

Abstract
In Alzheimer's disease and tauopathies, tau protein aggregates into neurofibrillary tangles that progressively spread to synaptically connected brain regions. A prion‐like mechanism has been suggested: misfolded tau propagating through the brain seeds neurotoxic aggregation of soluble tau in recipient neurons. We use transgenic mice and viral tau expression to test the hypotheses that trans‐synaptic tau propagation, aggregation, and toxicity rely on the presence of endogenous soluble tau. Surprisingly, mice expressing human P301Ltau in the entorhinal cortex showed equivalent tau propagation and accumulation in recipient neurons even in the absence of endogenous tau. We then tested whether the lack of endogenous tau protects against misfolded tau aggregation and toxicity, a second prion model paradigm for tau, using P301Ltau‐overexpressing mice with severe tangle pathology and neurodegeneration. Crossed onto tau‐null background, these mice had similar tangle numbers but were protected against neurotoxicity. Therefore, misfolded tau can propagate across neural systems without requisite templated misfolding, but the absence of endogenous tau markedly blunts toxicity. These results show that tau does not strictly classify as a prion protein.
Keywords: Alzheimer's disease, neurodegeneration, neurofibrillary tangles, P301L tau, prion‐like
Subject Categories: Neuroscience
Introduction
Tau aggregation and neurofibrillary tangle (NFT) formation are key features of Alzheimer's disease (AD) and other tauopathies (Hyman et al, 1984; Lee et al, 2001). Progressive tangle appearance in AD follows a highly consistent pattern: Starting in the entorhinal cortex (EC), NFT pathology then spreads to connected regions (Hyman et al, 1984; Braak & Braak, 1991), which subsequently undergo synaptic and neuronal loss leading to progressive cognitive failure. A prion‐like model of tau propagation has been proposed, in which aberrantly folded tau travels (propagates) trans‐synaptically, then recruits naive non‐misfolded tau—transgenic or endogenous—in recipient neurons, and templates its toxic aggregation, linking tau propagation, aggregation, and toxicity (Clavaguera et al, 2010; Walker et al, 2013; Sanders et al, 2014). Consistent with this idea, small amounts of mouse tau can co‐aggregate with human tau in tau transgenic mouse models (de Calignon et al, 2012; Van der Jeugd et al, 2012). In mice, restriction of human mutant P301Ltau expression to the EC (ECrTgTau model) mimics some aspects of the earliest disease stages in AD, and aged ECrTgTau mice revealed trans‐synaptic propagation of tau pathology to recipient neurons in the dentate gyrus (DG) (de Calignon et al, 2012; Harris et al, 2012; Liu et al, 2012). Mature silver stainable tangles in the DG formed gradually starting at the age of ~20 months. By analogy to the spreading of prion protein misfolding (Brandner et al, 1996b), the absence of endogenous tau would be expected to stop the propagation of misfolded tau—unless trans‐synaptic traveling of tau and accumulation in recipient cells is independent of recruitment and corruption of endogenous substrate.
In prion disease, the absence of endogenous PrPC prevents toxicity (Brandner et al, 1996a). In P301Ltau‐overexpressing mice, the continued presence of high levels of transgenic P301Ltau would be expected to be sufficient to cause tau toxicity, regardless of the presence or absence of endogenous tau. However, the mechanism of NFT toxicity is unclear; among the possibilities are a loss of soluble tau function upon aggregation, for example, its microtubule binding (Trinczek et al, 1995) or synapse recruitment (Ittner et al, 2010), or a gain of tau aggregate toxicity, for example, through the unfolded protein response and proteasome inhibition (Keck et al, 2003; Nijholt et al, 2012). Alternatively, soluble tau species rather than fibrillar aggregates have been implicated in tau toxicity (SantaCruz et al, 2005; Kopeikina et al, 2012; Lasagna‐Reeves et al, 2012; Tai et al, 2012).
To compare how closely tau neurobiology matches expectations based on analogy to prion behavior, we examined the effect of a tau‐null background (Mapt 0/0) in two separate tau transgenic models: The first, ECrTgTau, was designed to examine human mutant tau propagation. The second, rTg4510, is associated with marked tau toxicity. We further verified our findings using adeno‐associated virus‐mediated expression of tau in wild‐type and tau‐null mice. Our experiments show that tau propagation between neurons occurs independently of naïve endogenous tau, and thus, stable tau accumulation in recipient neurons does not rely exclusively on prion‐like templated misfolding. With regard to toxicity, however, the absence of endogenous tau leads to markedly diminished mutant tau‐induced neurotoxicity, gliosis, and brain atrophy (despite similar NFT formation), reinforcing the model that tau aggregation per se is not the crucial element underlying tau‐induced neurotoxicity.
Results
Transgenic mutant tau propagates without endogenous tau
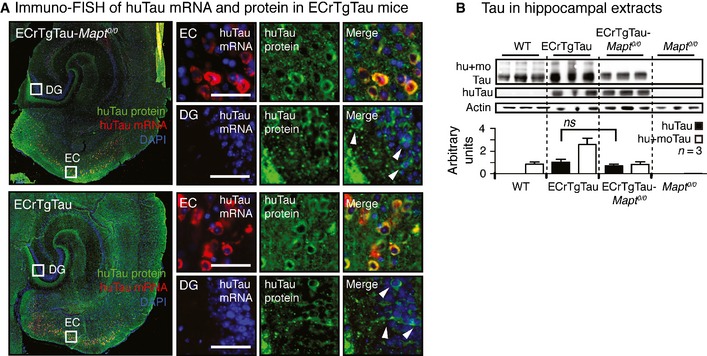
Transgenic tau propagation along the perforant pathway from transgene‐expressing neurons in layer II/III of the entorhinal cortex (EC) to transgene‐negative neurons in the dentate gyrus (DG) has previously been observed in aged ECrTgTau mice on a wild‐type background (de Calignon et al, 2012; Liu et al, 2012). To determine whether endogenous tau is needed for human transgenic tau to travel from neuron to neuron, we crossed ECrTgTau mice onto a tau‐null (Mapt 0/0) background (Figs 1A and EV1A). Surprisingly, immunostaining for human tau (using human tau N‐terminus‐specific antibodies Tau13 or TauY9) in horizontal brain sections from 18‐month‐old mice revealed robust propagation of transgenic tau to DG neurons in mice that expressed human P301Ltau in the EC in the absence (ECrTgTau‐Mapt 0/0) and presence (ECrTgTau) of endogenous mouse tau (Fig 1B–E).
Figure 1. Trans‐synaptic propagation of human tau in ECrTgTau mice in the absence of endogenous mouse tau.

- 3D brain model, horizontal brain section illustrating transgenic human P301Ltau expression in the entorhinal cortex (green, EC) of the ECrTgTau mouse lines, and the propagation of transgenic tau to the dentate gyrus (DG). Tau composition in ECrTgTau and control mouse lines investigated.
- Immunostained horizontal sections show the expression of human P301Ltau in EC neurons in the absence of endogenous mouse tau (ECrTgTau‐Mapt 0/0). Fluorescence in situ hybridization of human tau mRNA combined with immunofluorescence labeling (immuno‐FISH) of human tau protein (huTau) verifies P301Ltau transgene expression in the EC. Scale bars, 50 μm.
- Propagation of human tau protein to neurons in the DG (white arrowheads) in ECrTgTau‐Mapt 0/0 mice. Close‐ups show DG neurons from three ECrTgTau‐Mapt 0/0 mice (DG I‐III). Immuno‐FISH proofs the absence of human tau expression in DG neurons, which have huTau protein but no human tau mRNA. Scale bars, 50 μm.
- Immunostained horizontal sections of ECrTgTau mice show the expression of human P301Ltau in EC neurons in the presence of endogenous mouse tau. Immuno‐FISH proofs the absence of human tau expression in these DG neurons. Scale bars, 50 μm.
- Human P301Ltau propagation to DG neurons (white arrowheads) in the presence of endogenous mouse tau in ECrTgTau mice. Close‐ups show DG neurons from three ECrTgTau mice (DG I‐III). Scale bars, 50 μm.
- Human (huTau, antibody Tau13) and total tau (hu+moTau, DAKO) levels in entorhinal cortex (EC) extracts from 18‐month‐old mice show equal human P301Ltau expression in ECrTgTau and ECrTgTau‐Mapt 0/0 mice (Mean ± SEM, P = 0.201, n = 3 mice/group, one‐way ANOVA with Bonferroni correction).
- The number of human tau‐positive cell bodies in the DG (Mean ± SEM, P = 0.58, n = 4 sections and 3 mice/group) and human tau in hippocampal (HPC) extracts (P = 0.14, n = 3 mice/group) were similar in ECrTgTau‐Mapt 0/0 and ECrTgTau mice (two‐tailed Student's t‐test).
Source data are available online for this figure.
Figure EV1. Genotype confirmation of ECrTgTau(‐Mapt 0/0) mice by qPRC .

