

Abstract
Herpesvirus nucleocapsids escape from the nucleus in a process orchestrated by a highly conserved, viral nuclear egress complex. In human cytomegalovirus, the complex consists of two proteins, UL50 and UL53. We solved structures of versions of UL53 and the complex by X‐ray crystallography. The UL53 structures, determined at 1.93 and 3.0 Å resolution, contained unexpected features including a Bergerat fold resembling that found in certain nucleotide‐binding proteins, and a Cys3His zinc finger. Substitutions of zinc‐coordinating residues decreased UL50–UL53 co‐localization in transfected cells, and, when incorporated into the HCMV genome, ablated viral replication. The structure of the complex, determined at 2.47 Å resolution, revealed a mechanism of heterodimerization in which UL50 clamps onto helices of UL53 like a vise. Substitutions of particular residues on the interaction interface disrupted UL50–UL53 co‐localization in transfected cells and abolished virus production. The structures and the identification of contacts can be harnessed toward the rational design of novel and highly specific antiviral drugs and will aid in the detailed understanding of nuclear egress.
Keywords: Bergerat fold, crystal structure, interaction interface, nuclear egress complex, zinc finger
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
Like most DNA viruses, herpesviruses replicate and package their genomes in the nucleus. Their nucleocapsids, which are too large to pass through nuclear pores, then transit from the nucleus to the cytoplasm in a fascinating process known as nuclear egress (reviewed in Mettenleiter et al, 2006; Johnson & Baines, 2011). Nuclear egress encompasses multiple steps, including movement of nucleocapsids from the interior of the nucleus toward the nuclear rim, disruption of the nuclear lamina to permit access of the nucleocapsids to the inner nuclear membrane (INM), budding through the INM to form a primary enveloped particle, and fusion of the particle with the outer nuclear membrane to release the nucleocapsid into the cytoplasm. Thus, herpesviruses make use of multiple aspects of cell biology during nuclear egress. Interestingly, there is a somewhat similar nuclear envelope budding process in Drosophila muscle cells (Speese et al, 2012; Jokhi et al, 2013), although its mechanism remains obscure and differs from herpesvirus nuclear egress in several ways (Sharma & Coen, 2014; Sharma et al, 2014, 2015).
All three sub‐families (α, β, and γ) of herpesviruses of mammals, birds, and reptiles encode two highly conserved proteins that are central to the nuclear egress process, and combine to form a nuclear egress complex (NEC; reviewed in Mettenleiter, 2004; Mettenleiter et al, 2006). One of these proteins is anchored in the INM by a single C‐terminal membrane‐spanning segment, while the other is nucleoplasmic by itself and binds its NEC partner at the nuclear rim. Aside from its functions at the INM, the nucleoplasmic partner is important for DNA packaging and/or movement of nucleocapsids away from the interior of the nucleus (Chang et al, 1997; Granato et al, 2008; Popa et al, 2010; Pogoda et al, 2012; Funk et al, 2015). At the INM, both proteins are important for disruption of the nuclear lamina, evidently by recruiting host and/or viral protein kinases that phosphorylate lamins (Muranyi et al, 2002; Park & Baines, 2006; Sharma et al, 2014) and for budding through the INM. Indeed, various herpesvirus NECs and/or individual partners are sufficient for remodeling of nuclear membranes and even vesiculation in cells or in vitro (Klupp et al, 2007; Desai et al, 2012; Lee et al, 2012; Luitweiler et al, 2013; Bigalke et al, 2014; Lorenz et al, 2015). Interestingly, despite the strong conservation of NECs, there are important differences in their degree of importance for viral replication (more crucial for β‐ and γ‐herpesviruses than for α‐herpesviruses) and for specific functions, such as which protein kinases they recruit to the nuclear rim (e.g., viral vs. host) (Muranyi et al, 2002; Park & Baines, 2006; Sharma et al, 2014) and their ability to induce vesiculation in cells (Lee et al, 2012; Luitweiler et al, 2013).
We have focused on the structure and functions of the NEC of the β‐herpesvirus, human cytomegalovirus (HCMV), in part because HCMV is a major pathogen in immunosuppressed and immune‐naïve individuals, and may have roles in chronic diseases (Mocarski et al, 2013). There are no effective vaccines against HCMV, and drugs approved in the U.S.A. to treat HCMV infections have major drawbacks in terms of toxicities, pharmacokinetics, and drug resistance (Drew & Buhles, 1996; Gilbert et al, 2002; Villarreal, 2003; Lurain & Chou, 2010). Thus, there is considerable need for new anti‐HCMV drugs. As the NEC is essential for HCMV replication (Dunn et al, 2003; Yu et al, 2003; Sharma et al, 2014) and is not found in uninfected cells, it may be an attractive target for drug discovery. The HCMV NEC consists of UL50, the INM‐anchored protein, which is a homolog of herpes simplex virus‐1 (HSV‐1) and pseudorabies virus (PRV) UL34, and UL53, the nucleoplasmic binding partner, which is a homolog of HSV‐1 and PRV UL31. UL50 and UL53 are each essential for HCMV nuclear egress, lamina disruption, and recruitment of the viral protein kinase, UL97 (Sharma et al, 2014).
Clearly, high‐resolution structural information would greatly enhance our ability to understand NEC function and nuclear egress and would be valuable for drug discovery. Toward that end, we have previously expressed truncated forms of UL50 and UL53, and their murine CMV (MCMV) homologs (M50 and M53) that retain the most highly conserved segments (conserved cores), shown that cognate partners heterodimerize, and identified a long predicted helical segment in UL53 interrupted by a proline in which substitutions along one face affect heterodimerization (Sam et al, 2009; Leigh et al, 2015).
Recently, using nuclear magnetic resonance (NMR), we have solved the structure of the conserved core of M50, which adopts a strikingly intricate “β‐taco” fold (so named by E. Heldwein) that is unmatched in its entirety by any known protein (Leigh et al, 2015). Moreover, M50 includes a groove that contains a large cavity that binds a peptide corresponding to the long predicted helical segment of UL53 (Leigh et al, 2015). Single substitutions of UL50 residues corresponding to this region of M50 substantially decreased heterodimerization with UL53 in vitro and eliminated UL50–UL53 co‐localization, disruption of nuclear lamina, and viral replication in HCMV‐infected cells (Leigh et al, 2015). This work provided detailed structural information regarding this NEC subunit and suggested a possible drug target on UL50, but did not shed light on the structure of UL53 and its homologs or structural details regarding NEC subunit interactions.
Here, we report high‐resolution crystal structures of the conserved cores of HCMV UL53 and the NEC. Using these structures, we have constructed and analyzed mutant proteins for co‐localization in cells, and mutant viruses for viability. The results reveal novel and unexpected features of NEC structure and function, with implications for antiviral drug discovery.
Results
Construction and expression of HCMV UL53 for crystallization
Our initial efforts to crystallize the NEC or UL53 used UL53 residues 50–292 (UL5350−292), which contains the conserved core of the protein, and is well expressed in E. coli and readily purified (Sam et al, 2009). However, we failed to obtain well‐ordered crystals. Proteolysis studies on UL5350–292 either alone (Sam et al, 2009) or bound to UL501–169 (Appendix Supplementary Methods and Appendix Fig S1A) revealed a stable, proteolysis‐resistant domain from residues 84 to 292. We therefore constructed and expressed this protein (UL5384–292). This construct lacks the long predicted helical region important for binding UL50 (Sam et al, 2009). Thus, we also constructed and expressed UL5372–292, which includes about half of this helical region and residues known to be crucial for UL50 binding (Sam et al, 2009). A schematic of these constructs is provided in Appendix Fig S1B.
Structure determination of UL53
UL5372–292 crystallized in the space group P4 with one molecule in the asymmetric unit (a.s.u.). The structure was solved by sulfur single‐wavelength anomalous dispersion (S‐SAD) phasing and refined to an R/R free value of 0.2555/0.3088 at 3 Å resolution. Although a complete data set to 2.5 Å resolution was collected for UL5372–292 (Table 1) for S‐SAD phasing, it was assembled from 16 different crystals. We have chosen instead to report the structure based on data collected from a single native crystal, at 3 Å resolution. In the single‐crystal map, we found that electron density features for the less‐well‐ordered portions of the atomic model were more readily interpretable, and buildable as a single plausible conformation, which has facilitated structural comparisons. The model could be traced convincingly from residues 72 to 292 (Fig 1A). The data collection and structure refinement statistics are shown in Table 1. UL5384–292 crystallized in the P1 space group with two molecules (chains A and B) in the a.s.u. (Fig 1B). The structure was solved by molecular replacement using the UL5372–292 structure as a starting model. The structure refined to an R/R free value of 0.1855/0.2198 at 1.93 Å resolution. The models for chains A and B begin at residues 88 and 89, respectively, and end at 292. Using boundaries from 89 to 292 for all three structures, the Cα atoms of chains A and B of UL5384–292 aligned against UL5372–292 with an RMSD of 0.66 and 0.82 Å, respectively, indicating essentially the same structure, though slight differences were observed at the region just after the β3 strand (Figs 1A–C and 2A).
Table 1.
Data collection and refinement statistics of UL53 and the NEC
| Phasing S‐SAD UL5372–292 | Native UL5372–292 | Native UL5384–292 | Native NEC (UL50:UL53) | |
|---|---|---|---|---|
| Data collection | ||||
| Wavelength (Å) | 1.7712 | 1.0703 | 0.9792 | 0.9792 |
| Beamline | I04, DLS | NE‐CAT, APS | NE‐CAT, APS | NE‐CAT, APS |
| Space group | P 4 | P 4 | P 1 | C 2 |
| Unit cell (Å, °) |
a = b = 89.67; c = 33.78 |
a = b = 90.01; c = 33.94 |
a = 33.8; b = 50.4; c = 63 α = 91.42; β = 103.13; γ = 108.11 |
a = 142.25; b = 34.0; c = 93.0 β = 121.76 |
| No. of crystals | 16 | 1 | 1 | 1 |
| Resolution (Å) | 63.41–2.5 | 90–3.00 | 61.06–1.93 | 79.08–2.47 |
| Unique reflectionsa | 9,612 (700) | 5,574 (726) | 26,803 (3,037) | 13,720 (2,010) |
| R merge b | 0.265 (1.348) | 0.057 (0.54) | 0.03 (0.15) | 0.058 (0.583) |
| Multiplicitya | 140.8 (92.4) | 4.2 (4.1) | 3.7 (2.7) | 3.5 (3.28) |
| Completeness (%)a | 99.9 (99.6) | 97.8 (90.5) | 94.0 (72.6) | 97.5 (88.9) |
| <I/σI>a | 39.7 (6.9) | 14.2 (1.9) | 27.3 (5.9) | 14.47 (2.10) |
| Refinement | ||||
| Resolution (Å) | 60.00–3.00 | 61.11–1.93 | 79.07–2.47 | |
| R work/R free | 0.2555/0.3088 | 0.1855/0.2198 | 0.2542/0.2992 | |
| rmsd, bonds (Å) | 0.005 | 0.013 | 0.005 | |
| rmsd, angles (Å) | 1.101 | 1.437 | 1.019 | |
| <B> all (Å2) | 91.088 | 15.8 | 60.128 | |
| Ramachandran plot |
Favored: 85.8% Allowed: 13.2% Generously allowed: 0.5% Outliers: 0.5% |
Favored: 93.20% Allowed: 6.2% Generously allowed: 0% Outliers: 0.5% |
Favored: 95.1% Allowed: 4.6% Generously allowed: 0% Outliers: 0.3% |
|
Values for the highest resolution shell are indicated in parentheses.
