

Abstract
Studies over the last two decades have identified that amplified human epidermal growth factor receptor (HER‐2; c‐erbB‐2, neu) and its overexpression have been frequently implicated in the carcinogenesis and prognosis in a variety of solid tumours, especially breast cancer. Lots of painstaking efforts were invested on the HER‐2 targeted agents, and significantly improved outcome and prolonged the survival of patients. However, some patients classified as ‘HER‐2‐positive’ would be still resistant to the anti‐HER‐2 therapy. Various mechanisms of drug resistance have been illustrated and the alteration of HER‐2 was considered as a crucial mechanism. However, systematic researches in regard to the HER‐2 mutations and variants are still inadequate. Notably, the alterations of HER‐2 play an important role in drug resistance, but also have a potential association with the cancer risk. In this review, we summarize the possible mutations and focus on HER‐2 variants’ role in breast cancer tumourigenesis. Additionally, the alteration of HER‐2, as a potential mechanism of resistance to trastuzumab, is discussed here. We hope that HER‐2 related activating mutations could potentially offer more therapeutic opportunities to a broader range of patients than previously classified as HER‐2 overexpressed.
Keywords: breast cancer, HER‐2, HER‐2 mutation, variants, cancer risk, resistance
Introduction
HER‐2 is a member of the human epidermal growth factor receptor (HER) family, additionally comprised of epidermal growth factor receptor (EGFR), HER‐3, and HER‐4. These receptors regulate normal cell proliferation, survival, and differentiation via different signal transduction pathways 1. The gene encoding HER‐2 is located in chromosome 17, and codes for a 185‐kPa protein that functions as a transmembrane growth factor receptor 2. The intracellular domain of HER‐2 contains approximately 500 residues and composed of three parts: a cytoplasmic juxtamembrane linker, a tyrosine kinase (TK) domain and a carboxyl‐terminal tail 3, 4. The TK domain is more complicated than other parts of HER‐2 receptor, which contains several important loops: the C‐loop (residues 844–845), the αC‐helix (residues 761–775), the N‐loop (residues 727–732) and the activation loop (A‐loop residues 863–884), to form the enzyme active site 3.
Though HER‐2 point or insertion mutations were first described in 2004, researches efforts about them are not exhaustive compared with his family EGFR to date 5. According to the existing data, the probability of HER‐2 mutations is 1.67% in breast cancer, 1–4% in lung cancer and 2.9% in colorectal 6, 7, 8, 9, 10, 11, 12. Other human tumour types have also been reported to harbour HER‐2 mutations, including head and neck cancers, bladder cancers, gastric cancers, ovarian cancers, hepatic cancers 6, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21. Mutational activation of HER‐2 can result from three types of somatic molecular alterations: small insertions and missense mutations in the kinase domain, missense mutations in the extracellular domain, or large deletions of the extracellular domain which yield a truncated form of HER‐2 22, 23. More mutations are mainly located in the three exons (19–21) of the TK domain 24, and are encoded by the DNA sequences in the exons 18–23 25. HER‐2 kinase domain mutations have been described in lung carcinoma and breast cancer albeit at a lower frequency 26, 27, 28, 29. HER‐2 kinase domain mutations can be categorized as: missense point mutations, small in‐frame insertions or duplications which almost occurring in exon 20 and in frame deletions. Among these mutations, the in‐frame insertions or duplications in exon 20 are the most frequently encountered types of mutations 22, 30, 31, 32. In addition, we also take the HER‐2 splice variants into account, including p95HER‐2 and Δ16HER‐2.
The clinical success of gefitinib, an inhibitor of EGFR, in a subset of lung cancers with mutations in the TK domain of EGFR, holds a promise for the future of targeted therapy 33, 34, and also leads to the investigation of analogous mutations of HER‐2. With the application of HER‐2 fluorescent in situ hybridization and HER‐2 immunohistochemistry which are standard clinical tests for HER‐2 gene amplification 35, 36, HER‐2 gene amplification or protein overexpression has been extensively studied in breast cancer 37, 38, 39, 40, much less is known about genetic variants and mutations that might have an impact on the risk or therapy of breast cancer. It may be more challenged to successfully target HER‐2 mutations than EGFR mutation. More efforts are needed to translate this idea to clinic.
The HER‐2 mutations and variants
The HER‐2 mutations
These HER‐2 mutations are the common type found in the patients lacking HER‐2 overexpression and most of them were found in the TK domain (Fig. 1).
Figure 1.

The HER‐2 mutations. These HER‐2 mutations are the common type found in the patients lacking HER‐2 expression and most of them were found in the tyrosine kinase domain. HER: human epidermal growth factor receptor.
Mutations in TK domain
Human epidermal growth factor receptor‐2 gene amplification or protein overexpression has been identified as a mechanism of HER‐2 activation in breast cancer 1. However, HER‐2 activating mutations, another novel modus to activate HER‐2, have been reported 41, 42. Bose and his colleagues identified 16 HER‐2 somatic mutations though cancer genome sequencing in HER‐2 gene amplification‐negative breast cancer patients. Seven of these HER‐2 kinase domain mutations are activating and oncogenic, including G309A, D769H, D769Y, V777L, P780ins, V842I and R896C 23. Activating HER‐2 kinase domain mutations could also been found at low frequency in several other carcinomas, such as bladder cancer and lung cancer 23, 31, 43.
Human epidermal growth factor receptor‐2 gene with some kinase domain mutations shows the characteristics of constitutively activate kinase activity and increased oncogenicity compared to the wild‐type HER‐2 44. The positive effect of activating mutations on tumour growth has been demonstrated in vitro 23. The enhanced kinase activity promotes the formation of the dimmers. Meanwhile, the activating mutations particularly induce the phosphorylation of cellular signalling proteins 23.
The most prevalent activating mutations of HER‐2 involve the insertions within exon 20 and these mutations have stronger catalytic activity 44. These insertions are more potent in transphosphorylating EGFR compared with the wild‐type HER‐2. Conclusively, the mutant HER‐2 gene is more transforming and more capable to inhibit the effect of apoptosis 44.