qPCR using RNA extracted from fresh frozen brain tissue of 18‐month‐old tau knockout mice (Mapt 0/0), ECrTgTau, and ECrTgTau crossed to Mapt 0/0 mice (ECrTgTau‐Mapt 0/0, n = 4 mice/group) was transcribed into cDNA using primers recognizing human tau transgene (P301L tau, top) or genomic mouse tau (MapT, bottom). GAPDH mRNA was co‐transcribed as amplification control. ECrTgTau‐Mapt 0/0 and ECrTgTau mice showed human tau expression, and Mapt 0/0 and ECrTgTau‐Mapt 0/0 mice lacked mouse tau expression.
To confirm that that DG neurons containing human tau protein did not (aberrantly) express transgenic human tau mRNA due to promoter leakiness (de Calignon et al, 2012; Yetman et al, 2015), we employed fluorescent in situ hybridization of human tau mRNA and combined it with immunofluorescence labeling of human tau protein (immuno‐FISH; Figs 1B–E and EV2A). ECrTgTau‐Mapt 0/0 and ECrTgTau mice showed co‐localization of human tau mRNA and human tau protein (huTau) in transgene‐expressing neurons in the EC, whereas—in the same brain section—no human tau mRNA was found in any of the human tau protein‐containing DG neurons, convincingly demonstrating true propagation of human tau protein to DG neurons in both mouse lines.
Figure EV2. DG neurons do not express but have human tau in ECrTgTau(‐Mapt 0/0 ) mice.
- Representative images of the fluorescence in situ hybridization of transgenic human tau mRNA combined with immunofluorescence labeling of human tau protein (Tau13 antibody) shows human tau protein (green) in neuronal cell bodies in the EC and the DG (white arrowheads), but human tau mRNA only in EC neurons both in ECrTgTau‐Mapt 0/0 and ECrTgTau mice. n = 3 sections/mouse and 3 mice/group. Scale bars, 50 μm.
- Human tau (huTau, Tau13 antibody) and total tau (hu+moTau, DAKO antibody) levels in hippocampal extracts from 18‐month‐old mice are similar in ECrTgTau and ECrTgTau‐Mapt 0/0 mice. Mean ± SEM, n = 3 mice/group, two‐tailed Student's t‐test. ns, non‐significant.
Source data are available online for this figure.
In ECrTgTau‐Mapt 0/0 mice, which had equivalent levels of huTau expression in the EC (Fig 1F), the number of human tau‐containing neurons in the DG was similar to ECrTgTau mice, and Western blot analysis revealed comparable levels of transgenic tau in hippocampal (HPC) extracts (Figs 1G and EV2B). Co‐immunostaining of human tau with N‐ and C‐terminal antibodies suggested the propagation of full‐length tau (Fig EV3A), and co‐staining of human tau with Parvalbumin and GAD67, GFAP, and Iba1 showed only few inhibitory neurons in the DG tau has propagated to, but no glia cells (Fig EV3B and C).
Figure EV3. Propagation of full‐length tau to neurons not glia in the DG of ECrTgTau(‐Mapt 0/0 ) mice.

-
AHorizontal ECrTgTau‐Mapt 0/0 brain section co‐immunolabeled with Tau13 (mouse antibody recognizing the N‐terminal end of human not mouse tau; epitope: aa20–35; red) and DAKO (polyclonal rabbit antibody recognizing the C‐terminal half of all mouse and human tau; epitope: multiple sites in aa243–441; green). Human tau in cell bodies in both EC and DG neurons (white arrowheads) was recognized by both antibodies against the N‐terminus and the C‐terminal half, suggesting the trans‐synaptic propagation of full‐length tau. Scale bars, 50 μm.
-
B, CCo‐immunostaining of human tau with GAD67 and Parvalbumin suggest the propagation of tau to a few GABAergic interneurons (white arrowheads) in the DG of ECrTgTau‐Mapt 0/0 (B) and ECrTgTau (C) mice. Astrocytes (GFAP) and microglia (Iba1) did not have human tau in either mouse line. n = 4 sections/mouse, 3 mice/group. Scale bars, 50 μm.
Additionally, to further verify that human tau can propagate across synapses in tau‐null animals, we used a non‐transgenic approach, in which we injected adeno‐associated virus (AAV) encoding P301Ltau into the EC of adult mice (Fig 2A) (Siman et al, 2013). In this approach, EC neurons transduced with AAV eGFP‐2a‐huTauP301L (“tau donor neurons”) expressed eGFP and human P301Ltau as individual proteins (~95%; ~5% eGFP‐2a‐P310Ltau fusion protein; Fig 2B). “Tau recipient neurons,” which have P301Ltau but no eGFP, can readily be identified after immunostaining brain sections for human tau. Eight weeks after AAV injection into EC, tau recipient neurons could be found in the DG of Mapt 0/0 mice (Fig 2C). huTau propagation was also observed when AAV eGFP‐2a‐huTauP301L was injected into adult WT mice (Fig EV2D). In additional experiments, transfection of neurons in EC, DG, and HPC area CA1 (Fig 2D) led to long‐distance propagation of human tau aggregates into recipient neurons in the contralateral EC (Fig 2E).
Figure 2. P301Ltau propagation after viral expression in the entorhinal cortex.
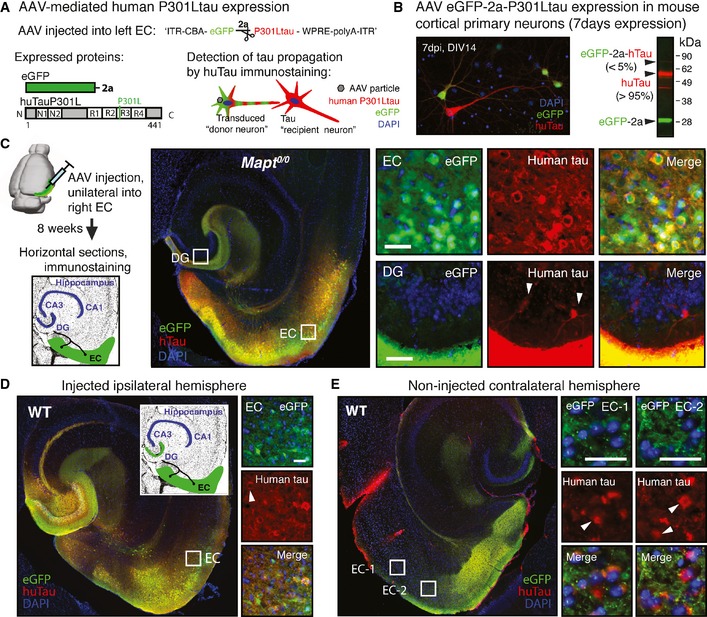
- Adeno‐associated virus (AAV) construct designed for expression of eGFP and human P301Ltau as individual proteins, separated by the self‐cleaving 2a peptide, under the CBA promoter (AAV8 CBA‐eGFP‐2a‐huTauP301L). AAV‐transduced “donor neurons” express eGFP and huTauP301L, and tau “recipient neurons” are identified after immunostaining for human tau as huTau+ but GFP − neurons.
- Primary cortical neuron cultures that were transduced with AAV eGFP‐2a‐P301Ltau at 7 DIV, and fixed and immunostained for GFP and human tau (Tau13 antibody) at 14 DIV, show tau donor (GFP +, huTau+; ˜10% neurons) and a small number of tau recipient neurons (GFP −, huTau+; ˜1% neurons). Western blot of whole cell lysates verified efficient cleavage (˜95%) of eGFP and P301Ltau by the 2a peptide (n = 3).
- Eight weeks after AAV injection into right EC of aged Mapt 0/0 mice (n = 3), immunostained brain sections showed that huTauP301L (red) propagated to a few DG “recipient neurons” (white arrowheads). Scale bar, 50 μm.
- Unilateral AAV‐mediated human P301L tau expression in the EC and DG of age‐matched WT mice (n = 3). Representative images of brain sections show donor neurons in the injected EC and DG, and a few tau recipient neurons (white arrowheads) adjacent to the AAV injection site. Scale bar, 50 μm.
- In the contralateral hemisphere of the same brain section as in (D), some tau recipient neurons (white arrowheads) were also present in the (non‐injected) axonal projection areas in the contralateral EC (GFP‐filled terminal ends). Scale bar, 100 μm.
These results show that tau propagation occurs in the absence of endogenous tau and has a similar propagation rate as in the presence of endogenous tau. These findings contrast with expectations from PrP‐prion behavior, in which propagation of misfolded prion protein (PrPSc) is facilitated through templated misfolding and thus calls for the presence of naïve endogenous prion protein (PrPC). The propagation of mutant tau—here reported in ECrTgTau mice and after AAV injection—instead can occur even in the absence of endogenous tau and relies on robust interneuronal transmission of preformed tau from the donor neurons.
Reduced tau misfolding and gliosis in the absence of endogenous tau
In the case of prion protein, the absence of endogenous PrPC prevents both misfolding and toxicity (Brandner et al, 1996a); we tested whether this paradigm also holds for tau, namely that P301Ltau‐induced neurotoxicity is absent or reduced in the absence of endogenous tau.
