regulation may be partially independent of its enzymatic activity. Further evidence of this is the finding that overexpression of EglN2 wild‐type (WT) or catalytic dead mutant (H358A) in breast cancer cells increases mitochondrial DNA content to similar levels. These findings suggest that under normoxia condition, EglN2 is an active enzyme affecting its substrate hydroxylation and stability. However, during cancer development, cancer cells become hypoxic with impaired EglN2 enzymatic activity, and EglN2 instead binds to NRF1 and affects mitochondrial function as a transcriptional activator. It is also important to point out that hypoxia, by using other mechanisms than HIF1α accumulation, induces EglN2 and NRF1 interaction to maintain mitochondrial function. This was supported by the observation that the interaction between NRF1 and EglN2 was not mitigated by HIF1α depletion in these cells under hypoxia. The detailed mechanism underlying hypoxia‐induced NRF1 and EglN2 interaction awaits further investigation. Another interesting observation is that HIF1α and ARNT motifs were enriched in both EglN2 positively and negatively regulated genes, suggesting that EglN2 might bind DNA via HIF and is involved in some signaling pathways other than mitochondrial functional regulation.
Hypoxia was shown to decrease oxygen consumption in many cell lines as well as in vivo (Denko, 2008). Our findings suggest that EglN2 overexpression under hypoxia induces mitochondrial function, while EglN2 depletion under this condition decreases oxygen consumption rate. Therefore, it is interesting to observe that even under hypoxic condition, oxygen consumption and mitochondrial function are still important for cancer cells. This phenomenon is supported by several published literatures. For example, glutamine‐driven oxidative phosphorylation is a major means of ATP production even in hypoxic cancer cells (Fan et al, 2013). Another example is that mitochondrial enzyme SHMT2 is induced upon hypoxia and is critical for maintaining NADPH production and redox balance to support cancer cell growth (Ye et al, 2014). In addition, previous research shows that hypoxia activates transcription via a mitochondria‐dependent signaling (Chandel et al, 1998). Our results suggest that by binding with NRF1 and PGC1α complex, EglN2 serves to maintain the mitochondrial function under hypoxia in ERα‐positive breast cancer.
It is also intriguing that FDXR, a mitochondrial flavoprotein that initiates the electron transport for cytochrome p450 receiving electrons from NDAPH, was able to regulate mtDNA levels in breast cancer cells. One possibility is that our metabolomics analysis showed the decreased glutamine to glutamate conversion upon FDXR depletion (Zhang J and Zhang Q, unpublished data). Since glutamine to glutamate conversion is important for generation of intermediates important for production of pyrimidine de novo synthesis (Newsholme et al, 2003; Pearce et al, 2013), FDXR depletion could lead to decreased pyrimidine synthesis and mtDNA reduction without affecting mitochondrial mass. The other possibility is that mitochondrial dNTP imbalance has been reported to decrease mtDNA content in various cells or mouse tissues (Song et al, 2003; Akman et al, 2008; Lopez et al, 2009). Our results show that FDXR depletion leads to unbalanced dNTP production (Zhang J and Zhang Q, unpublished data), which could contribute to the mtDNA reduction.
Development of EglN2 enzymatic inhibitors can potentially decrease cyclin D1 and breast tumorigenesis (Zhang et al, 2009; Zheng et al, 2014). Our study, however, suggests that EglN2's catalytic‐independent function could still contribute to mitochondrial functions in breast cancer cells, thus pointing to abrogating EglN2 expression as a potentially more effective therapy. With the maturing siRNA/shRNA delivery strategies in vitro and in vivo, as well as the emerging TALEN or CRISPR technologies, we can manipulate EglN2 expression in breast cancer in the near future to examine its therapeutic efficacy. It is worth noting that EglN2 knockout MEFs does not affect mtDNA or basal OCR phenotype and our unpublished data show that depletion of EglN2 in human mammary epithelial cells (HMECs) does not affect mtDNA content, which indicates the specificity of EglN2 regulation on mitochondrial function in breast cancer but not in normal cells. In addition, our current findings identified some downstream targets of EglN2 (such as FDXR) that are important for mediating mitochondrial function in ERα‐positive breast cancer. These targets will likely open new therapeutic avenues to modulate mitochondrial function and breast tumorigenesis in ERα‐positive breast cancer.
