

Abstract
Nanomedicine, the medical applications of devices based on nanotechnology, promises an endless range of applications from biomedical imaging to drug and gene delivery. The size range of the nanomaterials is strictly defined as 1–100 nm, although many marketed nanomedicines are in the submicron range of 100–1000 nm. The major advantages of using nanomaterials as a carrier for anticancer agents are the possibility of targeted delivery to the tumor; their physical properties such as optical and magnetic properties, which can be exploited for developing contrast agents for tumor imaging; their ability to hold thousands of molecules of a drug and deliver at the required site and also the ability to overcome solubility and stability issues. Currently, there are several nanotechnology-enabled diagnostic and therapeutic agents undergoing clinical trials and a few already approved by Food and Drug Administration. Targeted delivery of anticancer agents is achieved by exploiting a unique characteristic of the rapidly dividing tumor cells called “the enhanced permeability and retention effect.” Nanoparticles with mean diameter between 100 and 200 nm or even above 200 nm have also been reported to be taken up by tumor cells via the enhanced permeability and retention effect. In addition to this passive targeting based on size, the nanoparticle surface may be modified with a variety of carefully chosen ligands that would interact with specific receptors on the surface of the tumor cells, thus imparting additional specificity for active targeting. Regional release of a drug contained in a nanoparticulate system by the application of external stimuli such as hyperthermia to a thermosensitive device is another innovative strategy for targeted delivery. Nanoparticles protect the enclosed drug from rapid elimination from the body, keep them in circulation for prolonged periods and often evade expulsion by the efflux pump mechanisms, which also leads to avoidance of development of resistance. This review focuses on the science and technology of Food and Drug Administration–approved cancer nanomedicines such as Abraxane, Doxil, DaunoXome and those drug-delivery systems that have reached an advanced stage of clinical development utilizing liposomes, albumin nanospheres, thermosensitive devices and gold nanoshells.
Keywords: Cancer nanomedicines, approved products and in clinical development, multidrug resistance, targeted delivery, stimuli-sensitive release, liposomes, albumin-bound nanoparticles, gold nanoshells
Nanomaterials, with their unique size-dependent physical and chemical properties, have shown great promise as carriers of chemotherapeutic agents, vaccines and contrast agents for diagnostic imaging. One of the drawbacks of conventional chemotherapeutic agents for the treatment of cancer is their inability to deliver the drug in adequate quantities to the tumor site without undesirable side effects. This limitation is overcome to a great extent by enclosing or binding the anticancer agent to nanomaterials, which have already shown their potential in targeted delivery to the tumor. It was predicted by the National Science Foundation in early 2001 that nanotechnology will help prolong life, improve its quality and extend human physical capabilities and that half of pharmaceutical production, over 180 billion per year, would be dependent on nanotechnology. Nanomedicine is defined by the European Science Foundation as “the science and technology of diagnosing, treating and preventing diseases and traumatic injury, of relieving pain and of preserving and improving human health using molecular tools and molecular knowledge of the human body.”1,2 The National Institute of Health (NIH) refers to nanomedicine as a “highly specific medical intervention at the molecular scale for curing diseases or repairing damaged tissues such as bone, muscle or nerve.”3 According to the National Cancer Institute, the field of nanomedicine grew exponentially from 2000 to 2010, and the agency predicts that the United States workforce in nanomedicine will reach 2 million by 2015. According to a recent report from business communication company research, the global nanomedicine sector, which was 53 billion in 2009, is projected to surpass 100 billion in 2014.4 The nanomedicine market is divided into various therapeutic areas such as anticancer, central nervous system (CNS) product, anti-infective, anti-inflammatory, cardiovascular and others. Of these, the anticancer segment is projected to dominate the nanomedicine market (nanotechnology market forecast to 2013). The overall anticancer nanomedicine market is predicted to grow from US$5.5 billion in 2011 to US$12.7 billion by 2016.5 A limited number of nanopharmaceuticals, including anticancer drug-delivery systems and contrast agents for tumor imaging, are already in the market, and many have reached an advanced stage of development. Therefore, the physicians and pharmacists will sooner or later encounter these products, and this review is aimed at enhancing their knowledge of the science and technology of these products.
The prefix “nano” originally comes from the Greek word “nanos,” meaning dwarf. In scientific usage, “nano” means one-billionth of a meter. One nanometer is the length of 10 hydrogen atoms placed side by side. Cesium, the largest known atom has a diameter of 0.53 nm. The size of some materials in the nanoworld are as follows: DNA, 1–2 nm diameter; virus, 3–50 nm; insulin molecule, 3 nm and cytochrome, 4 nm. One million fullerenes, the smallest soccer ball made from 60 carbon atoms in a mix of hexa- and pentagonal structures, can fit into a grain of rice.
Why a big interest in such small things?
The size of materials does matter: materials in the nanosize range can exhibit very different behavior compared to their macro or bulk material. The electrical conductivity, color, strength, reactivity and melting point are subject to change. At the nanoscale, carbon is stronger than steel and six times lighter. The white and opaque zinc oxide becomes transparent, thus cosmetically appealing, and a gold colloid less than 100 nm appears intensely red. Nanoparticles (NPs) also have relatively huge surface areas per unit mass (above 1000 m2/g), a property that leads to some of their unexpected reactivity compared to their macro counterpart. One gram of graphene, consisting of single-layer carbon atoms in a honeycomb structure, has a surface area comparable to that of a football field. Most of the atoms of the nanoscale material will reside on the surface, hence the increased reactivity of nanomaterials.
Nanomedicine, the medical applications of devices based on nanotechnology, promises an endless range of applications from biomedical imaging to drug and gene delivery. Nanomedicines are produced by manipulation of atoms and molecules that can range in size from 1 to 100 nm. This size range, however, as pointed out by Bawa,6 is not critical from a drug formulation or delivery perspective. Enhanced solubility, improved bioavailability and reduced toxicity are important in drug formulation and delivery. This may be achieved in size range much greater than 100 nm. It is to be noted that marketed liposomal and albumin-bound nanoparticulate anticancer drug products are within the submicron size of 100–1000 nm.7 The nanomedicine, “Abraxane” (nanospheres of paclitaxel–albumin), has a particle size of 130 nm and lipoplatin (liposomal cisplatin) of size 110 nm. Nanoemulsion droplets range in size from 30 to 300 nm, solid lipid NPs between 80 and 300 nm and RNA nanotechnology–based nanomedicines from 20 to 40 nm.8,9 The size range of 1–100 nm is critical when there is a special interest in the color of light emitted from NPs as in nanophotonics (or nano-optics, the study of behavior of light on nanomaterials). The color of light emitted depends on particle size. The US National nanotechnology initiative refers to nanotechnology as the study of structures that are about 100 nm or less. The Food and Drug Administration (FDA) continues to use a similar definition.
The submicron size particles differ from their bulk counterpart due to the increased surface area. As the particle gets smaller and smaller, their surface area to volume ratio increases dramatically. They are more reactive and more soluble in water. In contrast to the macroscopic materials, the nanomaterials have tunable optical, electronic, magnetic and biologic properties. They can be engineered to have different sizes, shapes, chemical compositions, surface chemical properties and solid, hollow or porous structures. These properties are being exploited into new drug-delivery vehicles to deliver chemotherapeutic agents, recombinant proteins and vaccines, contrast agents and diagnostic devices. Nanomedicines have enormous potential in addressing some of the shortcomings of conventional drugs that could not be formulated effectively because of poor solubility, toxicity issues, poor bioavailability or lack of target specificity.6 In addition, NPs protect the enclosed drug from rapid elimination from the body, keep them in circulation for prolonged periods and often evade expulsion by the efflux pump mechanisms, which also leads to avoidance of development of resistance to chemotherapeutic agents.
NPs to overcome multidrug resistance
Multidrug resistance (MDR) is one of the biggest challenges in cancer chemotherapy. Development of cancer drug resistance is attributed to inefficient drug delivery to tumor cells, partly due to inefficient targeting and the removal of the drug from tumor cells by the efflux pump, P-glycoprotein (P-gp). This membrane transporter is known to be overexpressed in tumors compared to drug-sensitive parent cell lines. Approximately 50% of the anticancer drugs used clinically today are substrates of P-gp. Therefore, a plausible way to reduce MDR is targeted delivery of the drug to the tumor and reduction of MDR-based drug efflux. It has been suggested that NPs may be able to circumvent P-gp-mediated resistance by partially bypassing the efflux pump as they are internalized by endocytosis.10,11 For example, liposomes have been shown to inhibit P-gp efflux.12,13 The proposed mechanism included bypassing P-gp through an endocytosis pathway and direct interaction with P-gp.14 Polymer NPs and lipid NPs also have shown to overcome MDR.15–18 Even though the mechanisms to overcome MDR using the NPs are not fully understood, improved anticancer efficacy has been confirmed in vitro and in vivo.
Pathophysiological differences between normal and cancerous tissue: strategies for drug delivery
An imminent pathophysiological change that occurs in cancerous tissue in comparison to normal tissues is the display of uncontrolled growth which leads to the destruction of adjacent structures through malignant cell intrusion into nearby cells. As the disease process progresses, it leads to invasion and spread to other body locations and or sites. Facilitation of transformation of normal cells to cancer cells are mediated through genetic alterations that regulate cell growth and differentiation.