Immunostaining of brain sections revealed misfolded tau (Alz50) in EC and DG neurons of 18‐month‐old ECrTgTau, but not ECrTgTau‐Mapt 0/0 mice (Fig 3A). We then evaluated whether the reduced tau misfolding in the absence of mouse tau was accompanied by reduced neurotoxicity. As expected from previous characterization (de Calignon et al, 2012), 18‐month‐old ECrTgTau mice showed neither P301Ltau‐induced cell death in EC layer II/III, nor obvious synaptic protein (synapsin‐1) loss (Fig EV4A and B). However, gliosis serves as an early indirect sign of inflammation and likely neuronal damage (Serrano‐Pozo et al, 2011). We found a significantly higher number of microglia in the EC of ECrTgTau mice compared to WT mice, which were partially rescued in ECrTgTau‐Mapt 0/0 mice, which were partially rescued in ECrTgTau‐Mapt 0/0 mice (Fig 3B). The number of astrocytes in the HPC was similar across genotypes (Fig 3C).
Figure 3. Tau phosphorylation, misfolding, and gliosis in ECrTgTau(‐Mapt 0/0) mice.

-
ABrain sections from 18‐month‐old ECrTgTau‐Mapt 0/0 and ECrTgTau mice were co‐immuolabeled for human tau and misfolded tau (Alz50). Misfolded tau was only found in EC and DG neurons (white arrowheads) of ECrTgTau, but not ECrTgTau‐Mapt 0/0 animals (n = 4 sections/mouse, 3 mice/group). Scale bars, 50 μm.
-
B, CImmunofluorescence labeling and stereological counting of microglia in entorhinal cortex (B) and astrocytes in hippocampus (C) indicated early signs of neurodegeneration in ECrTgTau mice. The significantly increased number of Iba1‐positive microglia in the EC layer II/III of ECrTgtau mice (compared to WT) was partially rescued in ECrTgTau‐Mapt 0/0 mice (non‐significant). The number of GFAP‐positive astrocytes was similar across all genotypes (non‐significant). Mean ± SEM, n = 4 sections per mouse, 3 mice/group, one‐way ANOVA with Bonferroni correction. Scale bars, 100 μm.
Figure EV4. Increased phospho‐tau and axonal changes in ECrTgTau mice.
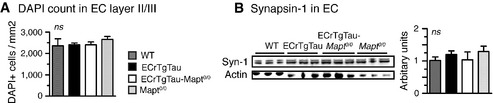
- Stereological counting of DAPI nuclei in EC layer II/III suggested no obvious neuronal loss in ECrTgTau(‐Mapt 0/0) mice at 18 months of age.
- Western blotting for pre‐synaptic marker synapsin‐1 (Syn‐1) showed similar levels, indicating no major synapse loss in ECrTgTau(‐Mapt 0/0) mice at 18 months of age.
These data reinforce the idea that glial response precedes overt neurodegeneration (Yoshiyama et al, 2007) and that the absence of endogenous tau attenuates early mutant tau‐induced pathological changes.
Tau knockout rescues P301Ltau‐induced neurotoxicity
Although tau aggregation and toxicity are frequently viewed as linked phenomena, recent data suggest the dissociation of tangle formation from acute neuronal death (de Calignon et al, 2010) or functional alterations (Kuchibhotla et al, 2014; Rudinskiy et al, 2014). Our results from ECrTgTau‐Mapt 0/0 mice suggested that removal of endogenous tau rescues early signs of neurodegeneration. We next explored the impact of mouse tau knockout on neurotoxicity and late‐stage tau aggregation in a more aggressive model with forebrain‐wide human P301Ltau expression (rTg4510) that develops robust NFT pathology and brain atrophy at 8–10 months of age (SantaCruz et al, 2005). We crossed this model onto a tau‐null background (rTg4510‐Mapt 0/0) and compared pathology of 9‐month‐old animals.
As expected, rTg4510‐ Mapt 0/0 had no mouse tau, similar amounts of human tau (huTau), and decreased total tau (hu+moTau) compared to rTg4510 mice (Fig 4A). We found that the whole brain weight of rTg4510 mice was reduced by ~16% (Fig 4B) mainly due to loss of cortical tissue, which decreased in thickness by ~26% compared to WT and Mapt 0/0 mice (Fig 4C). Impressively, this major loss of brain matter was virtually fully rescued in rTg4510‐Mapt 0/0 mice (brain weight: ~96%; CTX thickness: ~96%). Furthermore, in rTg4510 mice, the number of cortical neurons decreased by ~33% compared to WT mice, whereas rTg4510‐Mapt 0/0 mice showed no neuronal loss (Fig 4D). Neuron and volume loss in CA1 of rTg4510s, with cortex the most effected region in this model, were partially rescued in rTg4510‐Mapt 0/0 animals (Fig 4E).
Figure 4. Tau knockout rescues P301Ltau‐induced atrophy and neurodegeneration.
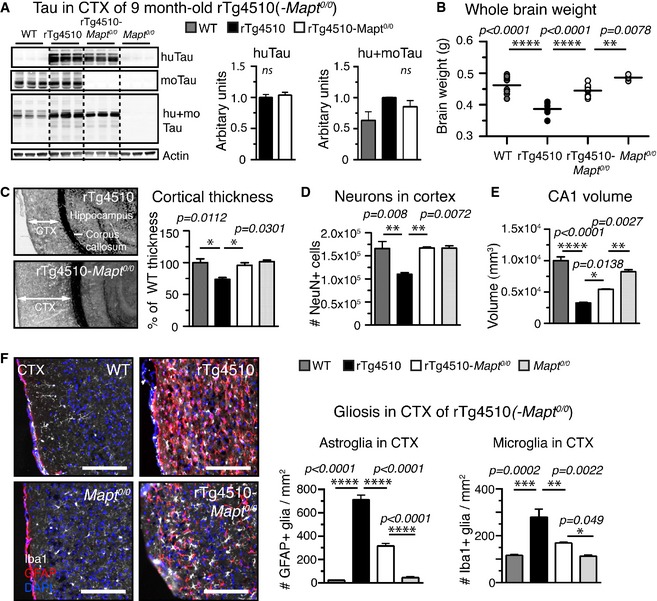
- Human, mouse, and total tau protein levels in cortical TBS‐extracts of rTg4510, rTg4510‐Mapt 0/0, and control mice: The amount of human tau (Tau13 antibody) was comparable in rTg4510 and rTg4510‐Mapt 0/0, moTau (Tau/5) was comparable in WT and rTg4510, and total tau levels (hu+moTau, DAKO antibody) were (expected) highest in rTg4510 mice. n = 3 mice/group, non‐significant.
- Whole brain weights of 9‐month‐old animals revealed pronounced brain matter loss in rTg4510 compared to WT mice (weight loss > 16%), which was rescued in rTg4510‐Mapt 0/0 mice to > 96%. n = 5 mice/group.
- Cortical thickness measured adjacent to HPC, from CTX surface to corpus callosum, was decreased in rTg4510 mice by ˜25% compared to WT mice. rTg4510‐Mapt 0/0 showed no CTX thinning compared to Mapt 0/0 or WT mice. n = 3 mice/group.
- The number of neurons (NeuN+ cells) in the cortex of rTg4510 mice was significantly reduced to ˜67% compared to both WT and rTg4510‐Mapt 0/0. n = 3 mice/group.
- The volume of hippocampal region CA1, with CT the most affected regions in rTg4510 mice, was significantly reduced in rTg4510 by ˜70% volume; rTg4510‐Mapt 0/0 had significantly larger CA1 volume left (reduced by only ˜40%). n = 3 mice/group.
- rTg4510 showed strong signs of neuroinflammation with extremely high numbers of activated astroglia (GFAP +, red) and microglia (Iba1+, white) in the CTX compared to WT mice. Both astro‐ and microgliosis were reduced by ˜50% in rTg4510‐Mapt 0/0 mice., n = 3 sections/mouse and 5 mice per/group. Scale bars, 100 μm.
An additional cohort of animals was aged further to explore whether the loss of endogenous tau persists to be protective, also in the face of a more severe tau‐induced neurodegeneration. As expected, brain weight, cortical thickness, and the number of cortical neurons decreased even further in 12‐month‐old rTg4510 mice (compared to WT: brain weight loss ~21%, decease in cortical thickness ~46%, loss of cortical neurons ~37%; Fig EV5). At this point, markedly reduced atrophy was observed in rTg4510‐Mapt 0/0 mice (compared to Mapt 0/0: brain weight loss ~15%, decease in cortical thickness ~29%, loss of cortical neurons ~12%).
Figure EV5. Reduced neurodegeneration in 12‐month‐old rTg4510‐Mapt 0/0 mice.

- Whole brain weights of 12‐month‐old animals show severe brain matter loss in rTg4510 compared to WT mice (weight loss > 23%), which was partially rescued in rTg4510‐Mapt 0/0 mice.
- Cortical thickness, measured from CTX surface to corpus callosum, decreased in rTg4510 mice at 12 months by ˜50% compared to WT, and rTg4510‐Mapt 0/0 showed CTX thinning of ˜30% at 12 months.