For the small insertion activating mutations, the basic mechanism is that these mutations lead to a conformational change of the autoinhibition, consequently keeping their inactive condition. The oncogenic insertions frequently alter the Adenosine triphosphate (ATP)‐binding cleft, which forms a conformational structure with many important structures surround the cleft, including phosphate‐binding and activation loops 45.These insertions induce a conformational change of the autoinhibitory αC‐β4 loop, thus, narrowing the ATP‐binding cleft, increasing both the ATP binding affinity and turnover number, and promoting the enhanced kinase activity that participates in the subsequent phosphorylation events 46. In addition, the HER‐2 insertions can potently transphosphorylate EGFR, even in the presence of EGFR tyrosine kinase inhibitors (TKI) 44. It induced transphosphorylation of kinase‐dead EGFR, and exhibited higher ligand‐independent tyrosine phosphorylation. Moreover, the insertions were more potent than wide‐type HER‐2 in associating with signal transducers that mediate proliferative and prosurvival responses 44.The activating missense mutations in kinase domain have also been reported. Most of the mutations were detected in the αC‐helix which is considered to play a critical role in the activation of HER‐2 gene 23. The structures of αC‐helix may be altered by the single missense mutations, and these altered structures might promote tumourigenesis and the phosphorylation of signalling proteins including (phospholipase γ C1, phospholipase Cγ (PLCγ) and mitogen‐activated protein kinase(MAP Kinase)) 25. Some other mutations, such as the HER‐ exon 19 in‐frame deletions 755–759, homologous to EGFR exon 19 in‐frame deletions, could potently increase phosphorylation of EGFR or HER‐3, and interact differently with its dimerization partners compared with other HER‐2 mutants 25. In addition to these activating mutations, some mutations are resistant to the targeted therapy, such as mutations located at codon 755 or 798. The underlying mechanisms of resistance will be discussed in the next sections.
HER‐2 mutations in other domain
Apart from the mutations in TK domain, recurring HER‐2 extracellular domain mutations in breast and lung cancer were also identified (e.g. S310F/Y, G309A/E, S335C) 23, 47, 48, 49, 50. Parts of these mutations that cluster in exon 8 are oncogenic and activated by two distinct mechanisms, characterized by elevated C‐terminal tail phosphorylation, such as S310F/Y, or covalent dimerization mediated by intermolecular disulfide bond formation, such as G309E and S335C 47. These extracellular domain mutations are also sensitive to small‐molecule inhibitors of HER‐2, more similar to the kinase domain mutations 23. Trastuzumab was shown to be effective against the cells expressing G309 and S310 mutations, giving hope to the patients harbouring these mutations 47.
Recently, novel transmembrane domain mutations were also reported in familial lung adenocarcinoma, including kinds of germline mutations (G660D, V659E and I655V). The V659E was first detected in a case report of Li‐Fraumeni syndrome, and was found to have a oncogenic role 51. G660D and V659E mutations in the transmembrane domain also correlate with the hereditary, sporadic lung adenocarcinomas 52. The G660D and V659E mutations, more stable than wild‐type genes, both can act as driver mutations in lung cancer, and have the capacity to activate Akt. Simultaneously, p38 was also activated to promote cell proliferation in lung adenocarcinoma 52. Additionally, I655V in the transmembrane domain was reported to increase the breast cancer risk.
The HER‐2 variants
These variants, different from the point mutations, can be defined as incomplete HER‐2 or fragments of HER‐2. They may even appear opposite functions due to their various constructions (Fig. 1).
Δ16HER‐2
Except for the small insertions or point mutations, some other resistant HER‐2 alterations have also been identified in HER‐2‐positive breast cancer, such as Δ16HER‐2 (a HER‐2 splice variant lacking exon 16) and p95HER‐2 (carboxy‐terminal HER‐2 fragments, mostly known as 611‐CTF; Fig. 2) 53, 54. Both of these alterations could explain the clinical failure of trastuzumab 55, 56, 57, 58, 59, 60. The Δ16HER‐2, a type of oncogenic variant caused by the in‐frame deletion of exon 16 in the extracellular domain of HER‐2, is reported to comprise 4–9% of total HER‐2 transcripts 56, 61. The Δ16HER‐2 variant, when expressed at high levels, harbours enhanced transforming activity compared with wild‐type HER‐2 54. The primary activating mechanism is that this conformation with removed relevant cysteine residues promotes intermolecular disulfide bonding and the generation of homodimers, thus transforming cells 62. Another study found that 44% of Δ16HER‐2‐expressing breast cancer showed the activated Src kinase, heralding the potential clinical implications of direct coupling of Δ16HER‐2 to Src kinase. The activated Src kinase is also associated with the metastatic tumour, suggesting that more aggressive therapeutic interventions are needed 54. ∆16HER‐2 has also been implicated in resistance of HER‐2 positive breast cancers to anti‐HER‐2 therapies. Thus, measurement of this variant may help predict the response to treatment of anti‐HER‐2 therapy. However, to our knowledge, no studies were conducted on the issue till now 63, 64.
Figure 2.
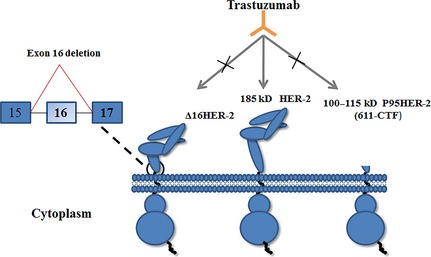
The structure of the ∆16HER‐2 and p95HER‐2 variants. The variants, different from the point mutations, can be defined as incomplete HER‐2 or fragments of HER‐2, including Δ16HER‐2 and p90HER‐2. HER: human epidermal growth factor receptor.
p95HER‐2
p95HER‐2 is a form of truncated HER‐2, which does not have the complete extracellular domain (Fig. 2). This CTF are yielded through at least two different mechanisms: proteolytic shedding of the extracellular domain of the full‐length HER‐2 receptor or translation of HER‐2 mRNA from alternate internal initiation codons (positions 611 and 678, respectively) 65, 66. Strikingly, in vitro studies of 611‐CTF (100–115 kD) revealed more rapid activation of multiple signalling pathways to promote tumour progression when compared with the full length receptor and 648‐CTF 67. p95HER‐2 is hyperactive and has been demonstrated to play a role in cancer progression, increased metastasis, poor prognosis and disease‐free survival when compared with patients that express the wild full‐length HER‐2 65, 68. Approximately 30% of HER‐2‐positive tumours express this HER‐2 fragment 66. The specific characteristic of p95HER‐2 is still unclear, but the overexpression of p95HER‐2 can promote the growth of tumour via forming homodimers by intermolecular disulfide bonds in subdomain IV, similar to dimers formed in extracellular domain mutations 69, 70.
Because p95 is thought to be sensitive to HER‐2 active TKI, measurement of quantitative p95 levels may have potential role of treatment decisions in future. Till now, at least two antibodies have been generated against the 611‐CTF form of p95 63, 64. One of the antibodies, D9, has been generated to build a quantitative p95 assay to identify a group of HER‐2 positive patients expressing p95HER‐2 that have a worse outcome while on trastuzumab.