, where I i(hkl) is the i th measurement of reflection hkl and <I(hkl)> is the weighted average of all measured reflections.
Figure 1. Crystal structures of UL53.
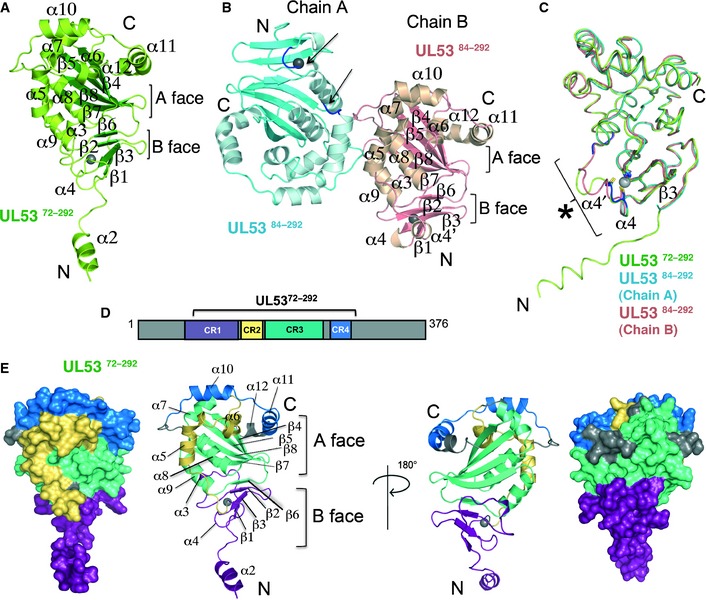
- Cartoon representation of UL5372–292‐encompassing residues 72–292 with the secondary structural elements labeled (β‐strands, green; other elements, limon). Two layers of β‐strands form an A and B face, as indicated. UL5372–292 binds zinc (shown as a gray sphere).
- UL5384–292 crystallized with two molecules in the asymmetric unit. The two molecules, chains A (β‐strands, blue; other elements, cyan) and B (β‐strands, salmon; other elements, beige), are shown. The secondary structural elements on chain B, shown in the same orientation (front view) as UL5372–292 in (A), are labeled. Residues 126–130 on chain A were omitted from the model due to poor corresponding electron density. The residues surrounding this deleted region are shown in dark blue and indicated by the arrows near the zinc ion (gray sphere). The A and B faces of β‐strands are indicated for chain B.
- Ribbon representation of chains A (cyan) and B (salmon) of UL5384–292 superpositioned onto UL5372–292 (limon). Residues coordinating the zinc ion (gray sphere) are conserved in all three structures. An area of structural variability is indicated by the asterisk (*), and the structural elements close by are labeled.
- Schematic representation of the sequence boundaries used in the UL5372–292 construct. The four regions conserved (CR1 to 4) among the herpesvirus subfamilies are shown color‐coded on the construct. CR1 is shown in purple, CR2 in yellow, CR3 in green‐cyan, and CR4 in blue.
- Front and back view (rotated 180°) of both space‐filling and ribbon representations of the UL5372–292 structure onto which CR1 to CR4 are mapped following the color scheme in (D).
Figure 2. Topology diagrams of the NEC subunits and conservation of the Bergerat fold.
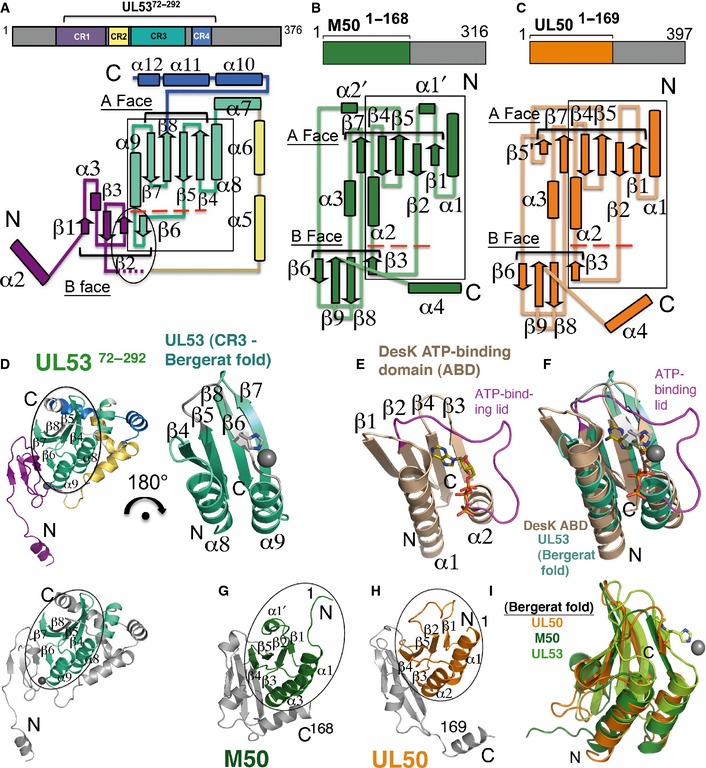
- Topology diagram of the structure of UL5372–292 with the CRs mapped and color‐coded according to the schematic above the structure. The structure begins at α2 (in purple) at the N‐terminus (N) and ends at α12 (in blue) at the C‐terminus (C). Secondary structure elements are numbered, with tubes representing helices and arrows representing strands. The A and B faces of strands are indicated. The region forming the zinc finger is indicated in the oval. The flexible region (observed across the different UL53 structures) encompassing α4 after β3 is indicated with small purple dashes. Conservation of the Bergerat fold is shown with a box. Below the large red dashes, UL53 has its β6 strand in place of an ATP‐binding loop/lid region found in other proteins with the Bergerat fold.
- Topology diagram of the structure of M50. The boundaries of the structure fully encompass the conserved core of the protein and are shown in green in the schematic above the topology diagram. Secondary structure elements are shown with tubes representing helices and arrows representing strands. To facilitate the comparison with UL50 (presented in C), the nomenclature used here for M50 [which differs from that described previously (Leigh et al, 2015)] is the same as that for UL50, with secondary structure insertions designated with a prime (‘). The A and B faces of β‐strands are indicated. Similar to (A), M50 has a β‐strand (β3) shown below the large red dashes in place of an ATP‐binding loop/lid region found in other proteins with the Bergerat fold.
- Topology diagram of the structure of UL501–169. The boundaries of the structure fully encompass the conserved core and are shown in orange in the schematic above the topology diagram. Secondary structure elements are shown with tubes representing helices and arrows representing strands and are numbered similarly to that in M50 (presented in B) with insertions designated with a prime (‘). The A and B faces of strands are indicated. The β3 strand shown below the large red dashes substituting for an ATP‐binding loop/lid region and completing the B face of the β‐taco fold observed in M50 (B) is also conserved in UL50.
- (Left) UL5372–292 with the Bergerat fold circled. The color scheme follows that in (A). (Right) 180° rotation of just the Bergerat fold (encompassing most of CR3). In place of an ATP‐binding loop/lid region is the β6 strand (shown in gray), with a histidine which coordinates zinc (gray sphere). (Bottom) UL5372–292 in the same orientation as the structure shown above and to the left but with the rest of the structure, except for the Bergerat fold shown in gray.
- The Bergerat fold in the DesK ATP‐binding domain (ABD) (Trajtenberg et al, 2010; PDB ID: 3EHG). The ATP‐binding loop/lid region is shown in magenta around the ATP molecule.
- Superposition of the Bergerat fold in UL5372–292 (green‐cyan, from D) onto that of DesK (brown, from E).
- M50 (in the same orientation as UL5372–292 in D) with the Bergerat fold circled (green) (Leigh et al, 2015; PDB ID: 5A3G).
- UL50 (in the same orientation as UL5372–292 in D) with the Bergerat fold circled (orange).
- Overlay of UL5372–292 (limon) onto M50 (green) and UL50 (orange). The histidine on the β6 strand is shown coordinating with zinc (gray sphere).
For UL5384–292, residues 126–130 in chain A could not be clearly resolved in the electron density map and were thus excluded from the final model (Fig 1B). Analysis of the neighboring molecules revealed unfavorable crystal packing contacts. The density in chain B and UL5372–292 at, and close to, this region, though weak, was present, and the backbone could be continuously traced in both these cases with two short helices (α4 and α4′) in chain B, but only one (α4) defined in UL5372–292 (Figs 1A–C and 2A). The variability of conformations that can be adopted under the influence of crystal packing contacts suggests an inherent flexibility at this region. Interestingly, these residues are located near residues C122 and C125 (Appendix Fig S2A), both of which are involved in coordination with a zinc ion (Fig 1A–C, see below).
The additional N‐terminal residues in UL5372–292 form a single helix (α2), as predicted (Sam et al, 2009), that extends out from the rest of the structure through a linker region (Fig 1A). In the crystal, each of these helices clusters at the fourfold rotation axis (Appendix Fig S1C). The structures pack with alternating layers of the helical cluster and the UL53 core (Appendix Fig S1D). While the electron density allowed for confident placement of the helices, density connecting the linker region to the helices was very diffuse, indicating a lack of consistency between the unit cells possibly due to flexibility in this region. The helices were thus assigned to a given UL53 core based on the length of the linker region.
UL53 has a unique overall topology that incorporates the Bergerat fold
Although the overall fold of UL53 is unique, it is somewhat reminiscent of the fold of M50 (Leigh et al, 2015), where the “spreading sandwich” of the β‐taco is formed by two sheets of β‐strands, each forming a “face” surrounded by helices on the wider end of the sandwich (Fig 1A and B). In UL53, the A face is formed by β4, β5, β7, and β8 and the B face by β1, β2, β3, and β6. As both UL5384–292 and UL5372–292 have essentially the same structures (Fig 1C), we describe the topology of the more complete protein UL5372–292 here and mention the slight differences observed between the two constructs.