A morphological change in blood vessels due to vascular reorganization has been observed in primary tumors.19–22 These changes are evident in the vasculature (blood vessels) and lymph channel (lymph nodes (LNs)) in the form of restructurings prior to the appearance of cancer cells.23–26 The key blood vessels in such LNs that are remodeled are high endothelial venules (HEV), which have been recognized to play a role in metastasis.27–29 This process is called lymphangiogenesis. In contrast to angiogenesis, which occurs in most blood vessels, lymphangiogenesis can be induced by interstitial fluid channeling. In this aspect, the alteration of lymph channels may facilitate the migration of cancer cells.30–33 Angiogenesis is a process whereby new vascular network is established. The orderly fashion of steps involved in the production of blood vessels through angiogenesis is considered a “normal” occurrence in most tissues needing repair due to inflammation and/or for wound healing. Angiogenesis and its role in malignancy/cancer are vital in initiating the outgrowth of new blood vessels from existing vessels. This is necessary for tumor growth beyond 1–2 mm3.34–36 Additionally, vessels in the tumor vicinity become abnormally tortuous.37 This abnormality extends beyond tumor boundaries and precedes vascular sprouting. Abnormal vascular tortuosity is associated not only with malignancy in animal models but also with a range of cancers in human patients.38,39 The etiology may be related to growth factor–related changes to the vessel wall, including alteration of the basement membrane, loss of pericytes and smooth muscle, and proliferation of endothelial cells.40 In animals, cancer-associated vessel tortuosity abnormalities are known to resolve during anti-angiogenic treatment.41–44 Abnormalities in vessel tortuosity appear early during tumor development and spread beyond the confines of tumor margins.45
Angiogenesis is crucial to the proliferation, as well as metastatic spread of, cancer cells. It is regulated by angiogenic activators and inhibitors, and their levels reflect the aggressiveness of tumor cells.44 Neovascularization reduces a tumor’s accessibility to chemotherapeutic drugs. A distinctive pathophysiological feature detected in normal tissues or organs is its capability for extensive angiogenesis, which leads to hypervasculature and consequently defective vascular architecture, impaired lymphatic drainage and recovery system. These changes are accompanied by an increase of permeability mediators such as bradykinin, nitric oxide and prostaglandins. A phenomenon observed to be universal in solid tumors is known as the enhanced permeability and retention (EPR) effect for lipid and macromolecular agents. Its principal mechanism is to enhance vascular permeability to maintain adequate supply of nutrients and oxygen for tumor growth. This offers huge opportunities for more selective targeting of lipid- or polymer-conjugated anticancer drugs, such as SMANCS (a conjugate of neocarzinostatin and poly (styrene-co-maleic acid) and PK-1(PK1 is a synthetic N-(2-hydroxypropyl) methacrylamide copolymer-doxorubicin (dox) conjugate) to the tumor.46,47 Its basic characteristic effects are in areas for the modulation and improvement of the delivery of macromolecular drugs to the tumor.48,49
The “EPR effect” and passage of NPs to the tumor
NPs selectively accumulate in tumor tissues by a purely physical phenomenon called EPR effect. At the early stages of tumor development, the lack of blood vessels results in the dependence of the growing tumor on its surrounding tissues for nutrients such as glucose and oxygen. Even then, the tumor core does not receive enough nutrients. This situation stimulates growth of new blood vessels that can carry the required oxygen and nutrients to the tumor for its rapid growth and replication. Because of the rapid growth of the blood capillaries, a process called angiogenesis, the new blood capillaries have a defective vasculature with large number of wide openings between the epithelial cells than healthy blood vessels and lack of the normal basement membrane. The gap size varies depending on the stage of development of the tumor and generally range from a few hundred nanometers to a few microns. In contrast, the pores of normal blood vessels are only 2–6 nm. In addition, there are no functional lymphatic vessels in solid tumors, which would normally be involved in transport of macromolecules. Another hallmark of solid tumors is the abnormal metabolic environment such as hypoxia and acidosis.50 The wider openings will permit passage of NPs from the blood into surrounding tumor tissues (extravasation). This results in the accumulation of NPs in the tumor as well as in the non-functional lymphatics leading ultimately to retention of the NPs within the tumor. This process, called EPR effect, explains the passive targeting of NPs to the tumor, which depends only on the size of the particles. It has been reported that drug carriers loaded with anticancer drugs of an average size of 200 nm or less is ideal for the EPR effect.51
Numerous nanoparticulate drug-delivery systems are developed to deliver their cargo to the tumor site by passive targeting. Additional selectivity in targeting (active targeting) can be obtained by suitable modification of the surface of NPs such as attachment of a ligand (e.g. a monoclonal antibody, folic acid or transferrin), that would specifically seek and attach to its receptor in the tumor tissue.
How is the particle size in the nanometer range measured?
The nano range is strictly defined as 1–100 nm. However, several marketed nanomedicines are in the range of 100–1000 nm, also called the submicron range. Measurement of particle size is a key element in the development of products containing nanosize-active pharmaceutical ingredient (API). Microscopic methods such as scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are electron microscopic methods used for particles of suitable size and electron density. The advantage is that one can visualize the particle size and morphology. Atomic force microscopy (AFM) is a very high resolution–type scanning probe microscopy used for imaging, measuring and manipulating matter at the nanoscale. Development of the precursor of the AFM, the scanning tunneling microscope, earned its developers the Noble Prize for Physics in 1986. Particles of colloidal size exhibit Brownian motion in dilute aqueous suspension. Tracking of the Brownian motion by a dynamic light scattering (DLS) instrument using laser (also called photon correlation spectroscopy) and applying the Stokes–Einstein equation enable us to measure the size of the colloidal particle. DLS methods also calculate the polydispersity index (PDI), which is a measure of the width of size distribution. The smaller the PDI, the more homogeneous is the particle size in the sample. Particle size determination by scattering of laser light has been around for quite some time. Sizing of particles less than 1 nm is now routinely accomplished for micelles, colloids, proteins and other nanoparticles by the modern sophisticated DLS methods.
FDA-approved nanomedicines and those at various stages of development
Doxil (liposomal doxorubicin)
Anthracyclines are widely used in oncology, but their use is limited by the risk of cumulative cardiac toxicity. Several strategies have been used to reduce the risk of toxicity such as reducing the total dose, administration in fractionated rather than single dose, use of protective agents and encapsulation in liposomes.52 Doxil (PEGylated doxorubicin) is the first FDA-approved (1995) nanodrug used to treat some types of cancers, including metastatic ovarian cancer and AIDS-related Kaposi’s sarcoma. It contains the chemotherapy drug doxorubicin (Adriamycin), an anthracycline, enclosed in a liposome. The mean diameter is in the range of 80–90 nm (Figure 1). The liposome formulation allows the doxorubicin to stay in the bloodstream longer, so that more of the drug reaches the cancer cells.53,54 Doxorubicin is believed to act on cancer cells by two different mechanisms: (1) intercalation into DNA and disruption of topoisomerase II–mediated DNA repair and (2) generation of free radicals and damage to cellular membranes, DNA and proteins.55 Doxorubicin is known to cause severe and possibly life-threatening heart problems (e.g. heart failure). These problems may occur during therapy or sometimes months to years after receiving doxorubicin. In some cases, heart problems are irreversible. The risk may be increased in patients using certain medicines that may affect heart function or have a history of heart problems, receiving radiation treatment to the chest area, or previous therapy with other anthracyclines (e.g. epirubicin; Table 1).
Figure 1.
Doxil.
Table 1.
Examples of nanomedicines for cancer approved by FDA and those undergoing clinical trials.
| Drug product | Active ingredient | Manufacturer | Indications | FDA approval date |
|---|---|---|---|---|
| Doxil (Caelyx) | PEGylated doxorubicin | Ortho Biotech, Schering-Plough | Ovarian/breast cancer | November 1995 |
| Abraxane | Albumin-bound paclitaxel nanospheres | Abraxis BioScience, AstraZeneca | Various cancers | January 2005 |
| Nab paclitaxel in combination with gemcitabine | Celgene | Metastatic pancreatic cancer | September 2013 | |
| Myocet | Liposome-encapsulated doxorubicin | Elan/Sopherion Therapeutics | Breast cancer | 2000, approved in Europe and Canada |
| DaunoXome | Liposome-encapsulated daunorubicin | Gilead Science | HIV-related Kaposi’s sarcoma | April 1996 |
| DepoCyt | Liposomal cytarabine | SkyePharma, Enzon Pharmaceuticals | Lymphomatous meningitis | April 1999 |
| Oncaspar | PEG asparaginase | Enzon Pharmaceuticals | Leukemia | February 1994 |
| Mylotarg | Gentuzumab-ozogamicin | Wyeth-Ayerst | Acute myeloid leukemia | 2000 |
| Onco TCS | Liposomal vincristine | INEX Pharmaceuticals | Non-Hodgkin lymphoma | In clinical phase 1/2 |
| LEP-ETU | Liposomal paclitaxel | Neopharma | Ovarian/breast/lung cancers | In clinical phase 1/2 |
| Aroplatin | Liposomal cisplatin analog | Antigenics, Inc. | Colorectal cancer | In clinical phase 1/2 |
| OSI-211 | Liposomal lurtotecan | OSI | Lung cancer/recurrent ovarian | In clinical phase 2 |
| SPI-77 | Stealth liposomal cisplatin | Alza | Head and neck cancer/lung cancer | In clinical phase 3 |
| EndoTAG-1 | Paclitaxel | Medigene/SynCore Biotechnology | Breast cancer/Pancreatic cancer | In clinical phase 2 |
| Marqibo | Vincristine | Talon Therapeutics, Inc. | Philadelphia chromosome–negative lymphoblastic leukemia | August 2012 |
| ThermoDox | Doxorubicin | Celsion | Hepatocellular carcinoma | In clinical phase III |
| Atragen | Liposomal all trans-retinoic acid | Aronex Pharmaceuticals | Acute promyelocytic leukemia | In clinical phase 2 |
| Lipoplatin | Liposomal cisplatin | Regulon | Pancreatic/head and neck/breast cancer | In clinical phase 3 |
| Aurimmune (CYT-6091) | TNF-α bound to colloidal gold nanoparticles | Cytimmune Sciences | Head and neck cancer | In clinical phase 2 |
| AuroShell | Gold nanoshells | Nanospectra Biosciences, Inc. | AuroLase Therapy of cancer | In clinical phase 1 |
| Genexal-PM | Paclitaxel-loaded polymeric micelle | Samyang | Breast cancer/small cell lung cancer | Marketed in Europe |
FDA: Food and Drug Administration; Nab: nanoparticle albumin bound; PEG: polyethylene glycol; TNF-α: tumor necrosis factor-α.
The risk of developing heart problems varies depending on the dose and condition. Given as liposomes, it has fewer side effects on healthy cells than regular doxorubicin.56–59 Liposomal doxorubicin is also called Doxil (Johnson & Johnson, USA), Caelyx (Janssen-Cilag, Europe), Evacet (The Liposome Company Inc.) and LipoDox (Sun Pharma). Doxorubicin hydrochloride liposome injection is currently on the FDA’s drug shortage list. The US FDA approved the first generic version of the cancer drug Doxil (doxorubicin hydrochloride liposome injection, February 2013) made by Sun Pharma Global FZE, a subsidiary of India’s Sun Pharmaceutical Industries Ltd, to ease drug shortage. LipoDox is the second generation of PEGylated liposomal doxorubicin composed of distearoyl phosphatidylcholine (DSPC) and cholesterol with surface coating of polyethylene glycol (PEG).60 LipoDox has a circulation half-life of 65 h. Moreover, due to the long circulation time of the PEGylated drug, stomatitis (inflammation of mucus lining) became the new dose-limiting toxicity. Doxil and LipoDox (both PEGylated) accumulate at tumor site by passive targeting mechanism. Unfortunately, both have more side effects than Myocet (non-PEGylated doxorubicin).
