

Abstract
During nuclear egress, herpesvirus capsids bud at the inner nuclear membrane forming perinuclear viral particles that subsequently fuse with the outer nuclear membrane, releasing capsids into the cytoplasm. This unusual budding process is mediated by the nuclear egress complex (NEC) composed of two conserved viral proteins, UL31 and UL34. Earlier, we discovered that the herpesvirus nuclear egress complex (NEC) could bud synthetic membranes in vitro without the help of other proteins by forming a coat‐like hexagonal scaffold inside the budding membrane. To understand the structural basis of NEC‐mediated membrane budding, we determined the crystal structures of the NEC from two herpesviruses. The hexagonal lattice observed in the NEC crystals recapitulates the honeycomb coats within the budded vesicles. Perturbation of the oligomeric interfaces through mutagenesis blocks budding in vitro confirming that NEC oligomerization into a honeycomb lattice drives budding. The structure represents the first atomic‐level view of an oligomeric array formed by a membrane‐deforming protein, making possible the dissection of its unique budding mechanism and the design of inhibitors to block it.
Keywords: herpesvirus, membrane budding, nuclear egress, UL31, UL34
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
The capacity of viruses to efficiently assemble and release new viral particles from host cells so that they can infect new ones is central to their ability to persist and cause disease. A detailed knowledge of viral replication mechanisms is critical for devising better strategies to combat them. Many viruses form infectious virus particles by enveloping themselves in the host cell membrane. These so‐called enveloped viruses typically acquire their lipid envelopes by capsid budding at the plasma membrane or at intracellular membranes such as ER, Golgi, or others, depending on the virus.
Herpesviruses are a large family of double‐stranded DNA, enveloped viruses that infect nearly all vertebrates and some mollusks. A total of 8 human herpesviruses cause lifelong latent infections from which viruses periodically reactivate, causing ailments such as skin lesions, encephalitis, and keratitis. Reactivations result not only in a substantial disease burden but also in a high rate of new infections. What sets herpesviruses apart from other enveloped viruses is that despite containing a single envelope, they bud twice: first time, at the inner nuclear membrane (INM) after being assembled in the nucleus and later at cytoplasmic membranes derived from Trans‐Golgi Network or the early endosomes (Mettenleiter et al, 2009; Johnson & Baines, 2011; Hollinshead et al, 2012) to be secreted by exocytosis. This also makes them the only known viruses to bud at the nuclear membrane. The envelope acquired during the first budding event does not end up in the mature viral particle. Only the second, and final, round of budding in the cytosol generates the single‐bilayer envelope of the mature virus. Instead, the unusual nuclear budding allows the viral capsids to escape from the nucleus. Herpesvirus genomes are replicated and packaged into capsids inside the nucleus. Most traffic in and out of the nucleus, which is surrounded by the nuclear envelope, occurs through the nuclear pores. Herpesvirus capsids are too large to exit through the nuclear pores, so nucleocapsids bud at the INM, forming immature perinuclear viral particles that then fuse with the outer nuclear membrane (ONM) releasing the naked capsids into the cytosol.
Efficient exit of nascent capsids from the nucleus, termed nuclear egress, requires the virally encoded nuclear egress complex (NEC) (reviewed in Johnson & Baines, 2011; Mettenleiter et al, 2013). The NEC consists of the conserved viral proteins UL31 and UL34. UL34 is anchored to the INM by a C‐terminal transmembrane helix with several residues extending into the perinuclear space (Shiba et al, 2000); its retention at the INM requires the presence of UL31 (Funk et al, 2015). UL31 is a nuclear phosphoprotein that localizes to the INM through interaction with UL34 (Chang & Roizman, 1993; Reynolds et al, 2002; Funk et al, 2015). In the absence of either UL31 or UL34, viral replication is impaired and most capsids are retained in the nucleus (Roller et al, 2000; Fuchs et al, 2002). The NEC is also sufficient to drive the vesiculation of the nuclear envelope in transfected cells in the absence of any other viral proteins (Klupp et al, 2007; Desai et al, 2012; Luitweiler et al, 2013). These results demonstrated that UL31 and UL34 are the only viral proteins necessary for nuclear envelope vesiculation but left the exact function of the NEC in membrane budding unclear.
Recently, by using purified NEC from herpes simplex virus type 1 (HSV‐1) and synthetic liposomes, we showed that the NEC has an intrinsic ability to vesiculate membranes in vitro (Bigalke et al, 2014). This finding was subsequently confirmed with the NEC from the related pseudorabies virus (PRV) (Lorenz et al, 2015). The NEC formed a coat‐like hexagonal lattice on the inner surface of the budded vesicles, which suggested that the NEC vesiculated membranes without the help of other proteins by oligomerizing on the membrane and creating a hexagonal scaffold inside the bud (reviewed in Bigalke & Heldwein, 2015).
To elucidate the structural basis of NEC‐mediated nuclear membrane deformation and vesiculation, here we determined the crystal structures of NEC from HSV‐1 and PRV. UL31 and UL34 have unique folds and form the NEC heterodimer through extensive interactions that involve residues distributed throughout the protein sequences. In crystals, HSV‐1 NEC packs into a hexagonal lattice that mimics the hexagonal NEC coats within budded vesicles. The 2.8‐Å crystal structure of the NEC lattice is the first atomic‐level view of an oligomeric array formed by a membrane‐deforming protein. Targeted mutagenesis of the oligomeric interfaces reduced NEC‐mediated budding in vitro, supporting the idea that NEC oligomerization drives capsid budding during nuclear egress of herpesviruses. The NEC structures provide a three‐dimensional road map to enable the dissection of the unique budding mechanism mediated by the NEC and the design of inhibitors to block it.
Results
Crystallization and structure determination
HSV‐1 NEC185Δ50 (UL31: 51–306; UL34: 15–185) and PRV NEC176Δ17 (UL31: 18–271; UL34: 1–176) were obtained by co‐expression in E. coli as described previously (Fig 1A) (Bigalke et al, 2014). HSV‐1 NEC185Δ50 crystallized in space group P6 with two heterodimers in the asymmetric unit and diffracted to 2.8 Å resolution (Table 1). PRV NEC176Δ17 crystallized in space group P43212 with two heterodimers in the asymmetric unit and diffracted to 2.7 Å resolution (Table 1). The crystal structure of the PRV NEC176Δ17 was determined by single anomalous dispersion using a selenomethionine derivative, and the crystal structure of HSV‐1 NEC185Δ50 was subsequently determined by molecular replacement using the PRV UL31 and UL34 structures as independent search models.
Figure 1. Crystal structures of NEC from HSV‐1 and PRV .
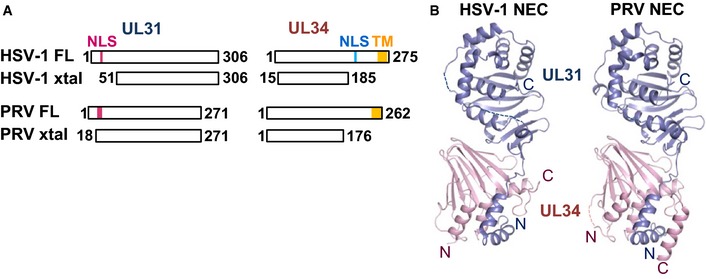
- UL31 and UL34 constructs from HSV‐1 or PRV used to obtain diffraction‐quality crystals are shown schematically next to the full‐length proteins. FL, full‐length protein; xtal, construct used for crystallization; NLS, nuclear localization signal; TM, transmembrane region.
- HSV‐1 and PRV NEC crystal structures strongly resemble each other. UL31 is shown in slate and UL34 in pink.
Table 1.
Data collection and refinement statistics
| PRV UL31 18–271/UL34 1–176 | HSV‐1 UL31 51–306/UL34 15–185 | |
|---|---|---|
| Data collectiona | ||
| Wavelength (Å) | 0.979200 | 1.600000 |
| Space group | P43212 | P6 |
| Unit cell (Å, °) | a = b = 125.456; c = 109.235 | a = b = 110.529; c = 155.850 |
| α = β = γ = 90.00 | α = β = 90.00; γ = 120.00 | |
| Resolution (Å) | 49.91–2.71 (2.80–2.71) | 47.86–2.77 (2.87–2.77) |
| Total reflections | 205,733 (30,237) | 519,936 (59,602) |
| Unique reflections | 24,223 (7,181) | 27,365 (2,663) |
| R merge | 0.087 (0.617) | 0.081 (0.559) |
| Redundancy | 4.55 (4.21) | 19.00 (22.38) |
| Completeness (%) | 99.36 (96) | 99.78 (97.80) |
| <I/σI> | 10.41 (2.02) | 34.37 (4.57) |
| Refinement | ||
| Number of non‐H atoms | 6,611 | 6,319 |
| Model content | A: UL34 4–22; 26–174 | A: UL34 –1–37; 39–105; 107–174 |
| B: UL31 19–100; 102–271 | B: UL31 55–130; 133–260; 269–306 | |
| C: UL34 3–23; 27–175 | C: UL34 –1–106; 108–175 | |
| D: UL31 18–100; 103–228; 232–271 | D: UL31 55–128; 134–262; 269–306 | |
| Number of water molecules | 38 | 74 |
| Solvent content (%) | 44.46 | 55.8 |
| R work/R free b | 0.217/0.269 (0.317/0.396) | 0.217/0.265 (0.271/0.344) |
| Rms deviationsc | ||
| Bond lengths (Å) | 0.007 | 0.006 |
| Bond angles (°) | 1.086 | 1.018 |
| <B> all (Å2) | 81.3 | 66.0 |
| <B> water (Å2) | 54.5 | 46.7 |
| Ramachandran plotd | ||
| Favored (%) | 98.04 | 96.80 |
| Allowed (%) | 1.96 | 3.20 |
Values in parentheses are for highest‐resolution shell.
R work and R free are defined as Σ||F obs|‐|F calc||/Σ|F obs| for the reflections in the working or the test set, respectively.
RMS, root mean square.
As determined using Molprobity (molprobity.biochem.duke.edu) (Davis et al, 2007).