Materials and Methods
Cell culture
293FT and MCF‐7 cells were cultured in DMEM containing 10% fetal bovine serum (FBS) plus 1% penicillin/streptomycin (Pen/Strep). T47D cells were maintained in RPMI medium containing 10% FBS plus 1% Pen/Strep. Following lentivirus infection, cells were maintained in the presence of hygromycin (200 μg/ml) or puromycin (2 μg/ml) depending on the vector. All cells were maintained at 37°C in 5% CO2 incubator.
Western blot analysis and antibodies
EBC buffer (50 mM Tris pH 8.0, 120 mM NaCl, 0.5% NP‐40, 0.1 mM EDTA, and 10% glycerol) supplemented with complete protease inhibitor (Roche Applied Biosciences) was used to harvest whole‐cell lysates from 5 × 105 indicated cells for immunoblots. Subcellular protein fractionation kits were obtained from Thermo Scientific. Cell lysate concentrations were measured by Bradford assay. Equal amount of cell lysates was resolved by SDS–PAGE. The experiments were repeated for three times with similar results. Rabbit EglN2 antibody (NB100‐310), HIF2α (NB100‐122), and HIF1α (NB100‐479) were from Novus Biological. Rabbit anti‐cyclin D1 was from Neomarker. Mouse anti‐HIF1α (610958), anti‐ARNT (611079), and anti‐cytochrome C (556433) were from BD Bioscience. Antibodies against vinculin (V9131), α‐tubulin (T9026), and MCU (HPA016480) were from Sigma. Mouse antibody against hemagglutinin (HA, MMS‐101P) was obtained from Covance. Mouse anti‐PGC1α antibody (St1202) and rabbit anti‐PDH Ser293 (AP1062) were from Calbiochem. Sheep anti‐EglN2 antibody (AF6394) was from R&D. Mouse anti‐NRF1 (ab55744), anti‐CLPP (ab124822), anti‐mitofusin‐1 (ab57602), and anti‐mitofusin‐2 (ab56889) were obtained from Abcam. Antibodies against AIF (#4642), Bcl‐XL (#2762), PDH (#2784), and VDAC (#4866) were obtained from Cell Signaling. Anti‐COX3 (G2413) was from Santa Cruz Biotechnology. Antibodies against ND1 (19703‐1‐AP), ND2 (19704‐1‐AP), ND5 (55410‐1‐AP), COX1 (55071‐1‐AP), COX2 (55070‐1‐AP), NDUFA9 (20312‐1‐AP), SDHA (14865‐1‐AP), and UQCRFS1 (18443‐1‐AP) were from Proteintech. Peroxidase‐conjugated goat anti‐mouse secondary antibody (31430) and peroxidase‐conjugated goat anti‐rabbit secondary antibody (31460) were purchased from Thermo Scientific.
Plasmids
Full‐length FLAG and HA double‐tagged EglN2 was amplified by PCR with a 5′ primer that introduced a FLAG tag and an HA tag with a BamHI site and a 3′ primer that introduced an EcoRI site. The PCR product was digested with BamHI and EcoRI and cloned into the pBABE‐puro vector cut with these two enzymes. pLenti6 FLAG‐EglN2 was described previously (Koivunen et al, 2012). pSG5‐TETr‐EglN2 fusion was created by ligating the full‐length EglN2 ORF (amplified with 5′ BamHI site and 3′ EcoRI site) into the pSG5‐TETr empty vector cut with BamHI/EcoRI. pSG5‐TETr‐CDK2, pSG5‐TETr‐E2F1, and pUHC 13‐3 reporter were described previously (Kim & Kaelin, 2001). GST‐NRF1 and HA‐NRF1 were described previously (Wang et al, 2006). The full‐length FOXO3a insert was cut with BamHI and NotI and ligated into pGex 4T.2 vector cut with these two enzymes to make the construct for GST‐FOXO3a. Full‐length HA‐tagged NRF1 was amplified by PCR with a 5′ primer that introduced a BamHI site and an HA tag and a 3′ primer that introduced a NotI site. The PCR product was digested with BamHI and NotI and cloned into pLenti‐UBC‐pGK‐Hyg (a modified version of pLL3.7) vector cut with these two enzymes. Full‐length HA‐tagged FDXR was amplified by PCR with a 5′ primer that introduced an XbaI site and an HA tag and a 3′ primer that introduced an XhoI site. The PCR product was digested with XbaI and XhoI and cloned into pLenti CMV GFP vector (addgene) digested with XbaI and SalI.