Myocet
Myocet is a non-PEGylated liposomal doxorubicin made by Enzon Pharmaceuticals for Cephalon in Europe and for Sopherion Therapeutics in the United States and Canada. The Myocet liposome is made up of egg phosphatidylcholine and cholesterol in a molar ratio of 55:45. Myocet is approved in Europe and Canada for treatment of metastatic breast cancer in combination with cyclophosphamide, but is not yet approved by the FDA for use in the United States. It is currently being studied in combination with Herceptin (trastuzumab) and Taxol (paclitaxel) for treatment of HER2-positive metastatic breast cancer. Myocet has a different pharmacokinetic profile from doxorubicin, resulting in an improved therapeutic index (less cardiotoxicity and equal anticancer activity). The clearance of doxorubicin in patients receiving Myocet was found to be 5–9 fold lower, and the volume of distribution about 10–25 fold lower than in patients receiving conventional doxorubicin. The half-life has been reported to be between 16 and 50 h, which is significantly longer than conventional doxorubicin. These findings are in agreement with the theoretical advantage of using a liposomal encapsulation for drug delivery.61–63
Liposomes are small, spherical vesicles composed of an outer lipid bilayer of phospholipids that wraps around an internal aqueous compartment which may contain a drug. They are classified as unilamellar (vesicles composed of one lipid bilayer) and multilamellar (consisting of several concentric lipid bilayers). Unilamellar vesicles have a diameter of 50–250 nm, and the multilamellar vesicles have a diameter of 1–5 µm. Liposomes can be prepared with different distribution characteristics in the body based on the lipid composition, size and surface charge. Hundreds of drugs, including anticancer agents, enzymes, proteins, vaccines, chelating agents and nutritional supplements have been encapsulated into the aqueous compartment or incorporated into the lipid bilayer. The liposomes carrying the anticancer agent eventually breakdown, releasing their contents in the tumor tissue to which it has migrated through the blood capillaries of the tumor. Several liposomal anticancer drugs are already available in the clinic for Kaposi’s sarcoma, ovarian cancer and breast cancer. The surface of the liposomes can be modified to achieve greater selectivity in targeting to the tumor. For example, the immunoliposomes contain a monoclonal antibody that is generated for a specific antigen on the tumor. It is known that transferrin is overexpressed in certain tumors. The nanocarrier surface is attached to transferrin, which will specifically bind to transferrin receptors on the tumor. Similarly, tumors over-expressing folate receptors can be attacked by folic acid–modified nanoparticles. In clinical studies, liposomes show improved pharmacokinetics and biodistribution of therapeutic agents to specific sites in the body, thus minimizing toxicity.64 The majority of the liposomal preparations are approved for intravenous use, while the surface antigens such as hepatitis A vaccine are administered by intramuscular route.65 Liposomal formulations are more expensive than the non-liposomal drugs. The average per dose of Doxil is approximately 10–20 fold higher compared to doxorubicin, but a corresponding increase in patient survival has not been demonstrated. Another concern is the toxicity of liposomal formulations especially that of the PEGylated liposomes, such as various skin reactions and hypersensitivity reactions.
PEGylated liposomes
Although the conventional liposomes are effective in decreasing the clearance of the encapsulated agent and in passively targeting to specific tissues, they are recognized as foreign body and are rapidly phagocytosed by the reticuloendothelial system (RES), thereby reducing the quantity of the drug available at the tumor site. The enclosed drug may also leak out while in circulation. The uptake by the phagocytes of liver and spleen may be advantageous to treat phagocyte-related diseases. In order to produce a long circulating liposome, the surface is modified by coating or conjugation with polyethylene glycol (PEG), a process called PEGylation. The product is called “stealth liposomes” that avoids detection and destruction by the phagocytes of the RES and are passively targeted to inflamed tissues and tumors. The longer circulation times achieved with PEGylation and their passive accumulation in tumors can result in tumor concentrations of 10–100 fold compared to the use of free drug.66,67 PEG is approved for human use by FDA because of its lack of toxicity and immunogenicity. The PEG density on the nanoparticle surface can be adjusted by selecting PEG of varying molecular weight (chain length). Longer PEG chains offer greater steric influence around the nanocarrier.68,69
ThermoDox
The new generation liposomes, called thermosensitive liposomes, are being developed in a manner that releases their encapsulated drug where local tissue temperatures are elevated to a clinically achievable temperature range (39°C–42°C). ThermoDox (Celsion), being evaluated for primary liver cancer, is a thermosensitive liposome. It releases doxorubicin at a tumor site where the temperature is elevated by application of radiofrequency, a technique referred to as radiofrequency ablation. The lipid components in the liposome undergo a gel to liquid transition, rendering it more permeable, thus releasing the drug. Application of local hyperthermia results in the blood vessels within tumors to leak, thus increasing accumulation of liposomes in the tumor. It is composed of a mixture of dipalmitoyl phosphatidylcholine (DPPC), monostearoyl phosphatidylcholine (MSPC) and PEG2000 DSPE (distearoylphosphatidylethanolamine) in 90:10:4 molar ratios. DPPC has a transition temperature of 41.5°C; therefore, it is good for temperature-triggered technology.70 For ThermoDox, this technology allows concentration of the drug up to 25 times more in the treatment area than intravenous (IV) doxorubicin, and several fold the concentration of other liposomally encapsulated doxorubicins.71
Hyperthermia
Hyperthermia is a type of cancer treatment in which body tissue is exposed to high temperatures (up to 113°F) to damage and kill cancer cells. The technique is almost always used with other forms of cancer therapy, such as radiation therapy and chemotherapy. Several methods of hyperthermia are currently under study, including local, regional and whole-body hyperthermia. Research has shown that high temperatures can damage and kill cancer cells, usually with minimal injury to normal tissues.72 Hyperthermia may make some cancer cells more sensitive to radiation or harm other cancer cells that radiation cannot damage. Hyperthermia is used in combination with other treatments, including radiotherapy, and the heat is generated with radiofrequency waves, microwaves or ultrasound. Hyperthermia can also enhance the effects of certain anticancer drugs.73,74 However, the difficulties involved in heating deep tumors to acceptable temperatures have limited the use of hyperthermia in cancer treatment. The use of nanoparticles for targeted, non-invasive thermal destruction, including gold and other metal nanoparticles, is an alternative to conventional hyperthermia treatment. Irradiation of metal nanoparticles such as gold nanoparticles with laser, which can be tuned to a specific wavelength to suit the size and shape of the nanoparticle, is a convenient method of applying heat within the tumor.
DaunoXome (liposomal daunorubicin)
DaunoXome
DaunoXome (Gilead sciences, now sold to Galen Pharmaceuticals) is the liposomal formulation of Daunorubicin (Daunomycin; Figure 2). The liposomes are not rapidly cleared from the plasma by the reticuloendothelial system, and release of daunorubicin continues in a sustained manner. Preclinical studies indicate increased tissue concentrations of daunorubicin in tumor, brain, liver, spleen and intestine following DaunoXome compared with free daunorubicin administration, but a reduced tissue concentration in cardiac tissue.75 In addition, studies using radio-labeled vesicles suggest selective uptake into tumor.76 The antitumor effects of Daunorubicin are due to intercalation into DNA and inhibition of topoisomerase II activity, resulting in decreased synthesis of both DNA and RNA. In addition, the free radical pathway that generates hydroxyl and superoxide radicals and which peroxidize lipids and damage cellular membranes also contribute to its antitumor activity. The liposomal formulation contains daunorubicin citrate, with a mean diameter of the particles of 45 nm and is made up of DSPC, cholesterol and daunorubicin in the molar ratio of 10:5:1. It was approved by FDA in 1996 for HIV-associated Kaposi’s sarcoma. Pharmacokinetic studies in pediatric patients revealed one-compartment elimination kinetics in all patients, and the PK parameters were not dose dependent. The total plasma clearance was 0.423 L/h/m2, elimination half-life of 5.5 h and a volume of distribution of 3.7 L/m2.77,78 These differences in the volume of distribution and clearance result in higher daunorubicin exposure (in terms of area under the curve (AUC)) than with conventional drugs. The liposomes are dispersed in an aqueous medium containing 2125 mg sucrose, 94 mg glycine and 7 mg calcium chloride dihydrate in a total volume of 25 mL/vial. The pH of the dispersion is between 4.9 and 6.0. The liposome dispersion is translucent and appears red in color.
Figure 2.
DaunoXome.
Abraxane (albumin-bound paclitaxel)
Paclitaxel is a semisynthetic diterpenoid taxane derivative with antineoplastic activity (Figure 3). Paclitaxel was initially obtained from the bark of Taxus brevifolia and later from the needles of Taxus baccata, the yew tree.
Figure 3.
Paclitaxel.
Taxanes generally have very low solubility in water. In order to solubilize paclitaxel, a derivative of castor oil called polyethoxylated castor oil (Cremophor) and ethanol is used. Cremophor itself causes hypersensitivity reactions (premedication with a steroid (oral dexamethasone) and antihistamine (diphenhydramine) was required) and is known to leach out plasticizers from injection tubing.79 The albumin-bound paclitaxel, Abraxane, however, does not contain Cremophor. Therefore, no premedication or special tubing is required to administer Abraxane. Abraxane (albumin-bound paclitaxel NPs (Abraxis Bioscience/Celgene)) was approved by the FDA in January 2005 for treatment of metastatic breast cancer and in October 2012 as a first-line treatment for advanced non-small-cell lung cancer in combination with carboplatin for patients who are not candidates for curative surgery or radiation therapy. Abraxane has also shown promise for treatment of advanced pancreatic cancer.80 Abraxane formulation uses nanotechnology to combine human albumin with paclitaxel allowing for the delivery of an insoluble drug in the form of nanospheres (130 nm in diameter). Paclitaxel exists in the NP in a non-crystalline amorphous state. Albumin is an ideal carrier for drug delivery because of its preferential uptake in tumor and inflamed tissue, ready availability, biodegradability and lack of toxicity and immunogenicity. Abraxane formulation has increased the bioavailability of paclitaxel and resulted in higher intra-tumor concentrations facilitated by albumin-receptor (gp60)-mediated endothelial transcytosis.81–83 Albumin-bound paclitaxel is the first biologic chemotherapeutic compound to exploit the gp60 receptor (albondin)-mediated pathway in endothelial cell walls of tumor microvessels to achieve enhanced intra-tumoral concentrations. NP albumin–bound (Nab) paclitaxel is administered as a suspension intravenously (260 mg/m2 as infusion for 30 min). Nab paclitaxel can be reconstituted in normal saline at concentrations of 2–10 mg/mL, compared with 0.3–1.2 mg/mL for paclitaxel. It undergoes biphasic elimination (two-compartment model of disposition) with a terminal half-life of 27 h (5.8 h for paclitaxel). The clearance is 43% slower (15 L/h/m2), and the mean volume of distribution is 632 L/m2 (indicating extensive extravascular distribution). The drug exposure (AUC) was proportional to the dose in the range of 80–375 mg/m2.