Overall molecular architecture of the NEC
The NEC has an elongated shape of approximately 80 Å × 40 Å × 40 Å. The two non‐crystallographic symmetry (NCS) mates in HSV‐1 and PRV have rmsd values of 0.42 and 0.60 Å, respectively, demonstrating the lack of any significant conformational differences (Appendix Fig S1). The HSV‐1 and PRV complexes resemble each other closely (overall rmsd of 1.420 Å), with UL34 structures being more similar (rmsd 0.62 Å) than the UL31 structures (rmsd 1.12 Å) (Fig 1B). UL34 has a globular fold and forms a pedestal. UL31Δ50 has a globular core that sits on top of the UL34 pedestal and an N‐terminal hook‐like extension that reaches the opposite end of the NEC while wrapping around one margin of UL34 (Fig 1B).
The crystallized HSV‐1 NEC is missing the first 50 amino acids of UL31, M134–D1434 of UL34, and E18634–L27534 of UL34, which includes residues necessary for membrane interactions in UL31 and UL34 and the transmembrane anchor of UL34 (Fig 1A). The crystallized PRV NEC is similarly missing residues M131–R1731 of UL31 and V17734–R26234 of UL34. In both structures, the last resolved residues abutting the membrane‐interacting regions in UL31 and UL34 are located near each other at the bottom of the complex as shown in Fig 1B. This would place the missing membrane‐interacting regions (HSV‐1 residues R4131–K5031 of UL31 and E18634–D22034 of UL34 Bigalke et al, 2014) at the bottom of the complex. From this, we infer that the UL34 pedestal is the membrane‐proximal end of the NEC structure, while the helical cap in UL31 is the membrane‐distal end.
UL31 has a novel fold
HSV‐1 UL31Δ50 is composed of a globular core and an N‐terminal V‐shaped “hook” (Figs 2 and EV1). The globular core (K8731–P30631) has a novel α/β fold that consists of two antiparallel β sheets, the 5‐stranded upper β sheet (β2‐β8‐β9‐β6‐β5), the 4‐stranded lower β sheet (β1‐β4‐β3‐β7), a helical “cap”, and two additional helices (Figs 2A, EV1 and EV2). Three β strands from each β sheet stack in a β‐sandwich manner, but the sheets twist away from each other generating the unusual fold (Fig 2C). The helical cap, which surrounds the upper β sheet, consists of six α helices and two 310 helices (α6, α7, α4, α5, α9, α10, η2, and η3). These helices are arranged in three layers, from the inner to the outer: layer 1 (α6‐α7), layer 2 (α4‐η2‐η3‐α5), and layer 3 (α9‐α10). PRV UL31Δ17 has an additional 310 helix η4, unresolved in the HSV‐1 UL31Δ50 structure, within layer 3 (Fig EV1). One margin of the lower β sheet is decorated with α helices α3 and α8 and a 310 helix η1. The V‐shaped hook (L5531–L8631) is composed of α helices α1 and α2 and wraps around UL34 such that α1 lies at the base of the NEC, perpendicular to the longest axis of the complex. According to DALI (Holm & Rosenstrom, 2010), there are no strong structural similarities to other known proteins. The top hit in the Dali search was the ATP‐binding domain of the histidine kinase response regulator DosS, PDB ID 3ZXO, with the Z score of 3.9 and an RMSD of 3.99 Å over 82 residues. By comparison, the Z scores between the NCS mates of UL31 of HSV‐1, HSV‐1 UL31 versus PRV UL31, and HSV‐1 UL31 versus HCMV UL53 (Lye et al, 2015) are 35.6, 30.1, and 24.2, respectively. The region in DosS that aligns with residues 181–262 of HSV‐1 UL31 corresponds to the Bergerat fold, an α‐β‐β‐α‐β‐β fold characteristic of the GHKL ATPase/kinase super‐family that includes diverse protein families such as DNA topoisomerase II, molecular chaperones Hsp90, DNA‐mismatch‐repair enzymes, and histidine kinases (Bergerat et al, 1997; Dutta & Inouye, 2000). Unlike the ATP‐binding proteins of the GHKL superfamily, UL31 has an additional β strand between the second strand and the second helix of the classic Bergerat fold, which results in α‐β‐β‐β‐α‐β‐β topology (Appendix Fig S2), and the region corresponding to the ATP‐binding site is lined with hydrophobic side chains that would not permit ATP binding. In light of these important differences, we refer to this structural element within UL31 as the Bergerat‐like fold.
Figure 2. Secondary structure assignment.
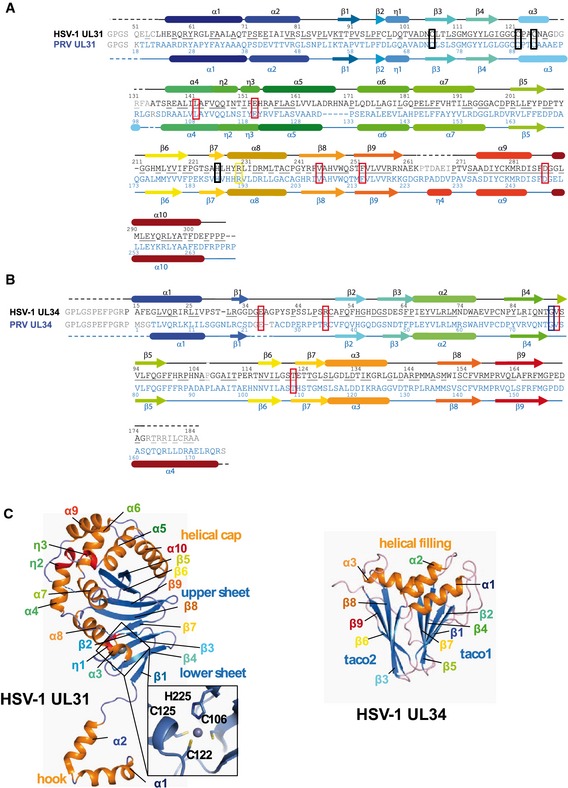
-
A, BUL31 sequences (A) and UL34 sequences (B). Unresolved residues are shown in gray and marked with dotted lines. Underlined residues are conserved in HSV‐1 and PRV. Secondary structure elements are shown as tubes for α‐helices and arrows for β‐sheets. Zn‐coordinating residues are boxed in black. Mutated residues are boxed in red, blue, or yellow, with red labeling mutants that show reduced budding, blue for no effect and yellow for an increase in budding.
-
CHSV‐1 UL31 and UL34 structures are shown separately. Structural elements are labeled and colored as in (A, B). The inlet shows the conserved UL31 Zn‐binding site with labeled coordinating residues.
Figure EV1. Secondary structure assignment in PRV UL31 and UL34.
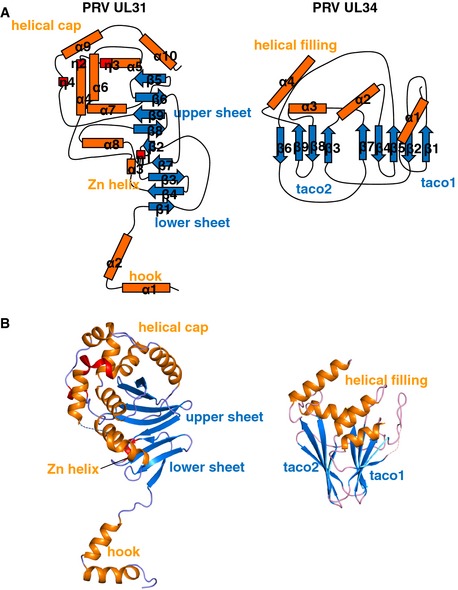
- Topology diagram for the PRV UL31 and UL34.
- PRV UL31 and UL34 structures are shown separately.
Figure EV2. Crystal structures of HSV‐1 and PRV UL31 and UL34.
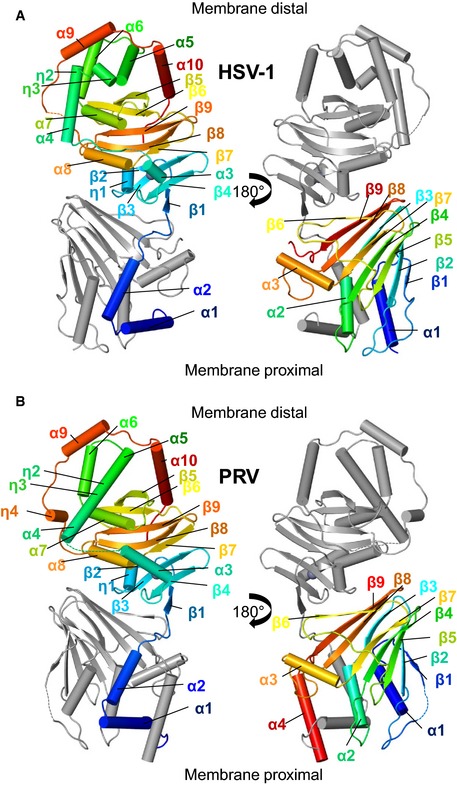
-
A, BCrystal structures of HSV‐1 (A) and PRV (B) UL31 and UL34, colored according to Fig 2A and B. The top of the NEC in the shown orientation represents the membrane‐distal end, whereas the bottom of the NEC represents the membrane‐proximal end.
Both HSV‐1 and PRV UL31 contain a CCCH‐type zinc‐binding site, where Zn2+ is coordinated by three cysteines and one histidine (HSV‐1: C10631, C12231, C12531, and H22531; PRV: C7331, C8931, C9231, and H18831) (Fig 2C, inset). C10631 and H22531 are located on β strands β3 and β7 within the lower β sheet, whereas C12531 and C12231 are located within helix α3 and the loop preceding it, respectively (Fig 2A and C). The Zn‐coordinating residues come from distant regions of UL31, and the Zn‐binding site does not form a domain such as a Zn‐finger domain. Instead, Zn coordination anchors the surface‐exposed helix α3 to the lower β sheet, probably to stabilize it. All four Zn‐coordinating residues are strictly conserved among UL31 sequences from α‐, β‐ and γ‐subfamilies along with only two other residues, P9531 and S11031, suggesting that the Zn‐binding site is conserved among herpesviruses and may play an important structural role.