Virus production and infection
293FT packaging cell lines were used for lentiviral amplification. Lentiviral infection was carried out similarly as previously described (Zhang et al, 2009). Briefly, post‐transfection with Lipofectamine 2000, viruses were collected twice after 48 and 72 h. After passing through 0.45‐μm filters, appropriate amount of viruses was used to infect target cells in the presence of 8 μg/ml polybrene. Subsequently, target cell lines underwent appropriate antibiotic selection.
siRNAs and lentiviral shRNA vectors
Non‐targeting siRNA no. 2 was obtained from Dharmacon (catalogue number: D0012100220). EglN2 (1) and EglN2 (4) siRNAs were described previously (Zhang et al, 2009). NRF1 smart pool siRNA was obtained from Dharmacon (catalogue number: L‐017924). PGC1α siRNAs were obtained from Dharmacon with the following targeting sequences:
PGC1α (#1): GCAGGUAACAUGUUCCCUA
PGC1α (#2): ACTCUCAGCUAAGUUAUAA
PGC1α (#3): GAAGAGCGCCGUGUGAUUU
PGC1α (#4): GAGAAUUCAUGGAGCAAUA
Lentiviral EglN2, HIF1α, ARNT, and FDXR shRNAs were obtained from Broad Institute TRC shRNA library. Target sequences are listed as follows:
Ctrl shRNA: AACAGTCGCGTTTGCGACTGG
EglN2 (325): GCTGCATCACCTGTATCTATT
EglN2 (326): GCCACTCTTTGACCGGTTGCT
EglN2 (327): ACTGGGACGTTAAGGTGCATG
EglN2 (328): CTGGGACGTTAAGGTGCATGG
HIF1α (3809): CCAGTTATGATTGTGAAGTTA
HIF1α (3810): GTGATGAAAGAATTACCGAAT
HIF2α (3804): CGACCTGAAGATTGAAGTGAT
ARNT (1770): GAGAAGTCAGATGGTTTATTT
FDXR (434): GCTCAGCAGCATTGGGTATAA
FDXR (435): CCATTTCTCCACACAGGAGAA
Real‐Time RT–PCR
Total RNA was isolated with RNeasy mini kit (Qiagen) from 2 × 105 indicated cells. First‐strand cDNA was generated with the iScript cDNA synthesis kit (Bio‐Rad). Real‐time PCR was performed in triplicate as described previously (Zhang et al, 2009). Real‐time RT–PCR primers used in this study are included in Table EV6. The experiments were repeated for three times with similar results.
GST protein purification and GST pull‐down
Glutathione S‐transferase plasmids were transformed with BL21 competent cells. Single colonies were picked from above and cultured in 50 ml LB medium containing ampicillin. After overnight culture, 5 ml LB medium was diluted in 500 ml LB medium for shaking at 37°C for 2–3 h until OD600 of 0.8–1.0 was reached. 0.2 mM IPTG was added to induce GST protein production for 4 h before harvesting pellets. Bacteria lysates were disrupted by the nanodebee homogenizer. Cleared bacteria lysates were purified by using glutathione–Sepharose 4B beads. About 20 μl of GST suspension proteins was incubated with either in vitro translated protein in 500 μl NETN buffer or cell lysates. After overnight incubation, bound complexes were washed with NETN buffer 8 times followed by boiling in SDS loading buffer and SDS–PAGE.
Immunoprecipitation
Cells were lysed in EBC lysis buffer supplemented with complete protease inhibitors (Roche Applied Bioscience). The lysates were clarified by centrifugation and then mixed with primary antibodies or 3F10 HA‐conjugated beads (Roche Applied Bioscience) overnight. For primary antibody incubation overnight, cell lysates were incubated further with protein G sepharose beads (Roche Applied Bioscience) for 2 h. The bound complexes were washed with NETN buffer for 8 times and were eluted by boiling in SDS loading buffer. Bound proteins were resolved in SDS–PAGE followed by Western blot analysis. The experiments were repeated for three times with similar results.
Luciferase reporter assay
For TETr‐fusion protein transcription assay, subconfluent 293FT or T47D cells (200,000 cells/24‐well plate) were transiently transfected with pCMV‐Renilla (30 ng), 100 ng of pUHC13‐3 reporter plasmid and indicated plasmids encoding TETr‐fusion plasmids or empty vector (100 ng unless indicated otherwise). Forty‐eight hours after transfection, luciferase assay were performed as described previously (Kim & Kaelin, 2001). The experiments were repeated for three times with similar results.