- The number of cortical neurons (NeuN+ cells) in 12‐month‐old rTg4510 mice was significantly reduced (˜63% of WT). The number of neurons in rTg4510‐Mapt 0/0 slightly decreased to ˜88% of Mapt 0/0 mice (ns).
We next evaluated the degree of gliosis in the presence and absence of endogenous tau. Immunostaining and stereological assessment revealed an increase in activated astrocytes (GFAP) and overall microglia (Iba1) in the cortex of rTg4510, which was dramatically reduced by ~50% in rTg4510‐Mapt 0/0 mice (Fig 4E). An excess of misfolded proteins, including P301Ltau, can overwhelm the unfolded protein response and proteasome, leading to an accumulation of ubiquitinated proteins (Canu et al, 2000; Tai et al, 2012). Accordingly, the amount of ubiquitin was increased both in rTg4510 and in rTg4510‐Mapt 0/0 compared to control mice (Fig 4G). The levels of CHOP, indicative of ER stress, were slightly increased only in rTg4510, but not in rTg4510‐Mapt 0/0 mice (Fig EV6).
Figure EV6. Elevated ER stress in the presence of mouse tau in rTg4510 mice.

Western blot analysis of extracts from 9‐month‐old mice revealed equally high ubiquitin levels in rTg4510 and rTg4510‐Mapt 0/0 mice compared to controls; P301ltau aggregates appear to expose a similar challenge to the proteasome both in the presence and in the absence of mouse tau. Levels of the endoplasmatic reticulum (ER) stress marker CHOP seemed elevated only in rTg4510 mice, suggesting less ER stress and subsequent triggering of apoptotic pathways in the absence of mouse tau. Mean ± SEM, n = 3 mice/group, one‐way ANOVA with Bonferroni. ns, not significant.Source data are available online for this figure.
NFTs accumulate, but are less toxic in tau knockout mice
Next, we evaluated whether the absence of endogenous tau also reduces pathological changes in tau—phosphorylation and aggregation into NFTs—in aged rTg4510 mice, which show increasing tangle accumulation from 8th month onwards. To test the hypothesis that the absence of endogenous mouse tau delays pathological alterations in tau, we analyzed tau phosphorylation using phospho‐tau‐specific antibodies CP13 (pS202/pS205; AT8 analog), PHF1 (pS395/pS404), 12E8 (pS262/pS356), and mature NFT formation using classic silver stains. All antibodies showed an increase in phospho‐tau in rTg4510 mice. Interestingly, phospho‐tau recognized by CP13 (early tau phosphorylation marker) and PHF1 (pathological tau marker) was reduced by ~20–30% (Fig 5A), in rTg4510‐Mapt 0/0, whereas 12E8 (a marker for tau dissociated from microtubules (Singh et al, 1996; Augustinack et al, 2002) was similar to rTg4510 mice. In contrast to the clear but modest differences observed with phospho‐tau antibodies, silver stains (Gallyas, 1971) of 12‐month‐old mice revealed a striking difference in the extent of mature tangles in the cortex (Fig 5B). Silver‐positive NFTs also developed in the cortex of rTg4510‐Mapt 0/0 mice, but there was a “healthier” neuropil (less axonal and dendritic silver staining) at 12 months, which can even be appreciated in thioflavine‐S stains of 9‐month‐old mice (Fig 5C).
Figure 5. Reduced P301Ltau and NFT neurotoxicity in the absence of endogenous tau.
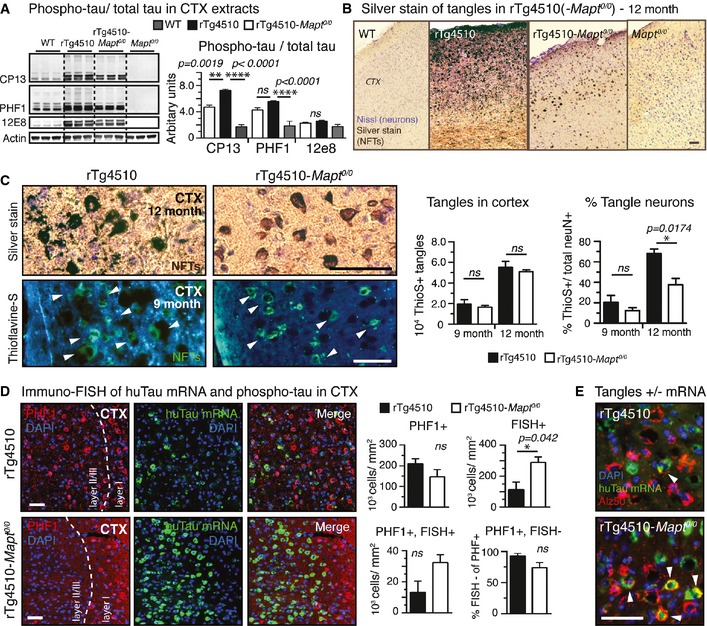
- Cortical extracts from rTg4510‐Mapt 0/0 brains had significantly less phospho‐tau (CP13, PHF1, 12E8) than rTg4510 extracts. Compared to WT mice, both transgenic tau lines had high levels of phospho‐tau. n = 3 mice/group.
- Representative images of gallyas silver‐stained aggregated tau in cortices from 12‐month‐old mice unravel stunning differences in the degree of tau pathology in rTg4510 compared to rTg4510‐Mapt 0/0 mice. n = 3 mice/group.
- Higher magnification images of silver (12‐month‐old) and thioflavine‐S (9‐month‐old)‐stained cortices show mature tangles (white arrowheads in Thio‐S stain) in rTg4510 and rTg4510‐Mapt 0/0 mice; enhanced pathological changes such as neuritic tau accumulation and neuropil vacuolation around NFTs are found only in rTg4510 mice. Stereological counting revealed similar numbers of cortical NFTs between rTg4510 and rTg4510‐Mapt 0/0 mice at 9 and 12 months of age. Because of the pronounced neuronal death only in rTg4510 mice, the percentage of tangle‐bearing neurons was ˜1.6‐ to 1.8‐fold higher in 9‐ and 12‐month‐old rTg4510 mice. n = 3 sections/mouse, 3 mice/group.
- Immuno‐FISH for huTau mRNA (green) and phospho‐tau (PHF1, red) shows obvious differences in the distribution of neurons in cortex layer II/III: 9‐month‐old rTg4510 mice had more neurons filled with NFT‐like phosphorylated tau (PHF1+, red), and rTg4510‐Mapt 0/0 mice had significantly more huTau mRNA‐positive neurons (FISH +). rTg4510‐Mapt 0/0 mice had also more neurons still expressing both PHF1 and huTau mRNA (PHF1+ FISH +), suggesting a reduced neurotoxicity of P301Ltau expression in rTg4510‐Mapt 0/0 mice. n = 3 sections per mouse, 3 mice/group.
- Immuno‐FISH showing EC neurons having both misfolded somatic tau (Alz50, red) and human tau mRNA (green; white arrowheads) in rTg4510‐Mapt 0/0 but rarely in rTg4510 mice. n = 3 sections, 2 mice/group.
In the Tg4510 model, neuronal loss parallels NFT formation (Spires et al, 2006). To assess the effect of tangle pathology on neuronal loss in the setting of the tau null cross, we determined the number of cortical NFTs stereologically. The number of tangles increased with age in both rTg4510 and rTg4510‐Mapt 0/0 mice between nine and 12 months of age. rTg4510 mice had slightly more cortical tangles than rTg4510‐Mapt 0/0 mice. Because rTg4510‐Mapt 0/0 mice had similar tangle numbers but reduced neuronal loss, the percentage of neurons having a tangle was reduced in rTg4510‐Mapt 0/0 animals. The onset of pathological changes caused by P301lTau aggregation, such as cortex thinning and neuronal loss, was shifted to older ages (Fig EV7A and B).
Figure EV7. Lack of mouse tau largely delays onset of NFT‐induced neurodegeneration.

-
A, BAverage ratios of (A) tangle number:neuron loss (=tangles/([number of neurons in WT or Mapt 0/0 controls]‐[number neurons in rTg4510 or rTg4510‐Mapt 0/0]) and (B) average rations of tangle number:cortex thinning (=tangles/([CTX thickness of WT or Mapt 0/0 controls]‐[CTX thickness of rTg4510 or rTg4510‐Mapt 0/0]) at 9 and 12 months of age highlight the improved neuronal survival at given tangle load in rTg4510‐Mapt 0/0 mice. Comparing the effect of NFTs on neuronal loss (A) and cortex thinning (B) between 9‐ and 12‐month‐old animals discovers a delayed onset of pathological changes—in the context of tangles—in tau‐null animals. n = 3 mice/group.
-
CRepresentative images of an immuno‐FISH for human tau protein (TauY9 antibody, pink) and human tau mRNA (green) on brain sections of 9‐month‐old rTg4510(‐Mapt 0/0) mice. Human tau transgene expression appears diminished in the outer CTX layers in rTg4510 but not rTg4510‐MapT 0/0 animals (low magnification images). In rTg4510‐MapT 0/0 animals, more EC neurons still express human tau mRNA while having human tau protein accumulated in the somata (white arrowheads) compared to rTg4510 mice; the neurotoxicity caused by P301Ltau expression, missorting into the soma, and aggregation seems to be reduced in the absence of mouse tau. n = 4 sections/mouse, 2 mice/group. Scale bars, 100 μm.