UL34 is a β sandwich of novel fold
HSV‐1 UL34 has a globular fold with three α helices and nine β strands (Figs 2, EV1 and EV2). The β strands assemble into two antiparallel β sheets with the topology β1‐β2‐β5‐β4‐β7 (ABEDG) and β3‐β8‐β9‐β6 (CHIF) that form a β sandwich (Fig 2C). This topology is novel and does not resemble known β folds such as the jellyroll, which is commonly found in viral capsid proteins. Along one edge of the β sandwich, the β sheets are splayed open such that UL34 resembles a taco, also observed in the NMR structure of the murine cytomegalovirus (MCMV) M50, a UL34 homolog (Leigh et al, 2015). Three α helices are arranged at the opening of the “taco”. In the complex, the UL34 taco is oriented upside down with the α helices α1‐α3 located at the membrane‐proximal end of the NEC. Like UL31, UL34 does not have close structural neighbors according to DALI (Holm & Rosenstrom, 2010). However, just like the DALI search with HSV‐1 UL31, the search with HSV‐1 UL34 yielded the same top hit, DosS, with Z score 3.4 and an RMSD of 5.28 Å over 92 residues. By comparison, the Z scores between the NCS mates of UL34 of HSV‐1, HSV‐1 UL34 versus PRV UL34, and HSV‐1 UL34 versus HCMV UL50 are 30.3, 24.5, and 14.7, respectively. Like UL31, UL34 contains the Bergerat‐like fold with a non‐canonical α‐β‐β‐β‐α‐β‐β topology, and the corresponding residues 181–262 of UL31 and residues 14–106 of UL34 can be overlayed with a Z score of 6.1 and an RMSD of 4.28 Å (Appendix Fig S2). Whether this structural similarity reflects an evolutionary relationship, for example, as the result of gene duplication, is unclear.
In the crystallized HSV‐1 NEC, the UL34 construct ends at residue A18534, but the last resolved residue is G17534 (Fig 2B). By contrast, in the crystallized PRV NEC, UL34 ends at residue S17634, which is equivalent to T19034 in HSV‐1 UL34. This means that PRV UL34 construct has 5 additional residues at its C‐terminus, and all except the last one are resolved in the crystal structure where residues A16034–R17334 form helix α4 (Figs 2B and EV1). No density was observed for this helix in HSV‐1 UL34 even though all but two of the equivalent residues A17434–Q18734 are present in the crystallized construct. It is possible that helix α4 is disordered in the crystallized HSV‐1 NEC because missing residues E18634–Q18734 could be essential for its stability. Alternatively, helix α4 may be longer in HSV‐1 UL34 and may require additional residues for stability beyond residue Q18734.
UL31 and UL34 interact extensively through multiple regions
UL31 and UL34 interaction buries a large accessible surface area, 1,757 and 1,894 Å2 in HSV‐1 and PRV structures, respectively (Figs 3 and EV3, Appendix Tables S1 and S2), which helps explain the stability of the complex. Previously, neither UL31 nor UL34 could be purified individually due to their tendency to precipitate after removal of a solubility tag (Bigalke et al, 2014). The structures suggest that in the absence of their respective binding partners, both UL31 and UL34 would expose hydrophobic patches, normally buried at the interface, which would lead to aggregation. Additionally, the “hook” in UL31 is likely misfolded in the absence of UL34. The interface between UL31 and UL34 can be divided into two sections based on whether the interactions are mediated by the V‐shaped hook composed of helices α1 and α2 of UL31 (interface 1) or by its globular core (interface 2) (Fig 3).
Figure 3. UL31 binds to UL34 via two distinct interfaces.
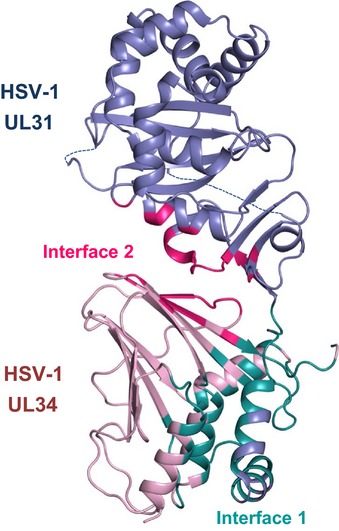
HSV‐1 UL31 is shown in slate and HSV‐1 UL34 in pink. Residues involved in interface 1 are colored deep teal and in interface 2 hot pink. A list of residues involved in the UL31/UL34 interactions can be found in Appendix Table S1. Similar interfaces can be observed in PRV NEC (Fig EV3).
Figure EV3. UL31 binds to UL34 using two distinct interfaces.
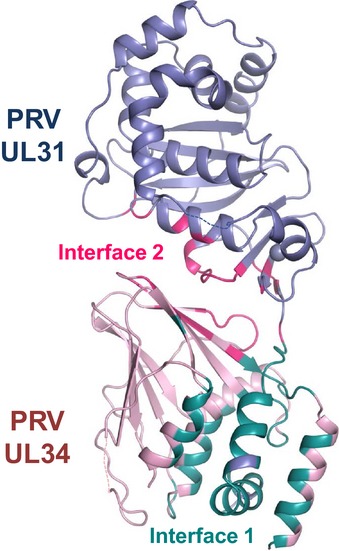
PRV UL31 is colored in light blue and PRV UL34 in light pink. Residues involved in interface 1 are shown in deep teal, and residues involved in interface 2 are shown in hot pink.
Interface 1 contributes approximately 68 and 73% of the contacts in HSV‐1 and PRV NEC, respectively. In HSV‐1 NEC, interface 1 is predominantly formed by hydrophilic interactions (62%), whereas interface 2 is slightly more hydrophobic (54%). In PRV NEC, interface 1 contains more hydrophobic than hydrophilic interactions (58%), whereas interface 2 has a more balanced ratio of hydrophilic and hydrophobic contacts (51 versus 49%). Interface 1 contains a single salt bridge common to both structures (HSV‐1: E7531–R2234, PRV: E4231–R834) (Appendix Fig S3). This salt bridge is likely important for NEC formation in all α‐herpesviruses because it is formed by conserved residues. In HSV‐1 NEC, interface 1 is stabilized by two additional salt bridges (R5831–E7834, R6231–D7534) that are missing from PRV NEC. Interface 2 has two similarly located salt bridges that are formed by residues conserved in all α‐herpesviruses (HSV‐1: D10431–R16734 and D23231–R15834; PRV: D7131–R15334 and D19531–R14434) (Appendix Fig S3). The extensive interdigitation of side chains along interface 1 suggests that it may be rigid. By contrast, interface 2 is relatively smooth and may permit some motion between the UL31 and UL34. Indeed, UL31 and UL34 within HSV‐1, PRV, and HCMV (Lye et al, 2015) structures have distinct relative orientations (Appendix Fig S4). Although some of the UL31/UL34 interactions are conserved among alphaherpesviruses, the majority of contacts between UL31 and UL34 appear species‐specific (Appendix Tables S1 and S2).
Previously, residues L6131–I9231 of HCMV UL53 (equivalent to residues Y6131–V9231 in HSV–1 UL31) (Sam et al, 2009; Schnee et al, 2012) and residues K13734–L18134 of HSV‐1 UL34 (Liang & Baines, 2005) were identified as essential for NEC complex formation and subsequently designated as the binding sites for their respective binding partners. These residues map to the UL31 “hook” and to one side of the taco (taco2: β8 and β9; Fig 2C) in UL34. Both regions are involved in UL31/UL34 interactions yet map to two distinct interfaces and hardly contact each other, which emphasizes the complexity of UL31/UL34 interactions that involve multiple regions throughout the protein sequence.
Several additional residues within UL34 have been identified as important for the NEC formation on the basis of mutagenesis (Bjerke et al, 2003; Bubeck et al, 2004; Roller et al, 2010; Milbradt et al, 2012; Passvogel et al, 2013, 2014, 2015). While some of these mutated residues, indeed, map to the UL31/UL34 interface, others are located within the core of either UL34 or UL31 and appear important for their structural stability. These latter mutants may be defective in NEC formation due to protein misfolding. Additionally, some mutations did not have an effect on HSV‐1 NEC formation in vitro, which suggests differences in complex formation between in vitro and in vivo experiments (Appendix Fig S5). The detailed analysis is presented in Appendix Table S4 and Appendix Fig S5.
Hexagonal lattice in HSV‐1 NEC crystals resembles NEC coats
The HSV‐1 NEC185Δ50 crystallized in space group P6 with two NEC heterodimers in the asymmetric unit, NECAB and NECCD. In the crystals, each NEC forms a hexagonal lattice resembling a honeycomb (Figs 4B and 5A) such that there are two lattices stacked on top of each other, one formed by multiple copies of NECAB, and the other by NECCD. Each hexagonal lattice is built from NEC hexamers. The hexamer‐to‐hexamer distance within the lattice is 110.5 Å, and the thickness of each lattice is 78.0 Å (Movie EV1). The hexameric rings are stacked head‐to‐head and tail‐to‐tail (head refers to the membrane‐distal end and tail refers to the membrane‐proximal end of the NEC) along the crystallographic c‐axis (Fig EV4). The individual NEC molecules are tilted with respect to the crystallographic c‐axis, and the NECAB and NECCD are related by two‐fold non‐crystallographic symmetry (Fig EV4). The head‐to‐head packing is mediated by interactions of residues within helices α6 (P17131–D17431) and α9 (S27231–R28131). The side chains of the NCS‐related residues C27831 in chains B and D may form a disulfide bond (Fig EV4). Additionally, there are two salt bridges between R28131 and D27531 of both chains and two hydrogen bonds between Q17331 and Y27731. The tail‐to‐tail packing is mediated by several residues within helix α1 of UL31 (R5831–T7131), and this interface is mostly hydrophobic (Fig EV4). There are two hydrogen bonds between Q7031 (chain D) and the backbone carbonyl oxygen of A6731 (chain B), and the backbone carbonyl of Q5931 (chain D) and R6231 (chain B). The head‐to‐head and the tail‐to‐tail interfaces bury a relatively small area, 305 and 316 Å2, respectively.
Figure 4. The NEC forms hexameric lattices in the presence of membranes or at high concentrations.
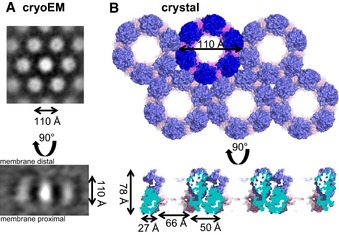
- Hexameric lattice as observed by cryoEM (Bigalke et al, 2014). The diameter of the hexameric rings is ˜110 Å, while the spikes are ˜110 Å in length.