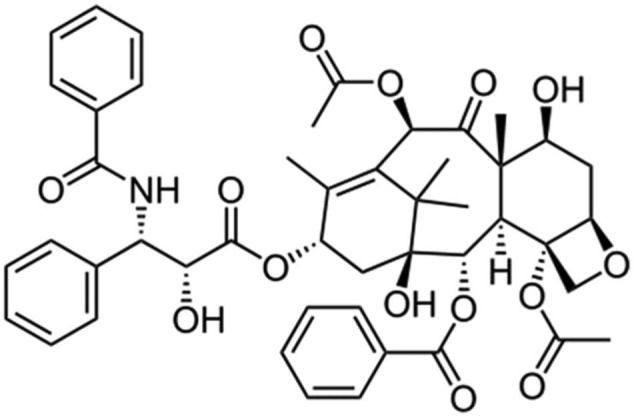
Another nanocarrier for paclitaxel is also being developed by Cell Therapeutics, called “paclitaxel polyglumex (PPX).” This product is also known as Xyotax and Opaxio or CT 2103. It is a large macromolecular conjugate of paclitaxel and poly-l-glutamic acid and contains 37% of paclitaxel. The polymer poly-l-glutamic acid is biodegradable. PPX, because of its large size, accumulates in tumor tissues by taking advantage of the enhanced permeability of tumor vasculature and lack of lymphatic drainage. The drug is released from the polymeric backbone by the lysosomal enzyme protease, cathepsin B, which is up-regulated in many tumor types.84,85 Preclinical studies in animal tumor models demonstrate that PPX is more effective than standard paclitaxel and is associated with prolonged tumor exposure to active drug while minimizing systemic exposure. Phase 1 and 2 clinical studies with PPX showed encouraging outcomes compared to standard taxanes with reduced neutropenia and alopecia and allowed a more convenient administration schedule without the need for routine premedications (Figure 4; Table 2).85
Figure 4.

Polyglumex.
Table 2.
Comparison of Taxol, polyglumex and Abraxane.
| Characteristic | Taxol | PPX (polyglumex) | Abraxane (albumin nanospheres) |
|---|---|---|---|
| Solubility | Requires solubilizing agent (Cremophor, alcohol) | Water soluble | Water soluble |
| Administration | 3–24 h infusions with routine premedications | 10–20 min infusion. No premedication | Intravenous infusion for 30 min. No premedication required |
| Systemic exposure | High exposure | Reduced Cmax. Gradual drug release from inactive drug conjugate | Reduced Cmax, minimize systemic exposure |
| Pharmacokinetics | Short elimination half-life (6 h) | Prolonged distribution phase and long terminal elimination half-life (130 h) | Gradual drug release, prolonged elimination half-life (27 h) |
| Tumor selectivity | No | Passive tumor accumulation, evade MDR efflux pump by pinocytotic uptake | High intra-tumor concentration facilitated by albumin-receptor mediated endocytosis |
PPX: paclitaxel polyglumex; MDR: multidrug resistance.
EndoTAG-1
MediGene AG (Germany) obtained US patent for EndoTAG®-1 for the treatment of triple-negative breast cancer. The clinical drug candidate EndoTAG-1 is an innovative composition of the established cytostatic drug paclitaxel combined with neutral and positive lipids. Due to the positively charged lipids, EndoTAG-1 interacts with newly developing, negatively charged endothelial cells, which are especially required for the growth of tumor blood vessels. The EndoTAG-1 paclitaxel component attacks the activated endothelial cells as they divide, thus targeting the blood supply to tumors, without affecting the supply to endothelial cells of healthy tissue. By doing this, EndoTAG-1 is expected to prevent the formation of new tumor blood vessels and to inhibit tumor growth.86
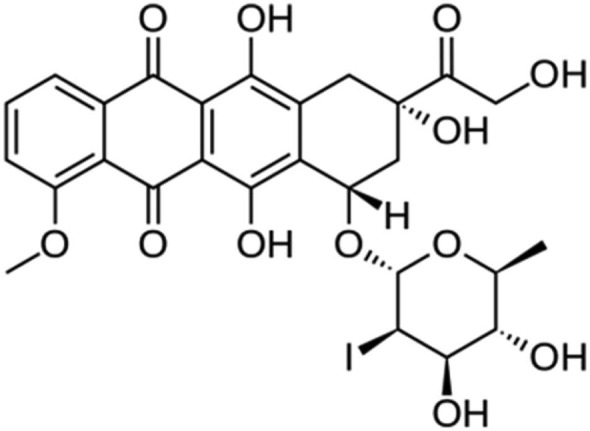
Lipoplatin (liposomal cisplatin)
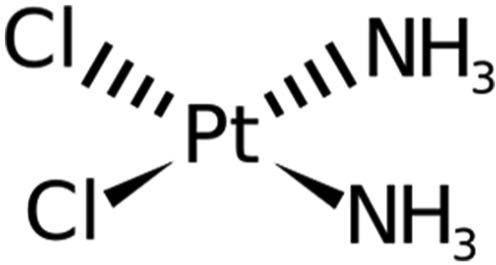
Lipoplatin is a liposomally encapsulated drug product of the FDA-approved cytotoxic agent cisplatin (Figure 5). The major concern about the use of cisplatin and other platinum compounds is nephrotoxicity. Patients receiving these agents need to be hydrated to prevent renal damage. In the lipoplatin product, cisplatin (cis-diamino-dichloro-platinum) is encapsulated in a liposome shell composed of dipalmitoyl phosphatidyl glycerol, soy phosphatidylcholine, cholesterol and methoxy PEG–distearoyl phosphatidylethanolamine lipid conjugate. The ratio of cisplatin to lipids is 8.9%:91.1% (w/w).87,88 Lipoplatin is used against pancreatic cancer in combination with gemcitabine as first-line treatment. Lipoplatin accumulates in the cancer tissues by the extravasation of the NPs through the defective vasculature of the tumor tissue during neoangiogenesis. Lipoplatin had mild hematological and gastrointestinal toxicity and did not show nephro-, neuro- or ototoxicity or any other side effects characteristic of cisplatin. The half-life of total platinum in the human plasma was 60–117 h compared to 6 h for cisplatin.87
Figure 5.
Lipoplatin (liposomal cisplatin).
Onco TCS (vincristine)
Vincristine (VCR) and vinblastine (VLB) are alkaloids obtained from the flowering plant Catharanthus roseus (periwinkle, also called the Madagascar periwinkle). Both have powerful anticancer activity. On 9 August 2012, the FDA granted accelerated approval for VCR sulfate liposome injection (VSLI; Marqibo®, made by Talon Therapeutics, Inc.) for the treatment of adult patients with Philadelphia chromosome–negative (Ph−) acute lymphoblastic leukemia (ALL). VCR administered as the liposomal formulation exhibits a lower clearance and higher AUC compared with conventional VCR.89 INEX Pharmaceuticals is developing a liposomal formulation of VCR (Onco TCS, vincacine, VSLI, VCR sulfate liposomes for injection) for the treatment of relapsed aggressive non-Hodgkin’s lymphoma (NHL) and other cancers (INEX Pharmaceuticals is a Canadian biopharmaceutical company developing and commercializing proprietary drugs and drug-delivery systems to improve treatment of cancer; Figure 6). VCR is being developed using INEX Pharmaceuticals’ proprietary drug-delivery technology platform called the “transmembrane carrier systems” (TCS). Liposomal VCR is expected to have certain advantages over the existing standard preparation because VCR in liposomes enables the drug to increase blood circulation time, increase the drug accumulation in the blood, increase drug accumulation in the tumor and be released over an extended period.
Figure 6.
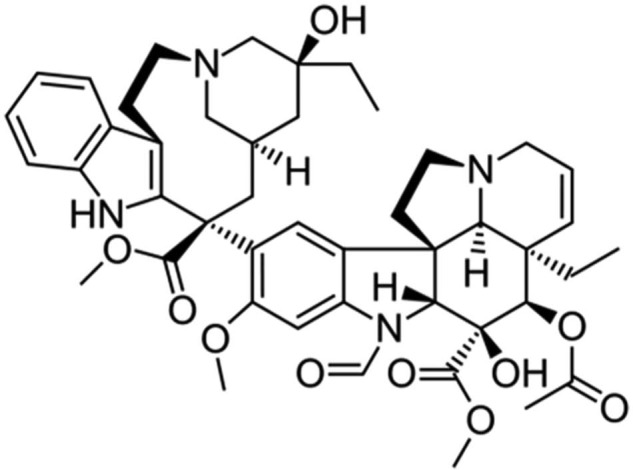
Onco TCS (vincristine).
Transmembrane pH gradient (inside acidic) liposomes preparation
A special technique called transmembrane pH gradient is used to prepare liposomes with very high encapsulation efficiency and increased stability of drugs such as doxorubicin and VCR. The method consists of preparing liposomes with the aqueous compartment containing a weak acid such as citric acid to maintain a pH of 4. VCR sulfate solution is then added to the vesicles and the pH raised to 7.0–7.2. The lipophilic VCR that is formed from the salt at this pH will permeate through the lipid membrane into the acidic internal compartment where it will remain as the cationic form. This method results in encapsulation efficiencies approaching 100% and a drug to lipid ratio is 200 fold higher than the conventional method. Drug entrapment and retention within the liposomes is dependent on the magnitude of the pH gradient between the inside aqueous compartment and the outside of the lipid membrane.90
Vinorelbine
Vinorelbine is a semisynthetic vinca alkaloid shown to be useful for treatment of a variety of malignancies, such as small-cell lung, breast, ovarian, head and neck, cervical and Kaposi’s sarcoma (Figure 7). A new formulation of vinorelbine, called Allocrest, consists of vinorelbine encapsulated in the aqueous core of a liposome (sphingomyelin-based liposomes called Optisome™). This formulation has been developed to achieve targeted delivery of the drug in high concentration in the tumor and also sustained release. In animal models, the Optisome technology resulted in prolonged plasma circulation (100-fold increased area under the concentration–time curve) and 9.5-fold enhanced cancer tissue drug penetration and accumulation as compared to that achievable with unencapsulated, standard vinorelbine. The high intra-tumoral vinorelbine concentration is expected to improve cancer cell killing beyond the capability of standard vinorelbine. The prospect of providing enhanced anticancer activity without enhanced toxicity is of particular importance in the treatment of elderly cancer patients and those with marginal performance status. Nano vinorelbine (NanoVNB) was found to be active against human breast cancer and lung cancer xenograft.91
Figure 7.