The conserved portions of UL53 and its homologs across the α‐, β‐, and γ‐herpesvirus sub‐families have been previously divided into four conserved regions (CRs)—CRs 1, 2, 3, and 4, comprising residues 58–125, 127–160, 163–243, and 254–282, respectively (Lotzerich et al, 2006) (Figs 1D and 2A). These CRs map onto the core domain of the UL5372–292 structure, with the exception of the α2 helix in CR1 (shown in purple) at the N‐terminus, which extends away from the core (Figs 1D and E, and 2A). The rest of CR1 is on the core domain and consists of β1, a short 310 helix (α3) β2, β3, and α4 (and an additional α4′ in UL5384–292) (Figs 1E and 2A). This is followed by CR2 (yellow) with two helices (α5 and α6) that form an L shape on one face of the structure (Figs 1E and 2A), which then leads into CR3 (green‐cyan), which forms much of UL53's core (Fig 2A). CR3 starts with α7 and α8, followed by β4, β5, β6, α9, β7, and β8 (Fig 2A). While β6 loops back toward CR1 (purple) at the N‐terminus to complete the B face of the β‐taco, the other four β‐strands in CR3 form an antiparallel β‐sheet that make up the A face of the taco (Figs 1E and 2A). A linker region after β8 then leads into CR4 (blue), which forms a crown atop the UL53 structure (Fig 1E) and encompasses the last three helices—α10, α11, and α12 (Fig 2A).
We searched for structural homologs of UL53 using the homology search database VAST (http://structure.ncbi.nlm.nih.gov/Structure/VAST/vastsearch.html), looking for matches with the lowest RMSD score that cover the largest continuous span of residues possible. We unexpectedly found a striking α‐β‐β‐α‐β‐β topological similarity between CR3 of UL53 (Fig 2D) with the ATP‐binding domain (ABD) within the catalytic core of the Bacillus subtilis DesK histidine kinase (Fig 2E). DesK is a transmembrane sensor protein that, as part of a two‐component regulatory system, relays signals to a response regulator (RR) through phospho‐transfer from the ABD to a histidine residue on a dimerization and histidine phosphotransfer domain, and finally to the RR to elicit a response to environmental stimuli (Gao & Stock, 2009). Overlaying the Cα atoms of the ABD domain [residues 276–365 (PDB ID 3EHG); Trajtenberg et al, 2010] and this portion of UL53 (residues 172–245; Appendix Fig S2A) in DaliLite (http://ekhidna.biocenter.helsinki.fi/dali_server) produces a Z‐score of 7.8 and an RMSD of 2.4 Å over 72 residues, indicating significant structural homology (Fig 2F). Alignment of just the conserved secondary structural elements produced an RMSD of 1.8 Å over 57 residues.
Comparison of the DesK ABD with UL53 also reveals several interesting differences. Although the α‐β‐β‐α‐β‐β topology is conserved in both proteins, UL53 has a short β‐strand (β6) (Fig 2D) with a histidine residue (H211) that is involved in the formation of a zinc finger (discussed in detail in the next section). Instead of a β‐strand, DesK has an ATP‐binding lid, which is a loop characteristic of histidine kinases that covers bound nucleotide (Fig 2E and F), in this region (Trajtenberg et al, 2010; Cho et al, 2013). Interestingly, a structurally homologous fold was also found within the N‐terminal portion of MCMV M50 (Fig 2B, G and I).
The ABD fold of the DesK histidine kinase is structurally homologous to a large family of nucleotide‐binding proteins including DNA gyrase, Hsp90, and MutL, from which the name GHKL domain (the letter in each protein that contributes to the name of the domain is underlined) was derived (Gao & Stock, 2009). Also known as the Bergerat fold (Bergerat et al, 1997; Dutta & Inouye, 2000), it is characterized both by its distinctive α‐β‐β‐α‐β‐β fold and by the structural properties of the linker regions between these conserved secondary elements, such as the ATP‐lid between the second β‐strand and helix (i.e., α1‐β1‐β2‐(ATP‐binding lid)‐α2‐β3‐β4). Variations in the structures of these linker regions have previously been observed when comparing the canonical DNA gyrase, Hsp90, and MutL structures with that of EnvZ histidine kinase (Dutta & Inouye, 2000).
To look for conservation or differences in the structures of proteins in this family, we aligned the portions of UL53 and M50 that encompass this fold with three members of the histidine kinase family, DesK, DosS, and EnvZ, and with a vesicle‐trafficking protein Bet3 previously identified as being partially homologous to M50 from structural homology searches (Leigh et al, 2015). Despite poor sequence homology among these proteins, the fold conservation was striking (Fig EV1). In concurrence with the differences observed in linker regions observed by Dutta & Inouye (2000), we observed major differences in the regions corresponding to the ATP‐lid as described earlier for UL53 and M50. Interestingly, like UL53, DosS also does not have a characteristic ATP‐lid (Cho et al, 2013), but has a shorter segment that coordinates zinc (Fig EV1). Both UL53 and UL50, and their homologs, lack conservation of residues important for ATP binding in the GHKL family of proteins.
Figure EV1. Structural comparison of the conserved Bergerat fold in UL50 and UL53 with that of members of the GHKL superfamily and Bet3.
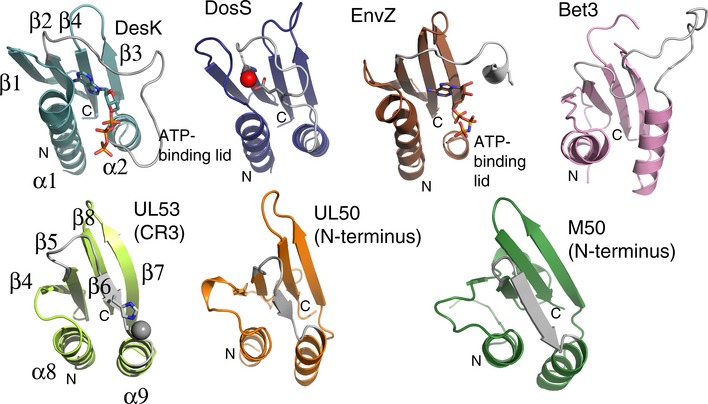
Ribbon representations of (from left to right in the top row) the ATP‐binding domains of three histidine kinase proteins (DesK, DosS, and EnvZ). The ATP molecules bound in DesK and EnV are shown in stick form, and the ATP‐binding lid is shown in gray. A short linker region (gray) on which a glutamate residue (in part) coordinates zinc (red sphere) takes the place of an ATP‐binding lid in DosS. The ATP‐binding lid is also missing in a portion of Bet3, a vesicle‐trafficking protein (top row, right), and the NEC subunits (bottom row, left to right) UL53 (comprising most of conserved region 3 (CR3)), and UL50 and M50, both comprising the N‐terminal portion of the proteins, where otherwise the Bergerat fold (α‐β‐β‐α‐β‐β topology) is fully conserved (the secondary elements of DesK and UL53 are labeled as an example of the fold sequence). The deviations observed between the structures at the ATP‐binding lid or equivalent region are shown in gray.
UL53 is a zinc metalloprotein
A second unexpected finding was that UL53 has a C3H zinc finger (Fig 3A) with a Cys‐X15‐Cys‐X2‐Cys‐Xn‐His sequence pattern (Appendix Fig S2A) where n > 60 residues. Strikingly, the cysteine and histidine residues are fully conserved across the α, β, and γ sub‐families, as are amino acids serine and glycine at positions +4 and +7 from the first cysteine (Appendix Fig S2A). The zinc ion is tetrahedrally coordinated by the cysteine residues located in CR1, with the first cysteine (C106) on β2 followed by two cysteines (C122 and C125) on the flexible region after β3, and a histidine residue (H211) on β6 in CR3 (Appendix Fig S2A, Fig 3A). Neither the purification buffers nor the crystallization solutions included zinc, suggesting endogenous zinc incorporation upon protein expression in E. coli. Elemental analysis conducted on UL53 using energy‐dispersive X‐ray spectroscopy (EDS) showed peaks at 8.62 and 9.56 keV, corresponding to Zn K‐α1 and K‐β1 emission line peaks, respectively (Appendix Fig S5).
Figure 3. Effects of zinc finger and NEC contact residue mutations on UL53 localization.
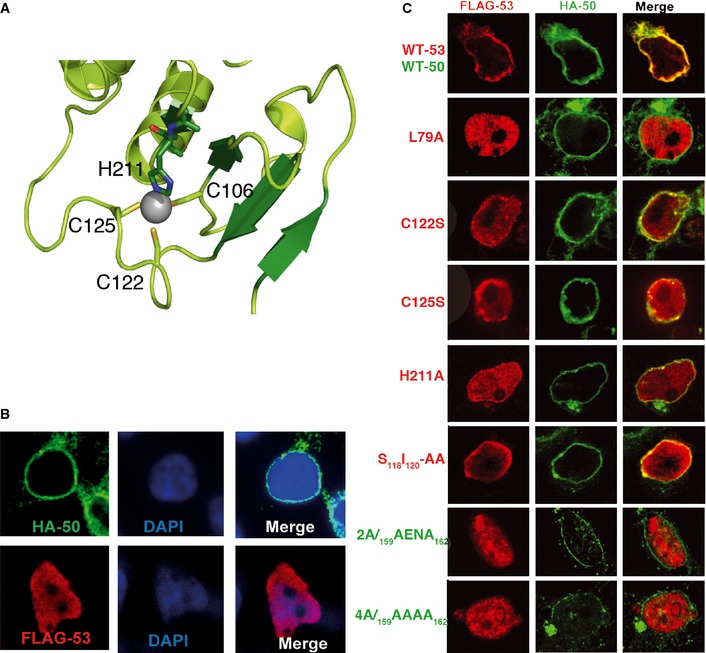
- Close‐up view of the zinc finger on UL53. The residues involved in coordinating the zinc ion (gray sphere) are shown and labeled.
- Localization of UL50 and UL53 when expressed by themselves. HEK293T cells transfected with DNA constructs expressing HA‐tagged WT UL50 (top) or FLAG‐tagged WT UL53 (bottom) were stained with DAPI and antibodies against the epitopes, and imaged using confocal fluorescence microscopy.
- Effects of mutations. HEK293T cells were co‐transfected with DNA constructs expressing FLAG‐tagged WT or mutant (L79A, C122S, C125S, H211A, S118I120‐AA) UL53 and HA‐tagged WT or mutant (Y159A and L162A (2A) or Y159A, E160A, N161A, L162A (4A)) UL50, stained with antibodies against the epitopes, and imaged using confocal fluorescence microscopy.