Some other variants of HER‐2
Other variants of HER‐2 with contrasting roles in tumour have also been detected, such as Herstatin (results from intron 8 retention) and p100 (results from intron 15 retention) 71. These variants can interfere with the oncogenic activity of wild‐type HER‐2, to inhibit tumour cell growth 56. Further exploration of p100 found a decrease in downstream signal induction. Besides, the protective Herstatin have also been reported to inhibit the activity of HER‐2 by interfering with the phosphorylation of dimmers (HER‐2/HER‐3 and HER‐2/EGFR) 56. Herein, considering of the less association with breast cancer risk or drug resistance, we will not focus on these two subsets of variants more.
The HER‐2 mutations associated with breast cancer risk
A breadth of literature describes the link between genetic variations and breast cancer risk. Single nucleotide polymorphisms (SNPs) are the commonest sources of human genetic variations that contribute to a susceptibility of tumour progression 72. A polymorphism of the HER‐2 gene that results in the substitution of isoleucine‐to‐valine atcodon 655 of the transmembrane domain (Ile655Val, rs1136201) has been extensively investigated as a risk factor for breast cancer 73. Since the initial case–control study on 700 Han chinese women from Xie et al. reported a significantly increased risk for carriers of this allele [odds ratio (OR) = 1.4] 74, many epidemiological studies have been conducted to reveal an association between the HER‐2 655V polymorphism and an increased risk of breast cancer 31. However, the results are inconsistent. Several meta‐analyses have been performed to investigate the association between the polymorphism and breast cancer. Due to the differential including studies and methodological issues, different results arise from these meta‐analyses. Lu et al. found a significant association among Africans and Asians, but not in Europeans 75. However, Ma et al. did not demonstrate any significant associations between HER‐2 codon 655 polymorphism and breast cancer susceptibility, either at the overall or the ethnicity analyses 76. Moreover, a novel updated meta‐analysis suggests that this polymorphism is marginally associated with breast cancer in worldwide populations with additive and dominant models, but not a recessive model 77. Thus, no confirmed associations could be identified between the polymorphism and an increased breast cancer risk among different ethnicities.
A stronger association was revealed in women both under the age of 45 years and with a family history, and the valine allele might not have any effect among women older than 60 78. Additionally, it also raised a possibility that the risk associated with carrying the HER‐2 valine allele might predominantly affect pre‐ or peri‐menopausal breast cancer 78. Consistent with the above investigations, another study also suggested that that V/V or V/I genotype have a twofold increased risk compared with I/I genotype among women who were both younger than 45 years of age and reported a positive family history of breast cancer (OR = 2.3, 95% CI = 1–5.3) 79. However, some conflicting results emerged, revealing that HER‐2 I655V polymorphism may be a biomarker for breast cancer susceptibility among older women 80. Furthermore, a rare HER‐2 variant Ile654Val is also associated with an increased familial breast cancer risk, which revealed an oncogenic role for carriers of the heterozygous Val654 allele (OR = 2.56, 95% CI = 1.08–6.08, P = 0.028) 81, meanwhile, it is linked with the more frequent Val655 to form two consecutive valines instead of two isoleucine residues 81.
Human epidermal growth factor receptor‐2 is considered an orphan receptor since it is the only receptor of HER family in the absence of an identified ligand (Fig. 3) 82, 83. However, HER‐2‐containing heterodimers could function as the most active signalling complex of the HER family 84, 85. A strong pro‐tumourigenic signalling cascade is initiated by the overexpression of HER‐2, and leads to the generation of dimmers 86, 87. A subsequent activation of HER‐2 cytoplasmic kinase activity is needed for the downstream PI3K/AKT signalling pathways to promote cell proliferation and the effect of apoptosis (Fig. 3) 85, 87, 88. The basic activating mechanism of HER‐2 variants is to enhance the kinase activity and cell transformation by increasing the formation of active HER‐2 heterodimmers 72. The implication of HER‐2 polymorphism in tumour progression may preferentially occur through the modification of function rather than the amplification of protein 72. Furthermore, the independent genetic variant in growth factors signalling appear to have the stronger influence on breast cancer risk via combining with other variant gene, including variant fibroblast growth factor 1 (FGF1), FGF2 and neuregulin 2 (NRG2), interacted with SNPs in platelet‐derived growth factor B (PDGFB), EGFR, HER‐2 and FGFR2 89.
Figure 3.
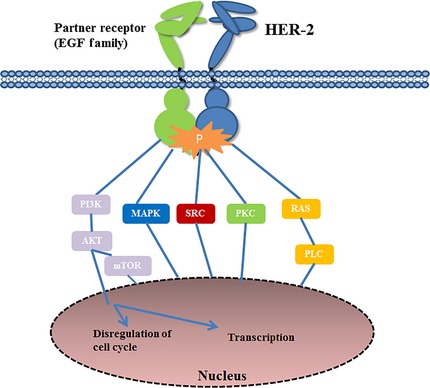
Working model for the human epidermal growth factor receptor (HER)‐2 oncogenic activities in breast cancer development and progression.
The HER‐2 mutations associated with breast cancer resistance
In 1987, HER‐2 amplification and overexpression were first reported. These molecular alterations could be found in approximately 20–30% of breast cancer 90. Since then, HER‐2 was considered as a significant targeted point due to its distinctive role in tumour cell proliferation and metastasis, facilitating the development of HER‐2 targeted agents, which have shown a tremendous success 91. However, the efficacy of anti‐HER‐2 therapeutics such as trastuzumab or small molecule HER‐2 TKI (lapatinib) is limited by the occurrence of therapeutic resistance 92, 93. Major mechanisms of primary or acquired resistance against these targeted agents include 94: (i) Alteration in binding sites or TK receptor domain. (ii) Up‐regulation of alternative ErbB ligands and dimerization of receptors to counteract for receptor inhibition. (iii) Dimerization/interaction with other receptors. (iv) Downstream controllers‐deficient tumours. (v) Activation of downstream signalling and survival pathways. Here we mainly discuss the alteration of HER‐2 gene (Table 1).
Table 1.