- Hexameric lattice in the HSV‐1 NEC crystal. The lattice for NECCD is depicted. The diameter of each hexameric ring is 110 Å, while the length of the spikes is 78 Å. The difference in length can be accounted for by regions absent from the crystallization construct but present in the construct used in budding assays and cryoEM.
Figure 5. The two NCS mates in the HSV‐1 NEC crystal form two types of hexameric lattices.
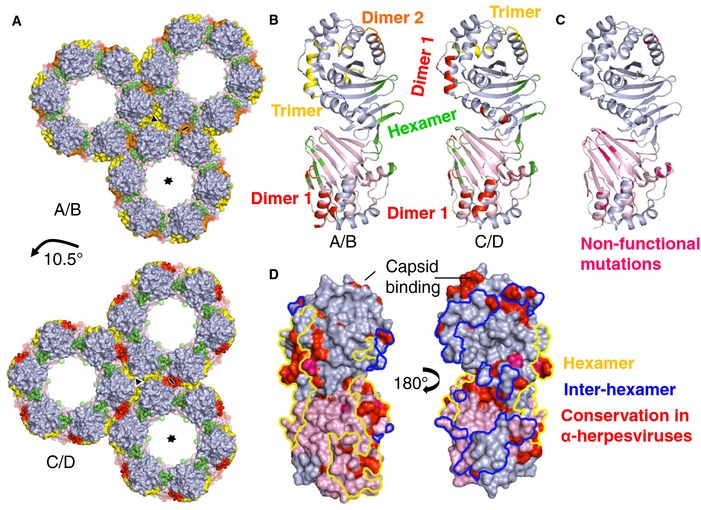
- The hexameric contacts are largely the same in both NECAB and NECCD (Appendix Table S3), but inter‐hexameric contacts differ. Hexameric interfaces are colored green, trimeric interfaces yellow, and dimeric interfaces red and orange. The lattice is shifted by 10.5° in NECCD versus NECAB.
- A detailed comparison of NECAB and NECCD and the oligomeric contacts. Color scheme is the same as in (A).
- Previously described non‐functional mutations, shown in hot pink, are mapped onto NECCD. Mutations that map to the UL34 interior likely disrupt the structural stability of the protein. Mutations that map to the oligomeric interfaces probably interfere with proper lattice formation, which explains the non‐functional phenotype of these mutants.
- Conserved residues in α‐herpesviruses are shown in red, and strictly conserved residues are shown in hot pink. Hexameric contact patches are outlined in yellow and inter‐hexameric patches in blue. Most conserved and surface‐exposed residues are located at the hexameric interface. A proposed conserved capsid‐binding site is located at the top of UL31 on the membrane‐distal side of the NEC.
Figure EV4. Crystal packing for HSV‐1 NEC .
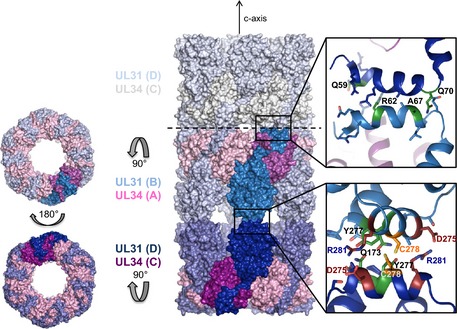
UL31 molecules are colored in shades of blue and UL34 molecules in shades of pink. The two different hexagonal lattices are stacked on top of each other, but because the individual NEC molecules are tilted toward the crystallographic c‐axis, the two different lattices can be accommodated. One asymmetric unit is shown in bold colors, visualizing that the only contacts mediating the NCS interface are provided by residues from helices α6 and α9 in UL31. The interactions are shown in detail next to the interface. The proposed disulfide bond is shown in yellow, hydrogen bonds in green, and salt bridges in firebrick. Other residues that are involved in hydrophobic interactions are not labeled, but the side chains are shown. The tail‐to‐tail interactions link the next layer (gray) to the lattice. These interactions are mediated by helix α1 from UL31. A detailed view is shown on the top right. Again, hydrogen bonds are colored green and the side chains of other residues involved in the interaction are shown.
The NEC hexagonal lattice observed in the crystals is strikingly similar to the hexagonal NEC coats previously visualized by cryoEM on the inner surface of the budded vesicles obtained in vitro (Fig 4A) (Bigalke et al, 2014). Both the crystal lattice and the membrane coat share hexagonal symmetry that results in a honeycomb array with inter‐hexamer distances of ~110 Å. The membrane‐distal spherical density corresponds to the globular domain of UL31, while the stalk is formed primarily by UL34. The previously proposed building block of the NEC coat, which was depicted as a cylinder topped with a sphere (Bigalke et al, 2014), corresponds to a NEC trimer.
NEC coats are composed of a curved honeycomb lattice whereas in the crystals, the lattice is flat. Although the crystals were reproducibly obtained, they formed thin, fragile plates (2D crystals), consistent with limited contacts between honeycomb layers and, perhaps, indicating that interactions that mediate crystal lattice formation may be able to produce a curved honeycomb array. The main difference between the two honeycomb lattices, the crystal and the membrane coat, is their thickness. While the crystal lattice is 78 Å thick, the thickness of the NEC coat in budded vesicles, excluding the lipid bilayer, is ~110 Å (Bigalke et al, 2014), leaving ~30‐Å‐thick density in the vicinity of the membrane unaccounted for by the crystal structure (Fig 4). Unlike the crystallized HSV‐1 NEC185Δ50, the NEC220 construct that forms inner coats on the in vitro budded vesicles contains 50 additional residues at the N‐terminus of UL31, 14 additional residues at the N‐terminus of UL34, and 35 additional residues at the C‐terminus of UL34. Given the location of the residues adjacent to these missing regions in the crystal structure, all three regions are expected to co‐localize at the membrane‐proximal end of the NEC. Thus, the additional density seen at the membrane‐proximal end of the spikes forming the membrane coats can be attributed to the N‐terminus of UL31 and the N‐ and C‐termini of UL34. These regions would extend the NEC spike by ~30 Å toward the membrane producing a characteristic fence‐like pattern in side‐view cryoEM projections of the NEC membrane coats (Fig 4A) (Bigalke et al, 2014).
The arrangement of the NEC in the crystal lattice agrees very well with the geometry and dimensions of the NEC coats formed on membranes. Such similar molecular organization strongly suggests that the NEC crystal lattice recapitulates the membrane coats in the budded vesicles and that the NEC/NEC interactions observed in the crystals are relevant to the NEC‐mediated budding.
Analysis of interactions within the honeycomb crystal lattice
The hexagonal honeycomb crystal lattice is composed of NEC hexamers (Fig 5A, Movie EV1), which are formed by UL34/UL34 and UL34/UL31 interactions (Appendix Table S3). UL31/UL31 interactions are not involved in the hexamer formation, but only mediate contacts between individual hexamers. The two NEC molecules in the asymmetric unit, NECAB and NECCD, form nearly identical hexamers, involving the same set of residues at the interface (Fig 5A, Appendix Table S3). Each UL34/UL31 and UL34/UL34 interface within the hexamer (shown in green in Fig 5A and B) is predominantly hydrophobic and buries 588 and 276 Å2 of accessible surface area, respectively. The involved residues are listed in Appendix Table S3. About 37% of all residues at the hexameric interface are conserved among α‐herpesviruses, suggesting that the ability to form hexamers is a common property, at least, among α‐herpesviruses (Fig 5D, Appendix Table S3). The extensive interactions that form the hexamer support the idea that it is the building block of the honeycomb lattice.
Interestingly, hexamers formed by either NECAB or NECCD are arranged differently within their respective lattices (Fig 5A and Appendix Table S3). That is, they form different dimeric and trimeric interfaces (at local two‐fold and three‐fold symmetry axes) while utilizing largely the same residues (Appendix Table S3). A close comparison of NECAB and NECCD (Appendix Fig S1) reveals a small shift of helices α4 and η2 in UL31. These helices participate in three‐fold symmetry contacts within the NECAB lattice but two‐fold symmetry contacts within the NECCD lattice. These two modes of hexamer packing may be dictated by the head‐to‐head/tail‐to‐tail stacking of hexamers along the c dimension. Although the two hexameric rings sit on top of each other, the NECAB hexamer is rotated about the six‐fold symmetry axis of the crystal by approximately 10.5° relative to the NECCD hexamer, and this could explain why the lateral interactions between the NECAB and the NECCD hexamers are different.
The contact area between the hexamers (NECAB: 629 Å2 versus NECCD: 877 Å2) is similar in size to the contact area within the hexamers (NECAB: 841 Å2 versus NECCD: 864 Å2), although the individual interfaces at the dimeric and trimeric symmetry axes are smaller (Appendix Table S3). While many conserved residues from both UL31 and UL34 are located at the hexameric interface, the inter‐hexameric interactions are less conserved. In both NEC lattices, interactions at the trimeric interface are mediated exclusively by UL31 residues, some of which are conserved (Fig 5D), while the dimeric interface also involves few non‐conserved UL34 residues (Appendix Table S3).
The biological significance of the ability of the NEC hexamers to pack in two different ways, as observed in the crystals, is yet unclear. Yet, this ability suggests that there is flexibility in how the hexamers can be arranged and that the hexamers may potentially be capable of interacting in more than the two ways seen in the crystal lattice.
Known mutation that blocks capsid budding maps to the hexameric interface
A number of UL31 and UL34 mutants defective in viral replication yet forming the NEC have been reported (Bjerke et al, 2003; Roller et al, 2010; Passvogel et al, 2013, 2014). Although some of these mutations (Bjerke et al, 2003; Passvogel et al, 2013, 2014) target residues inaccessible to solvent (Appendix Table S4) and probably destabilize the NEC structure, others target solvent‐accessible residues at the NEC hexameric interfaces (Fig 5C) and likely disrupt NEC function by perturbing its oligomerization (Bjerke et al, 2003; Roller et al, 2010; Passvogel et al, 2014). Previously, we showed that a mutant NEC containing a double point mutation D35A/E37A in HSV‐1 UL34 (DN), which blocks capsid nuclear budding in a dominant‐negative manner (Roller et al, 2010), was defective both in in vitro budding and in forming the hexagonal coats on membranes (Bigalke et al, 2014) (Fig 6C). In the crystal structure, residues D3534 and E3734 are located within a flexible loop but only E3734 maps to the hexameric interface (Fig 6A), and we hypothesized that the E37A34 mutation alone is responsible for the dominant‐negative non‐budding phenotype. By destabilizing the NEC hexamer formation, the E37A34 mutation hinders the correct lattice assembly and, when present in sufficient amounts, can “poison” the formation of the NEC coat even in the presence of the WT UL34, which explains the dominant‐negative effect.