Effects of substituting zinc‐coordinating residues
Given the unexpected finding of a zinc finger, we investigated the effects of substituting residues coordinating the zinc ion on UL50‐53 co‐localization in co‐transfection studies, or on viability and spread of HCMV in human foreskin fibroblasts (HFFs). Immunofluorescence assays of HEK293T cells transfected with plasmids expressing either epitope‐tagged, wild‐type (WT) versions of UL50 or UL53 alone revealed that UL50 localized to the nuclear rim, while UL53 localized to the nucleoplasm (Fig 3B), as reported previously (Milbradt et al, 2007; Camozzi et al, 2008; Sam et al, 2009). In contrast, in cells co‐transfected with both plasmids, both proteins co‐localized at the nuclear rim, with very little UL53 in the nucleoplasm (Fig 3C, top), as previously observed (Milbradt et al, 2007; Camozzi et al, 2008; Sam et al, 2009). Co‐expression of WT UL53 with a UL50 mutant, L79A, known to be unable to bind to UL53 (Sam et al, 2009), led to no detectable co‐localization (Fig 3C). However, serine substitution of C122 (C122S) or C125 (C125S) or alanine substitution of H211 (H211A) led to an intermediate phenotype with discrete areas of co‐localization of UL53 with UL50 at the nuclear rim, and much of the UL53 in the nucleoplasm (Fig 3C). To assess these co‐localization phenotypes more quantitatively, we calculated Manders M1 coefficients for each of the co‐transfections. Co‐transfection of plasmids expressing WT UL50 with ones expressing UL53 zinc finger substitutions resulted in intermediate coefficients (0.22–0.48) relative to the high coefficient (~0.8) of the plasmid expressing WT UL53 protein (~0.8) and the low coefficient (0.03) of the L79A mutant (Fig EV2).
Figure EV2. Quantitation of UL50 and UL53 co‐localization in cells.
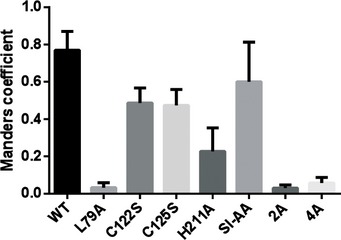
Co‐localization analysis between wild‐type and mutant versions of HA‐tagged UL50 or FLAG‐UL53 in cells using Fiji/ImageJ. The Manders M1 coefficients were calculated for multiple cells of each sample within a defined region of interest (ROI) proximal to the nucleus and summarized in a bar graph. The values represent the mean and the error bars depict the standard deviation (±SD).
We also constructed HCMV bacterial artificial chromosomes (BACs) encoding UL53 substitutions of the zinc‐coordinating residues C106 (to serine; C106S) and H211A. The BACs and viruses derived from them express GFP in human cells (Strang et al, 2012). Following electroporation of wild type (and certain other mutant BACs) into HFFs, spread of GFP signal into neighboring cells was observed by day 5 post‐electroporation, followed by further spread and cytopathic effect (CPE), while electroporation of the C106S and H211A BACs resulted in only individual cells expressing GFP (Fig EV3), which eventually disappeared. Rescued derivatives of both these viruses (C106SR and H211AR, respectively) displayed spread of GFP signal by day 5 (Fig EV3) and eventually throughout the monolayer, similar to WT virus spread. Thus, substitutions of the zinc finger motif ablated virus replication.
Figure EV3. Effects of mutations on the zinc finger and the amphiphatic helices of UL53 on HCMV viability.
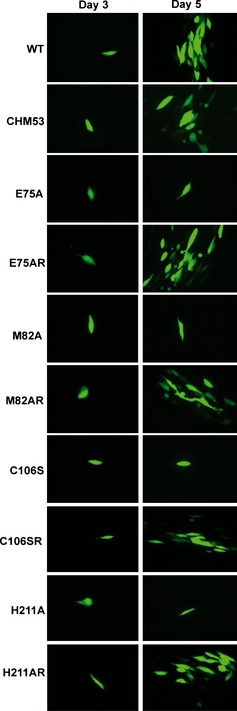
Human foreskin fibroblasts (HFFs) electroporated with HCMV BACs that express GFP, and either WT UL53, UL53 mutants, or their rescued derivatives as indicated to the left of each panel, were imaged at day 3 or 5 post‐electroporation to monitor the spread of the virus infection in the cell monolayer.
Construction and expression of the HCMV NEC for crystallization
UL5372–292 did not form a complex with UL50, as assessed by gel filtration chromatography at 200 and 600 μM of UL53 and UL50, respectively (Fig EV4A). Efforts to obtain well‐ordered crystals of longer versions of UL53, including UL5361–292, bound to UL50 did not succeed. Given the ability of a peptide corresponding to the predicted helical region of UL53 to bind to MCMV M50 (Leigh et al, 2015), we investigated whether replacing residues 61–70 of UL5361–292 with the corresponding residues (111–120) of the MCMV homolog, M53, would permit formation of an NEC amenable to crystallography. We call this protein, which effectively substitutes H62, D63, I67, R69, and E70 of UL53 with S, E, V, Q, and R, respectively, Chm53 (Appendix Figs S1B, S2A and S3). Consistent with the modest effects of substitutions affecting residues 61–70 of UL53 (Sam et al, 2009), in isothermal titration calorimetry (ITC) experiments (Fig EV3B), Chm53 bound to UL501–169 with a dissociation constant (K d) of 1.0 ± 0.08 μM, only ~2.5‐ to 3‐fold higher than that of UL5350–292 (current study (0.42 ± 0.05 μM) and Sam et al, 2009 (0.29 ± 0.03 μM)). Interestingly, inclusion of the full‐length N‐terminal region (UL531–292) decreased the binding affinity of UL53 with UL501–169 (K d of 3.8 ± 0.8 μM) compared to the N‐terminal truncation mutants (Fig EV4C), possibly implying a regulatory mechanism of the N‐terminal portion on UL53 activity. Attempts to obtain the binding affinity of UL5361–292 (which has the same boundaries as Chm53) with UL50 were unsuccessful as UL5361–292 was unstable in the ITC buffers used for all the other samples.
Figure EV4. The N‐terminal amphiphatic helices on UL53 and the C‐terminal helix of UL50 are required for NEC formation.
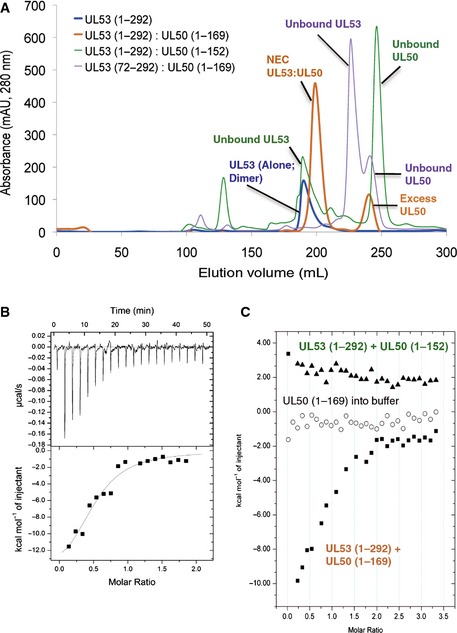
- Column chromatography testing the ability of UL50 and UL53 truncation mutants to form the NEC. UL53‐encompassing residues 1–292 (UL531–292) alone elute as a homodimer (blue). Incubation of a 1:3 ratio of UL531–292 with UL501–169 results in complete binding of UL53 to form a heterodimer with excess UL50 eluting as a monomer (orange). A C‐terminal truncation mutant of UL50 (UL501–152), however, is unable to bind to UL531–292, and each protein elutes separately, with UL531–292 as a homodimer and UL50 as a monomer (green). A similar result is observed with UL5372–292, an N‐terminal truncation mutant, with both proteins eluting separately as unbound monomeric proteins (purple). Unlike UL531–292, UL5372–292 is a monomer, similar to UL5384–292 (Sam et al, 2009).
- ITC studies of binding of Chm53 to UL501–169. The K d derived was 1.0 ± 0.08 μM.
- ITC studies of binding of UL531–292 with UL501–169 (labeled in orange) or UL501–152 (labeled in green), the binding isotherms of which are shown as squares and triangles, respectively. The isotherm for UL501–169 titrated into buffer is shown in circles. The K d derived for UL531–292 with UL501–169 was 3.8 ± 0.8 μM.
Electroporation of a BAC containing HCMV encoding Chm53 gave rise to a spreading infection in HFFs (Fig EV3), which progressed to full cytopathic effect. Supernatants from the electroporation contained infectious virus, as measured by a plaque assay, and sequencing of the viral DNA confirmed the presence of the Chm53 substitutions. Thus, Chm53 is functional in the context of HCMV infection.
The NEC crystal structure
Chm53 forms a stable complex with UL501–169 that can be isolated by gel filtration chromatography (Materials and Methods). The complex crystallized in the C2 space group with one molecule in the a.s.u. The crystal structure of UL5384–292 and the NMR structure of MCMV M50 (Leigh et al, 2015) were used as templates for molecular replacement to solve the structure of the NEC. The structure was refined to an R/R free value of 0.2542/0.2992 at 2.47 Å, and the data collection and structure refinement statistics are shown in Table 1. The model for the NEC could be traced from residues 4 to 168 for UL50, and 61–289 on Chm53 (Fig 4A). Similar to chain B of UL5384–292, only one short helix (α4) after β1 could be traced into the density for Chm53 (Appendix Fig S2A). Interestingly, unlike the three other UL53 structures, density corresponding to the C‐terminal helices in CR4 was not as well defined for Chm53, though a helical character was present (Fig EV5A). The general placement of the helices was similar to that of the UL53 monomers, though side chain density was more diffuse, thus suggesting an inherent mobility in this helical region.
Figure 4. Structure of the NEC and conformational changes of the subunits upon NEC formation.
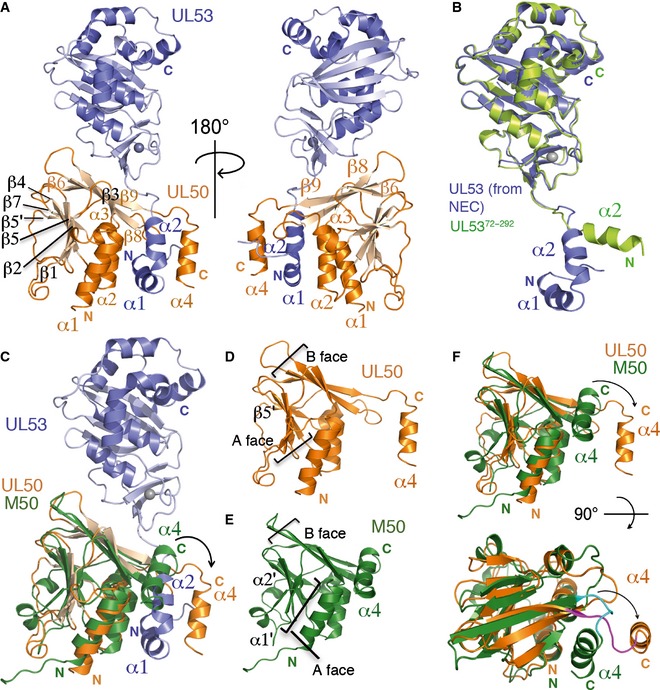
- (Left) Front view of the NEC composed of UL53 (purple) and UL50 (orange). The zinc ion is shown as a gray sphere. The secondary structural elements of UL50 are labeled on the structure, with those involved in the NEC interface labeled with orange font. The N‐terminal amphiphatic helices (α1 and α2) of UL53 are indicated in purple font. (Right) Back view of the NEC structure.