Cell proliferation assays
T47D cells were plated, in triplicate, in 96‐well plates (3,000 cells/well) in appropriate growth medium. At indicated time points, cells were replaced with 90 μl fresh growth medium supplemented with 10 μl MTS reagents (Promega) followed by incubation at 37°C for 2 h. The OD absorbance value was measured at 490 nm using a 96‐well plate reader. The experiments were repeated for three times with similar results.
Oxygen consumption rate (OCRs) measurement in cells
The extracellular oxygen consumption was determined by OCRs using the Seahorse XF24 extracellular flux analyzer (Seahorse Bioscience). About 1 × 105 of indicated cells were seeded into XF24 cell culture microplate 24 h before the assay. For OCR, the baseline mitochondrial respiration was established by recording extracellular oxygen concentration at several time points. Respiration not linked to mitochondrial ATP synthesis was measured after adding 1 μM oligomycin through an automated injection port of XF24. Uncoupled respiration measured was obtained after adding 1 μM FCCP. Hansa‐tech oxygen electrode was used to measure the total cellular oxygen consumption of indicated cell lines under hypoxia (for T47 cell lines: 2 × 106, for MCF‐7 cell lines: 1 × 106) according to the manufacturer's instructions and previous literature (Zhang et al, 2012). For each experiment, equal numbers of cells suspended in 1 ml respiration buffer were pipetted into the calibrated oxygen electrode chamber and the temperature was maintained at 37°C for measurement. The experiments were performed for three times with similar results.
Measurement of mitochondrial DNA content
Archive‐quality DNA was extracted with Gentra Puregene cell kit (Qiagen) according to the manufacturer's instructions from 2 × 105 indicated cells. Mitochondrial DNA content was measured by the relative values of mtDNAs (within D‐loop, Cox1, or mt‐ND2) versus nuDNA (within the TBP nuclear regions on chromosome 6) by qRT–PCR (Moiseeva et al, 2009). See Table EV6 for the primer sequences used for nuDNA, D‐loop, Cox1, and mt‐ND2. The experiments were repeated for three times with similar results.
ChIP, ChIP‐Seq, and gene expression microarray analysis
ChIP was performed with HA antibody (Santa Cruz) or NRF1 antibody (Abcam, a55744) as previously described for T47D (3 × 108) cells (Chen et al, 2008). The ChIP‐Seq library was prepared using ChIP‐Seq DNA sample preparation kit (Illumina) according to the manufacturer's instructions. All of ChIP‐Seq peaks were identified by using MACS package with a P‐value cutoff of 1 × 10−5 (Zhang et al, 2008). For gene expression microarray, cells (2 × 105) were treated with indicated conditions followed by total RNA extraction by using RNeasy kit with on column DNase digestion (Qiagen); Biotin‐labeled cRNA was prepared from 1 mg of total RNA, fragmented, and hybridized to Affymetrix human gene 1.0 ST expression array. All gene expression microarray data were normalized and summarized using RMA. The differentially expressed genes were identified using Limma. The ChIP‐Seq and microarray data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series Accession Number GSE59937.
Anchorage‐independent growth assay
Cells were plated at a density of 5,000 cells per ml in complete medium with 0.4% agarose, onto bottom layers composed of medium with 1% agarose followed by incubation at 4°C for 10 min. Afterward, cells were moved to 37°C incubator. For every 4 days, three drops of complete media were added onto the plate. After 2 weeks, the extra liquid on the plate was aspirated, 1 ml medium was added into each well, and colonies were stained by 100 μg/ml iodonitrotetrazoliuim chloride solution. Cell culture plates were put back in the incubator overnight followed by counting of foci number. The experiments were repeated for three times with similar results.
Orthotopic tumor growth
Six‐week‐old female NOD/SCID gamma mice (NSG, Jackson laboratory) were randomly distributed for xenograft studies. FDXR knockdown (sh434 or sh435) and control cells were injected bilaterally in order to have fair comparison. Approximately 4 × 106 viable T47D breast cancer cells were resuspended in 100 μl growth factor reduced matrigel (BD biosciences) and injected orthotopically into the mammary gland as described previously (Zhang et al, 2009). Bioluminescence imaging was performed as described previously (Zhang et al, 2009). Seven mice were included in the group (FDXR sh434/Ctrl) and six mice were included in the group (FDXR sh435/Ctrl). For each mouse, total photons from mammary fat pad injected with cells expressing FDXR shRNA were divided by total photons from the contralateral fat pad with cells expressing control shRNA and normalized to the ratio for that mouse on the day 3 post‐implantation of cancer cells. Mice were sacrificed 4 weeks after the first imaging, as specified in the figure legends. The total mass of tumors was presented as mean ± SEM and evaluated statistically using the unpaired two‐tail Student's t‐test. All animal experiments were complied with National Institutes of Health guidelines and were approved by the University of North Carolina at Chapel Hill Animal Care and Use Committee.