Figure 6. Mutational analysis of hexameric lattice formation.
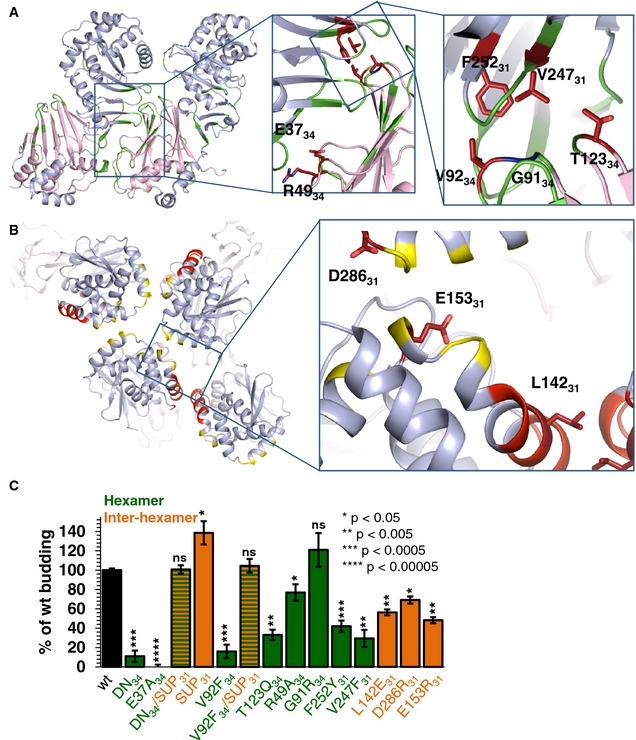
- Overview of mutations designed to perturb hexamer formation. Mutated residues that reduced budding are colored in firebrick while those that did not significantly affect budding are colored in blue.
- Three mutants were designed to perturb the inter‐hexamer interface. Mutated residues that reduced budding are colored in firebrick.
- Quantification of budding events. Budding efficiency is shown compared to wild‐type (wt). Mutants designed to interfere with hexamer formation are colored green while mutants designed to interfere with inter‐hexamer formation are colored orange. The reported values represent averages of the results of at least two individual experiments. Error bars represent the standard errors of measurement from at least two individual experiments, with a count of at least 75 GUVs per sample and experiment. The statistical analysis used is the Student's t‐test, indicating the significance compared to wt. *P‐value < 0.05, **P‐value < 0.005, ***P‐value < 0.0005, ****P‐value < 0.00005. DN budding data have been shown previously (Bigalke et al, 2014). DN, dominant‐negative non‐budding UL34 mutant containing D35A34/E37A34. DN/SUP is DN mutant that additionally contains mutation R222L31 in UL31 and has a wt phenotype. Raw average values of all mutants are listed in Appendix Table S5.
We generated the NEC220 containing the E37A34 mutation and tested it in an in vitro budding assay with fluorescently labeled GUVs, as previously described (Bigalke et al, 2014). The mutant was defective in membrane budding (0% of WT), suggesting that E3734 is a critical residue at the hexameric interface (Fig 6C). The nuclear budding defect due to the NEC‐DN mutation can be overcome by a suppressor mutation R229L in UL31 (SUP) (Roller et al, 2010). This mutation also restores the defect in in vitro budding (DN/SUP; Fig 6C). In the structure, residue R22931 is located at the dimeric interface in NECCD. In NECAB, R22931 does not mediate any inter‐hexamer contacts but is located near the inter‐hexamer interface and could make contacts in the curved NEC lattice. We hypothesized that if the E37A34 mutation interferes with NEC oligomerization by destabilizing the hexamers, the R229L31 mutation compensates by reinforcing contacts between the hexamers and stabilizing the NEC scaffold. To test whether the mutation R229L31 alone could improve the budding efficiency, we expressed and purified the NEC220 containing the R229L31 mutation only (SUP) and tested it for membrane budding (Fig 6C). The mutant shows an increase in budding efficiency (139%), which explains how it can restore WT budding efficiency in the DN mutant without being in the vicinity of these residues.
NEC oligomerization is required for budding
The hexagonal coats observed on the inner surface of budding vesicles suggested that oligomerization is the driving force for NEC‐mediated budding (Bigalke et al, 2014). To test this hypothesis and to determine the contribution of different interfaces to budding, we designed mutations to destabilize the hexagonal lattice by perturbing either conserved contacts within the hexamers (R49A34, G91R34, V92F34, T123Q34, V247F31, and F252Y31) or the contacts between the hexamers (L142E31, E153R31, and D286R31) (Fig 6A and B). All targeted residues are solvent‐exposed residues, in order to minimize the chance of misfolding. Nine mutant NEC220 complexes were purified and evaluated for membrane binding prior to being tested in the in vitro budding assay with fluorescently labeled GUVs. NEC formation and WT‐like membrane binding, indicated that the protein complexes were folded correctly. All mutants formed stable NEC when expressed in E. coli. Only mutant L142E31 displayed largely reduced membrane binding, 56% of the WT, even though the mutated residue is located in the membrane‐distal globular core of UL31 (Figs 6B and EV5). Out of nine mutants, six had significantly reduced budding ranging between 16 and 56% of the WT, while two showed only a slightly reduced budding efficiency (69 and 77% of the WT) and one displayed no significant change in budding (121%) (Fig 6C).
Figure EV5. Cosedimentation assay with NEC mutants and multilamellar vesicles (MLVs).
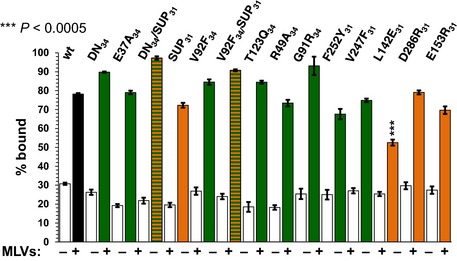
Most mutants bind to membranes comparable to WT NEC220, except for L142E31, which shows reduced binding. The reported values represent averages of the results of three individual experiments. Error bars represent the standard errors of measurement. The statistical analysis used is the Student's t‐test. ***P‐value < 0.005. The asterisks above L142E31 represent the significance compared to WT. All other samples do not show significant changes in membrane binding compared to WT. Coloring corresponds to Fig 6C, with hexamer mutants in green and inter‐hexamer mutants in orange.
Three mutations, V92F34, T123Q34, and V247F31 reduced budding the most (16, 33 and 30% of WT, respectively). All three residues are located within the same hexameric interface (Fig 6A); by introducing bulky residues, these mutations disrupted the hexamer formation, which reduced budding. Two other mutations targeted the same interface but reduced budding to a lesser extent or did not affect it (G91R34 and F252Y31). G91R34, resulted in essentially WT budding (121% of WT). This result was surprising because the bulky side chain of arginine was expected to disrupt the interface. But arginine side chains can adopt a range of conformations. We hypothesize that due to an inherent side chain flexibility, an arginine at this position can be accommodated without any steric effect, whereas a bulky residue such as phenylalanine is much more disruptive. On the other side of the same interface, mutation F252Y31 resulted in a modest yet significant decrease in budding. Mutation F252Y31 reduced budding to 42% of WT, which is a significant defect. The R49A34 also targeted the hexameric interface, but at a different location than the mutations discussed above, in the vicinity of E3734, which we described earlier (Fig 6A). This mutation reduced the NEC budding activity to a modest yet significant extent, 77% of WT. Taken together, our results demonstrate that the disruption of the UL34/UL31 hexameric interface through steric hindrance leads to reduced budding. Therefore, this interface plays an important role in the NEC oligomerization that drives budding.
To test whether the R229L31 (SUP) mutation could rescue the budding defect caused by mutations at the hexameric interface, we tested the budding activity of the double mutant V92F34/R229L31. The presence of SUP mutation restored budding from 16 to 105% of the WT (Fig 6C). Thus, SUP mutation can restore the budding defect caused by the disruption of the UL34/UL31 hexameric interface by mutations at two different locations, E37A34 or V92F34. We conclude that reinforcing contacts between the hexamers can stabilize the NEC lattice regardless of where the hexameric interface is perturbed.
Three single mutants were designed to destabilize the inter‐hexamer interactions by simultaneously disrupting salt bridges (E153R31 and D286R31) or by disrupting hydrophobic interactions (L142E31) (Fig 6B). Although mutations E153R31 and D286R31 reduced budding to 48 and 69% of WT, respectively, the budding defect due to either mutation was not as pronounced as with the mutations at the hexameric interface, arguing that the inter‐hexamer contacts appear more tolerant of mutations. Mutation L142E31 reduced both membrane binding and budding to the same extent (56% of WT), suggesting that reduced binding is responsible for the reduced budding. The L142E31 mutation is located far from the membrane‐proximal region of the NEC and is unlikely to affect membrane interactions directly. Instead, L14231 could contribute to membrane binding through a cooperative effect.
Discussion
Herpesviruses are unusual among enveloped viruses because they bud twice yet acquire a single envelope. They are also the only known viruses that bud across the nuclear envelope. Previously, we demonstrated that the NEC alone could bud synthetic membranes in vitro by forming a hexagonal scaffold inside the budding membrane. This provided the first evidence that the unique nuclear budding of herpesvirus capsids is mediated by the virally encoded NEC and may not need the help of any host proteins. Here, we present the crystal structures of the NEC from HSV‐1 and PRV, which provide insights into how the NEC functions and serve as a three‐dimensional template for a detailed exploration of its membrane‐budding mechanism.