The HER‐2 mutations and variations associated with breast cancer resistance
| Mutations | Primary tumour | Number of patients screened | TNM stage | Drug resistance | Reference |
|---|---|---|---|---|---|
| L726I | Cell study | NA | NA | Gefitinib | 104 |
| L726F | NA | 76 | NA | Lapatinib | 42 |
| L726F | Cell study | NA | NA | Lapatinib | 101 |
| L755S | Invasive ductal | 94 | IIIA | Lapatinib | 7 |
| L755S | Invasive ductal | 94 | IIA | Lapatinib | 7 |
| L755S | Lobular | 193 | NA | Lapatinib | 28 |
| L755S | TNBC | 104 | NA | Lapatinib | 29 |
| L755S | Lobular | 1499 | IIA | Lapatinib | 23 |
| L755S | Ductal | 1499 | I | Lapatinib | 23 |
| L755P | Cell study | NA | NA | Lapatinib | 103 |
| P780L | Cell study | NA | NA | Lapatinib | 101 |
| S783P | Cell study | NA | NA | Lapatinib | 101 |
| L785F | Cell study | NA | NA | Lapatinib | 101 |
| T798M | Cell study | NA | NA | Lapatinib/Trastuzumab | 97 |
| T798M | Cell study | NA | NA | Lapatinib | 103 |
| T798I | Cell study | NA | NA | Lapatinib | 101 |
| T798I | Cell study | NA | NA | Lapatinib | 102 |
| p95HER‐2 | NA | 483 | NA | Trastuzumab | 68 |
| Δ16HER‐2 | NA | NA | NA | Trastuzumab | 62 |
NA: not available; TNBC: triple negative breast cancer; HER: human epidermal growth factor receptor.
The point or small insertion resistance mutations
To our knowledge, HER‐2 point or small insertions were found predominantly in patients lacking HER‐2 amplification to date 24. It suggests that the mutant gene may not be associated with real amplification 95. In fact, insufficient evidence about the mutations in HER‐2 ‘positive’ can be searched. Among these limited researches, rare patients harbouring HER‐2 amplification were tested for the presence of mutations. Hence, it is an arduous work to obtain any conclusions about relative characteristics. Herein, we conclude some possible reasons that could explain this low frequency event, including: one possible reason is that these mutations may be acquired only after the utilization of anti‐HER‐2 agents, suggesting that the anti‐HER‐2 agents might be a trigger to the generation of resistance alterations. Another possible explanation is that these mutations only occupy small parts of the amplified HER‐2, thereby, may not be detected by the DNA sequencing methods due to below the limits of sensitivity 95. Further investigations are needed to clarify the specific mechanisms.
These unusual intrinsic mutations in HER‐2 overexpressed patients occur with an inconspicuous probability. It is reported that a 52‐year‐old man diagnosed with stage IV non‐small cell lung cancer (NSCLC), was detected to overexpress HER‐2 and harbour an L869R mutation. Later he achieved a partial response to lapatinib, but showed no response to trastuzumab alone. The mutation in this patient is analogue to the previously reported L861 mutation in EGFR 7, 96. But whether the resistance is caused by the mutation or not is still unknown. Another study revealed that the HER‐2 gene‐amplified breast cancer cells, which harbour the T798M mutant alleles, acquired a resistance to both lapatinib and trastuzumab alone. However, after the treatment of a simultaneous blockade of HER‐2 and EGFR, an effective response was shown, hinting a possible connection between increased EGFR ligand production and drug resistance 97. Additionally, it should be noted that T798 is a gatekeeper residue, analogous to the gatekeeper EGFR T790M 98, ABLT315I 99 and cKITT670I 100 mutations 97, which are all related to the clinical drug resistance. All above results indicate that the amino acids L755 and T798 in HER‐2 are critical residues, enable to determine lapatinib sensitivity. Strong lapatinib resistance caused by L755S, L755P and T798M, T798I have been reported 101, 102. The frequency of T798 mutations is higher than other mutations 23, 103. Despite the resistance mutation, activating mutation (D769H) have also been detected in HER‐2 positive patients. But whether some associations exist between activating mutations and HER‐2‐positive patients is unclear yet 23.
Apart from the above conditions, the mutations could also occur after the treatment of anti‐agents. The wild‐type HER‐2 could acquire the L755S and T862A mutation after the exposure of lapatinib, suggesting that kinase domain mutations may cause a secondary resistance in patients with wild type HER‐2 103. Bose and his colleagues identified that L755S mutation was the most common subtype of mutant HER‐2 in breast cancer. In his study, six of total 27 patients with mutant HER‐2 were detected with the L755S mutation 23. Similar condition occurred after the treatment of other agents, such as gefitinib or iressa, a selective epidermal growth factor receptor TKI, primarily for NSCLC, also respond to breast cancer with positive HER‐2. A subset of breast cancer cell lines overexpressing the activated HER‐2 was treated with 5 Mmol/l gefitinibt. Then a novel point mutation‐L726I in the ATP‐binding pocket was found, enabling these cells insensitive to gefitinib 104.
Most HER‐2 mutations associated with lapatinib resistance locate in the ATP‐binding and hinge region. Herein, we mainly discuss the mechanism of L755 and T798 due to their vital roles. Mutations at L755 can conclusively stabilize the active conformation of the HER‐2 kinase. They may not directly affect inhibitor binding, but form a conformation where the αC‐helix is fixed in to inhibit lapatinib binding and influence the structure of the active state 103. As the HER‐2 ‘gatekeeper’, T798 is located in exon 20 within hinge region, which is the most prominent site of resistance mutations. Mutations at T798 may lead to the obstacle of TKI by directly interfering with the steric structure 101. One mechanism is to increase the affinity of HER‐2‐T798M towards ATP, similar to the T790M mutation in EGFR. Another mechanism is that lapatinib binds the inactive conformation preferentially, then incapable to bind the active conformation in T798M 103. Additionally, another studies revealed that cells harbouring T798M mutation showed increased expressions of the EGFR ligands EGF, transforming growth factor‐α, amphiregulin and proheparin‐binding epidermal growth factor (HB‐EGF), leading to the resistance of TKI 97. Besides, the ability of HER‐2YVMA's mutation to amplify their transforming potential and modify tumour microenvironment through induction of growth factors was recently demonstrated 105.
The resistance variants of HER‐2
Human epidermal growth factor receptor‐2 variants were identified in patients with HER‐2 amplification, and conferred resistance to targeted therapy. These variants mainly include Δ16HER‐2 and p95HER‐2.