Statistical analysis
The unpaired two‐tail Student's t‐test was used for experiments comparing two sets of data. Data represent mean ± SEM from three independent experiments. *, **, and *** denote P‐value of < 0.05, 0.01, and 0.005, respectively. NS denotes not significant.
Author contributions
QZ, WGK, and XSL conceived and supervised the project. QZ, WC, JZ, and XSL analyzed the data. JZ and ChengyW performed most of experiments and genomic data analysis. XC performed most of ChIP‐Seq experiments. CF performed the patient data analysis. MT and XZ helped with animal experiments and biochemical experiments. HW, YL, KMA, ChenguW, and RGP provided essential reagents and key advices on the project. QZ wrote the paper together with help from JZ, WGK, ChengyW, and XSL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File
Acknowledgements
The authors thank members of Zhang and Kaelin laboratory for helpful discussions; W.Kimryn Rathmell, Kimberly Briggs, Li Qian, Gang Greg Wang, Jiandong Liu, and Kevin Byrd for critical readings and suggestions. The authors thank Ella Liberzon for providing pSG‐TETr‐EglN2 fusion plasmid. The authors also want to thank UNC Lineberger comprehensive cancer center functional genomic core facility for technical help. This work has been partially supported by K99/R00 (CA160351) award from NIH (QZ), 2R01CA68490 (WGK), University Cancer Research Fund (QZ) from University of North Carolina at Chapel Hill, Kimmel Scholar Award (QZ), The V Foundation Scholar Award (QZ) and Susan G. Komen Career Catalyst Award (QZ). [Correction added on 2 December 2015 after first online publication: The last sentence has been added to the Acknowledgements section.]
The EMBO Journal (2015) 34: 2953–2970
References
- Akman HO, Dorado B, Lopez LC, Garcia‐Cazorla A, Vila MR, Tanabe LM, Dauer WT, Bonilla E, Tanji K, Hirano M (2008) Thymidine kinase 2 (H126N) knockin mice show the essential role of balanced deoxynucleotide pools for mitochondrial DNA maintenance. Hum Mol Genet 17: 2433–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia‐inducible factor. J Biol Chem 279: 38458–38465 [DOI] [PubMed] [Google Scholar]
- Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez‐Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A et al (2008) Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet 40: 170–180 [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J (2003) HIF prolyl‐hydroxylase 2 is the key oxygen sensor setting low steady‐state levels of HIF‐1alpha in normoxia. EMBO J 22: 4082–4090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bost F, Sahra IB, Le Marchand‐Brustel Y, Tanti JF (2012) Metformin and cancer therapy. Curr Opin Oncol 24: 103–108 [DOI] [PubMed] [Google Scholar]
- Brown JM, Wilson WR (2004) Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 4: 437–447 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT (1998) Mitochondrial reactive oxygen species trigger hypoxia‐induced transcription. Proc Natl Acad Sci USA 95: 11715–11720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117 [DOI] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Group M, Langerod A, Green A, Provenzano E et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8: 705–713 [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54 [DOI] [PubMed] [Google Scholar]
- Evans MJ, Scarpulla RC (1989) Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter. Characterization of upstream NRF‐1, ATF, and intron Sp1 recognition sequences. J Biol Chem 264: 14361–14368 [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD (2013) Glutamine‐driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol 9: 712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N, Markova D, Anderson DG, Chiba K, Toyama Y, Shapiro IM, Risbud MV (2012) Expression of prolyl hydroxylases (PHDs) is selectively controlled by HIF‐1 and HIF‐2 proteins in nucleus pulposus cells of the intervertebral disc: distinct roles of PHD2 and PHD3 proteins in controlling HIF‐1alpha activity in hypoxia. J Biol Chem 287: 16975–16986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891–899 [DOI] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli‐Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E (2011) Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25: 460–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli‐Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD, White E (2013) Autophagy suppresses progression of K‐ras‐induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27: 1447–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M, Leedman P, Zu X, Russell V (2002) Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF‐7 breast cancer cells. Biochem J 364: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, Mieczkowski P, Lieb JD, Zhao K, Brown M, Liu XS (2010) Nucleosome dynamics define transcriptional enhancers. Nat Genet 42: 343–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmuhamedov E, Jahangir A, Bienengraeber M, Lewis LD, Terzic A (2003) Deletion of mtDNA disrupts mitochondrial function and structure, but not biogenesis. Mitochondrion 3: 13–19 [DOI] [PubMed] [Google Scholar]