Both UL31 and UL34 have unique folds and do not share structural similarity with any viral capsid or matrix proteins as would be expected based on the ability of the NEC to form coats and deform membranes. Thus, the evolutionary origin of the NEC remains unclear. Extensive interactions between UL31 and UL34 imply that the affinity between UL31 and UL34 is likely high and that at the INM of infected or transfected cells UL31 and UL34 mostly exist as a complex. Most previously reported mutations that interfere with complex formation (Bjerke et al, 2003; Bubeck et al, 2004; Sam et al, 2009; Roller et al, 2010; Milbradt et al, 2012; Passvogel et al, 2013, 2014, 2015) map to the UL31/UL34 interface although a few appear to disrupt the complex by perturbing the fold of either UL31 or UL34. When expressed individually in E. coli, UL31 and UL34 tended to precipitate and could not be purified, probably due to exposure of hydrophobic patches normally buried at the NEC interface and the misfolding of the N‐terminal hook in UL31, which is likely flexible in the absence of UL34. In mammalian cells, UL31 and UL34 may engage chaperones to avoid aggregating or getting degraded when the respective binding partner is not present.
In crystals, HSV‐1 NEC packs into a hexagonal lattice with dimensions and geometry similar to the hexagonal NEC coats within budded vesicles. Thus, the crystal structure of the NEC lattice reveals atomic‐level interactions that form the NEC scaffold. Moreover, the structure provides the first high‐resolution view of an oligomeric array formed by a membrane‐deforming protein (Movie EV1). Mutations that disrupt hexameric or inter‐hexameric contacts reduce budding in vitro, which demonstrates that the assembly of the NEC scaffold is necessary for budding (Fig 7). While inter‐hexamer contacts appear more tolerant of mutations, additional mutagenesis is necessary to evaluate fully the relative contributions of intra‐hexamer and inter‐hexamer interactions to budding.
Figure 7. Model of NEC‐mediated budding.
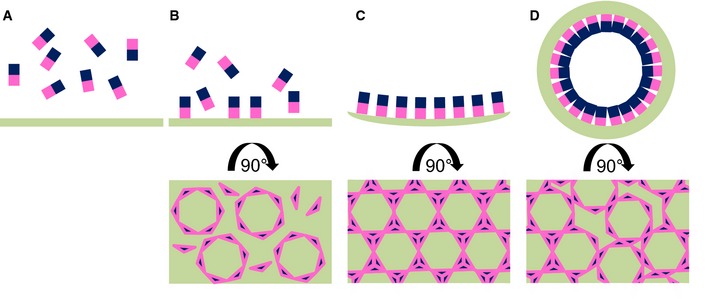
-
A–DThe NEC is represented by rectangles with UL31 in blue and UL34 in pink. Upon membrane binding (B), individual NEC heterodimers assemble into hexameric rings. These represent the individual building blocks of the lattice. (C) Once the hexamers are linked to each other, a negative curvature is induced. (D) The NEC‐lattice forms a coat enabling budding independently of other factors. Flaws in the hexameric lattice are required to form a spherical object, but these have not yet been visualized.
Recently, PRV UL31 has been reported to vesiculate synthetic liposomes in the absence of UL34 (Lorenz et al, 2015). UL31 was N‐terminally tagged with a tandem His‐EGFP tag and used with Ni‐NTA‐containing liposomes. Authors concluded that UL34 was necessary for UL31 membrane recruitment but not for membrane budding and that UL34 function could be bypassed by membrane tethering of UL31 (Lorenz et al, 2015). These observations are difficult to reconcile with the fact that the NEC lattice is formed by interactions that involve both UL31 and UL34 and with our data showing that mutations in UL34 at the hexameric interface reduce budding without reducing membrane binding. Furthermore, they do not explain why certain UL34 mutants, described previously, are non‐functional despite being able to form the NEC and to localize to the INM (Bjerke et al, 2003; Roller et al, 2010). In the structure, these UL34 mutations map to the oligomerization interfaces, and their non‐functional phenotype can be explained by the disruption of the NEC honeycomb lattice and, thus, budding. Additional studies are necessary to resolve these apparent discrepancies.
The hexagonal crystal lattice formed by the NEC is flat, whereas the honeycomb coats are spherical. While strictly symmetrical hexagonal packing works within a flat array, formation of a curved scaffold requires distortions, or defects, in hexagonal packing. A closed spherical lattice is typically achieved through a regular inclusion of pentagons, which generates a polyhedral particle, for example, an icosahedron. In some cases, however, the hexameric lattice is closed by incorporation of irregular defects, such as observed in the immature HIV capsids formed by Gag (Briggs et al, 2009; Schur et al, 2015) and the early poxvirus envelope (Heuser, 2005) formed by D13 (Hyun et al, 2011). The NEC coats observed by cryoEM lack obvious polyhedral symmetry (Bigalke et al, 2014), and curvature could arise from the incorporation of such irregular defects into a hexagonal lattice. The two equivalent yet different inter‐hexamer arrangements observed in the crystals suggest that the lateral packing of hexamers has some flexibility and that the hexamers may potentially pack in more than the two ways seen in the crystal lattice.
Such flexibility could provide a way for introducing lattice disruptions so that a perfectly hexagonal but flat lattice becomes an imperfect curved lattice and the spherical NEC coats are formed (Fig 7). For example, hexamers may have to rotate slightly while the curved lattice is being formed. Perfectly hexagonal patches of NEC lattice would be interrupted by irregularities resulting in a curved array (Fig 7). The weaker contacts between hexamers may also permit an easy disassembly.
Higher resolution cryoEM images will be needed to visualize the geometry of the NEC scaffold in detail, but in line with this hypothesis, we have previously observed occasional heptameric rings by cryoEM. Interestingly, in both reported cases of a spherical hexagonal lattice with irregular defects, the coats are transient and are not retained in mature viral particles. The immature HIV capsid is converted into a mature capsid characterized by “broken” polyhedral symmetry (Briggs et al, 2009). The early poxvirus envelope is formed by D13, which is thought to drive the formation of membrane crescents and their coalescence into a spherical particle. This D13 coat is disassembled shortly afterward (Condit et al (2006). Likewise, the NEC coat is disassembled during de‐envelopment (Skepper et al, 2001). It is tempting to speculate that hexagonal coats containing irregularities possess characteristics that make them susceptible to modifications that may ease their disassembly.
The major difference between the HSV‐1 and PRV NEC structures lies within the C‐terminal helix α4 that is well ordered in PRV NEC but unresolved in HSV‐1 NEC (Appendix Fig S6). The crystallized PRV UL34 construct is 5 amino acids longer than its HSV‐1 homologue, and these 5 residues may be required for the stability of α4. Although we do not know whether helix α4 is formed in a longer HSV‐1 NEC construct, 6 out of 14 residues in this helix are conserved between HSV and PRV UL34, suggesting that helix α4 is present in longer HSV‐1 UL34 constructs. Moreover, this helix was also observed in the NMR structure of MCMV UL50, a UL34 homolog (Leigh et al, 2015) and in the crystal structure of HCMV NEC (Lye et al, 2015). Whereas HSV‐1 NEC formed a hexagonal crystal lattice, no hexagonal symmetry was observed in the PRV NEC crystals. Analysis of crystal packing revealed that helix α4 in PRV NEC would be incompatible with the hexagonal lattice formed by NECCD due to steric hindrance at the dimeric interface (Appendix Fig S7). But, it would not affect the hexagonal lattice formed by NECAB. It is currently unclear whether both honeycomb lattices observed in crystals are formed during budding. The contact area between the hexamers is larger in the NECCD lattice (NECCD: 877 Å2 versus NECAB: 629 Å2), which favors the NECCD lattice as the biologically relevant lattice. This would implicate helix α4 in UL34 as an important regulatory element that blocks oligomerization and must relocate or unravel to permit oligomerization and budding. In individually expressed MCMV M50, a UL34 homolog, a corresponding helix packs against the core of M50, effectively replacing helix α2 of M53, a UL31 homolog (Leigh et al, 2015). This supports the idea that helix α4 has conformational flexibility that could be involved in the regulation of budding. Alternatively, if both honeycomb lattices observed in crystals form during budding, the regulatory role of helix α4 is less clear‐cut.
Both in vitro and in transfected cells, the NEC has powerful membrane vesiculation activity. Yet, empty perinuclear vesicles are not typically observed during infection, and mature rather than immature capsids primarily bud into the INM (Klupp et al, 2011). This means that in infected cells, the intrinsic budding potential of the NEC is likely regulated to ensure productive budding and to avoid non‐productive budding. Given that NEC oligomerization is the driving force for the vesiculation, formation of the NEC lattice would need to be inhibited until the mature capsid comes along. The hexagonal honeycomb lattice observed in the crystals of HSV‐1 NEC185Δ50, which lacks membrane‐interacting regions, attests to its intrinsic ability to self‐assemble in solution, at least, at high protein concentration achieved in crystal setups. By contrast, HSV‐1 NEC220, used in the in vitro budding assay, oligomerizes only in the presence of membranes. These observations suggest that the membrane‐interacting regions within the NEC could inhibit its ability to oligomerize correctly in the absence of membranes; their displacement (in NEC220 upon membrane binding) or removal (in NEC185Δ50) enables self‐assembly of the NEC honeycomb coat. Membrane‐interacting regions could therefore be a part of the regulatory mechanism that controls NEC‐mediated budding. Another component of this inhibitory mechanism could be helix α4 in UL34, which is adjacent to the membrane‐binding region of UL34. The presence of this helix is incompatible with one of the hexagonal arrays and could function as a “brake” by preventing either the premature NEC oligomerization at the membrane or a premature membrane deformation. A triggering signal would then enable oligomerization by either displacing the helix or causing it to unravel.
How the budding activity of NEC is inhibited in infected cells and how this inhibition is relieved in the presence of the capsid is unclear. The fact that primarily mature capsids bud into the INM (Klupp et al, 2011) is consistent with NEC oligomerization being triggered by proteins present on mature but not on immature capsids. The NEC is thought to recruit capsids to the INM (Yang & Baines, 2011) and has been reported to interact with capsids by using UL31 to bind either the accessory capsid protein UL25 (Yang & Baines, 2011) or the major capsid protein VP5 (Yang et al, 2014). A mature capsid, with multiple binding sites for the NEC that would create avidity effects, could provide a major driving force for the formation of an enveloping vesicle containing a coat composed of extended patches of NEC hexamers. A surface patch in helix α9 in UL31 at the membrane‐distal end of the NEC, which is conserved in α‐herpesviruses, could potentially be the capsid‐binding site. Transmitting the signal from the membrane‐distal region to the membrane‐proximal region would require large conformational changes within the NEC. Alternatively, capsids could trigger oligomerization indirectly by inactivating an inhibitor that blocks NEC oligomerization. Phosphorylation of the HSV‐1 NEC by the viral kinase US3 may play a role in inhibition of its budding activity (Mou et al, 2009), while dephosphorylation could serve as a trigger for oligomerization. Another question is how the hexagonal coat gets disassembled for the de‐envelopment step in the perinuclear space. US3 may also be involved in this process because it is present in the perinuclear viral particles and because in its absence, these particles get retained in the perinuclear space (Reynolds et al, 2002). Phosphorylation of the NEC after primary budding may lead to structural rearrangements that disrupt the hexameric lattice, thereby enabling de‐envelopment. By interfering with oligomerization, phosphorylation of the NEC could both inhibit budding in the absence of the capsid and disassemble the NEC coat during de‐envelopment.