In rTg4510 mice, which express transgenic human tau under the CamKIIα promoter, almost all excitatory cortical neurons in layer II/III and V show intense P301Ltau expression (Ramsden et al, 2005; SantaCruz et al, 2005). Neurons that express the transgene are, intuitively, the first to develop tangles and are thought to die first in this model. We hypothesized that the dissociation between tangle formation and neuronal death in the rTg4510 mice compared to the rTg4510‐Mapt 0/0 mice might lead to paradoxical preservation of neurons expressing the P301L transgene, as they are apparently spared from neuronal death in the rTg4510‐Mapt 0/0 mice. To evaluate this possibility, we detected transgenic human tau mRNA using FISH and co‐immunostained for pathological phospho‐tau (PHF1), representative for tangles if accumulated in cell bodies (Fig 5D).
Comparing the huTau mRNA expression and tangle pattern in 9‐month‐old mice, we found an obvious difference (Figs 5D and EV7C): rTg4510 showed sparse P301L mRNA expression, in comparison with rTg4510‐Mapt 0/0 cortex, which had innumerable remaining P301L mRNA‐positive neurons. Stereological quantification confirmed this impression: The overall number of PHF1‐positive tangle‐like tau inclusions was slightly reduced in rTg4510‐Mapt 0/0 compared to rTg4510 mice, not reaching statistical significance, whereas the number of human tau mRNA‐positive neurons still expressing transgenic human tau was significantly higher in rTg4510‐Mapt 0/0 mice. Moreover, the number of neurons with phospho‐tau accumulation in the soma (PHF1+) that (still) express human tau mRNA (PHF1+, FISH+) was higher in rTg4510‐Mapt 0/0. Assuming that active transgene expression indicates the functionality of a healthy neuron, the higher number of actively P301Ltau‐expressing neurons in tau‐null animals suggested a reduced toxicity of P301Ltau expression and accumulation. Accordingly, ~95% of PHF1‐positive neurons in rTg4510 showed no transgene expression (~75% in rTg4510‐MapT 0/0), likely due to the strong neurotoxicity of P301Ltau accumulation in this model. Similarly, EC neurons filled with misfolded tau (Alz50‐positive) retained human tau expression in rTg4510‐Mapt 0/0, but not in rTg4510 mice (Fig 5E). Interestingly, in both rTg4510 and rTg4510‐Mapt 0/0 mice, many neurons with somatic PHF1 accumulation lacked transgene expression (PHF+, FISH−), suggesting that “spread” of P301Ltau from transgene‐positive to transgene‐negative neurons occurs in the cortex of rTg4510 mice, analogous to our observations in the ECrTgTau model; this spread appeared not overtly affected by the presence or absence of endogenous tau. These findings further support the idea of reduced P301Ltau neurotoxicity and delayed NFT formation in the absence of endogenous tau and reveal a surprising segregation of NFTs from human tau transgene‐expressing neurons.
Pathological tau from tau‐null animals has increased detergent solubility and less seeding activity
We further examined the biochemical and biophysical properties of tau from the two preparations. The lack of endogenous tau did not appreciably change TBS‐soluble tau, but significantly more Triton X‐100 detergent‐soluble tau was observed in rTg4510‐Mapt 0/0 animals (Fig 6A). Non‐reducing gel electrophoresis (Native PAGE) showed subtle differences in higher molecular weight species of tau (Fig 6B). We hypothesized that tau from rTg4510 mice would favor tau aggregation and have higher seeding activity than tau from rTg4510‐Mapt 0/0 mice. We used a highly sensitive cell‐based seeding assay (Holmes et al, 2014), in which HEK293 cells stably express both the mutant repeat domain of tau fused to CFP (TauRDP301S‐CFP) and to YFP (TauRDP301S‐YFP). Treating these cells with brain lysate from 9‐month‐old rTg4510 mice resulted in significant more intracellular FRET‐positive aggregates than treatment with lysate from rTg4510‐Mapt 0/0 animals (Fig 6C).
Figure 6. Differences in tau oligomers and reduced seeding activity in rTg4510‐Mapt 0/0 mice.
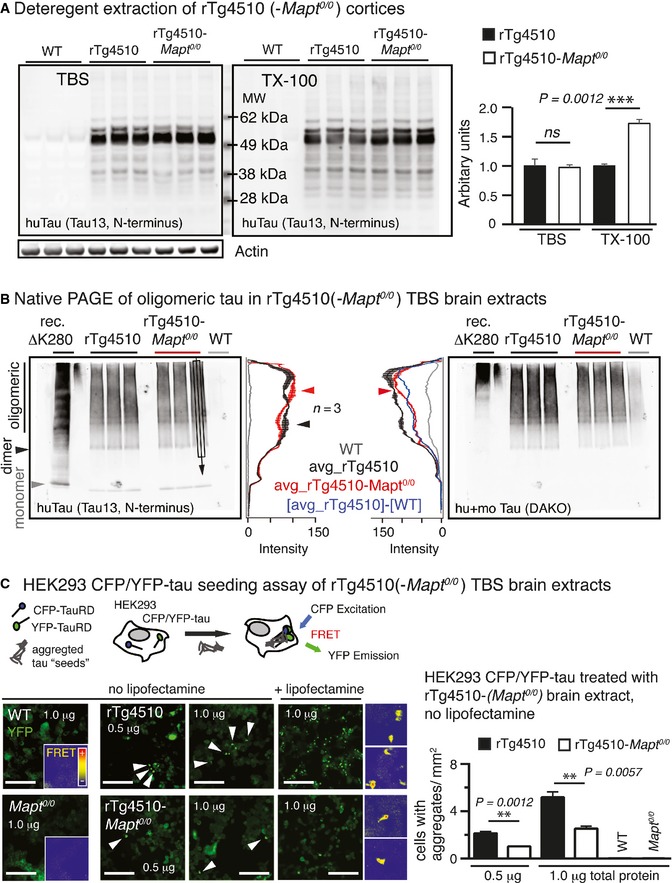
- Extraction of cortices revealed similar human tau (Tau13) in TBS‐extracts (not significant) but significantly more human tau in Triton X‐100 (TX‐100) extracts of rTg4510‐Mapt 0/0 compared to rTg4510 mice. Mean ± SEM, n = 3 mice/group, two‐tailed Student's t‐test, ns, non‐significant.
- Native gel electrophoresis of cortical TBS‐extracts showed small differences in HMW (oligomeric) human tau between rTg4510 and rTg4510‐Mapt 0/0 brains. Western blot lanes were averaged across ˜2/3 of the width (black rectangular and arrow in Tau13 blot). The mean ± SEM (n = 3 mice/group) of these averages was plotted as longitudinal lane profiles. Differences in HMW tau are indicated by red and black arrowheads.
- TBS‐brain extracts were applied to a HEK293 cell tau aggregation seeding assay (Holmes et al, 2014; Sanders et al, 2014), in which TauRDP301S‐CFP and TauRDP301S‐YFP are co‐expressed intracellularly. The formation of intracellular fluorescent TauRDP301S aggregates leads to Foerster resonance energy transfer (FRET) activity between co‐aggregated CFP and YFP‐tags and correlates with the tau aggregation seeding activity of the applied brain extracts. After 24 h, cells treated with extract (0.5 and 1.0 μg total protein per 96 well) from 9‐month‐old rTg4510 had significantly more intracellular YFP‐positive (white arrowheads) aggregates compared to rTg4510‐Mapt 0/0 extracts; FRET activity of TauRDP301S aggregates appeared similar for both rTg4510 and rTg4510‐Mapt 0/0. WT and Mapt 0/0 extracts never showed seeding activity. Addition of lipofectamine corrected for differences in cellular uptake of tau and led to similar differences in seeding activity between rTg4510 and rTg4510‐Mapt 0/0 mice. Two‐tailed Student's t‐test, mean ± SEM, n = 3 replicates. ns, not significant. Insets, 100 μm. Scale bars, 50 μm.
Discussion
The hypothesis that tau has prion‐like properties has evolved from recent work on tau protein aggregation seeding, toxicity, and propagation. Tau has been suggested to be “prionoid” mainly based on its ability to trigger the aggregation of naïve transgenic tau after cerebral inoculation of pre‐aggregated “seeds” (Clavaguera et al, 2010; Iba et al, 2013; Jucker & Walker, 2013; Sanders et al, 2014) and the observation, in vivo and in vitro, of trans‐synaptic spread of misfolded tau (de Calignon et al, 2012; Liu et al, 2012; Calafate et al, 2015; Iba et al, 2015). We now directly test parallels between tau and the well‐established behavior of PrP: (i) that PrPSc “propagates” only in the setting of endogenous PrPC based on template misfolding, and (ii) that PrPSc toxicity is directly linked to the presence of endogenous PrPC. Our findings for tau show that the propagation and accumulation of misfolded tau in recipient neurons are preserved in the absence of endogenous tau, while P301Ltau toxicity is markedly reduced but present. This shows that the suggested analogy between tau and PrP is not exact. One should keep in mind that tau propagation studied in overexpressing transgenic and virus‐based systems likely overestimates the naturally occurring tau propagation speed and may introduce further artifacts. The rather low overexpression of human P301Ltau in ECrTgTau mice (~threefold compared to endogenous mouse tau) may thus correlate with the slow tau propagation rate in this model (~15–18 months for single synapse connection from EC to DG), compared to the fast P301Ltau propagation (~8 weeks) in our strongly expressing AAV model.