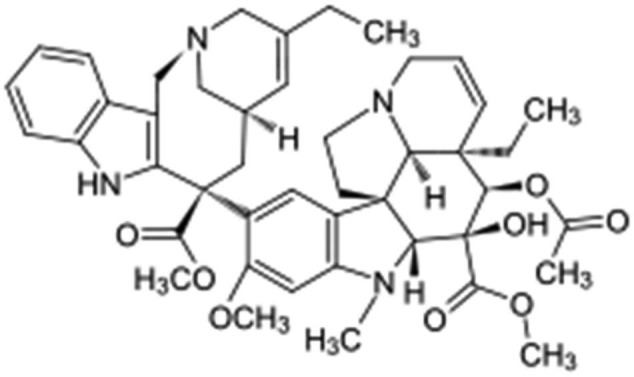
Vinorelbine.
DepoCyt (liposomal cytarabine)
DepoCyt is a sustained-release liposomal formulation of cytarabine, prepared by a unique proprietary technology called DepoFoam (Figure 8). Unlike the usual unilamellar or multilamellar liposomes, the DepoFoam is a multivesicular system containing hundreds of water-filled compartments separated by a lipid bilayer. This structure allows encapsulation of large quantities of drugs and ensures prolonged release.92–94
Figure 8.
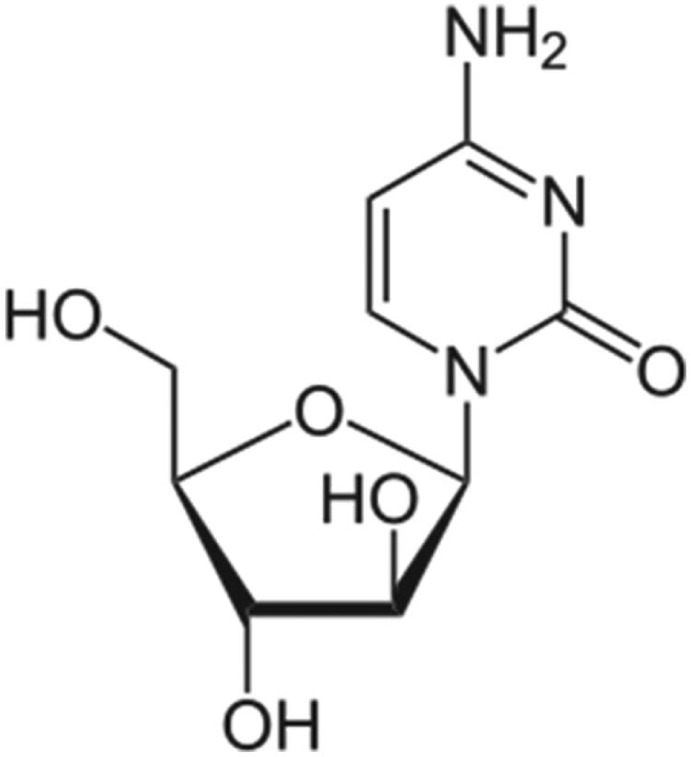
Cytarabine.
Cytarabine liposome injection (DepoCyt, Enzon Pharmaceuticals) was given full approval by the FDA in April 2007 for the treatment of lymphomatous meningitis, a life-threatening complication of lymphoma. It was originally approved in April 1999 under the accelerated approval regulations based on increased response rate compared to the unencapsulated drug. It is the only liposomal drug for intrathecal use and should be administered only under supervision of a qualified physician. Systemic exposure to cytarabine is negligible when the liposome is given intrathecally. Cytarabine is a cell-cycle-specific antineoplastic agent affecting cells only in the s-phase of cell division. DepoFoam-encapsulation has been shown to result in a sustained-release lasting several days to weeks (DepoCyt has a half-life of up to 82.4 h compared to 3.4 h for the unencapsulated drug) after non-vascular administration. DepoCyt is well distributed throughout the cerebrospinal fluid to provide continuous exposure of the tumor cells to cytarabine (the routes of administration most viable for delivery of drugs via DepoFoam formulations include intrathecal, epidural, subcutaneous, intramuscular (IM), intra-articular and intraocular). DepoFoam particles are distinguished structurally from unilamellar vesicles, multilamellar vesicles and niosomes in that each particle comprises a set of closely packed non-concentric vesicles. The particles are tens of microns in diameter and have large trapped volume, thereby affording delivery of large quantities of drugs in the encapsulated form in a small volume of injection. A number of methods based on a manipulation of the lipid and aqueous composition can be used to control the rate of sustained release from a few days to several weeks.
The liposome contains dioleoyl phosphatidylcholine, dipalmitoyl phosphatidyl glycerol, cholesterol and triolein. It is a long-acting, preservative-free formulation. The pharmacokinetic advantage of this formulation was that the terminal half-life was 40 times longer than that of standard cytarabine.95 Chemical arachnoiditis, manifested primarily by nausea, vomiting, headache and fever, has been a common adverse event following administration of DepoCyt. The incidence and severity of arachnoiditis can be reduced by the co-administration of dexamethasone. Therefore, it is recommended that all patients receiving DepoCyt should be treated concurrently with dexamethasone. Cytarabine can cause fetal harm if a pregnant woman is exposed to the drug systemically. However, the concern for fetal harm following intrathecal DepoCyt administration is low because systemic exposure to cytarabine is negligible.96
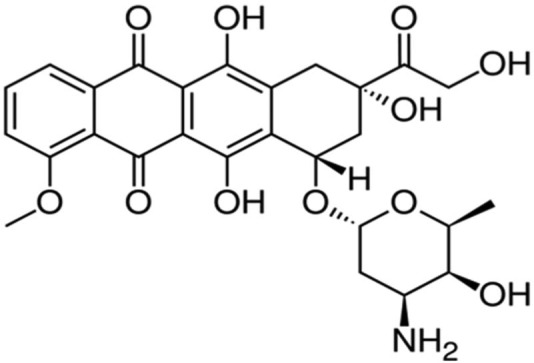
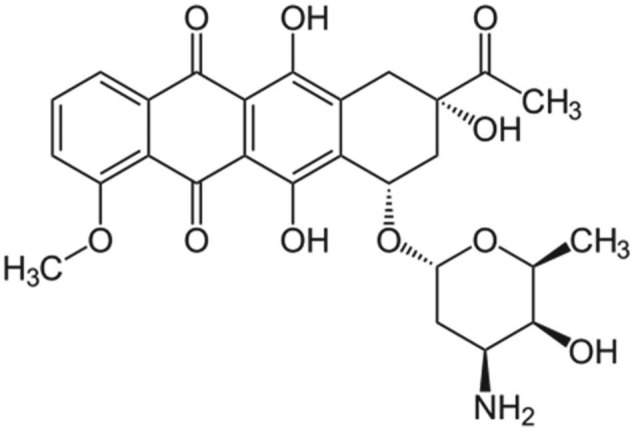
Annamycin
Annamycin is a semisynthetic doxorubicin analog developed at the MD Anderson Cancer Institute in Houston, Texas (Figures 9 and 10). Annamycin was initially investigated as an antineoplastic without the MDR related to P-gp. The P-gp is a major factor limiting the efficacy of anthracycline group of drugs.97 As shown in the structure above, annamycin do not have the amino group in the sugar moiety as in doxorubicin. Removal of the amino group seems to reduce the cardiac toxicity without changing its antitumor properties. Annamycin intercalates into DNA and inhibits topoisomerase II. This results in the inhibition of DNA replication and repair as well as RNA and protein synthesis. Liposomal annamycin is less toxic and shows improved antitumor activity compared to annamycin and is indicated for breast cancer.98
Figure 9.
Annamycin.
Figure 10.
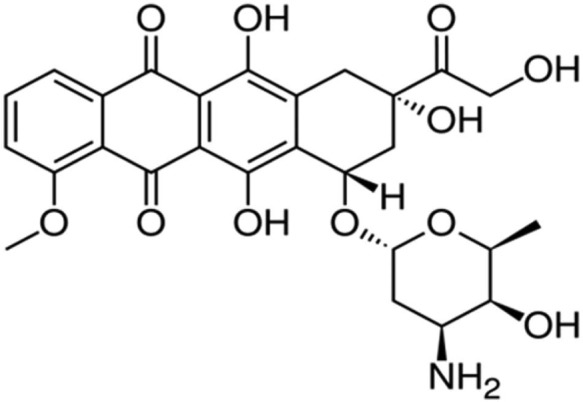
Doxorubicin.
Oncaspar (asparaginase)
Leukemic cells are unable to synthesize asparagine due to lack of asparagine synthetase and are dependent on exogenous source of asparagine for survival. Asparaginase is the enzyme that depletes the amino acid asparagine. Depletion (starving the leukemic cells) of asparagine ultimately results in leukemic cell death. Normal cells are less affected because of their ability to synthesize asparagine. l-asparaginase has been an important component in the treatment of ALL.99 Oncaspar is a modified form of the enzyme, l-asparaginase (Figure 11). The tetrameric enzyme, derived from Escherichia coli, is covalently conjugated with monomethoxy polyethylene glycol (mPEG). Approximately 69–82 molecules of mPEG are linked to l-asparaginase. The molecular weight of each mPEG molecule is 5 kDa. PEG asparaginase (Enzon Pharmaceuticals) was approved by FDA in July 1994 for use in ALL. Patients with allergy to the drug were unable to receive l-asparaginase. The use of Oncaspar in place of l-asparaginase, markedly reduced the number of drug injections required from 21 injections of Elspar (l-asparaginase), to three injections with Oncaspar over the 20-week course of treatment. Through the process of PEGylation, the half-life of l-asparaginase is significantly increased (approximately 6 days), and the l-asparaginase activity is sustained.100 Oncaspar provides patients the full benefits of asparaginase therapy with an enhanced convenience over native l-asparaginase (non-PEGylated form). Oncaspar can be administered through IM injection or IV infusion. When utilized as a component of induction therapy for ALL, a single dose of Oncaspar achieved similar levels of asparagine depletion as nine doses of native l-asparaginase.101
Figure 11.
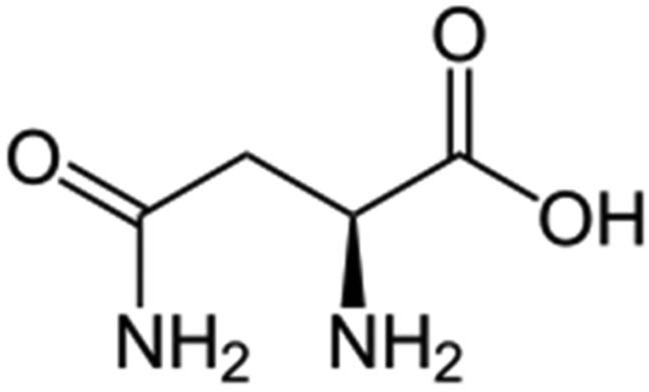
Asparagine.