The patients harbouring this Δ16HER‐2 are also refractory to the treatment of trastuzumab. The mechanism of this clinical failure still needs exploration. The potential oncogenic properties were mediated through a direct interaction between Δ16HER‐2 and Src kinase. Treatment with single‐agent TKI dasatinib overcame the resistance to trastuzumab, and suppressed tumourigenicity. In addition, the capacity of stabilizing HER‐2 homodimers and the phosphorylated state of phosphatase and tensin homolog (PTEN) may also contribute to the resistance 54. Surprisingly, trastuzumab was even identified to promote the growth and invasion of tumour cell 54. Interestingly, it also demonstrated that 89% of patients with the expressed Δ16HER‐2 were locally disseminated node‐positive breast cancer, indicating that more attention should be put on this subtype 56.
p95HER‐2 is hyperactive, and was described as a truncated form of HER‐2 lacking the antibody's binding region. It has been demonstrated to be associated with cancer progression, metastasis, poor prognosis and disease‐free survival when compared with patients expressing the wild full‐length HER‐2 65, 68. Additionally, patients with breast cancer harbouring the expression of p95HER‐2 exhibit less response to trastuzumab compared to patients without p95HER‐2. The lack of outer‐cell attachment domain containing the binding site for trastuzumab results in the failure of drug binding. Besides, actively signalling protein generated by ectopic expression of p95HER‐2, also promotes trastuzumab resistance that has been demonstrated in preclinical and clinical studies 95.
The therapy methods to these HER‐2 mutations
Recently, more researches about the HER‐2 mutations have been investigated. Most of the mutations were described in patients without HER‐2 overexpression or amplification. These have brought us a profound change to the traditional targeted therapy for patients with mutant HER‐2. Studies about HER‐2 mutations, both in vitro and in vivo, have proved that the applicability of anti‐HER‐2 agents, such as inhibitor combinations, lapatinib plus trastuzumab, or afatinib plus rapamycin, are the most effective therapy in HER‐2‐mutant cancers 106, 107. Patients with HER‐2‐mutant NSCLC were also reported to have achieved disease control with anti‐HER‐2 therapy 108, 109. Similar outcome of HER‐2‐targeted therapy has also been achieved in breast cancer patients with HER‐2‐mutantions 103, 110. Notably, in Bose's research, all HER‐2 mutations including mutation L755S which is resistant to lapatinib, exhibited sensitivity to the irreversible HER‐2 inhibitor, neratinib 23. All these findings validate that patients with HER‐2‐mutant could benefit from existing HER‐2 targeted therapy, particularly irreversible inhibitors, such as neratinib. More preclinical and clinical trials should be designed to investigate the application of HER‐2 targeted therapy in HER‐2 mutation positive patients. To date, a prospective, multicenter clinical trial is being launched to screen patients with metastatic breast cancer for HER‐2 mutation and investigate the clinical outcome of HER‐2 targeted therapy (NCT01670877).
Previous researches have revealed that tumours contain p95HER‐2 are resistant to trastuzumab, but still sensitive to the TKI, such as lapatinib 53. In addition, some studies also reported that these TKIs could also provide an effective solution for the patients expressing mutant Δ16HER‐2, suggesting that the TKI may be an alternative therapy for HER‐2 mutations 56. Furthermore, another study indicated that p95HER‐2 could be a possible biomarker to evaluate the efficacy of therapeutic regimens including lapatinib and chemotherapy, and overcome the clinical failure of trastuzumab monotherapy. The effect of lapatinib, as single agents or in combination with other drugs, may be equal in patients regardless of the p95HER‐2 expression 70. Interestingly, a study revealed that tumours expressing these p95HER‐2 fragments, also respond to trastuzumab plus chemotherapy, advocating that p95HER‐2 is also a predictive biomarker for the patients treated with trastuzumab and chemotherapy 53. However, no inhibitors were directly targeted against p95HER‐2. To improve the outcome of patients harbouring these two variants, new researches are still on the way.
Conclusion
During the past decades, the application of trastuzumab, which targeted against HER‐2, has significantly improved the outcome and prognosis of HER‐2‐overexpressing breast cancer. However, despite the clinical success, the resistance of HER‐2 targeted agents occurred. Primary activating mutations and acquired secondary mutations were detected to play a critical role in breast cancer progression and drug resistance, revealing a sophisticated challenge for the effective treatment of HER‐2 targeted therapy. Lots of preclinical data suggest that the combination of multi‐points HER‐2 targeted therapy may break the drug resistance. This will require an ‘individualized diagnosis and treatment’ based on the detailed molecular analysis of tumours both before and after progression on primary HER‐2 targeted therapy. Future works are still needed to evaluate the role of altered HER‐2 as future prognostic and predictive factors, as well as potential therapeutic targets, and providing ‘an individualized strategy’ for patients with HER‐2 mutant.
Conflicts of interest
The authors do not have any financial interests to publish this article.
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (no. 81272252 and 81470357), a Foundation for Clinical Medicine Science and Technology Special Project of the Jiangsu Province, China (no. BL2014071 to X. G).
Zijia Sun and Yaqin Shi should be regarded as joint first authors for their equal contributions.
References
- 1. Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014; 2014: 852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brandt‐Rauf PW, Pincus MR, Carney WP. The c‐erbB‐2 protein in oncogenesis: molecular structure to molecular epidemiology. Crit Rev Oncog. 1994; 5: 313–29. [DOI] [PubMed] [Google Scholar]
- 3. Telesco SE, Radhakrishnan R. Atomistic insights into regulatory mechanisms of the HER2 tyrosine kinase domain: a molecular dynamics study. Biophys J. 2009; 96: 2321–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010; 141: 1117–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stephens P, Hunter C, Bignell G, et al Intragenic ERBB2 kinase mutations in tumours. Nature. 2004; 431: 525–6. [DOI] [PubMed] [Google Scholar]
- 6. Yan M, Parker BA, Schwab R, et al HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev. 2014; 40: 770–80. [DOI] [PubMed] [Google Scholar]