In the case of tau, the current work demonstrates that mutant P301Ltau propagates trans‐synaptically and remains stably present in recipient neurons in the absence of endogenous tau, indicating that tau neuron‐to‐neuron transfer does not obligatorily depend on templated misfolding, but instead largely reflects apparent stability of tau in recipient neurons. The idea of a prion‐like pathology spreading of “tau‐prions” appears oversimplified, and a modification of this hypothesis seems necessary; one should also consider the different nature of extracellularly accessible membrane‐bound PrPC versus intracellular soluble tau.
On the other hand, importantly, the association between tau aggregation and neurotoxicity appears to be changed in the context of a mouse tau‐null phenotype: Mapt 0/0 animals were protected against tau misfolding and earliest indications of neuronal damage (gliosis) in ECrTgTau mice, and against late‐stage neuronal loss and gliosis in the setting of widespread NFT pathology in rTg4510 mice. Although tau‐null mice had similar NFT numbers, the severity of tau aggregation was largely blunted and the onset of neuronal damage strongly delayed (compare 9‐month‐old rTg4510 with 12‐month‐old rTg4510‐Mapt 0/0) in tau‐null animals. Tangles per se appeared to be not as toxic in rTg4510‐Mapt 0/0, and the robust neuroprotection in the face of continued tau aggregation in rTg4510‐Mapt 0/0 mice supports recent in vivo and in vitro experimental data (Kuchibhotla et al, 2014; Kumar et al, 2014; Rudinskiy et al, 2014) dissociating NFT formation from neurotoxicity. Furthermore, human cases with innumerable tangles but little or no neuronal loss and cognitive impairment (Perez‐Nievas et al, 2013) underline the relevance of this finding for clinical AD.
Reducing endogenous tau is protective against neurotoxic insults associated with seizures and amyloid‐beta (Rapoport et al, 2002; Roberson et al, 2007; Ke et al, 2012; DeVos et al, 2013), although the mechanisms remain unknown. In the current setting, reducing endogenous tau appears to be protective against toxicity associated with overexpression of P301Ltau. We considered the possibility that overall reduction of tau could reduce tau misfolding and tau‐associated neuronal stress. However, in rTg4510 mice with ~10‐fold overexpression of human tau (SantaCruz et al, 2005), the impact of mouse tau knockout on total tau levels is relatively small and does not change the number of tangles, yet is robustly neuroprotective. The strong overexpression of human P301Ltau in the rTg4510 model, as well as the lack of mouse tau during development in MapT 0/0 mice, may as well cause undesired artifacts such as changes in neuronal metabolism or gene profile. However, overexpression of human mutant tau appears to be necessary for achieving robust NFT pathology in mice (Duff et al, 2000; Spires & Hyman, 2005; Gilley et al, 2012), an essential when studying NFT toxicity. Future experiments involving tau knockdown strategies in adult mice, for example, using antisense oligonucleotides (DeVos et al, 2013), will have to proof the benefits of endogenous tau reduction on tangle toxicity. Alternatively, because NFTs appear less toxic without mouse tau, endogenous tau may increase tau aggregate toxicity, perhaps by supporting the formation of more toxic tau species (Lasagna‐Reeves et al, 2011) or increasing their seeding activity. For example, conformational changes associated with co‐aggregation of mouse with human P301Ltau may create toxic “strains” of tau aggregates (Sanders et al, 2014). Supporting this hypothesis, we found that brain extracts from rTg4510‐Mapt 0/0 mice had more detergent‐soluble tau, a slightly different composition of TBS‐soluble high molecular weight “oligomeric” tau (by Native PAGE), and significantly lower potential to trigger tau aggregation in a cell‐based tau seeding assay (Holmes et al, 2014); this could explain a faster pathology progression in the presence of endogenous tau. However, it is also possible that the process of conversion—from naïve to misfolded mouse tau—rather than the aggregates themselves are responsible for neurotoxicity in the presence of endogenous tau. Furthermore, one may also consider neurotoxicity‐mediating functions of soluble endogenous tau (but not P301Ltau) that, for example, trigger ER stress [elevated CHOP in rTg4510; (Hoozemans & Scheper, 2012)] and gliosis.
In sum, the current data revealed some features of tau that are analogous to and some features that are discrepant from expectations based on a “prion‐like” model of tau propagation and toxicity. In particular, in contrast to PrP, in three separate models of tau propagation (ECrTgTau, rTg4510, and AAV‐mediated P301Ltau expression), tau could be released by donor neurons and taken up and be detected weeks to months later in recipient neurons, even if endogenous tau was missing. Therefore, aggregated tau remains for substantial periods of time in the recipient cells without a requirement for templated misfolding of endogenous tau to “maintain” the aggregate, as is necessary in case of PrP. A second prediction of the PrP analogy is that the absence of endogenous tau protein would be neuroprotective, presumably by halting the spread of P301Ltau misfolding. However, although tau‐null animals did not alter tau propagation and had mature NFT pathology, the neurotoxic phenotype of P301Ltau overexpression was markedly blunted in the absence of endogenous tau. These paradoxical results suggest caution in drawing too close a parallel between PrP and tau‐related neurotoxicity. They provide, however, a potentially important, but unexpected, avenue toward neuroprotective therapies in tau‐related neurodegenerative diseases. Understanding the mechanism of neuroprotection by tau reduction and/or the role of endogenous tau for increased neurotoxicity will be critical to determine whether therapeutic strategies aimed at tau reduction (e.g. tau immunotherapy) will be beneficial in AD.
Materials and Methods
Animals
ECrTgTau mice were produced by crossing (C57BL/6J)B6.TgEC‐tTA mice expressing the tetracycline‐controlled transactivator (tTa) exclusively in EC‐II (Yasuda & Mayford, 2006) with FVB‐Tg(tetO‐TauP301L)4510 mice (SantaCruz et al, 2005) as previously described (de Calignon et al, 2012). Mice having both tTa and tau transgene expressed human mutant P301Ltau (0N4R) in the EC, mice lacking the tau transgene served as controls (WT). ECrTgTau mice were crossed to mice lacking endogenous mouse tau [B6.Mapt tm1(EGFP)Kit mice (Mapt 0/0), from the Jackson Laboratory (Tucker et al, 2001)] to obtain ECrTgTau‐Mapt 0/0 animals. We analyzed gender‐mixed 18‐month‐old littermates. The CK‐tTA transgene, driving expression in forebrain excitatory neurons (Mayford et al, 1995), was transferred to the B6.Mapt 0 background, enabling production of rTg4510 and rTg4510‐Mapt 0/0 mice. We analyzed gender‐mixed 9‐ and 12‐month‐old animals. Animal experiments were performed in accordance with United States National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committees of Massachusetts General Hospital and McLaughlin Research Institute.
RT–PCR analysis
To verify mouse genotypes, RNA was extracted from frozen brain tissue with Trizol (Sigma) and RNA quality was tested using Agilent RNA 6,000 Pico kit (Agilent Technologies). For RT–PCR, 1 μg RNA was transcribed into cDNA with SuperScript® III Reverse Transcriptase (LifeSciences) using primers targeting the tau transgene product and mouse tau exon 7. Transcript number of human and mouse tau RNA was quantified with RT–qPCR Primer Assay and normalized to GAPDH7 (Fig EV1A). Two‐tailed Student's t‐test.
Cortical primary neuron cultures
Primary neuronal cultures were derived from cerebral cortex of embryonic days 15–19 wild‐type mice (CD1, Charles River Laboratories). Briefly, cortices were dissected, gently minced, neurons dissociated using a papain kit (Worthington Biochemicals Corp.), and then washed with 1× HBSS. Neurons were seeded into culture dishes that were pre‐treated with poly‐D‐lysine (100 μg/ml) for > 1 h room temperature. Cultures were maintained at 37°C with 5% CO2, supplemented with neurobasal medium with 2% B27 supplement, 2 mM l‐glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). At 7 days in vitro (DIV), neurons were transduced with AAV eGFP‐2a‐P301L to achieve ~5–10% transfection rate. At 14 DIV, neuron cultures were washed briefly with PBS, then fixed for 15 min with 4%‐PFA/PBS, and immunostained for GFP (chicken anti‐GFP, anti‐chicken Alexa488), human tau (mouse Tau13, anti‐mouse Cy3), and DAPI. Images were taken on a Zeiss AxioImager Microscope with a 20× objective.