Camptothecines
Lurtotecan is a potent semisynthetic derivative of camptothecin (Figures 12 and 13.). This analog is water-soluble. Lurtotecan is a topoisomerase inhibitor used for epithelial ovarian cancer. Lurtotecan has been formulated into unilamellar liposomes. Pharmacokinetic studies in nude mice have demonstrated increased plasma residence time and 1500-fold increase in AUC.102,103 Another agent of this group, irinotecan, is being developed by Nektar Therapeutics as a PEGylated liposomal formulation for the treatment of colorectal cancer.
Figure 12.
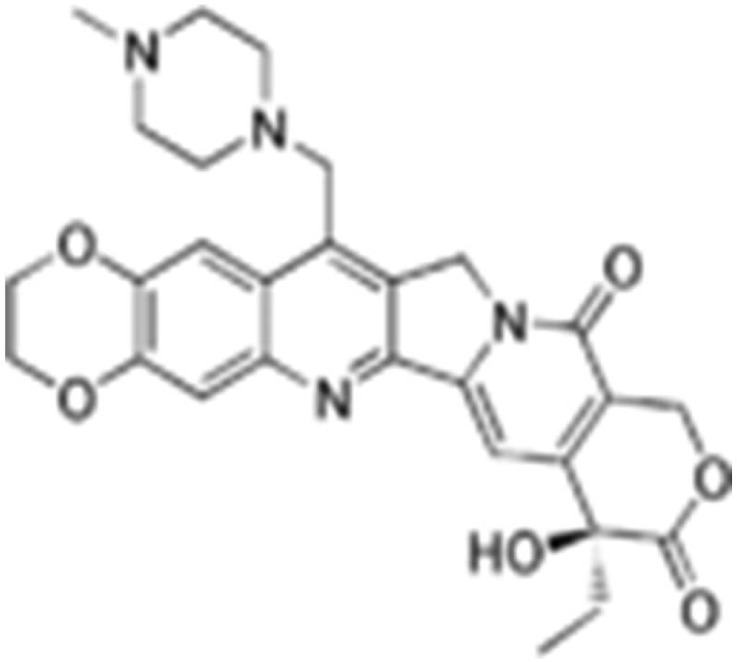
Lurtotecan.
Figure 13.
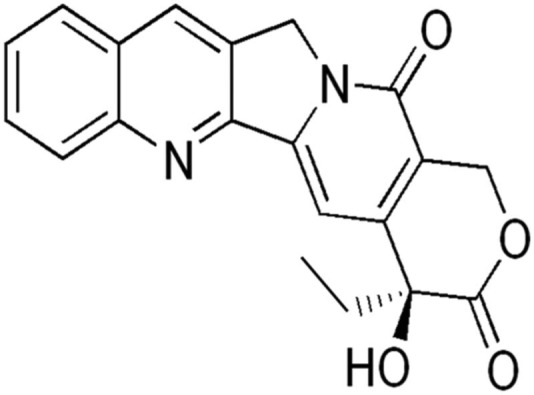
Camptothecine.
All trans retinoic acid
All trans retinoic acid (ATRA) or Atragen is an all trans-retinoic acid, a relative of vitamin A (Figure 14). Retinoids are involved in normal cell growth, cell differentiation and cell death. ATRA is used to treat acute promyelocytic leukemia (APL or APML) in patients who have not responded to other treatments. Retinoids are relatively new types of anticancer drugs that have been used alone or in combination to treat a variety of cancers. The liposomal delivery system of ATRA alters the drug’s pharmacokinetics to improve its tissue distribution. Following IV injection, this formulation is able to bypass the hepatic clearance mechanism that metabolizes the oral formulation of ATRA. One of the disadvantages of oral ATRA is its very poor bioavailability. It is almost insoluble in aqueous medium with highly variable absorption from the intestine. The use of IV liposomal formulation decreases toxicities associated with oral tretinoin doses. Additionally, in vitro studies have shown that liposomal ATRA has a greater antiproliferative effect on neoplastic cells as compared to free ATRA. It can inhibit the proliferation of lymphoma cells in a dose-dependent manner by inducing apoptosis.104,105 It has also been shown that the IV administration of liposomal retinoic acid in human subjects resulted in a 13–15 fold higher plasma concentration than retinoic acid, and the side effects were similar to that of oral dose of the free drug.106
Figure 14.
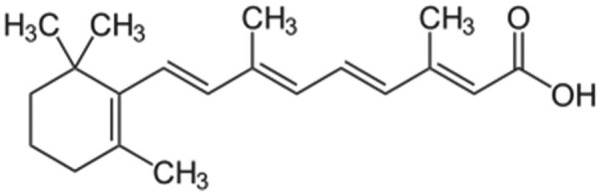
All trans retinoic acid.
Gold NPs
One of the most studied NPs is the gold NPs (GNPs). GNPs are potential drug carriers, photothermal agents, radiosensitizers and contrast agents. They also have shown promise for cancer therapy. There are several methods for the preparation of gold NPs. One of the methods involves reduction of chloroauric acid with sodium citrate. Varying the molar ratio of citrate to chloroauric acid results in variation of GNP sizes from 50 to 150 nm. The NPs are stabilized by coating with PEG to which anticancer drugs can be attached. GNPs exhibit unique physicochemical properties: their optical properties and ability to bind to amino and thiol groups permit surface functionalization for various biomedical applications. Tumor-specific ligands such as transferrin, folic acid, monoclonal antibodies and tumor necrosis factor (TNF) have been attached to surface of gold NPs combined with a chemotherapeutic agent for specific delivery to tumor. Aurimmune (Cytimmune Sciences) is a 27 nm gold NP coated with PEG and attached to recombinant human TNF-α, which has the dual ability to accumulate in the tumor and exert its antitumor activity. Prior to aurimmune, attempts to use TNF-α in adequate doses for its anticancer response were not successful due to dose-limiting toxicity. A recent study reported the amplification of biochemical action of AuTNF-α by laser-induced photothermal effect in the target tumor.107 Another device that has advanced to clinical trials is a NP of silica coated with a thin layer of gold, called AuroShell (Nanospectra Bioscience, Inc.). Photothermal therapy of head and neck cancer is possible when the AuroShell is irradiated with near infra-red rays from a laser. Gold nanoshells are more efficient in converting the incident light in to heat than NPs. This is referred to as AuroLase Therapy.108–112
RNA NPs
RNA nanotechnology is becoming increasingly popular because of its potential in the treatment of not only cancer but also viral infections and genetic diseases. RNA can be manipulated using a variety of biochemical methods to incorporate therapeutic and imaging functionalities and the resulting nanomedicines have several advantages: the “true” nanosize of 20–40 nm that displays favorable EPR effect but does not accumulate in normal organs, targeted delivery for lung, ovarian and liver cancers, antigenicity-free since they are protein free and the promise for long-term treatment.113–115 An interesting area of development is the use of small pieces of nucleic acid known as short interfering RNAs, or siRNAs that can turn off the production of specific proteins. This property has shown great therapeutic potential for diseases caused by abnormal gene overexpression or mutation. The gene silencing effect makes them new classes of anticancer drugs in development. Currently, there are six phase 1 cancer clinical trials underway using NP-based siRNA delivery based on polymers or liposomes.116
Conclusion
Nanotechnology-based nanomedicines have the potential to overcome the limitations of conventional cancer chemotherapy by their ability to selectively target the cancer cells without doing too much damage to healthy tissue. Properly designed NPs have the ability to accumulate in tumors either by passive or active targeting and enhance the cytotoxic effects of antitumor agents. Several anticancer drugs in nano formulations have been evaluated and a few are already approved for clinical use and others are undergoing phase 2/3 clinical trials. Although the nanomedicines have numerous advantages compared to conventional chemotherapy, there are concerns about their potential for toxicity to patients and to the environment in addition to the high cost of production and premature drug release. Liposomal formulations are reported to be less toxic compared to their non-liposomal drugs. On the other hand, nanotechnology offers the opportunity to reformulate the discontinued drugs because of poor bioavailability, lack of selectivity to desired target or extreme toxicity. It is also known that drug-loaded NPs evade the efflux mechanism, maintain a high concentration within tumor cells and therefore avoid development of resistance.
Footnotes
Declaration of conflicting interests: The authors declare that there is no conflict of interest.