- Superposition of UL5372–292 (limon) onto UL53 (purple) from the NEC. The N‐terminal amphiphatic helices (α1 and α2) of UL53 are labeled with purple font. UL5372–292 does not encompass α1 and begins instead at α2 (labeled with limon font).
- Superposition of M50 (green; Leigh et al, 2015; PDB ID: 5A3G), which represents unbound UL50, onto UL50 (orange) in the NEC (UL53 shown in purple). In UL50, α4 (orange) swings out from the rest of the UL50 structure to accommodate α1 and α2 of UL53.
- Structure of UL50 from the NEC shown alone. The A and B faces of β‐strands are indicated by the brackets. UL50 has an additional β‐strand (β5′ labeled) in the A face not found in M50. The α4 helix involved in binding to UL53 is labeled using orange font.
- Structure of M50. The A and B faces of β‐strands are indicated by the brackets. M50 has two additional α helices (α1′ and α2′ labeled) compared to UL50. The α4 helix which associates with the B face in an unbound state is labeled using green font.
- (Top) Side view of the B face of UL50 (orange) from the NEC superpositioned onto M50 (green). The C‐terminal helices are labeled using their respective colors. (Bottom) Top view of the B face. To form the NEC, the C‐terminal helix in the unbound state (α4, green) swings out (α4, orange) as indicated by the curved arrows to form a gap for binding UL53.
Figure EV5. Crystal packing of UL53 and the NEC .
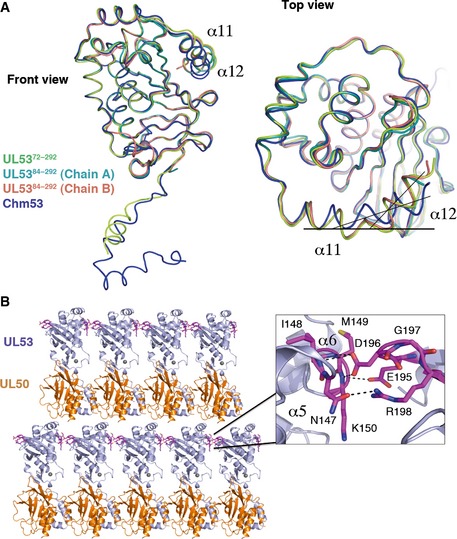
- Comparison of the C‐terminal helices in the UL53 structures. The C‐terminus tail (α11 and α12) of the UL53‐only structures comprising residues 72–292 (UL5372–169, limon), which crystallized in the P4 space group, and UL5384–292 [both chains A (cyan) and B (salmon)], which crystallized in the P1 space group, overlay almost perfectly (front view and top view). These structures have a bend of ˜145° between α11 and α12. In contrast, however, the bend flattens to ˜170° between α11 and α12 of Chm53 (blue) from the NEC structure, which crystallized in the C2 space group (top view).
- Lateral contacts mediating NEC crystal packing. A close‐up view of the extensive hydrogen bonding interactions (dashed black lines) between the residues on α5 and α6 on one molecule (left), with residues between β4 and β5 of a neighboring molecule (right). The interacting residues are shown in magenta.
Notably, the crystal packing contacts between neighboring UL53 molecules in the NEC crystals were very similar to those in the other three crystal forms, involving a strong interaction between residues 147 and 150 (NIMK) between α5 and α6 in CR2 on one molecule with residues 195–198 (EDGR) between β4 and β5 in CR3 of the neighboring molecule (Appendix Fig S2A, Fig EV5B).
Comparison of the M50 monomer with UL50 in the heterodimeric NEC
UL50 exhibits high structural homology with M50 (Fig 2B and C) with conservation of the β‐taco fold. Both proteins begin with an N‐terminal α helix (α1) followed by a β‐strand (β1). There is then a six residue helical insertion (α1′) in M50 whose corresponding residues are not as structured in UL50 (Fig 2B and C). The structures proceed with two successive β‐strands (β2 and β3), followed by a helix (α2) and two β‐strands (β4 and β5). After β5, UL50 has an additional short‐strand (β5′) insertion (not present in M50), which is composed of three residues that complete the edge of the A face. Instead, M50 has an additional short‐helix insertion (α2′) that follows β5. The proceeding secondary elements in both proteins are then β6, β7, α3, β8, β9, and α4 at the C‐terminus. Similar to M50, the β‐strands in UL50 align into two sheets to form the A and B face of the taco (Fig 4D and E). The Bergerat fold is also perpetuated in UL50 (Fig 2C and H). A major structural difference between the monomeric M50 and UL50 in the NEC is the orientation of their C‐terminal α4 helix (Fig 4C–E). In the M50 structure, this helix interacts directly with the B face of the remainder of the protein (Fig 4E and F). In the UL50 structure in the complex, however, this helix has moved away from the B face to accommodate the two N‐terminal helices of UL53 (Fig 4C, D and F). Excluding the C‐terminal helix, the remainder of the conserved elements of the two proteins (residues 152–169 on UL50 and 154–168 on M50) align with an RMSD of 2.8 Å over 148 residues.
The NEC interaction interface
The NEC structure revealed several expected and unexpected features regarding the UL50–UL53 interface. As predicted (Sam et al, 2009), the N‐terminal ~20 residues of UL53 form two amphipathic helices (α1 and α2), interrupted by a proline, that form extensive contacts with UL50 (Fig 5A). Additional binding interactions are mediated by residues near the zinc finger on the core domain as well as the linker region connecting the helices to the core (Fig 5A). These extensive contacts are mapped on the structure‐based sequence alignment of UL53 and UL50 with its homologs across the α‐, β‐, and γ‐herpesvirus sub‐families (Appendix Fig S2A and B).
Figure 5. The NEC interaction interface is extensive and mediated by hydrophobic and electrostatic interactions.
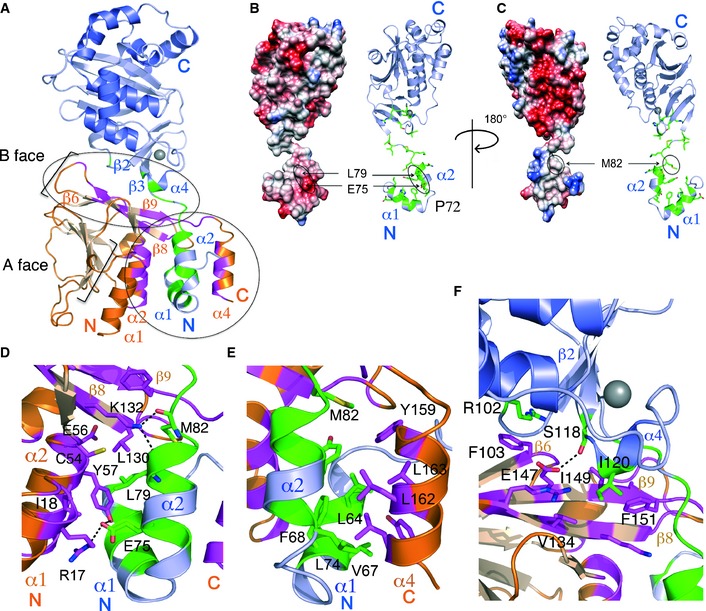
- The NEC with the interaction interface mapped onto UL53 (purple) in green and UL50 (orange) in pink. The two main sites of interaction are indicated by the circles. The secondary elements involved, or close to the residues involved in the interface, are labeled. The A and B faces of strands are indicated. The zinc ion is shown in gray.
- (Left) Electrostatic surface potential (red indicates acidic properties while blue indicates basic properties) on one side of UL53 on which residues L79 and E75 lie on the α2 helix (shown also in the ribbon representation on the right). The proline (P72) that separates α1 and α2 is labeled on the structure.
- (Left) Electrostatic surface potential on the other side (180° rotation compared to B) of UL53 that has residue M82 just above the α helix (shown also in the ribbon representation on the right).
- Close‐up view of the interactions between the main body of UL50 (orange with the interacting residues shown in magenta) with residues on α1 and α2 of UL53 (purple with interacting residues in green). Hydrogen bond interactions are represented by black dotted lines.
- Close‐up view of hydrophobic interactions between α4 of UL50 (orange with interacting residues shown in magenta) with residues on α1 and α2 of UL53 (purple with the interacting residues in green).
- Interaction of residues close to the zinc finger on UL53 (purple with interacting residues in green) with the B face of UL50 (orange with interacting residues shown in magenta). Hydrogen bond interactions are represented by black dotted lines.
The interaction involving the UL53 α1 and α2 helices is particularly fascinating. These two helices extend out and insert between the UL50 core and the C‐terminal α4 helix (Figs 2C and H, and 5A). Separated by a ~45° angle mediated by the proline residue (P72), the helices resemble the letter V, with distinct chemical properties on each side. On one side, the V interacts with α1, α2 (both part of the Bergerat fold on UL50), and β8 (Fig 5B and D), and on the other side, it interacts with α4 of UL50 (Fig 5C and E). The NEC is formed as α4 of UL50, which acts as the moveable jaw of a vise, swings away from the B face (Fig 4C and F) to accommodate the UL53 V, and then clamps shut to hold it against the fixed jaw formed by the UL50 core (Fig 4C). Notably, this fixed jaw contains a large amphipathic groove/cavity originally identified on M50 (Leigh et al, 2015) (Fig 5D).
The side of the V that binds to the fixed jaw is partially hydrophobic and negatively charged (Fig 5B), and the jaw provides a complementary binding surface. This surface of the V includes residues E75, L79, and M82 on UL53 α2, which previously were identified as critical for NEC formation (Appendix Table S1; Sam et al, 2009). L79 and M82 mediate direct contacts at the junction between the UL50 B face and the groove/cavity of the fixed jaw. In particular, K132 on β8 of UL50 forms hydrogen bonds with the carbonyl groups of both M82 and L79 (Fig 5D). Additionally, the side chain of L79 is buried in the groove/cavity, and clusters with other hydrophobic residues on UL50 including Y57 on α2, and L130 on β8 (Appendix Fig S2B, Fig 5D). Both UL50 Y57 and L130 are very important for binding to UL53, and L130 is crucial for viral replication (Leigh et al, 2015; Appendix Table S1). E75 forms a salt bridge with R17 on the α1 helix of UL50 and hydrogen bonds with Y57 (Fig 5D). Also located in this region is E56 on α2 of UL50, which is crucial for binding to UL53 and for viral replication (Milbradt et al, 2012; Leigh et al, 2015). This side chain, which is buried in a pocket, makes no direct contacts with Chm53 in the structure. We think that it is probably important for stabilizing the local structure of UL50 and/or ordinarily makes a contact with a basic residue of UL53 such as H62 that is substituted in Chm53 (Appendix Figs S2A and S3A).