AAV injection
AAV encoding eGFP, the translation interrupting 2a peptide (Szymczak et al, 2004), and full‐length human mutant P301Ltau (AAV8‐CBA‐eGFP‐2a‐P301Ltau; titer: 6 × 1013 particles/ml; produced at MEEI vector core, Boston) was stereotactically injected into 14‐ to 16‐month‐old WT and Mapt 0/0 mice for unilateral expression of eGFP and P301Ltau in the left EC/HPC (bregma: −4.5 mm posterior, −4.5 mm lateral, depth from brain surface: 1.7 mm). Eight weeks after AAV injection, mice were perfused with 4% PFA/PBS and brains cut into 30‐μm‐thick horizontal sections. Brain sections were immunostained for GFP, human tau, and DAPI (as described for cryosections).
Immunohistochemistry
Mice were killed and intracardially perfused with PBS, and brain hemispheres were drop‐fixed in 4% PFA/PBS for 3 days, cryoprotected in 30% sucrose/PBS, frozen embedded in M1 mounting medium, cut into 10‐μm‐thick horizontal sections, mounted on glass slides, and stored at −80°C. Sections were permeabilized with 0.1% TX‐100/PBS and blocked in 5% normal goat serum (NGS)/PBS, and primary antibodies diluted in 5% NGS were applied over night at 4°C. After washing in PBS, secondary antibodies (Cy3 or Alexa647‐labeled anti‐mouse or anti‐rabbit IgGs) were applied for 1 h at room temperature, sections were mounted using mounting medium with DAPI (VectaShield), and images were recorded on a Zeiss AxioImager microscope equipped with a Coolsnap digital camera and Axio‐VisionV4.8.
Antibodies
For IHC and Western blot analyses, the following antibodies were used: rabbit anti‐human tau (N‐terminus) TauY9 (Enzo Lifescience), mouse anti‐human tau (N‐terminus) Tau13 (Covance/BioLegend), rabbit anti‐mouse and human tau (C‐terminus; DAKO), mouse anti‐mouse tau Tau/5 (Johnson&Johnson), mouse anti‐phospho‐tau CP13 (pS202/sT205; all phosho‐tau antibodies detect mouse and human tau), PHF1 (pS396/pS404) (courtesy of Peter Davies), and 12E8 (pS262/pS356; Elan Pharmaceuticals), rabbit anti‐”misfolded” tau Alz50 (1:500, Peter Davies), mouse anti‐glial fibrillary acidic protein (GFAP; Abcam), rabbit anti‐Iba1 (for ICC or WB; WAKO), mouse anti‐NeuN (Millipore), mouse anti‐synapsin‐1 (EMD), mouse anti‐CHOP (Cell Signaling), and rabbit anti‐Ubiquitin (Millipore).
Fluorescence immuno/in situ hybridization (immuno‐FISH)
FISH combined with immunohistochemistry was performed as previously described (de Calignon et al, 2012). Digoxigenin‐labeled RNA probes matching human Mapt (NM_016835; nucleotides 2,773–3,602) were generated by RT–PCR and detected with anti‐digoxigenin horseradish peroxidase‐labeled antibodies (Roche) and Alexa568‐labeled tyramide (Invitrogen) (Schaeren‐Wiemers & Gerfin‐Moser, 1993). Human tau was immunolabeled using primary antibody TauY9‐ and Alexa647‐conjugated goat anti‐rabbit secondary antibody.
Cell counting, stereology, and cortical thickness
Astrocytes (GFAP), microglia (Iba1), nuclei (DAPI), neurons (NeuN), and tangles (ThioS) were stereologically assessed in respective brain regions of 3–4 10‐μm‐thick sections per mouse in 3 mice/group. Sections were spaced each by 100 μm, spanning a total depth of 4 × 100 μm = 400 μm brain tissue per mouse. We determined cell densities (number/mm2) using ImageJ and CAST. Investigators were blinded for all staining and counting procedures.
Brain extracts and TX‐100 extraction
For the ECrTgTau cohort, EC and HPC were dissected from frozen brains and homogenized in RIPA buffer (Sigma) including protease (Complete, Roche) and phosphatase (PhosphoStop, Roche) inhibitors. For the rTg4510 cohort, CTX were dissected from frozen brains and homogenized in TBS (20 mM Tris, 150 mM NaCl, 1 mM EGTA, pH 7.8) including protease and phosphatase inhibitors. Homogenates were spun down at 104 g for 30 min (TBS‐extract =supernatant). For TX‐100 extraction, supernatant was replaced by equal amount of TBS + 1% TX‐100 and samples were incubated for 20 min at RT. Homogenates were spun down again (TX‐100‐extract = supernatant).
Brain weight, SDS–PAGE, native PAGE, and Western blotting
Wet weight of whole brains including cerebellum and olfactory bulb from 9‐ and 12‐month‐old rTg4510, rTg4510‐Mapt 0/0, WT, and Mapt 0/0 mice was determined after intracardial perfusion with 15 ml PBS. For analysis of brain extracts, 10–30 μg total protein was loaded onto 4–12% or 10% Bis‐Tris SDS–polyacrylamide gels (Life Technologies) and separated using MOPS or MES buffer. For Native PAGE of rTg4510 extracts, TBS‐extracts (40 μg protein) were mixed with Tris‐glycine Native loading buffer (Life Technology), loaded on a 4–16% Bis–Tris Native PAGE gel, and ran at 150 V for 4 h; 20 μg pre‐aggregated recombinant full‐length human mutant ΔK280 tau was loaded as a tau standard. After blotting onto nitrocellulose membranes and blocking in Odyssey blocking buffer (LI‐COR), primary antibodies were applied overnight at 4°C. After washing in 0.05% (v/v) Tween‐20 in TBS, blots were incubated with goat anti‐rabbit‐IRDye680 and anti‐mouse‐IRDye800 (Rockland) 1 h at room temperature. Protein bands were visualized using Odyssey imaging system (LI‐COR), and band intensities were analyzed using ImageJ (http://rsb.info.NIH.gov/nih-image/).
Gallyas silver and thioflavine‐S staining of tangles
Silver staining of tangles in 12‐month‐old mice (rTg4510 cohort) was done in 30‐μm‐thick coronal sections of PFA‐perfused brains as described previously (Gallyas, 1971). After silver staining, sections were mounted on glass slides and neurons (Nissl substance) counterstained with cresyl violet and mounted in mounting media. For thioflavine‐S (ThioS) stain of tangles (Sun et al, 2002), 10‐μm‐thick cryosections of PFA‐fixed brains were rinsed in de‐ionized water and incubated in 0.05% (w/v) ThioS in 50% (v/v) ethanol in the dark for 8 min, and ThioS staining was differentiated by two 10‐s washes in 80% ethanol and rinsed in de‐ionized water (mounting medium without DAPI).
HEK293 tau aggregation assay
HEK293 cells stably expressing CFP‐/YFP‐TauRDP301S [TauRD: repeat domain aa244–372 of 2N4R human tau, P301S mutation (Holmes et al, 2014)] were plated in a 96‐well plates. After 24 h, TBS‐soluble brain extracts were applied at 0.5 or 1.0 μg protein/well in a total of 40 μl Opti‐MEM (Gibco). After 30 h, cells were fixed with 4% PFA and the number of cells with aggregates was counted. Each condition (genotype and concentration) was performed in triplicate. For studies using lipofectamine, cells were plated in 8‐chambered coverslip dishes and treated with brain extracts containing 1.0 μg protein and 0.5 μl lipofectamine (Invitrogen) in 50 μl Opti‐MEM for 25 h.
Statistical analysis
To compare cell numbers, we determined the average cell densities (cell number/mm2) per mouse from 3 to 4 brain sections, 3 mice/group. For Western blots, 3 mice/group were analyzed. For statistical analysis of differences between groups, normal distribution was assumed and analysis was performed using GraphPad Prism 6; multi‐comparisons were made applying one‐way ANOVA with Bonferroni corrections; when comparing two groups, non‐paired two‐tailed Student's t‐tests with confidence intervals of 95% were used. All values are given as mean ± SEM. Sample size was chosen to minimize number of animals but be able to carry out statistical analysis (n = 3–5 animals per group) and no data was excluded.
Author contributions
SW and BTH, with the help of TLS‐J, AMP, and GAC, wrote the manuscript. SW and EAM performed experiments and analysis. MJK, LS, AR, SLD, SN, ZF, ST, OC‐G, and CMW helped with experiments. RP and GAC performed mouse work.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1F
Source Data for Figure 4A
Source Data for Figure 5A
Source Data for Expanded View
Acknowledgements
We thank Dr. Marc Diamond for the HEK293 CFP‐/YFP‐TauRD cell line. AAVs were produced by Ry Xiao at MEEI vector core in Boston. This research was funded by Massachusetts General Hospital, NIH AG 26249, the German Research Association (DFG), Alzheimer's Research UK, and the Chief Scientist's Office, Scotland. Mapt 0/0 congenic strains were produced with funding by NIH program project grant NS41997.