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
References
- 1. European Science Foundation (ESF). Nanomedicine. An ESF—EuropeanMedical Research Councils (EMRC) Forward Look report (p. 11), ESF, Strasbourg cedex, November 2004. [Google Scholar]
- 2. Martin CR. Welcome to nanomedicine. Nanomedicine 2006; 1(1): 5–9. [Google Scholar]
- 3. National Institute of Health Roadmap for Medical Research. Nanomedicine, 2006, http://nihroadmap.nih.gov/nanomedicine/
- 4. BCC Research. Nanotechnology in medical applications: the global market, 2010, http://www.bccresearch.com/market-research/healthcare/nanotechnology-medical-applications-hlc069a.html
- 5. Heger M. Amgen deal triggers watchful waiting in targeted nanomedicine. Nat Nanomedicine 2013; 19: 120. [DOI] [PubMed] [Google Scholar]
- 6. Bawa R. Patents and nanomedicine. Nanomedicine 2007; 2: 351–374. [DOI] [PubMed] [Google Scholar]
- 7. Matheolabakis G, Riga B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine 2012; 7(10): 1577–1590. [DOI] [PubMed] [Google Scholar]
- 8. Wang L, Tabor R, Easto J, et al. Formation and stability of nanoemulsions with mixed ionic and non-ionic surfactants. Phys Chem Chem Phys 2009; 11(42): 9772–9778. [DOI] [PubMed] [Google Scholar]
- 9. Wong HL, Bendayan R, Rauth AM, et al. Chemotherapy with anticancer drugs encapsulated in solid-lipid nanoparticles. Adv Drug Deliv Rev 2007; 59(6): 491–504. [DOI] [PubMed] [Google Scholar]
- 10. Hu CM, Zhang L. Therapeutic nanoparticles to combat cancer drug resistance. Curr Drug Metab 2009; 10: 836–841. [DOI] [PubMed] [Google Scholar]
- 11. Xiaowei D, Russel JM. Nanomedicinal strategies to treat multidrug resistant tumors: current progress. Nanomedicine 2010; 5(4): 597–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sadava D, Coleman A, Kane SE. Liposomal daunorubicin overcomes drug resistance in human breast, ovarian and lung carcinoma cells. J Liposome Res 2002; 12(4): 301–309. [DOI] [PubMed] [Google Scholar]
- 13. Rahman A, Husain SR, Sidiqqui J, et al. Liposome-mediated modulation of multidrug resistance in human HL-60 leukemia cells. J Natl Cancer Inst 1992; 84(24): 1909–1915. [DOI] [PubMed] [Google Scholar]
- 14. Meyer LD, Shabbits JA. The role of liposomal drug delivery in molecular and pharmacological strategies to overcome multidrug resistance. Cancer Metastasis Review 2001; 20(1–2): 87–93. [DOI] [PubMed] [Google Scholar]
- 15. Ford JM, Hait WN. Pharmacology of drugs that alter multidrug resistance in cancer. Pharmacol Rev 1990; 42(3): 155–199. [PubMed] [Google Scholar]
- 16. Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharmacol 2005; 2(5): 373–383. [DOI] [PubMed] [Google Scholar]
- 17. Chavanpatil MD, Khadair A, Gerard B, et al. Surfactant-polymer nanoparticles overcome P-glycoprotein-mediated drug efflux. Mol Pharmacol 2007; 4(5): 730–738. [DOI] [PubMed] [Google Scholar]
- 18. Batrakova EV, Li S, Alakhov VY, et al. Effect of pluronic P-85 on ATPase activity of drug efflux transporters. Pharm Res 2004; 21(12): 2226–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elizabeth B, Nancy UL, Smith KJ, et al. Blood vessel morphological changes as visualized by MRA during treatment of brain metastases: a feasibility study. Radiology 2007; 245(3): 824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folkman J. Tumor angiogenesis: therapeutic implications. New Engl J Med 1971; 285: 1182–1186. [DOI] [PubMed] [Google Scholar]
- 21. Li CH, Shan S, Huang Q, et al. Initial stages of tumor cell-induced angiogenesis: evaluation via skin window chambers in rodent models. J Natl Cancer Inst 2000; 92: 143–147. [DOI] [PubMed] [Google Scholar]
- 22. Burger PC, Scheithauer BW, Vogel FS. Surgical pathology of the nervous system and its coverings. New York: Churchill Livingstone, 1991. [Google Scholar]
- 23. Lenander C, Holmgren L. A novel method of visualizing vessels in human tumor biopsies. Angiogenesis 1999; 3: 291–293. [DOI] [PubMed] [Google Scholar]
- 24. McDonald MD, Baluk P. Significance of blood vessel leakiness in cancer. Cancer Res 2002; 62: 5381–5385. [PubMed] [Google Scholar]
- 25. Bullitt EZ, Gerig G, Alyward S, et al. Vessel tortuosity and brain tumor malignancy: a blinded study. Acad Radiol 2005; 12(10): 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maeda H, Wu J, Sawa T, et al. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release 2000; 65(1–2): 271–284. [DOI] [PubMed] [Google Scholar]
- 27. Fang J, Sawa T, Maeda H, et al. Factors and mechanism of “EPR” effect and the enhanced antitumor effects of macromolecular drugs including SMANCS. Adv Exp Med Biol 2003; 519: 29. [DOI] [PubMed] [Google Scholar]
- 28. Qian C, Berghuis B, Tsarfaty G, et al. Preparing the “soil”: the primary tumor induces vasculature reorganization in the sentinel lymph node before the arrival of metastatic cancer cells. Cancer Res 2006; 66: 10365–10376. [DOI] [PubMed] [Google Scholar]
- 29. Chung MK, Do IG, Jung E, et al. Lymphatic vessels and high endothelial venules are increased in the sentinel lymph nodes of patients with oral squamous cell carcinoma before the arrival of tumor cells. Ann Surg Oncol 2012; 18(5): 1595–1601. [DOI] [PubMed] [Google Scholar]
- 30. Nathanson SD. Insights into the mechanisms of lymph node metastasis. Cancer 2003; 98: 413–423. [DOI] [PubMed] [Google Scholar]
- 31. Martinet L, Garrido I, Filleron T, et al. Human solid tumors contain high endothelial venules: association with T- and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer Res 2011; 71: 5678–5687. [DOI] [PubMed] [Google Scholar]
- 32. Lee SY, Nan Qian C, Seng A, et al. Changes in specialized blood vessels in lymph nodes and their role in cancer metastasis. J Transl Med 2012; 10: 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boardman K, Swartz M. Interstitial flow as a guide for lymphangiogenesis. Circ Res 2003; 92: 801–808. [DOI] [PubMed] [Google Scholar]
- 34. Halin C, Detmar M. An unexpected connection: lymph node lymphangiogenesis and dendritic cell migration. Immunity 2006; 24: 129–131. [DOI] [PubMed] [Google Scholar]
- 35. Boscacci RT, Pfeiffer F, Gollmer K, et al. Comprehensive analysis of lymph node stroma-expressed Ig superfamily members reveals redundant and nonredundant roles for ICAM-1, ICAM-2, and VCAM-1 in lymphocyte homing. Blood 2010; 116: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishida N, Hirohisa Yano H, Nishida T, et al. Angiogenesis in cancer. Health Risk Manag 2006; 2(3): 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Achen MG, Jeltsch M, Kukk E, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA 1998; 95: 548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Amioka T, Kitadai Y, Tanaka S, et al. Vascular endothelial growth factor-C expression predicts lymph node metastasis of human gastric carcinomas invading the submucosa. Eur J Cancer 2002; 38: 1413–1419. [DOI] [PubMed] [Google Scholar]
- 39. Awada A, de Castro G. An integrated approach for tailored treatment in breast cancer. Ann Oncol 2005; 16: 203–208. [DOI] [PubMed] [Google Scholar]
- 40. Baldwin ME, Catimel B, Nice EC, et al. The specificity of receptor binding by vascular endothelial growth factor-D is different in mouse and man. J Biol Chem 2001; 276: 19166–19171. [DOI] [PubMed] [Google Scholar]
- 41. Bellomo D, Headrick JP, Silins GU, et al. Mice lacking the vascular endothelial growth factor-B gene (Vegfb) have smaller hearts, dysfunctional coronary vasculature, and impaired recovery from cardiac ischemia. Circ Res 2000; 86: 29–35. [DOI] [PubMed] [Google Scholar]
- 42. Andre T, Kotelevets L, Vaillant JC, et al. Vegf, Vegf-B, Vegf-C and their receptors KDR, FLT-1 and FLT-4 during the neoplastic progression of human colonic mucosa. Int J Cancer 2000; 86: 174–181. [DOI] [PubMed] [Google Scholar]
- 43. Bottaro DP, Liotta LA. Cancer: out of air is not out of action. Nature 2003; 423: 593–595. [DOI] [PubMed] [Google Scholar]
- 44. Boehm T, Folkman J, Browder T, et al. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997; 390: 404–407. [DOI] [PubMed] [Google Scholar]
- 45. Enholm B, Paavonen K, Ristimaki A, et al. Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by serum, growth factors, oncoproteins and hypoxia. Oncogene 1997; 14: 2475–2483. [DOI] [PubMed] [Google Scholar]
- 46. Boocock CA, Charnock-Jones DS, Sharkey AM, et al. Expression of vascular endothelial growth factor and its receptors fit and KDR in ovarian carcinoma. J Natl Cancer Inst 1995; 87: 506–516. [DOI] [PubMed] [Google Scholar]
- 47. Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology 2005; 19(4 Suppl. 3): 7–16. [PubMed] [Google Scholar]
- 48. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 2005; 307(5706): 58–62. [DOI] [PubMed] [Google Scholar]
- 49. Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998; 95: 4607–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Harris AL. Hypoxia: a key regulatory factor in tumor growth. Nature Rev Cancer 2002; 2: 38–47. [DOI] [PubMed] [Google Scholar]
- 51. Kong G, Braun RD, Dewhirst MW. Hyperthermia enables tumor-specific nanoparticle delivery: effect of particle size. Cancer Res 2000; 60(16): 4440–4445. [PubMed] [Google Scholar]
- 52. Weiss AJ, Manthel RW. Experience with the use of adriamycin in combination with other anticancer agents using a weekly schedule, with particular reference to lack of cardiac toxicity. Cancer 1977; 40: 2046–2052. [DOI] [PubMed] [Google Scholar]
- 53. Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale and clinical applications, existing and potential. Int J Nanomedicine 2006; 1(3): 297–315. [PMC free article] [PubMed] [Google Scholar]
- 54. Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999; 57: 727–741. [DOI] [PubMed] [Google Scholar]
- 55. Park JW. Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res 2002; 4(3): 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gordon AN, Granai CO, Rose PG, et al. Phase II study of liposomal doxorubicin in platinum- and paclitaxel-refractory epithelial ovarian cancer. J Clin Oncol 2000; 18: 3093–3100. [DOI] [PubMed] [Google Scholar]
- 57. Gordon AN, Fleagle JT, Guthrie D, et al. Recurrent epithelial ovarian carcinoma: a randomized phase III study of PEGylated liposomal doxorubicin versus topotecan. J Clin Oncol 2001; 19: 3312–3332. [DOI] [PubMed] [Google Scholar]
- 58. Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac sparing effect of liposomal formulation. Int J Nanomedicine 2007; 2(4): 567–583. [PMC free article] [PubMed] [Google Scholar]
- 59. O’Brian ME, Wigler N, Inbar M, et al. ; CAELYX Breast Cancer Study Group. Reduced cardiotoxicity and comparable efficacy in a phase III trial of PEGylated liposomal doxorubicin HCL (CAELYX/DOXIL) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann Oncol 2002; 5(3): 440–449. [DOI] [PubMed] [Google Scholar]
- 60. Hong RL. Liposomal anticancer drug researches the myth of long circulation. J Chinese Oncol Soc 2004; 20: 10–21. [Google Scholar]