- 7. Lee JW, Soung YH, Seo SH, et al Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin Cancer Res. 2006; 12: 57–61. [DOI] [PubMed] [Google Scholar]
- 8. Sasaki H, Shimizu S, Endo K, et al EGFR and erbB2 mutation status in Japanese lung cancer patients. Int J Cancer. 2006; 118: 180–4. [DOI] [PubMed] [Google Scholar]
- 9. Ding L, Getz G, Wheeler DA, et al Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455: 1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minami Y, Shimamura T, Shah K, et al The major lung cancer‐derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI‐272. Oncogene. 2007; 26: 5023–7. [DOI] [PubMed] [Google Scholar]
- 11. Gilmer TM, Cable L, Alligood K, et al Impact of common epidermal growth factor receptor and HER2 variants on receptor activity and inhibition by lapatinib. Cancer Res. 2008; 68: 571–9. [DOI] [PubMed] [Google Scholar]
- 12. Suzuki M, Shiraishi K, Yoshida A, et al HER2 gene mutations in non‐small cell lung carcinomas: concurrence with her2 gene amplification and her2 protein expression and phosphorylation. Lung Cancer. 2015; 87: 14–22. [DOI] [PubMed] [Google Scholar]
- 13. De Greve J, Teugels E, Geers C, et al Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer. 2012; 76: 123–7. [DOI] [PubMed] [Google Scholar]
- 14. Del Campo J, Hitt R, Sebastian P, et al Effects of lapatinib monotherapy: results of a randomised phase II study in therapy‐naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer. 2011; 105: 618–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bang Y‐J, Van Cutsem E, Feyereislova A, et al Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet. 2010; 376: 687–97. [DOI] [PubMed] [Google Scholar]
- 16. Lin W‐L, Kuo W‐H, Chen F‐L, et al Identification of the coexisting HER2 gene amplification and novel mutations in the HER2 protein‐overexpressed mucinous epithelial ovarian cancer. Ann Surg Oncol. 2011; 18: 2388–94. [DOI] [PubMed] [Google Scholar]
- 17. Lassus H, Sihto H, Leminen A, et al Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J Mol Med. 2006; 84: 671–81. [DOI] [PubMed] [Google Scholar]
- 18. Bekaii‐Saab T, Williams N, Plass C, et al A novel mutation in the tyrosine kinase domain of ERBB2 in hepatocellular carcinoma. BMC Cancer. 2006; 6: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tschui J, Vassella E, Bandi N, et al Morphological and molecular characteristics of HER2 amplified urothelial bladder cancer. Virchows Arch. 2015; 466: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kubo T, Kuroda Y, Shimizu H, et al Resequencing and copy number analysis of the human tyrosine kinase gene family in poorly differentiated gastric cancer. Carcinogenesis. 2009; 30: bgp206. [DOI] [PubMed] [Google Scholar]
- 21. Cohen EE, Lingen MW, Martin LE, et al Response of some head and neck cancers to epidermal growth factor receptor tyrosine kinase inhibitors may be linked to mutation of ERBB2 rather than EGFR. Clin Cancer Res. 2005; 11: 8105–8. [DOI] [PubMed] [Google Scholar]
- 22. Arcila ME, Chaft JE, Nafa K, et al Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res. 2012; 18: 4910–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bose R, Kavuri SM, Searleman AC, et al Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013; 3: 224–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matthew J. Mutational analysis of breast cancer: Guiding personalized treatments. Breast 2013; 22: S19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee JW, Soung YH, Kim SY, et al ERBB2 kinase domain mutation in the lung squamous cell carcinoma. Cancer Lett. 2006; 237: 89–94. [DOI] [PubMed] [Google Scholar]
- 26. Tomizawa K, Suda K, Onozato R, et al Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer. 2011; 74: 139–44. [DOI] [PubMed] [Google Scholar]
- 27. Kelly RJ, Carter CA, Giaccone G. HER2 mutations in non–small‐cell lung cancer can be continually targeted. J Clin Oncol. 2012; 30: 3318–9. [DOI] [PubMed] [Google Scholar]
- 28. Shah SP, Morin RD, Khattra J, et al Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009; 461: 809–13. [DOI] [PubMed] [Google Scholar]
- 29. Shah SP, Roth A, Goya R, et al The clonal and mutational evolution spectrum of primary triple‐negative breast cancers. Nature. 2012; 486: 395–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Buttitta F, Barassi F, Fresu G, et al Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer. 2006; 119: 2586–91. [DOI] [PubMed] [Google Scholar]
- 31. Shigematsu H, Takahashi T, Nomura M, et al Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005; 65: 1642–6. [DOI] [PubMed] [Google Scholar]
- 32. Shimamura T, Ji H, Minami Y, et al Non‐small‐cell lung cancer and Ba/F3 transformed cells harboring the ERBB2 G776insV_G/C mutation are sensitive to the dual‐specific epidermal growth factor receptor and ERBB2 inhibitor HKI‐272. Cancer Res. 2006; 66: 6487–91. [DOI] [PubMed] [Google Scholar]
- 33. Lynch TJ, Bell DW, Sordella R, et al Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small‐cell lung cancer to gefitinib. N Engl J Med. 2004; 350: 2129–39. [DOI] [PubMed] [Google Scholar]
- 34. Mitsudomi T, Morita S, Yatabe Y, et al Gefitinib versus cisplatin plus docetaxel in patients with non‐small‐cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010; 11: 121–8. [DOI] [PubMed] [Google Scholar]
- 35. Pauletti G, Godolphin W, Press MF, et al Detection and quantitation of HER‐2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene. 1996; 13: 63–72. [PubMed] [Google Scholar]
- 36. Kamoshida S. Challenges of immunohistochemistry for individualized cancer chemotherapy. Rinsho Byori. 2014; 62: 710–8. [PubMed] [Google Scholar]
- 37. Perez EA, Romond EH, Suman VJ, et al Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2–positive breast cancer: planned joint analysis of overall survival from NSABP B‐31 and NCCTG N9831. J Clin Oncol. 2014; 32: 3744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slamon DJ, Leyland‐Jones B, Shak S, et al Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344: 783–92. [DOI] [PubMed] [Google Scholar]
- 39. Romond EH, Perez EA, Bryant J, et al Trastuzumab plus adjuvant chemotherapy for operable HER2‐positive breast cancer. N Engl J Med. 2005; 353: 1673–84. [DOI] [PubMed] [Google Scholar]
- 40. Baselga J, Cortés J, Im S‐A, et al Biomarker analyses in CLEOPATRA: a phase III, placebo‐controlled study of pertuzumab in human epidermal growth factor receptor 2–positive, first‐line metastatic breast cancer. J Clin Oncol. 2014; 32: 3753–61. [DOI] [PubMed] [Google Scholar]