- Hwang PM, Bunz F, Yu J, Rago C, Chan TA, Murphy MP, Kelso GF, Smith RA, Kinzler KW, Vogelstein B (2001) Ferredoxin reductase affects p53‐dependent, 5‐fluorouracil‐induced apoptosis in colorectal cancer cells. Nat Med 7: 1111–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr, Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402 [DOI] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC (2004) Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368 [DOI] [PubMed] [Google Scholar]
- Kim TY, Kaelin WG Jr (2001) Differential control of transcription by DNA‐bound cyclins. Mol Biol Cell 12: 2207–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF‐1‐mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–185 [DOI] [PubMed] [Google Scholar]
- Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, Ligon KL, Verhaak RG, Yan H, Kaelin WG Jr (2012) Transformation by the (R)‐enantiomer of 2‐hydroxyglutarate linked to EGLN activation. Nature 483: 484–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Ortiz JA, Taing L, Meyer CA, Lee B, Zhang Y, Shin H, Wong SS, Ma J, Lei Y, Pape UJ, Poidinger M, Chen Y, Yeung K, Brown M, Turpaz Y, Liu XS (2011) Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol 12: R83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez LC, Akman HO, Garcia‐Cazorla A, Dorado B, Marti R, Nishino I, Tadesse S, Pizzorno G, Shungu D, Bonilla E, Tanji K, Hirano M (2009) Unbalanced deoxynucleotide pools cause mitochondrial DNA instability in thymidine phosphorylase‐deficient mice. Hum Mol Genet 18: 714–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzen E, Berchner‐Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig‐Burgel T, Jelkmann W, Acker H, Fandrey J (2003) Intracellular localisation of human HIF‐1 alpha hydroxylases: implications for oxygen sensing. J Cell Sci 116: 1319–1326 [DOI] [PubMed] [Google Scholar]
- Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr (2008) Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111: 3236–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva O, Bourdeau V, Roux A, Deschenes‐Simard X, Ferbeyre G (2009) Mitochondrial dysfunction contributes to oncogene‐induced senescence. Mol Cell Biol 29: 4495–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Sanchez R, Rodriguez‐Enriquez S, Marin‐Hernandez A, Saavedra E (2007) Energy metabolism in tumor cells. FEBS J 274: 1393–1418 [DOI] [PubMed] [Google Scholar]
- Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima RA, Paggi MG, Pedersen PL (1984) Contributions of glycolysis and oxidative phosphorylation to adenosine 5'‐triphosphate production in AS‐30D hepatoma cells. Cancer Res 44: 5702–5706 [PubMed] [Google Scholar]
- Newsholme P, Procopio J, Lima MM, Pithon‐Curi TC, Curi R (2003) Glutamine and glutamate–their central role in cell metabolism and function. Cell Biochem Funct 21: 1–9 [DOI] [PubMed] [Google Scholar]
- Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti‐diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348(Pt 3): 607–614 [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC (2006) HIF‐1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197 [DOI] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, Jones RG (2013) Fueling immunity: insights into metabolism and lymphocyte function. Science 342: 1242454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak MN (2012) Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov 2: 778–790 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Alvarez R, Martinez‐Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, Sartini M, Rubin E, Lisanti MP, Sotgia F (2013) Mitochondrial dysfunction in breast cancer cells prevents tumor growth: understanding chemoprevention with metformin. Cell Cycle 12: 172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC (2006) Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem 97: 673–683 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2012) Hypoxia‐inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci 33: 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Krop I, Porter D, Polyak K (2002) Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression). Oncogene 21: 836–843 [DOI] [PubMed] [Google Scholar]
- Shannon AM, Bouchier‐Hayes DJ, Condron CM, Toomey D (2003) Tumour hypoxia, chemotherapeutic resistance and hypoxia‐related therapies. Cancer Treat Rev 29: 297–307 [DOI] [PubMed] [Google Scholar]