- 61. Klaus M, Niemann B, Massing U, et al. Pharmacokinetics of liposomal doxorubicin (TLC-D99; Myocet in patients with solid tumors: an open-label, single-dose study. Cancer Chemother Pharmacol 2004; 54(6): 514–524. [DOI] [PubMed] [Google Scholar]
- 62. Lao J, Madani J, Puértolas T, et al. Liposomal Doxorubicin in the treatment of breast cancer patients: a review. J Drug Deliv 2013; 2013: 456409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Swenson CE, Bolcsak LE, Batist G, et al. Pharmacokinetics of doxorubicin administered i.v. as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs 2003; 14(3): 239–246. [DOI] [PubMed] [Google Scholar]
- 64. Slingerland M, Guchelaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today 2012; 17(3): 160–166. [DOI] [PubMed] [Google Scholar]
- 65. Bovier PA. Epaxal: a virosomal vaccine to prevent hepatitis A infection. Expert Rev Vaccines 2008; 7(8): 1141–1150. [DOI] [PubMed] [Google Scholar]
- 66. Kaul G, Amiji M. Long-circulating polyethylene glycol modified gelatin nanoparticles for intracellular delivery. Pharm Res 2002; 19: 1061–1067. [DOI] [PubMed] [Google Scholar]
- 67. Milton HJ, Martin NE, Modi M. PEGylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet 2001; 40(7): 539–551. [DOI] [PubMed] [Google Scholar]
- 68. Gref R, Domb A, Quellec P, et al. The controlled intravenous delivery of drugs using PEG-coated sterically stabilized nanospheres. Adv Drug Deliv Rev 1995; 16: 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stolnik S, Dunn SE, Garnett MC, et al. Surface modification of poly (lactide-co-glycolide) nanospheres by biodegradable poly (lactide)-poly (ethylene glycol) copolymers. Pharm Res 1994; 11: 1800. [DOI] [PubMed] [Google Scholar]
- 70. Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochi Biophys Acta 2005; 1716: 77–96. [DOI] [PubMed] [Google Scholar]
- 71. Yarmolenko PS, Zhao Y, Landon C, et al. Cooperative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumors. Int J Hyperthermia 2010; 26(5): 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Van der Zee J. Heating the patient: a promising approach? Ann Oncol 2002; 13(8): 1173–1184. [DOI] [PubMed] [Google Scholar]
- 73. Wust P, Hildebrandt B, Sreenivasa G, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol 2000; 3(8): 487–497. [DOI] [PubMed] [Google Scholar]
- 74. Landon CD, Park JY, Needham D, et al. Nanoscale drug delivery and hyperthermia: the materials design and preclinical and clinical testing of low temperature-sensitive liposomes used in combination with mild hyperthermia in the treatment of local cancer. Open Nanomed J 2011; 3: 38–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Forssen EA, Ross ME. DaunoXome treatment of solid tumours: preclinical and clinical investigations. J Liposome Res 1994; 4: 481. [Google Scholar]
- 76. Forssen EA, Coulter DM, Proffitt RT. Selective in vivo localization of daunorubicin small unilamellar vesicles in solid tumors. Cancer Res 1992; 52: 3255–3261. [PubMed] [Google Scholar]
- 77. Lowis S, Lewis I, Elsworth A, et al. A phase 1 study of intravenous liposomal daunorubicin (DaunoXome) in pediatric patients with relapsed or resistant solid tumors. Br J Cancer 2006; 95: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gill PS, Espina BM, Muggia F, et al. Clinical and pharmacokinetic evaluation of liposomal Daunorubicin. J Clin Oncol 1995; 13: 996–1003. [DOI] [PubMed] [Google Scholar]
- 79. Guarneri V, Dieci MV, Conte P. Enhancing intracellular taxane delivery: current role and perspectives of nanoparticle albumin—bound paclitaxel in the treatment of advanced breast cancer. Expert Opin Pharmacother 2012; 13(3): 395–406. [DOI] [PubMed] [Google Scholar]
- 80. Von Hoff DD, Ramanathan R, Borad M, et al. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol 2011; 29(34): 4548–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Desai N, Teieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin Cancer Res 2006; 12(4): 1317–1324. [DOI] [PubMed] [Google Scholar]
- 82. Desai N, Trieu V, Yao Z, et al. Increased endothelial transcytosis of nanoparticle albumin-bound paclitaxel (ABI-007) by endothelial gp60 receptors: a pathway inhibited by Taxol. In: Proceedings of SABCS, San Antonio, TX, December 13–16, 2004, abstract no. 1071. [Google Scholar]
- 83. Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother 2006; 7(8): 1041–1053. [DOI] [PubMed] [Google Scholar]
- 84. Singer JW. Paclitaxel poliglumex (XYOTAX™, CT-2103): a macromolecular taxane. J Control Release 2005; 109(1–3): 120–126. [DOI] [PubMed] [Google Scholar]
- 85. Chipman SD, Oldham FB, Gabriella Pezzoni G, et al. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer–drug conjugate. Int J Nanomedicine 2006; 1(4): 375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Eichhorn ME, Ischenko I, Luedemann S, et al. Vascular targeting by EndoTAG-1 enhances therapeutic efficacy of conventional chemotherapy in lung and pancreatic cancer. Int J Cancer 2010; 126(5): 1235–1245. [DOI] [PubMed] [Google Scholar]
- 87. Strathopoulos GP, Boulikias T. Lipoplatin formulation, review. J Drug Deliv 2012; 2012: 581363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Boulikias T. Clinical overview of Lipoplatin, a successful liposomal formulation of cisplatin. Expert Opin Investig Drugs 2009; 18(8): 1–22. [DOI] [PubMed] [Google Scholar]
- 89. Mayer LD, Bally MB, Loughrey H, et al. Liposomal vincristine preparations which exhibit decreased drug toxicity and increased activity against murine LI 210 and P 388 tumors. Cancer Res 1990; 50: 575–579. [PubMed] [Google Scholar]
- 90. Igor VZ, Norbert M, Quet-Fah A, et al. Liposome-encapsulated vincristine, vinblastine and vinorelbine: a comparative study of drug loading and retention. J Control Release 2005; 104(1): 103–111. [DOI] [PubMed] [Google Scholar]
- 91. Yang SH, Lin CC, Lin ZZ, et al. A phase 1 pharmacokinetic study of liposomal vinorelbine in patients with advanced solid tumor. Invest New drugs 2012; 30: 282–289. [DOI] [PubMed] [Google Scholar]
- 92. Mantripragada S. A lipid based depot (DepoFoam® technology) for sustained release drug delivery. Prog Lipid Res 2002; 41(5): 392–406. [DOI] [PubMed] [Google Scholar]
- 93. Angst MS, Drover DR. Pharmacology of drugs formulated with DepoFoam: a sustained release drug delivery system for parenteral administration using multivesicular liposome technology. Clin Pharmacokinet 2006; 45(12): 1153–1176. [DOI] [PubMed] [Google Scholar]
- 94. Benesch M, Urban C. Liposomal cytarabine for leukemic and lymphomatous meningitis: recent developments. Expert Opin Pharmacother 2008; 9(2): 301–309. [DOI] [PubMed] [Google Scholar]
- 95. Chamberlain MC, Kormanik P, Howell SB, et al. Pharmacokinetics of intralumbar DTC-101 for the treatment of leptomeningeal metastases. Arch Neurol 1995; 52: 912–917. [DOI] [PubMed] [Google Scholar]
- 96. Kim S, Chatelut E, Kim JC, et al. Extended CSF cytarabine exposure following intrathecal administration of DTC 101. J Clin Oncol 1993; 11: 2186–2219. [DOI] [PubMed] [Google Scholar]
- 97. Pearce HL, Winter MA, Beck WT. Structural characteristics of compounds that modulate p-glycoprotein-associated multidrug resistance. Adv Enzyme Regul 1990; 30: 357. [DOI] [PubMed] [Google Scholar]
- 98. Boozer DJ, Esteva FJ, Rivera E, et al. Phase II study of annamycin in the treatment of doxorubicin resistant breast cancer. Cancer Chemother Pharmacol 2002; 1: 6–8. [DOI] [PubMed] [Google Scholar]
- 99. Alfieri DR. Pegaspargase. Pediatr Nurs 1995; 21(5): 471–474. [PubMed] [Google Scholar]
- 100. Avramis VI, Tiwari PN. Asparaginase (native ASNase or PEGylated ASNase) in the treatment of acute lymphoblastic leukemia. Int J Nanomedicine 2006; 1(3): 241–254. [PMC free article] [PubMed] [Google Scholar]
- 101. Zeidan A, Wang ES, Wetzler M. Pegasparaginase: where do we stand? Expert Opin Biol Ther 2009; 9(1): 111–119. [DOI] [PubMed] [Google Scholar]
- 102. Emerson DL, Bendele R, Brown E, et al. Antitumor efficacy, pharmacokinetics, and biodistribution of NX 211: a low clearance liposomal formulation of lurtotecan. Clin Cancer Res 2000; 6: 2903–2912. [PubMed] [Google Scholar]
- 103. Dark GG, Calvert AH, Grimshaw R, et al. Randomized trial of two intravenous schedules of the topoisomerase inhibitor liposomal lurtotecan in women with relapsed epithelial ovarian cancer: a trial of the national cancer institute of Canada clinical trials group. J Clin Oncol 2005; 23: 1859–1866. [DOI] [PubMed] [Google Scholar]
- 104. Nastruzzi C, Walde P, Menegatti E, et al. Liposome-associated retinoic acid. Increased in vitro antiproliferative effects on neoplastic cells. FEBS Lett 1990; 259: 293–296. [DOI] [PubMed] [Google Scholar]
- 105. Parthasarathy R, Sacks PG, Harris D, et al. Interaction of liposome-associated all-trans-retinoic acid with squamous carcinoma cells. Cancer Chemother Pharmacol 1994; 34: 527–534. [DOI] [PubMed] [Google Scholar]
- 106. Ozpolat B, Lopez-Berestein G, Adamson P, et al. Pharmacokinetics of intravenously administered liposomal all-trans-retinoic acid (ATRA) and orally administered ATRA in healthy volunteers. J Pharmaceut Sci 2003; 6(2): 292–301. [PubMed] [Google Scholar]
- 107. Shawo J, Griffin RJ, Galanzha EI, et al. Photothermal nanodrugs: potential of TNF- gold nanospheres for cancer theranostics. Sci Rep 2013; 3: 1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Day ES, Zhang L, Thompson PA, et al. Vascular-targeted photothermal therapy of an orthotopic murine glioma model. Nanomedicine 2012; 7(8): 1133–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Morton JG, Day ES, Halas NJ, et al. Nanoshells for photothermal cancer therapy. Methods Mol Biol 2010; 624: 101–117. [DOI] [PubMed] [Google Scholar]
- 110. Schwartz JA, Shetty AM, Price RE, et al. Feasibility study of particle-assisted laser ablation of brain tumors in orthotopic canine model. Cancer Res 2009; 69(4): 1659–1667. [DOI] [PubMed] [Google Scholar]
- 111. Stern JM, Stanfield J, Kabbani W, et al. Selective prostate cancer thermal ablation with laser activated gold nanoshells. J Urol 2008; 179(2): 748–753. [DOI] [PubMed] [Google Scholar]
- 112. O’Neal DP, Hirsch LR, Halas NJ, et al. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett 2004; 209(2): 171–176. [DOI] [PubMed] [Google Scholar]
- 113. Guo P. The emerging field of RNA nanotechnology. Nat Nanotechnol 2010; 5(12): 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Khaled A, Guo S, Li F, et al. Controllable self-assembly of nanoparticles for specific delivery of multiple therapeutic molecules to cancer cells using RNA technology. Nano lett 2005; 5(9): 1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Guo S, Tschammer N, Mohammed S, et al. Specific delivery of therapeutic RNA to cancer cells via the dimerization mechanism of phi29 motor RNA. Hum Gen Ther 2005; 16(9): 1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Lee JM, Yoon TJ, Cho YS. Recent developments in nanoparticle-based siRNA delivery for cancer therapy. Biomed Res Int 2013; 213: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]