The EMBO Journal (2015) 34: 3028–3041
See also: JC Polanco & J Götz (December 2015)
References
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol 103: 26–35 [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82: 239–259 [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A (1996a) Normal host prion protein necessary for scrapie‐induced neurotoxicity. Nature 379: 339–343 [DOI] [PubMed] [Google Scholar]
- Brandner S, Raeber A, Sailer A, Blattler T, Fischer M, Weissmann C, Aguzzi A (1996b) Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci USA 93: 13148–13151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calafate S, Buist A, Miskiewicz K, Vijayan V, Daneels G, de Strooper B, de Wit J, Verstreken P, Moechars D (2015) Synaptic contacts enhance cell‐to‐cell tau pathology propagation. Cell Rep 11: 1176–1183 [DOI] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires‐Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464: 1201–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez‐Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires‐Jones TL, Hyman BT (2012) Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73: 685–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canu N, Barbato C, Ciotti MT, Serafino A, Dus L, Calissano P (2000) Proteasome involvement and accumulation of ubiquitinated proteins in cerebellar granule neurons undergoing apoptosis. J Neurosci 20: 589–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Goedert M, Tolnay M (2010) Induction and spreading of tau pathology in a mouse model of Alzheimer's disease. Med Sci 26: 121–124 [DOI] [PubMed] [Google Scholar]
- DeVos SL, Goncharoff DK, Chen G, Kebodeaux CS, Yamada K, Stewart FR, Schuler DR, Maloney SE, Wozniak DF, Rigo F, Bennett CF, Cirrito JR, Holtzman DM, Miller TM (2013) Antisense reduction of tau in adult mice protects against seizures. J Neurosci 33: 12887–12897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P (2000) Characterization of pathology in transgenic mice over‐expressing human genomic and cDNA tau transgenes. Neurobiol Dis 7: 87–98 [DOI] [PubMed] [Google Scholar]
- Gallyas F (1971) Silver staining of Alzheimer's neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung 19: 1–8 [PubMed] [Google Scholar]
- Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, Misgeld T, Brion JP, Anderton B, Hanger DP, Coleman MP (2012) Age‐dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging 33: 621e1–621e15 [DOI] [PubMed] [Google Scholar]
- Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu GQ, Masliah E, Mucke L (2012) Human P301L‐mutant tau expression in mouse entorhinal‐hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS ONE 7: e45881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, Belaygorod L, Cairns NJ, Holtzman DM, Diamond MI (2014) Proteopathic tau seeding predicts tauopathy in vivo . Proc Natl Acad Sci USA 111: E4376–E4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoozemans JJ, Scheper W (2012) Endoplasmic reticulum: the unfolded protein response is tangled in neurodegeneration. Int J Biochem Cell Biol 44: 1295–1298 [DOI] [PubMed] [Google Scholar]
- Hyman BT, Van Hoesen GW, Damasio AR, Barnes CL (1984) Alzheimer's disease: cell‐specific pathology isolates the hippocampal formation. Science 225: 1168–1170 [DOI] [PubMed] [Google Scholar]
- Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's‐like tauopathy. J Neurosci 33: 1024–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM (2015) Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC's afferent and efferent connections. Acta Neuropathol 130: 349–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J (2010) Dendritic function of tau mediates amyloid‐beta toxicity in Alzheimer's disease mouse models. Cell 142: 387–397 [DOI] [PubMed] [Google Scholar]
- Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke YD, Suchowerska AK, van der Hoven J, De Silva DM, Wu CW, van Eersel J, Ittner A, Ittner LM (2012) Lessons from tau‐deficient mice. Int J Alzheimer's Dis 2012: 873270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck S, Nitsch R, Grune T, Ullrich O (2003) Proteasome inhibition by paired helical filament‐tau in brains of patients with Alzheimer's disease. J Neurochem 85: 115–122 [DOI] [PubMed] [Google Scholar]
- Kopeikina KJ, Hyman BT, Spires‐Jones TL (2012) Soluble forms of tau are toxic in Alzheimer's disease. Transl Neurosci 3: 223–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Wegmann S, Kopeikina KJ, Hawkes J, Rudinskiy N, Andermann ML, Spires‐Jones TL, Bacskai BJ, Hyman BT (2014) Neurofibrillary tangle‐bearing neurons are functionally integrated in cortical circuits in vivo . Proc Natl Acad Sci USA 111: 510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Tepper K, Kaniyappan S, Biernat J, Wegmann S, Mandelkow EM, Muller DJ, Mandelkow E (2014) Stages and conformations of the tau repeat domain during aggregation and its effect on neuronal toxicity. J Biol Chem 289: 20318–20332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild‐type mice. Mol Neurodegener 6: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna‐Reeves CA, Castillo‐Carranza DL, Sengupta U, Sarmiento J, Troncoso J, Jackson GR, Kayed R (2012) Identification of oligomers at early stages of tau aggregation in Alzheimer's disease. FASEB J 26: 1946–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121–1159 [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans‐synaptic spread of tau pathology in vivo . PLoS ONE 7: e31302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Wang J, Kandel ER, O'Dell TJ (1995) CaMKII regulates the frequency‐response function of hippocampal synapses for the production of both LTD and LTP. Cell 81: 891–904 [DOI] [PubMed] [Google Scholar]
- Nijholt DA, van Haastert ES, Rozemuller AJ, Scheper W, Hoozemans JJ (2012) The unfolded protein response is associated with early tau pathology in the hippocampus of tauopathies. J Pathol 226: 693–702 [DOI] [PubMed] [Google Scholar]
- Perez‐Nievas BG, Stein TD, Tai HC, Dols‐Icardo O, Scotton TC, Barroeta‐Espar I, Fernandez‐Carballo L, de Munain EL, Perez J, Marquie M, Serrano‐Pozo A, Frosch MP, Lowe V, Parisi JE, Petersen RC, Ikonomovic MD, Lopez OL, Klunk W, Hyman BT, Gomez‐Isla T (2013) Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain 136: 2510–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH (2005) Age‐dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25: 10637–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta ‐amyloid‐induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364–6369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce‐Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316: 750–754 [DOI] [PubMed] [Google Scholar]
- Rudinskiy N, Hawkes JM, Wegmann S, Kuchibhotla KV, Muzikansky A, Betensky RA, Spires‐Jones TL, Hyman BT (2014) Tau pathology does not affect experience‐driven single‐neuron and network‐wide Arc/Arg3.1 responses. Acta Neuropathol Commun 2: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, Miller TM, Grinberg LT, Seeley WW, Diamond MI (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82: 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: 476–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren‐Wiemers N, Gerfin‐Moser A (1993) A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin‐labelled cRNA probes. Histochemistry 100: 431–440 [DOI] [PubMed] [Google Scholar]
- Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: a006189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Lin YG, Malthankar‐Phatak G, Dong Y (2013) A rapid gene delivery‐based mouse model for early‐stage Alzheimer disease‐type tauopathy. J Neuropathol Exp Neurol 72: 1062–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundke‐Iqbal I, Iqbal K (1996) Calcium/calmodulin‐dependent protein kinase II phosphorylates tau at Ser‐262 but only partially inhibits its binding to microtubules. FEBS Lett 387: 145–148 [DOI] [PubMed] [Google Scholar]
- Spires TL, Hyman BT (2005) Transgenic models of Alzheimer's disease: learning from animals. NeuroRx 2: 423–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Orne JD, SantaCruz K, Pitstick R, Carlson GA, Ashe KH, Hyman BT (2006) Region‐specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol 168: 1598–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun A, Nguyen XV, Bing G (2002) Comparative analysis of an improved thioflavin‐s stain, Gallyas silver stain, and immunohistochemistry for neurofibrillary tangle demonstration on the same sections. J Histochem Cytochem 50: 463–472 [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA (2004) Correction of multi‐gene deficiency in vivo using a single ‘self‐cleaving’ 2A peptide‐based retroviral vector. Nat Biotechnol 22: 589–594 [DOI] [PubMed] [Google Scholar]
- Tai HC, Serrano‐Pozo A, Hashimoto T, Frosch MP, Spires‐Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin‐proteasome system. Am J Pathol 181: 1426–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinczek B, Biernat J, Baumann K, Mandelkow EM, Mandelkow E (1995) Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol Biol Cell 6: 1887–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4: 29–37 [DOI] [PubMed] [Google Scholar]
- Van der Jeugd A, Hochgrafe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D'Hooge R, Mandelkow EM (2012) Cognitive defects are reversible in inducible mice expressing pro‐aggregant full‐length human Tau. Acta Neuropathol 123: 787–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, Hyman BT (2013) Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol 70: 304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Mayford MR (2006) CaMKII activation in the entorhinal cortex disrupts previously encoded spatial memory. Neuron 50: 309–318 [DOI] [PubMed] [Google Scholar]
- Yetman MJ, Lillehaug S, Bjaalie JG, Leergaard TB, Jankowsky JL (2015) Transgene expression in the Nop‐tTA driver line is not inherently restricted to the entorhinal cortex. Brain Struct Funct doi:10.1007/s00429‐015‐1040‐9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53: 337–351 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1F
Source Data for Figure 4A
Source Data for Figure 5A
Source Data for Expanded View