- Shin H, Liu T, Manrai AK, Liu XS (2009) CEAS: cis‐regulatory element annotation system. Bioinformatics 25: 2605–2606 [DOI] [PubMed] [Google Scholar]
- Simon MC (2006) Coming up for air: HIF‐1 and mitochondrial oxygen consumption. Cell Metab 3: 150–151 [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3 [DOI] [PubMed] [Google Scholar]
- Song S, Wheeler LJ, Mathews CK (2003) Deoxyribonucleotide pool imbalance stimulates deletions in HeLa cell mitochondrial DNA. J Biol Chem 278: 43893–43896 [DOI] [PubMed] [Google Scholar]
- Sotgia F, Whitaker‐Menezes D, Martinez‐Outschoorn UE, Salem AF, Tsirigos A, Lamb R, Sneddon S, Hulit J, Howell A, Lisanti MP (2012) Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle 11: 4390–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff A, Pientka FK, Mockel S, Kettelhake A, Hartmann E, Kohler M, Depping R (2009) Cellular oxygen sensing: Importins and exportins are mediators of intracellular localisation of prolyl‐4‐hydroxylases PHD1 and PHD2. Biochem Biophys Res Commun 387: 705–711 [DOI] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli‐Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E (2013) Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E‐driven lung tumors. Cancer Discov 3: 1272–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, White E (2014) Autophagy promotes BrafV600E‐driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 10: 384–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH (2006) Placental but not heart defects are associated with elevated hypoxia‐inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol 26: 8336–8346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomlinson RH, Gray LH (1955) The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer 9: 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- To KK, Huang LE (2005) Suppression of hypoxia‐inducible factor 1alpha (HIF‐1alpha) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol Chem 280: 38102–38107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12: 685–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Li Z, Lu Y, Du R, Katiyar S, Yang J, Fu M, Leader JE, Quong A, Novikoff PM, Pestell RG (2006) Cyclin D1 repression of nuclear respiratory factor 1 integrates nuclear DNA synthesis and mitochondrial function. Proc Natl Acad Sci USA 103: 11567–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Posener K, Negelein E (1924) Über den Stoffwechsel der Carcinomzelle. Biochem Zeitschr 152: 309–344 [Google Scholar]
- Warburg O (1925) Über den Stoffwechsel der Carcinomzelle. Klin Wochenschr 4: 534–536 [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS (2010) Mitochondrial metabolism and ROS generation are essential for Kras‐mediated tumorigenicity. Proc Natl Acad Sci USA 107: 8788–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS (2014) Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 3: e02242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Fan J, Venneti S, Wan YW, Pawel BR, Zhang J, Finley LW, Lu C, Lindsten T, Cross JR, Qing G, Liu Z, Simon MC, Rabinowitz JD, Thompson CB (2014) Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov 4: 1406–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Shi Y, Wei X, Yang Y, Zhou Y, Hao X, Zhang N, Niu R (2007) Depletion of mitochondrial DNA by ethidium bromide treatment inhibits the proliferation and tumorigenesis of T47D human breast cancer cells. Toxicol Lett 170: 83–93 [DOI] [PubMed] [Google Scholar]
- Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M (2006) Metformin is an AMP kinase‐dependent growth inhibitor for breast cancer cells. Cancer Res 66: 10269–10273 [DOI] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL (2007) HIF‐1 inhibits mitochondrial biogenesis and cellular respiration in VHL‐deficient renal cell carcinoma by repression of C‐MYC activity. Cancer Cell 11: 407–420 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, Polyak K, Brown M, Richardson AL, Hahn WC, Kaelin WG Jr, Bommi‐Reddy A (2009) Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell 16: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Wisidagama DR, Setoguchi K, Hong JS, Van Horn CM, Imam SS, Vergnes L, Malone CS, Koehler CM, Teitell MA (2012) Measuring energy metabolism in cultured cells, including human pluripotent stem cells and differentiated cells. Nat Protoc 7: 1068–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, Geisen C, Chakraborty AA, Moslehi JJ, Smalley DM, Wei X, Chen X, Chen Z, Beres JM, Zhang J, Tsao JL, Brenner MC, Zhang Y, Fan C, DePinho RA et al (2014) Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev 28: 1429–1444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, Guppy M (2004) Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun 313: 459–465 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Review Process File