- 41. Weigelt B, Reis‐Filho JS. Activating mutations in HER2: neu opportunities and neu challenges. Cancer Discov. 2013; 3: 145–7. [DOI] [PubMed] [Google Scholar]
- 42. Boulbes DR, Arold ST, Chauhan GB, et al HER family kinase domain mutations promote tumor progression and can predict response to treatment in human breast cancer. Mol Oncol. 2014; 9: 586–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ross JS, Wang K, Gay LM, et al A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin Cancer Res. 2014; 20: 68–75. [DOI] [PubMed] [Google Scholar]
- 44. Wang SZE, Narasanna A, Perez‐Torres M, et al HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell. 2006; 10: 25–38. [DOI] [PubMed] [Google Scholar]
- 45. Fang Y, Jiang Y, Wang X, et al Somatic mutations of the HER2 in metastatic breast cancer. Tumour Biol. 2014; 35: 11851–4. [DOI] [PubMed] [Google Scholar]
- 46. Herter‐Sprie GS, Greulich H, Wong K‐K. Activating mutations in ERBB2 and their impact on diagnostics and treatment. Front Oncol. 2013; 3: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Greulich H, Kaplan B, Mertins P, et al Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci USA. 2012; 109: 14476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kan ZY, Jaiswal BS, Stinson J, et al Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010; 466: 869–U103. [DOI] [PubMed] [Google Scholar]
- 49. Vornicova O, Hershkovitz D, Yablonski‐Peretz T, et al Treatment of metastatic extramammary Paget's disease associated with adnexal adenocarcinoma, with anti‐HER2 drugs based on genomic alteration ERBB2 S310F. Oncologist. 2014; 19: 1006–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Serra V, Vivancos A, Puente XS, et al Clinical response to a lapatinib‐based therapy for a Li‐Fraumeni syndrome patient with a novel HER2V659E mutation. Cancer Discov. 2013; 3: 1238–44. [DOI] [PubMed] [Google Scholar]
- 52. Yamamoto H, Higasa K, Sakaguchi M, et al Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J Natl Cancer Inst. 2014; 106: djt338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Parra‐Palau JL, Morancho B, Peg V, et al Effect of p95HER2/611CTF on the response to trastuzumab and chemotherapy. J Natl Cancer Inst. 2014; 106: dju291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mitra D, Brumlik MJ, Okamgba SU, et al An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009; 8: 2152–62. [DOI] [PubMed] [Google Scholar]
- 55. Barić M, Kulić A, Sirotković‐Skerlev M, et al Circulating her‐2/neu extracellular domain in breast cancer patients‐correlation with prognosis and clinicopathological parameters including steroid receptor, her‐2/neu receptor coexpression. Pathol Oncol Res. 2014; 21: 1–7. [DOI] [PubMed] [Google Scholar]
- 56. Jackson C, Browell D, Gautrey H, et al Clinical significance of HER‐2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol. 2013; 2013: 973584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev. 2008; 34: 378–90. [DOI] [PubMed] [Google Scholar]
- 58. Fang L, Barekati Z, Zhang B, et al Targeted therapy in breast cancer: what's new. Swiss Med Wkly. 2011; 141: w13231. [DOI] [PubMed] [Google Scholar]
- 59. De Mattos‐Arruda L, Cortes J. Advances in first‐line treatment for patients with HER‐2+ metastatic breast cancer. Oncologist. 2012; 17: 631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Thery J‐C, Spano J‐P, Azria D, et al Resistance to human epidermal growth factor receptor type 2‐targeted therapies. Eur J Cancer. 2014; 50: 892–901. [DOI] [PubMed] [Google Scholar]
- 61. Marchini C, Gabrielli F, Iezzi M, et al The human splice variant Delta16HER2 induces rapid tumor onset in a reporter transgenic mouse. PLoS ONE. 2011; 6: e18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Castiglioni F, Tagliabue E, Campiglio M, et al Role of exon‐16‐deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006; 13: 221–32. [DOI] [PubMed] [Google Scholar]
- 63. Parra‐Palau JL, Pedersen K, Peg V, et al A major role of p95/611‐CTF, a carboxy‐terminal fragment of HER2, in the down‐modulation of the estrogen receptor in HER2‐positive breast cancers. Cancer Res. 2010; 70: 8537–46. [DOI] [PubMed] [Google Scholar]
- 64. Sperinde J, Jin X, Banerjee J, et al Quantitation of p95HER2 in paraffin sections by using a p95‐specific antibody and correlation with outcome in a cohort of trastuzumab‐treated breast cancer patients. Clin Cancer Res. 2010; 16: 4226–35. [DOI] [PubMed] [Google Scholar]
- 65. Arribas J, Baselga J, Pedersen K, et al p95HER2 and breast cancer. Cancer Res. 2011; 71: 1515–9. [DOI] [PubMed] [Google Scholar]
- 66. Tural D, Akar E, Mutlu H, et al P95 HER2 fragments and breast cancer outcome. Expert Rev Anticancer Ther. 2014; 14: 1089–96. [DOI] [PubMed] [Google Scholar]
- 67. Pedersen K, Angelini P‐D, Laos S, et al A naturally occurring HER2 carboxy‐terminal fragment promotes mammary tumor growth and metastasis. Mol Cell Biol. 2009; 29: 3319–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sáez R, Molina MA, Ramsey EE, et al p95HER‐2 predicts worse outcome in patients with HER‐2‐positive breast cancer. Clin Cancer Res. 2006; 12: 424–31. [DOI] [PubMed] [Google Scholar]
- 69. Duchnowska R, Sperinde J, Chenna A, et al Quantitative measurements of tumoral p95HER2 protein expression in metastatic breast cancer patients treated with trastuzumab: independent validation of the p95HER2 clinical cutoff. Clin Cancer Res. 2014; 20: 2805–13. [DOI] [PubMed] [Google Scholar]
- 70. Andrade de Mello R, de Vasconcelos A, Ribeiro RA, et al Insight into p95HER2 in breast cancer: molecular mechanisms and targeted therapies. Recent Pat DNA Gene Seq. 2012; 6: 56–63. [DOI] [PubMed] [Google Scholar]
- 71. Azios NG, Romero FJ, Denton MC, et al Expression of herstatin, an autoinhibitor of HER‐2/neu, inhibits transactivation of HER‐3 by HER‐2 and blocks EGF activation of the EGF receptor. Oncogene. 2001; 20: 5199–209. [DOI] [PubMed] [Google Scholar]
- 72. AbdRaboh NR, Shehata HH, Ahmed MB, et al HER1 R497K and HER2 I655V polymorphisms are linked to development of breast cancer. Dis Markers. 2013; 34: 407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Millikan RC, Hummer AJ, Wolff MS, et al HER2 codon 655 polymorphism and breast cancer: results from kin‐cohort and case–control analyses. Breast Cancer Res Treat. 2005; 89: 309–12. [DOI] [PubMed] [Google Scholar]
- 74. Xie D, Shu X‐O, Deng Z, et al Population‐based, case‐control study of HER2 genetic polymorphism and breast cancer risk. J Natl Cancer Inst. 2000; 92: 412–7. [DOI] [PubMed] [Google Scholar]
- 75. Lu S, Wang Z, Liu H, et al HER2 Ile655Val polymorphism contributes to breast cancer risk: evidence from 27 case‐control studies. Breast Cancer Res Treat. 2010; 124: 771–8. [DOI] [PubMed] [Google Scholar]