Materials and Methods
Plasmid cloning
Codon‐optimized genes encoding PRV UL31 and UL34 were synthesized (Invitrogen) and cloned into the prokaryotic expression vector pET24b (Invitrogen) modified to include a sequence encoding a His6‐SUMO tag followed by a PreScission cleavage site and pGEX‐6P1 (GE Healthcare), respectively. Primers used to generate UL31 18–271 and UL34 1–176 were as follows: 5′‐aaaaaggatccaagacgctgacgcgcgcg‐3′ (UL31 fw), 5′‐aaaaagaattctcacgggcgaggagggc‐3′ (UL31 rev), 5′‐aaaaaaggatccatgagcggcaccctggtcc‐3′ (UL34 fw), and 5′‐aaaaagaattctcacgagcgctgccgcagctc‐3′ (UL34 rev). Restriction sites are underlined. Cloning of plasmids encoding HSV‐1 UL31 51‐306 and UL34 15–185 is described elsewhere (Bigalke et al, 2014). Site‐directed mutagenesis of mutations in UL31 (L142E, V247F, D286R, E153R, F252Y) and UL34 (E37A, R49A, E67A, Y68A, V79A, V92F, T123Q, G91R) was performed using the splicing‐by‐overlap‐extension PCR using primers (L142E fw 5′‐cgcgaggccctaatcgaggccttcgtgcagcag‐3′, rev 5′‐ctgctgcacgaaggcctcgattagggcctcgcg‐3′; V247F fw 5′‐cggttcgtcgcccacttttggcagagcacgtttg‐3′, rev 5′‐caaacgtgctctgccaaaagtgggcgacgaaccg‐3′; D286R fw 5′‐agggacatcagcttccgcggggggctcatgctag‐3′, rev 5′‐ctagcatgagccccccgcggaagctgatgtccct‐3′; E153R fw 5′‐gatcaacacgatattcaggcatcgcgccttcctg‐3′, rev 5′‐caggaaggcgcgatgcctgaatatcgtgttgatc‐3′; F252Y fw 5′‐gtgtggcagagcacgtatgtgctcgtggtccgg‐3′, rev 5′‐ccggaccacgagcacatacgtgctctgccacac‐3′; E37A fw 5′‐gggcggggacggggcgggcccc‐3′; rev 5′‐ggggcccgccgccccgtccccgccc‐3′;; R49A fw 5′‐ccctccagcctcccctccgcgtgcgcctttcag‐3′; rev 5′‐ctgaaaggcgcacgcggaggggaggctggaggg‐3′; E67A fw 5′‐gggtccgacgagtcgtttcccatcgcgtatgtactgcggcttatgaacg‐3′, rev 5′‐cgttcataagccgcagtacatacgcgatgggaaacgactcgtcggaccc‐3′; Y68A fw 5′‐gggtccgacgagtcgtttcccatcgaggctgtactgcggcttatgaacg‐3′, rev 5′‐cgttcataagccgcagtacagcctcgatgggaaacgactcgtcggaccc‐3′; V79A fw 5′‐tccgaggagatcgccatcgcgcgctcgctctcggtgcccctg‐3′, rev 5′‐ caggggcaccgagagcgagcgcgcgatggcgatctcctcgga‐3′; V92F fw 5′‐catacagaacaccggcttttcggtgctgtttcagg‐3′, rev 5′‐cctgaaacagcaccgaaaagccggtgttctgtatg‐3′; T123Q fw 5′‐gtgatcctggggtcccaagagacgacggggttg‐3′, rev 5′‐caaccccgtcgtctcttgggaccccaggatcac‐3′; G91R fw 5′‐cgcatacagaacacccgcgtgtcggtgctgttt‐3′, rev 5′‐aaacagcaccgacacgcgggtgttctgtatgcg‐3′).
Expression and purification of recombinant HSV‐1 UL31 51–306/UL34 15–185 and PRV UL31 18–271/UL34 1–176
Plasmids encoding HSV‐1 or PRV UL31 or UL34 were co‐transformed into Escherichia coli BL21(DE3) Rosetta cells (Novagen) and expressed at 18°C for 16 h after induction with 0.3 mM IPTG. All purification steps were performed at 4°C. All complexes were purified in lysis buffer (50 mM HEPES pH 7.0, 500 mM NaCl, 10% glycerol, and 1 mM TCEP) unless noted otherwise. Cells were resuspended in lysis buffer in the presence of Complete protease inhibitor (Roche) and lysed using a Microfluidizer. The cleared cell lysate was first passed over a Ni‐NTA sepharose (GE Healthcare); the column was washed with lysis buffer containing 20–40 mM imidazole, and bound proteins were eluted with lysis buffer containing 250 mM imidazole and loaded onto a glutathione sepharose to obtain the NEC free from excess His6‐SUMO‐UL31. After washing with lysis buffer, His6‐SUMO and GST tags were cleaved on the glutathione sepharose column for 16 h using PreScission protease produced in house using a GST‐PreScission fusion protein expression plasmid. Cleaved proteins were eluted from the GSH column with lysis buffer. Protein‐containing fractions were applied onto a Talon column (HiTrap, GE Healthcare). The flow‐through containing the NEC was collected. As the final purification step, proteins were purified by size‐exclusion chromatography using a Superdex 75 column (GE Healthcare) equilibrated with 20 mM HEPES, pH 8.0, 100 mM NaCl, and 1 mM TCEP. The column was calibrated using blue dextran (~2,000 kDa), conalbumin (75 kDa), carbonic anhydrase (29 kDa), ribonuclease A (13.7 kDa), and aprotinin (6.5 kDa). The NEC complexes were purified to homogeneity as assessed by 12% SDS–PAGE and Coomassie staining. Fractions containing the NEC were concentrated up to ~10 mg/ml and stored at −80°C to avoid aggregation and degradation at 4°C. Protein concentration was determined by absorbance measurements at 280 nm. The typical yield was 5 mg/l LB culture.
Selenomethionine containing PRV NEC was produced using minimal autoinduction media (Studier, 2005). Purification was conducted as above, but with 2 mM TCEP in each buffer. Incorporation of selenomethionine was confirmed by mass spectrometry (David King, UC Berkeley).
Crystallization and data collection
Crystals of native HSV‐1 NEC and SeMet PRV NEC were grown by vapor diffusion at 20°C in hanging drops with 0.5 μl protein and 0.5 μl reservoir solution (HSV‐1: 0.1 M Na citrate pH 5.6, 5 mM NiCl2, 10% PEG8000; PRV NEC: 0.3 M NaSCN, 18% PEG3350, 0.3 M NaCl). Hexagonal HSV‐1 NEC crystals appeared after 2 days and grew to their final size in 1 week. Tetragonal PRV NEC crystals appeared after 1 day and reached their final size after 2 days. Crystals were flash‐frozen in solution identical to the well solution and supplemented with cryoprotectant, 25% glycerol (HSV‐1 NEC) or 15% meso‐erythritol (PRV NEC). A native dataset of HSV‐1 NEC was collected at the wavelength of 1.6000 Å at 100 K at beamline X25 at the National Synchrotron Radiation Source and processed to 2.8 Å resolution using XDS (Kabsch, 2010) (Table 1). Crystals took space group P6 with a = b = 110.529 Å, c = 155.850 Å, α = β = 90°, γ = 120°. A SeMet SAD dataset of PRV NEC was collected at the peak wavelength of 0.9792 Å at 100 K at beamline 24ID‐C at Advanced Photon Source and processed to 2.7 Å resolution with XDS (Kabsch, 2010) (Table 1) in space group P422 with a = b = 125.456 Å, c = 109.235 Å, and α = β = γ = 90°.
Structure determination of PRV NEC
A total of 19 out of 24 selenium sites were found using direct methods as implemented in hkl2map in the SHELX suite (Pape & Schneider, 2004; Sheldrick, 2010) and refined using AutoSol (Adams et al, 2010). At this point, the space group ambiguity was resolved in favor of P43212. There are two NEC heterodimers in the asymmetric unit, NECAB and NECCD. After phase improvement by density modification, including two‐fold NCS averaging, solvent flattening, and histogram matching as implemented in Autosol (Adams et al, 2010), the experimental electron density map allowed tracing and sequence assignment for over 90% of the ordered polypeptide chain using Coot (Emsley et al, 2010). Prior to refinement, 8% of the data were set aside for cross‐validation. Model refinement included gradient minimization refinement of coordinates, individual thermal parameters, and TLS parameters, all as implemented in phenix.refine (Adams et al, 2010). After several rounds of refinement, rebuilding, and the addition of solvent molecules, the R work was 21.7% and the R free was 26.9% (Table 1). In the final model, NECAB contains residues 19–271 of UL31, chain B (unresolved residue 101), and residues 4–174 of UL34, chain A (unresolved residues 23–25). NECCD contains residues 18–271 of UL31, chain D (unresolved residues 101–102 and 229–231), and residues 3–175 of UL34, chain C (unresolved residues 24–26). A total of 38 water molecules were placed as well. According to Molprobity (Davis et al, 2007), 98.04% of residues lie in the most favored and 1.96% in the additionally allowed regions of the Ramachandran plot. The structure was deposited in the Protein Data Bank with the ID 4Z3U. All software was installed and maintained by SBGrid (Morin et al, 2013).