On the moveable jaw side of the V‐shaped UL53 heterodimerization domain is a hydrophobic interaction surface formed by a cluster of residues including L74 on UL53 α2, and L64, F68, and V67 (ordinarily I67; Appendix Fig S3B) on α1 (Appendix Fig S2A, Fig 5C and E). Substitution of L74 with alanine has only a slight effect on complex formation in vitro, but substitutions of L64, and F68 have 10‐ to 30‐fold effects (Sam et al, 2009; Appendix Table S1). M82, which is C‐terminal to α2 on the linker between α2 and β1 of UL53, also contributes to this hydrophobic surface (Fig 5C and E). These residues are all snugly buried by the hydrophobic face of the UL50 α4 helix that includes residues Y159, L162, and L163 (Fig 5E).
Another site of UL50–UL53 interaction involves residues on the top of the B face of UL50 and residues close to the zinc finger of UL53 (Fig 5A and F). The end of the UL50 B face that is more distant from the V‐shaped helices makes close contacts with R102, located just before β2 in UL53 (Fig 5A and F). Interestingly, R102 forms cation‐pi interactions with F103 on β7 of UL50 (Fig 5F). The end of the UL50 B face that lies closer to the V‐shaped helices interacts with residues of UL53 that follow β3 (S118 and I120). S118 forms hydrogen bonds with E147 on β10, while I120 inserts into a hydrophobic pocket formed by V134 on β9, and I149 and F151 on and just after β10 on UL50 (Fig 5F).
Effects of substitutions affecting the interaction interface
Previous work on the UL50–UL53 interaction has focused primarily on the roles of UL53 residues in the amphiphatic heterodimerization domain and residues on the fixed jaw side of UL50 (Sam et al, 2009; Leigh et al, 2015; Appendix Table S1). To investigate other UL53 residues that contact UL50, we tested the effect of a double alanine substitution of two residues near the zinc finger, Ser118 and Ile120 (Appendix Fig S2A, Fig 5F), on UL50–UL53 co‐localization in transfected cells, as above for the zinc finger substitutions. We observed that, like the zinc finger substitutions, this mutant exhibited a mixed UL53 distribution, with some co‐localization of UL50 at the rim and some in the interior of the nucleus (S118I120‐AA; Fig 3C), and an intermediate Manders M1 coefficient (Fig EV2). Thus, these two residues do not seem to be absolutely required for the interaction.
We next investigated the contribution of residues on α4 of UL50 to NEC formation. First, we tested whether this helix is required for heterodimerization by mixing 200 μM purified UL531–292 with 600 μM of either UL501–169 or a truncated protein UL501–152 that lacks the residues comprising the helix, and assessing complex formation by column chromatography. As shown in Fig EV4A, UL531–292 formed a complex with UL501–169, and no free UL53 was detected, while the truncated protein did not form a complex, giving rise to free UL53. Thus, the helix is required for heterodimerization in vitro. This was supported by ITC studies which showed no detectable binding between UL531–292 and UL501–152, whereas the K d of UL531–292 to UL501–169 was 3.8 ± 0.8 μM (Fig EV4C). These results, plus the retention of helical character in both the bound and unbound states, are consistent with the finding that residues 137–181 of HSV‐1 UL34 that include this helix are both necessary and sufficient for interacting with UL31 (Liang & Baines, 2005).
We then investigated whether specific residues in the helix that make contacts with UL53 are important for co‐localization of the two proteins in transfected cells (Fig 5E). Co‐transfection of plasmids encoding UL50 with either a quadruple substitution of Y159‐L162 to alanine (4A) or a double substitution of Y159 and L162 to alanine (2A) with a UL53 plasmid led to no detectable co‐localization of the two proteins (Fig 3C) and very low Manders M1 coefficients (Fig EV2). We also constructed two independent isolates of the 2A mutant in different HCMV BACs (Appendix Table S3A). Following electroporation into cells, neither BAC gave rise to a spreading infection, while a rescued derivative of one of these did. Thus, these residues in helix α4 of UL50 are required for co‐localization in transfected cells and virus viability.
Finally, we examined three alanine substitutions in α2 of UL53—E75A, M82A, and L79A (Fig 5D)—that were previously shown to drastically decrease NEC formation in vitro, with L79A and M82A also decreasing co‐localization with UL50 in co‐transfected cells (Sam et al, 2009). To investigate the effects of these substitutions on virus viability, GFP‐expressing BACs containing HCMV harboring these substitutions were electroporated into HFFs. While wild type (and certain other mutant BACs) showed observable spread of GFP expression within 5 days post‐electroporation (Fig EV3), followed by further spread and CPE, even 10 days after electroporation of the three mutant BACs, only individual cells expressing GFP were observed (Fig EV3), and those cells eventually disappeared, indicating the mutant viruses are non‐viable.
To ensure that non‐viability was not due to unwanted changes in the viral genome, we constructed an independent isolate of one of the mutants, L79A, and rescued derivatives for all three mutants (Appendix Table S3A). The independent L79A mutant also failed to spread in cell culture and, when the cultures were harvested, no infectious virus was detected by plaque assay. The rescued derivative of L79A, L79AR, did spread in cell culture, and the virus recovered replicated with kinetics indistinguishable from those of WT virus (Appendix Fig S4 and Appendix Supplementary Methods). The rescued derivatives of E75A and M82A (E75AR and M82AR) showed spread of GFP signal by day 5 post‐electroporation, similar to WT (Fig EV3), and eventually led to complete cytopathic effect. Thus, single substitutions in the amphiphatic helical region on UL53 that abrogate the interaction with UL50 are lethal for HCMV.
Discussion
The crystal structures of UL53 and the NEC not only explain data from previous studies and verify previous predictions but also reveal unexpected features. One unexpected feature is the Bergerat fold observed in UL50 and M50, and in CR3 of UL53 (Fig 2I). Additionally, the organization of two sheets of β‐strands into an A and a B face is conserved in both NEC subunits.
A second unexpected result is the zinc finger in UL53. We found that UL53 mutants containing substitutions of its zinc‐coordinating residues result in partial defects in subunit interactions in transfected cells and, when incorporated into the HCMV genome, ablate viral replication. Notably, the availability of four structures of UL53 in different crystal forms suggests a stable core domain with variability/flexibility near the zinc finger as well as in the C‐terminal helices that compose CR4. The dynamic nature of these regions suggests a role in binding interactions. For the region close to the zinc finger, we have shown that one such binding role is participation in NEC formation.
A third unexpected feature is a striking mechanism underlying complex formation. In particular, the structures and our biochemical, cell biological, and virological studies identify the key players at the NEC interface as a vise formed by the UL50 B face and helices α1 and α2 on one side, and the α4 helix on the other, which moves away from the B face to open the vise and then clamps the V‐shaped α1 and α2 helices to the B face. The jaws of the vise and the V‐shaped heterodimerization domain have complementary surfaces that stabilize the complex.
The Bergerat fold
The discovery of the Bergerat fold in UL53 and UL50 was remarkable. Neither these proteins nor their homologs have any known nucleotide binding functions, and they lack key conserved residues involved in ATP binding and the ATP‐binding lid, unlike all known members of the Bergerat fold‐encompassing GHKL superfamily, which have diverse functions ranging from protein folding to signal transduction that entail binding of ATP (Bergerat et al, 1997; Dutta & Inouye, 2000; Li & Buchner, 2013). The NEC proteins are an unusual addition to the family. If the NEC proteins do bind nucleotides, they do so in a manner different from that of the GHKL proteins. We speculate that herpesviruses have adopted, duplicated, and adapted this fold for several known functions of the NEC and possibly for other yet‐to‐be defined purposes. It is tempting to speculate that ancestral versions of these proteins had protein kinase activity, which then became unnecessary as later versions evolved to recruit viral and/or host protein kinases for functions such as lamina disruption. Another speculative possibility would be that ancestral forms used ATP in the budding process, but that this became superseded by a different mechanism. Regardless, in UL50, at least one of the two helices of the Bergerat fold has residues critical for the interaction with UL53. In UL53, this fold does not directly participate in NEC formation; though interestingly, in our crystals, it is involved in forming contact points between monomers of the complex to form lateral strands of NEC, as detailed in the next section. We thus raise the possibility of evolution of this fold into a protein–protein interaction site not only for NEC formation as is the case for UL50, but also to mediate strand formation at the membrane surface.
Implications from the multiple structures and the crystal behavior of UL53 and the NEC
The availability of multiple structures of UL53 in different crystal forms allows us to glean insight into properties of these proteins. We were struck by the lack of well‐resolved density for the C‐terminal helical region (encompassing α11–α12) of CR4 of UL53 in the NEC, which was well defined for both non‐crystallographic symmetric monomers of UL5384–292. A similar, though less pronounced, trend was also observed for UL5372–292, where the temperature factors (B‐factors) for this helical region were higher than those for the rest of the structure. Interestingly, the HSV‐1 and PrV‐1 structures whose overall folds are conserved with the HCMV NEC (Bigalke & Heldwein, 2015) showed well‐ordered helices in this region, though the B‐factors for the C‐terminal helices in PrV were slightly higher than those for the rest of the structure. In UL53 in the NEC, the densities for these helices were fairly poor, and the main chain trace indicated a different conformation of helices compared to the other UL53 structures and needed to be manually rebuilt. The result is a more straightened conformation of the two helices compared to that in all the other UL53 structures, which have a bend of ~145° between the α11 and α12 helices (Fig EV5A). This places the helices closer to α6 of CR2 and the backside of the β‐strands of the Bergerat fold in CR3. This placement is an average of the actual conformation as it accounts for a heterogenous population due to the mobility of these helices. The variability in the degree of order observed in all these different UL53 structures and its homologs in HSV‐1 and PrV‐1 in different crystal packing environments consolidate the biological implication that these helices are capable of conformational change for binding interactions. Indeed, certain mutations affecting CR4 of the MCMV homolog of UL53 (M53) result in a dominant negative phenotype (Mettenleiter & Minson, 2006; Popa et al, 2010), which implies an involvement of this region in an interaction.
The companion paper by Bigalke and Heldwein shows that the HSV‐1 NEC forms hexagonal arrays in the crystal lattice that are remarkably similar to those observed in membrane bound NEC, which they have proposed to perform the function of inducing curvature in the inner nuclear membrane during the budding step of nuclear egress. Moreover, substitutions predicted to interrupt inter‐NEC contacts in their crystal lattice reduce budding in vitro (Bigalke & Heldwein, 2015). These hexagonal arrays were not present in the PRV NEC reported in the companion paper and are also lacking in the HCMV NEC and UL53 structures. Interestingly, the interactions between UL53 molecules in the different UL53 crystal forms, and in the NEC, involve strong interactions that are conserved in all four observed structures, despite differences in the crystal packing environments. Though we think this unlikely, it is possible that not all NECs form hexagonal coats on the membrane. Another possibility is that the inter‐NEC contacts important for forming such coats at the membrane may not be conserved across the herpesvirus sub‐families, which would be consistent with virus‐specific differences in membrane remodeling and vesiculation in cells and in vitro (Klupp et al, 2007; Desai et al, 2012; Lee et al, 2012; Luitweiler et al, 2013; Bigalke et al, 2014; Lorenz et al, 2015).