- 76. Ma Y, Yang J, Zhang P, et al Lack of association between HER2 codon 655 polymorphism and breast cancer susceptibility: meta‐analysis of 22 studies involving 19,341 subjects. Breast Cancer Res Treat. 2011; 125: 237–41. [DOI] [PubMed] [Google Scholar]
- 77. Chen W, Yang H, Tang WR, et al Updated meta‐analysis on HER2 polymorphisms and risk of breast cancer: evidence from 32 studies. Asian Pac J Cancer Prev. 2014; 15: 9643–7. [DOI] [PubMed] [Google Scholar]
- 78. Rutter JL, Chatterjee N, Wacholder S, et al The HER2 I655V polymorphism and breast cancer risk in ashkenazim. Epidemiology. 2003; 14: 694–700. [DOI] [PubMed] [Google Scholar]
- 79. Millikan R, Eaton A, Worley K, et al HER2 codon 655 polymorphism and risk of breast cancer in African Americans and whites. Breast Cancer Res Treat. 2003; 79: 355–64. [DOI] [PubMed] [Google Scholar]
- 80. Kruszyna L, Lianeri M, Roszak A, et al HER2 codon 655 polymorphism is associated with advanced uterine cervical carcinoma. Clin Biochem. 2010; 43: 545–8. [DOI] [PubMed] [Google Scholar]
- 81. Frank B, Hemminki K, Wirtenberger M, et al The rare ERBB2 variant Ile654Val is associated with an increased familial breast cancer risk. Carcinogenesis. 2005; 26: 643–7. [DOI] [PubMed] [Google Scholar]
- 82. Tai W, Mahato R, Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J Controlled Release. 2010; 146: 264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kümler I, Tuxen MK, Nielsen DL. A systematic review of dual targeting in HER2‐positive breast cancer. Cancer Treat Rev. 2014; 40: 259–70. [DOI] [PubMed] [Google Scholar]
- 84. GrausPorta D, Beerli RR, Daly JM, et al ErbB‐2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997; 16: 1647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Alimandi M, Romano A, Curia MC, et al Cooperative signaling of Erbb3 and Erbb2 in neoplastic transformation and human mammary carcinomas. Oncogene. 1995; 10: 1813–21. [PubMed] [Google Scholar]
- 86. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001; 2: 127–37. [DOI] [PubMed] [Google Scholar]
- 87. Holbro T, Beerli RR, Maurer F, et al The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003; 100: 8933–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Citri A, Yarden Y. EGF‐ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006; 7: 505–16. [DOI] [PubMed] [Google Scholar]
- 89. Slattery ML, John EM, Stern MC, et al Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: the Breast Cancer Health Disparities Study. Breast Cancer Res Treat. 2013; 140: 587–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Slamon DJ, Clark GM, Wong SG, et al Human‐breast cancer ‐ correlation of relapse and survival with amplification of the her‐2 neu oncogene. Science. 1987; 235: 177–82. [DOI] [PubMed] [Google Scholar]
- 91. Nielsen DL, Andersson M, Kamby C. HER2‐targeted therapy in breast cancer. Monoclonal antibodies and tyrosine kinase inhibitors. Cancer Treat Rev. 2009; 35: 121–36. [DOI] [PubMed] [Google Scholar]
- 92. Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008; 14: 6730–4. [DOI] [PubMed] [Google Scholar]
- 93. Spector NL, Blackwell KL. Understanding the mechanisms behind trastuzumab therapy for human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol. 2009; 27: 5838–47. [DOI] [PubMed] [Google Scholar]
- 94. Tortora G. Mechanisms of resistance to HER2 target therapy. J Natl Cancer Inst Monogr. 2011; 2011: 95–8. [DOI] [PubMed] [Google Scholar]
- 95. Rexer BN, Arteaga CL. Intrinsic and acquired resistance to HER2‐targeted therapies in HER2 gene‐amplified breast cancer: mechanisms and clinical implications. Crit Rev Oncog. 2012; 17: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wu JY, Yu CJ, Chang YC, et al Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non‐small cell lung cancer. Clin Cancer Res. 2011; 17: 3812–21. [DOI] [PubMed] [Google Scholar]
- 97. Rexer BN, Ghosh R, Narasanna A, et al Human breast cancer cells harboring a gatekeeper T798M mutation in HER2 overexpress EGFR ligands and are sensitive to dual inhibition of EGFR and HER2. Clin Cancer Res. 2013; 19: 5390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pao W, Miller VA, Politi KA, et al Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005; 2: 225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Shah NP, Nicoll JM, Nagar B, et al Multiple BCR‐ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002; 2: 117–25. [DOI] [PubMed] [Google Scholar]
- 100. Antonescu CR, Besmer P, Guo TH, et al Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005; 11: 4182–90. [DOI] [PubMed] [Google Scholar]
- 101. Trowe T, Boukouvala S, Calkins K, et al EXEL‐7647 inhibits mutant forms of ErbB2 associated with lapatinib resistance and neoplastic transformation. Clin Cancer Res. 2008; 14: 2465–75. [DOI] [PubMed] [Google Scholar]
- 102. Li G, Wang X, Hibshoosh H, et al Modulation of ErbB2 blockade in ErbB2‐positive cancers: the role of ErbB2 mutations and PHLDA1. PLoS ONE. 2014; 9: e106349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kancha RK, von Bubnoff N, Bartosch N, et al Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS ONE. 2011; 6: e26760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Piechocki MP, Yoo GH, Dibbley SK, et al Breast cancer expressing the activated HER2/neu is sensitive to gefitinib in vitro and in vivo and acquires resistance through a novel point mutation in the HER2/neu. Cancer Res. 2007; 67: 6825–43. [DOI] [PubMed] [Google Scholar]
- 105. Wang SE, Yu Y, Criswell TL, et al Oncogenic mutations regulate tumor microenvironment through induction of growth factors and angiogenic mediators. Oncogene. 2010; 29: 3335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Falchook GS, Janku F, Tsao AS, et al Non‐small‐cell lung cancer with HER2 exon 20 mutation regression with dual HER2 inhibition and anti‐VEGF combination treatment. J Thorac Oncol. 2013; 8: E19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Perera SA, Li D, Shimamura T, et al HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci USA. 2009; 106: 474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mazieres J, Peters S, Lepage B, et al Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013; 31: 1997–U307. [DOI] [PubMed] [Google Scholar]
- 109. Cappuzzo F, Bemis L, Varella‐Garcia M. HER2 mutation and response to trastuzumab therapy in non‐small‐cell lung cancer. N Engl J Med. 2006; 354: 2619–21. [DOI] [PubMed] [Google Scholar]
- 110. Endo Y, Dong Y, Yoshimoto N, et al HER2 mutation status in Japanese HER2‐negative breast cancer patients. Jpn J Clin Oncol. 2014; 44: 619–23. [DOI] [PubMed] [Google Scholar]