Structure determination of HSV‐1 NEC
The correct molecular replacement solution for HSV‐1 NEC was obtained using PRV UL31 and UL34 as separate search models in Phaser‐MR (McCoy et al, 2007; Adams et al, 2010). There are two NEC heterodimers in the asymmetric unit, NECAB and NECCD. The resulting electron density map allowed tracing and sequence assignment for over 90% of the ordered polypeptide chain using Coot (Emsley et al, 2010). Prior to refinement, 5% of the data were set aside for cross‐validation. Model refinement included one round of rigid body refinement, gradient minimization refinement of the coordinates, individual thermal parameters, and TLS parameters, all as implemented in phenix.refine (Adams et al, 2010). After several rounds of refinement, rebuilding, and the addition of solvent molecules, the R work was 21.7% and the R free was 26.5% (Table 1). In the final model, NECAB contains residues 55–306 of UL31, chain B (unresolved residues 131–132 and 261–268), and residues ‐1–174 of UL34, chain A (unresolved residue 106). NECCD contains residues 55–306 of UL31, chain D (unresolved residues 129–133 and 263–268), and residues ‐1–175 of UL34, chain C (unresolved residue 107). A total of 74 water molecules were placed as well. According to Molprobity (Davis et al, 2007), 96.80% of residues lie in the most favored and 3.20% in the additionally allowed regions of the Ramachandran plot. The structure was deposited in the Protein Data Bank with the ID 4ZXS.
Structure analysis
Interfaces were analyzed using PISA (Krissinel & Henrick, 2004) and APBS (Baker et al, 2001). Secondary structure was assigned using DSSP (Kabsch & Sander, 1983). All structure figures were made using PyMOL (www.pymol.org).
Cosedimentation assay
Liposomes were prepared as described previously (Bigalke et al, 2014). Three micrograms of protein was centrifuged at 16,000 g for 20 min at 4°C to get rid of non‐specific aggregates and debris. The supernatant was incubated with or without 15 μg of freshly prepared MLVs at 20°C for 30 min. The samples were centrifuged again at 16,000 g for 20 min at 4°C. Aliquots of input fractions, protein/MLV pellet, and protein supernatant were analyzed by 12% SDS–PAGE and Coomassie staining. The amount of protein that pelleted with MLVs was determined by densitometry analysis of gels imaged using GBox (Syngene) and quantified using manufacturer's software GeneTools. For each protein, band intensities of the pelleted protein were integrated and expressed as a percentage of the total integrated intensity of all bands, that is, protein pelleted with MLVs plus un‐pelleted protein. Each experiment was done in triplicate and repeated at least twice, and the average value and the standard error of measurement are reported.
GUV budding assay
A total of 10 μl of GUVs containing POPE‐Atto594 at a concentration of 0.2 μg/μl was mixed with a final concentration of 0.8 μM protein and incubated for 5 min at 20°C. The total volume of the samples during imaging was 100 μl, and imaging was performed in a 96‐well chambered cover glass. Images were acquired using a Nikon A1R Confocal microscope at the Tufts Imaging facility of the Center for Neuroscience Research at Tufts University School of Medicine. A 60× oil immersion lens was used. Image analysis was performed using ImageJ (Schneider et al, 2012). A total of 0.2 mg/ml Cascade Blue Hydrazide (Life Technologies) was added to diluted GUVs. Quantification was performed by counting all vesicles in 17 random frames of each sample. The experiments were repeated at least twice independently. The number of vesicles counted in total is as follows: no protein (104, 111, 106, 111, 111, 123, 104, 108, 101, 136, 135), WT (83, 75, 119, 120, 128, 104, 111, 111, 105), V92F (72, 103, 176), T123Q (103, 150), G91R (97, 109), L142E (108, 108), V247F (88, 101, 101), D286R (104, 159), E153R (132, 109), F252Y (90, 86), DN (77, 136), DN/SUP (79, 92), SUP (104, 110, 111), E37A (110, 112), R49A (117, 117), and V92F/SUP (106, 109). The background was subtracted from the raw values, averaged, and normalized to WT (100%). The standard error of measurement is reported for each sample.
Author contributions
JMB performed experiments. JMB and EEH designed experiments, analyzed data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Review Process File
Acknowledgements
We thank James Schiemer for help with the HSV‐1 mutant analysis and Xuanzong Guo for expert technical assistance; the staff at the NE‐CAT (Advanced Photon Source) and A. Héroux (National Synchrotron Light Source) for help with collecting X‐ray diffraction data; David King (University of California, Berkeley) for mass spectrometry experiments; Richard Roller for the gift of the anti‐HSV‐1‐UL34 polyclonal antibody; and Peter Cherepanov for the gift of the GST‐PreScission protease expression plasmid. This work was funded by the NIH grants 1R21AI097573 and 1R01GM111795 (E.E.H.), the Burroughs Wellcome Fund (E.E.H.), and by the postdoctoral fellowship from the Deutsche Forschungsgemeinschaft GZ: BI 1658/1‐1 (J.M.B.). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract no. DE‐AC02‐98CH10886. All software was installed and maintained by SBGrid (Morin et al, 2013). This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the NIH grant P41 GM103403. The Pilatus 6M detector on 24‐ID‐C beam line is funded by a NIH‐ORIP HEI grant (S10 RR029205). [Correction added on 2 December 2015 after first online publication: The last two sentences have been added to the Acknowledgements section.]
The EMBO Journal (2015) 34: 2921–2936
See also: MF Lye et al (December 2015)
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P (1997) An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386: 414–417 [DOI] [PubMed] [Google Scholar]
- Bigalke JM, Heuser T, Nicastro D, Heldwein EE (2014) Membrane deformation and scission by the HSV‐1 nuclear egress complex. Nat Commun 5: 4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heldwein EE (2015) The great (nuclear) escape: new insights into the role of the nuclear egress complex of herpesviruses. J Virol 89: 9150–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ (2003) Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol 77: 7601–7610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG (2009) Structure and assembly of immature HIV. Proc Natl Acad Sci USA 106: 11090–11095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck A, Wagner M, Ruzsics Z, Lotzerich M, Iglesias M, Singh IR, Koszinowski UH (2004) Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol 78: 8026–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YE, Roizman B (1993) The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J Virol 67: 6348–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condit RC, Moussatche N, Traktman P (2006) In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66: 31–124 [DOI] [PubMed] [Google Scholar]
- Davis IW, Leaver‐Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB III, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J (2012) Reconstitution of the Kaposi's sarcoma‐associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86: 594–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25: 24–28 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC (2002) The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host‐cell nucleus and represent components of primary enveloped but not mature virions. J Virol 76: 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C, Ott M, Raschbichler V, Nagel CH, Binz A, Sodeik B, Bauerfeind R, Bailer SM (2015) The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by its N‐terminal domain. PLoS Pathog 11: e1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser J (2005) Deep‐etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J Cell Biol 169: 269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollinshead M, Johns HL, Sayers CL, Gonzalez‐Lopez C, Smith GL, Elliott G (2012) Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31: 4204–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res 38: W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyun JK, Accurso C, Hijnen M, Schult P, Pettikiriarachchi A, Mitra AK, Coulibaly F (2011) Membrane remodeling by the double‐barrel scaffolding protein of poxvirus. PLoS Pathog 7: e1002239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Baines JD (2011) Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9: 382–394 [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen‐bonded and geometrical features. Biopolymers 22: 2577–2637 [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010) Xds. Acta Crystallogr D 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC (2007) Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci USA 104: 7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Mettenleiter TC (2011) Nuclear envelope breakdown can substitute for primary envelopment‐mediated nuclear egress of herpesviruses. J Virol 85: 8285–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, Henrick K (2004) Secondary‐structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D 60: 2256–2268 [DOI] [PubMed] [Google Scholar]
- Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H (2015) Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci USA 112: 9010–9015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Baines JD (2005) Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol 79: 3797–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia‐Saez AJ, Mettenleiter TC, Antonin W (2015) A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290: 6962–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, McCaffery JM, Desai PJ (2013) Interactions of the Kaposi's sarcoma‐associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J Virol 87: 3915–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM (2015) Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34: 2937–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H (2009) Herpesvirus assembly: an update. Virus Res 143: 222–234 [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Muller F, Granzow H, Klupp BG (2013) The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15: 170–178 [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, Marschall M (2012) Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem 287: 24004–24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, Sliz P (2013) Collaboration gets the most out of software. eLife 2: e01456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou F, Wills E, Baines JD (2009) Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3‐encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83: 5181–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Schneider TR (2004) HKL2MAP: a graphical user interface for macromolecular phasing with SHELX programs. J Appl Crystallogr 37: 843–844 [Google Scholar]
- Passvogel L, Trube P, Schuster F, Klupp BG, Mettenleiter TC (2013) Mapping of sequences in pseudorabies virus pUL34 that are required for formation and function of the nuclear egress complex. J Virol 87: 4475–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Janke U, Klupp BG, Granzow H, Mettenleiter TC (2014) Identification of conserved amino acids in pUL34 which are critical for function of the pseudorabies virus nuclear egress complex. J Virol 88: 6224–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passvogel L, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC (2015) Functional characterization of nuclear trafficking signals in pseudorabies virus pUL31. J Virol 89: 2002–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD (2002) Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76: 8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Zhou YP, Schnetzer R, Ferguson J, DeSalvo D (2000) Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74: 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Bjerke SL, Haugo AC, Hanson S (2010) Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol 84: 3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, Hogle JM (2009) Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol 83: 2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnee M, Wagner FM, Koszinowski UH, Ruzsics Z (2012) A cell free protein fragment complementation assay for monitoring the core interaction of the human cytomegalovirus nuclear egress complex. Antiviral Res 95: 12–18 [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JA (2015) Structure of the immature HIV‐1 capsid in intact virus particles at 8.8 A resolution. Nature 517: 505–508 [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D 66: 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y (2000) The UL34 gene product of herpes simplex virus type 2 is a tail‐anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81: 2397–2405 [DOI] [PubMed] [Google Scholar]
- Skepper JN, Whiteley A, Browne H, Minson A (2001) Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment –> deenvelopment –> reenvelopment pathway. J Virol 75: 5697–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW (2005) Protein production by auto‐induction in high density shaking cultures. Protein Expr Purif 41: 207–234 [DOI] [PubMed] [Google Scholar]
- Yang K, Baines JD (2011) Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc Natl Acad Sci USA 108: 14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Wills E, Lim HY, Zhou ZH, Baines JD (2014) Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex‐specific complex. J Virol 88: 3815–3825 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Review Process File