Relating structure to NEC activities and mutant phenotypes
As reviewed in the Introduction, NEC subunits have several known activities including subunit interactions, recruitment of protein kinases, remodeling of membranes, and, for the nucleoplasmic subunit, DNA packaging and/or relocalization of capsids from the nuclear interior to the nuclear periphery. The structures presented here provide a detailed three‐dimensional basis for understanding the roles of conserved regions and specific residues of herpesvirus NECs in these activities.
As the Results section details the molecular basis for subunit interactions, we will focus here on other functions. For UL50 and its homologs, one such function has been defined by MCMV UL50 and HSV‐1 UL34 mutations that lie in the vicinity of the β1‐strand and the loop that follows it (Bjerke et al, 2003; Bubeck et al, 2004; Rupp et al, 2005; Roller et al, 2010). Interestingly, a HSV‐1 UL34 dominant negative mutant in this segment affects vesiculation in vitro (Bigalke et al, 2014; Bigalke & Heldwein, 2015) and can be suppressed by a substitution in UL31, R229L (Roller et al, 2010). Together with other data, this extragenic suppression suggested a second, direct interaction between the two subunits (Roller et al, 2010). However, in our structure, the residue that corresponds to R229 of HSV‐1 UL31 is quite far from this surface on UL50. Without a major conformational change to bring these two surfaces together, the structural and mutational analyses can be reconciled by the mutations affecting interactions between neighboring NECs (although we did not observe this surface participating in crystal contacts), which is consistent with effects of the UL34 mutation on hexamer formation on membranes (Roller et al, 2010; Bigalke et al, 2014), or by the surfaces interacting with a common third partner, which could be the inner nuclear membrane.
For UL53 and its homologs, the four CRs were hypothesized to mediate different functions (Lotzerich et al, 2006). From our results, most of CR1 plays a role in interacting with the nucleoplasmic subunit. However, CR1 likely plays other roles, too, especially as it contributes to the zinc finger. Indeed, studies of the Kaposi's sarcoma herpesvirus (KSHV) homolog have shown that an in‐frame deletion of four residues in CR1 including a cysteine from the zinc finger does not abolish subunit interactions, but does prevent vesiculation in cells (Luitweiler et al, 2013). This is consistent with our finding that substitution of zinc‐coordinating residues only partially decreases subunit co‐localization, while ablating viral replication.
Less is known about the biochemical roles of CR2‐CR4. Dominant negative mutants affecting CR2 or CR4 of MCMV M53 suggest that surfaces of these CRs interact with other proteins that are important for DNA packaging and/or localization of nucleocapsids to the nuclear periphery (Popa et al, 2010; Pogoda et al, 2012). The apparent flexibility of CR4 may be germane for such protein–protein interactions. The structure of UL53 should abet studies to discover and dissect the functions of these CRs.
Implications of the mechanism of heterodimer formation for drug discovery
There is an urgent need for new therapeutics against HCMV to address the issues that plague current therapeutics such as acute and long‐term toxicities, and, for some, poor oral bioavailability. As most therapeutics target the HCMV polymerase, cross‐resistance among existing drugs is also a problem, so identification of new targets is important. Our studies here build on previous studies (Sam et al, 2009; Leigh et al, 2015) that show that subunit interactions of the HCMV NEC are an attractive drug target as point mutations that abrogate these interactions are lethal, and the binding interface is both structurally characterized and has features that make it amenable for drug discovery. In particular, our study identifies binding pockets on UL50, and amino acid side chains that fit into those pockets with both chemical and shape complementarity, which could guide the design of small‐molecule inhibitors. Additionally, the mechanism by which the UL50 α4 helix opens and closes on the UL53 helices is informative for drug discovery. This mechanism suggests that a small molecule that would bind to the grooved fixed jaw of UL50, and also bind to the α4 helix, anchoring it to the rest of UL50, could be particularly potent and efficacious. These considerations could guide a structure‐based design approach to discover inhibitors of NEC subunit interactions with anti‐HCMV activity.
Materials and Methods
Plasmids, protein expression and purification, and analytical column chromatography
Full‐length and near full‐length HCMV UL50 and UL53 polypeptides exhibit poor expression and solubility (Sam et al, 2009). Truncated versions of UL50‐ and UL53‐encompassing residues conserved across the herpesvirus sub‐families (Sam et al, 2009) were thus used as starting templates for construct design. The gene sequences encoding UL50 from residues 1–169 (UL501–169) and UL53 residues 50–292 (UL5350–292) from plasmids described previously (Sam et al, 2009) were cloned into a pET15b and pGEX6P expression vector, respectively, using the primers listed in Appendix Table S2. Modifications were made to the UL501–169 and UL5350–292 plasmid using a QuikChange Site‐Directed Mutagenesis kit (Stratagene) to generate the UL5361–292, UL5372–292, UL5384–292, and Chm53 plasmids (Appendix Fig S1B, Appendix Table S2). The Chm53 plasmid, in which sequences encoding residues 61–70 of UL53 are replaced with sequences encoding the homologous MCMV M53 residues (111–120), was used for NEC formation. The plasmids were each transformed into E. coli BL21 (DE3) RIPL (Stratagene) cells, grown in 2YT media to an optical density of ~0.6, and protein expression was induced overnight using 0.3 mM isopropyl‐β‐D‐thiogalactopyranoside (Sigma‐Aldrich) at 20°C. Cells were lysed using a sonicator (5 min of 5 s on and 9 s off pulses with a duty cycle of 65% on a Branson Sonifier Cell Disruptor Model 250) on ice in 50 mM Hepes pH 7.5, 1 M NaCl, and 10% glycerol with protease inhibitors (SigmaFAST Protease Inhibitor Cocktail Tablets EDTA‐Free). To purify polyhisidine‐tagged UL501–169, the lysate was loaded onto a nickel affinity chromatography column (Sigma‐Aldrich) and washed with 50 mM Hepes pH 7.5 and 1 M NaCl, and the tag was removed using thrombin (500 units/ml) (BioPharm Laboratories) after elution from the column using a step gradient of imidazole to a final concentration of 350 mM. To purify glutathione S‐transferase‐tagged UL53 constructs, lysates were loaded onto glutathione‐agarose (GE Heathcare Life Sciences), and the tag was removed on‐column with PreScission protease (Genway Biotech) after washing with 50 mM Hepes, pH 7.5, and 1 M NaCl. The protein was eluted in the same buffer used in the washing step. Both UL50 and UL53 were subjected to size‐exclusion chromatography on a Superdex S200 10/300 GL column (GE) as a final purification step in 50 mM Hepes pH 7.5, 200 mM NaCl, and 5 mM DTT. The NEC was formed by incubating Chm53 with UL501–169 overnight at 4°C and the complex separated from unbound protein on the Superdex S200. Superdex 200 was also used for analytical column chromatography studies, where different species of UL50 and UL53 were identified by comparison with marker proteins and by analysis on SDS–polyacrylamide gels.
Crystallization
Crystals of UL5372–292, UL5384–292, and the NEC were grown at room temperature using the hanging drop vapor diffusion method. The UL5372–292 construct crystallized in 0.1 M Tris pH 7.5, 0.1 M NaCl, 20–30% polyethylene glycol (PEG) 3350 (w/v), and 10% glycerol, while the UL5384–292 construct crystallized in 0.1 M Hepes pH 7.5, 10–15% PEG 3350, and 0.15 M ammonium acetate. The NEC crystallized in 0.1 M Tris pH 7.9, 0.1 M NaCl, 10–15% PEG 3350 (w/v), and 0.3–0.5 M CaCl2. The crystals were flash‐frozen in liquid nitrogen and cryoprotected in a solution containing their respective crystallization compositions, except with a final PEG 3350 concentration of 35%.
Structure determination
Native diffraction data for UL5372–292, UL5384–292, and the NEC were collected at 100 K on the NE‐CAT 24‐ID‐C and E beamlines at the Advanced Photon Source at Argonne National Laboratory and processed in space groups P4, P1, and C2 (Table 1), respectively, with XDS (Kabsch, 2010) and XSCALE. Attempts to obtain phase information through heavy atom procedures were unsuccessful, and the UL53 protein was either very poorly expressed, or insoluble when grown in the presence of seleno‐methionine. Experimental phase information was thus obtained through sulfur‐SAD phasing on UL5372–292 at 100 K on the I‐04 beamline at the Diamond Light Source, UK following the general protocol of El Omari et al (2014). The diffraction images were processed with XIA2 (Winter, 2010) in the P4 space group. Sixteen sulfur sites were identified using HKL2MAP (Pape & Schneider, 2004), and the phases were calculated using Phenix_autosol (Adams et al, 2010). The maps were clearly interpretable with defined β‐strand and helical regions, and Phenix_autobuild (Adams et al, 2010) successfully built in approximately 60% of the model. This was then used in Phaser (McCoy et al, 2007) as a template for UL5384–292 in the P1 crystal form and the higher resolution maps allowed for complete building and refinement of the model in Coot (Emsley et al, 2010) and Refmac (Murshudov et al, 2011), respectively. The UL5384–292 model was then used for molecular replacement with a native 3 Å data set of UL5372–292 and Phaser returned a clear solution with density for the α2 helix that was not present in the UL5384–292 structure. The UL5384–292 and the M50 models were used to solve the structure of the NEC, as the sulfur‐SAD diffraction data collected on the NEC in the C2 space group was insufficient to solve the structure de novo, and molecular replacement performed using Phaser (McCoy et al, 2007) produced a clear solution. Electron density corresponding to UL50 was very weak, however. Thus, placement of the molecule into the maps was done manually using a truncated model with only the secondary structure elements. Anomalous difference Fourier maps were then calculated to locate the sulfur positions in the map to validate the placement of both the UL50 and Chm53 proteins. Rebuilding and refinement of the structures were done using Coot (Emsley et al, 2010), Refmac (Murshudov et al, 2011), and Phenix (Adams et al, 2010). TLS was applied with restrained refinement in Refmac (Murshudov et al, 2011), and the final models were validated using PROCHECK (Laskowski et al, 1993). The data collection and refinement statistics are summarized in Table 1.
Elemental analysis by energy‐dispersive X‐ray spectroscopy (EDS)
To identify the metal ion present in the protein, frozen protein solution in a Hampton Research CryoLoop was sent to the NE‐CAT 24‐ID‐C beamline at APS. An Amptek X‐123SDD Silicon Drift Diode detector was used for elemental analysis by energy‐dispersive X‐ray spectroscopy (EDS). The detector was calibrated using emission lines from several known metals, and the gain was set to 75% corresponding to an energy range of 0–16.7 keV. The sample was centered so the 50 micron X‐ray beam was aligned with the center of the sample drop. The incident X‐ray energy was set to 12.66 keV for the duration of the experiment.
Isothermal Titration Calorimetry (ITC)
ITC studies were conducted using a VP‐ITC calorimeter (MicroCal Inc.). Prior to analysis, the samples were subjected to gel filtration in 50 mM Hepes pH 7.5, 200 mM NaCl, and 0.5 mM TCEP (Tris(2‐carboxyethyl) phosphine hydrochloride (Sigma)). The titrations were carried out as described previously (Sam et al, 2009) with UL50 as the titrant (concentrations ranging from 50 to 100 μM) in the syringe, and UL53 in the reservoir (concentrations ranging from 1 to 10 μM). The binding isotherms were fit to a one‐site model using Origin.
Bacterial artificial chromosome (BAC) construction
The BACs used in this study are summarized in Appendix Table S3A. BACs carrying UL53 mutations E75A, L79A, M82A, Chm53, C106S, and H211A or the UL50 mutations 2A and 4A were generated by using the PCR primers listed in Appendix Table S3B, and the two‐step Red recombination method of Tischer and coworkers (Tischer et al, 2006, 2010) in pBADGFP or 53‐F BADGFP, as described previously in Sharma et al (2014). To construct the rescued derivative BACs—E75AR, L79AR, M82AR, and 2AR pBADGFP, wild‐type sequences were restored to the respective mutant BACs, using the relevant PCR primers listed in Appendix Table S3B, and the same methodology.
Viruses
pBADGFP‐based BACs were electroporated into HFFs as described previously (Sharma et al, 2014) to test for infectivity. The cells were monitored for spread of the GFP signal, and infectious viruses were harvested when the entire monolayer showed complete cytopathic effect.
Immunofluorescence
HEK293T cells were transfected with pcDNA plasmid constructs expressing FLAG‐UL53 and/or HA‐UL50 WT or the same constructs carrying mutations in the UL50‐ or UL53‐coding sequences, and stained and imaged as described previously (Sam et al, 2009). Co‐localization analysis was performed using Fiji/ImageJ. Images of UL53 were first background subtracted using the measured background intensity in the nucleus of an untransfected HEK293T cell. A region of interest (ROI) proximal to the nucleus was defined by thresholding the DAPI channel and applying a dilation of 12 pixels to the resulting nuclear mask. The Manders M1 coefficient was then calculated within this ROI using the Coloc2 plugin, with the UL50 image as the reference and UL53 image as the variable. The values represent mean ± SD.
Author contributions
JMH, DMC, MFL, and MS conceived and designed the studies; MFL, MS, KEO, and JPS acquired data; all authors interpreted data; MFL, JMH, MS, DJF, JPS, and DMC drafted the manuscript; and all authors critically revised the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We are grateful for the assistance of staff and the availability of equipment at the Nikon Imaging Center at Harvard Medical School for acquisition and analysis of immunofluorescence data, and for the assistance of staff at the Taplin Mass Spectrometry Facility, Hunter Eliott at the Imaging and Data Analysis Core for analysis of confocal images, and at the Northeastern Collaborative Access Team (NE‐CAT) and Diamond Light Source. We also thank Mona Bodenhoefer and William Polachek for experimental assistance, My Sam Mansueto and Kendra Leigh for helpful discussions, and Janna Bigalke and Katya Heldwein for sharing their data and manuscript prior to submission. This work was supported by NIH grants R01 AI026077 to D.M.C. and J.M.H., and P41 GM103403 to the NE‐CAT.
The EMBO Journal (2015) 34: 2937–2952
See also: JM Bigalke & EE Heldwein (December 2015)
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergerat A, de Massy B, Gadelle D, Varoutas PC, Nicolas A, Forterre P (1997) An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386: 414–417 [DOI] [PubMed] [Google Scholar]
- Bigalke JM, Heuser T, Nicastro D, Heldwein EE (2014) Membrane deformation and scission by the HSV‐1 nuclear egress complex. Nat Commun 5: 4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigalke JM, Heldwein EE (2015) Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34: 2921–2936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ (2003) Effects of charged cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J Virol 77: 7601–7610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck A, Wagner M, Ruzsics Z, Lotzerich M, Iglesias M, Singh IR, Koszinowski UH (2004) Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J Virol 78: 8026–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camozzi D, Pignatelli S, Valvo C, Lattanzi G, Capanni C, Dal Monte P, Landini MP (2008) Remodelling of the nuclear lamina during human cytomegalovirus infection: role of the viral proteins pUL50 and pUL53. J Gen Virol 89: 731–740 [DOI] [PubMed] [Google Scholar]
- Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B (1997) The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J Virol 71: 8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho HY, Lee YH, Bae YS, Kim E, Kang BS (2013) Activation of ATP binding for the autophosphorylation of DosS, a Mycobacterium tuberculosis histidine kinase lacking an ATP lid motif. J Biol Chem 288: 12437–12447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J (2012) Reconstitution of the Kaposi's sarcoma‐associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86: 594–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew WL, Buhles WC (1996) Cytomegalovirus: nucleosides and foscarnet: clinical applications In Antiviral Drug Resistance. Richman DD. (ed.), pp 594–598. Chichester, UK: John Wiley & Sons; [Google Scholar]
- Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F (2003) Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci USA 100: 14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta R, Inouye M (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25: 24–28 [DOI] [PubMed] [Google Scholar]
- El Omari K, Iourin O, Kadlec J, Fearn R, Hall DR, Harlos K, Grimes JM, Stuart DI (2014) Pushing the limits of sulfur SAD phasing: de novo structure solution of the N‐terminal domain of the ectodomain of HCV E1. Acta Crystallogr D Biol Crystallogr 70: 2197–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C, Ott M, Raschbichler V, Nagel CH, Binz A, Sodeik B, Bauerfeind R, Bailer SM (2015) The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by its N‐terminal domain. PLoS Pathog 11: e1004957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Stock AM (2009) Biological insights from structures of two‐component proteins. Annu Rev Microbiol 63: 133–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, Bestman‐Smith J, Boivin G (2002) Resistance of herpesviruses to antiviral drugs: clinical impacts and molecular mechanisms. Drug Resist Updat 5: 88–114 [DOI] [PubMed] [Google Scholar]
- Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, Faggioni A, Delecluse HJ (2008) Deletion of Epstein‐Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J Virol 82: 4042–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Baines JD (2011) Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9: 382–394 [DOI] [PubMed] [Google Scholar]
- Jokhi V, Ashley J, Nunnari J, Noma A, Ito N, Wakabayashi‐Ito N, Moore MJ, Budnik V (2013) Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep 3: 988–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) Xds. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC (2007) Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci USA 104: 7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26: 283–291 [Google Scholar]
- Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR (2012) The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus‐associated membrane for the maturation of Epstein‐Barr Virus. PLoS Pathog 8: e1002904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H (2015) Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci USA 112: 9010–9015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Buchner J (2013) Structure, function and regulation of the hsp90 machinery. Biomed J 36: 106–117 [DOI] [PubMed] [Google Scholar]
- Liang L, Baines JD (2005) Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J Virol 79: 3797–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia‐Saez AJ, Mettenleiter TC, Antonin W (2015) A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290: 6962–6974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotzerich M, Ruzsics Z, Koszinowski UH (2006) Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J Virol 80: 73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, McCaffery JM, Desai PJ (2013) Interactions of the Kaposi's Sarcoma‐associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J Virol 87: 3915–3929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurain NS, Chou S (2010) Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev 23: 689–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ (2007) Phaser crystallographic software. J Appl Crystallogr 40: 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettenleiter TC (2004) Budding events in herpesvirus morphogenesis. Virus Res 106: 167–180 [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Klupp BG, Granzow H (2006) Herpesvirus assembly: a tale of two membranes. Curr Opin Microbiol 9: 423–429 [DOI] [PubMed] [Google Scholar]
- Mettenleiter TC, Minson T (2006) Egress of alphaherpesviruses. J Virol 80: 1610–1611; author reply 1611–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Marschall M (2007) Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J Gen Virol 88: 2642–2650 [DOI] [PubMed] [Google Scholar]
- Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, Marschall M (2012) Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J Biol Chem 287: 24004–24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Shenk T, Griffiths PD, Pass RF (2013) Cytomegaloviruses In Fields Virology, Knipe DM, Howley PM. (eds), 6th edn, pp 1960–2014. Philadelphia, PA, USA: Wolters Kluwer Lippincott Williams & Wilkins; [Google Scholar]
- Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH (2002) Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297: 854–857 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr 67: 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape T, Schneider TR (2004) HKL2MAP. J Appl Crystallogr 37: 843–844 [Google Scholar]
- Park R, Baines JD (2006) Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80: 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoda M, Bosse JB, Wagner FM, Schauflinger M, Walther P, Koszinowski UH, Ruzsics Z (2012) Characterization of conserved region 2‐deficient mutants of the cytomegalovirus egress protein pM53. J Virol 86: 12512–12524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa M, Ruzsics Z, Lotzerich M, Dolken L, Buser C, Walther P, Koszinowski UH (2010) Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J Virol 84: 9035–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roller RJ, Bjerke SL, Haugo AC, Hanson S (2010) Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol 84: 3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp B, Ruzsics Z, Sacher T, Koszinowski UH (2005) Conditional cytomegalovirus replication in vitro and in vivo . J Virol 79: 486–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sam MD, Evans BT, Coen DM, Hogle JM (2009) Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J Virol 83: 2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Coen DM (2014) Comparison of effects of inhibitors of viral and cellular protein kinases on human cytomegalovirus disruption of nuclear lamina and nuclear egress. J Virol 88: 10982–10985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM (2014) Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88: 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM (2015) Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J Virol 89: 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V (2012) Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149: 832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strang BL, Boulant S, Chang L, Knipe DM, Kirchhausen T, Coen DM (2012) Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J Virol 86: 2089–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer BK, von Einem J, Kaufer B, Osterrieder N (2006) Two‐step red‐mediated recombination for versatile high‐efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40: 191–197 [DOI] [PubMed] [Google Scholar]
- Tischer BK, Smith GA, Osterrieder N (2010) En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634: 421–430 [DOI] [PubMed] [Google Scholar]
- Trajtenberg F, Grana M, Ruetalo N, Botti H, Buschiazzo A (2010) Structural and enzymatic insights into the ATP binding and autophosphorylation mechanism of a sensor histidine kinase. J Biol Chem 285: 24892–24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal EC (2003) Current and potential therapies for the treatment of herpes‐virus infections. Prog Drug Res 60: 263–307 [DOI] [PubMed] [Google Scholar]
- Winter D (2010) xia2: an expert system for macromolecular crystallography data reduction. J Appl Cryst 43: 186–190 [Google Scholar]
- Yu D, Silva MC, Shenk T (2003) Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc Natl Acad Sci USA 100: 12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File