

Abstract
RNase P is a conserved endonuclease that processes the 5′ trailer of tRNA precursors. We have isolated mutations in Rpp30, a subunit of RNase P, and find that these induce complete sterility in Drosophila females. Here, we show that sterility is not due to a shortage of mature tRNAs, but that atrophied ovaries result from the activation of several DNA damage checkpoint proteins, including p53, Claspin, and Chk2. Indeed, we find that tRNA processing defects lead to increased replication stress and de‐repression of transposable elements in mutant ovaries. We also report that transcription of major piRNA sources collapse in mutant germ cells and that this correlates with a decrease in heterochromatic H3K9me3 marks on the corresponding piRNA‐producing loci. Our data thus link tRNA processing, DNA replication, and genome defense by small RNAs. This unexpected connection reveals constraints that could shape genome organization during evolution.
Keywords: Drosophila, dysgenesis, heterochromatin, oogenesis, transposon
Subject Categories: Development & Differentiation; DNA Replication, Repair & Recombination; RNA Biology
Introduction
tRNAs are non‐coding RNAs (~75 nt) transcribed by the RNA polymerase III and perform critical functions in protein synthesis (Dieci et al, 2007; Durdevic & Schaefer, 2013). In eukaryotes, tRNAs are encoded by a multigene family containing up to 500 genes in humans and 300 genes in Drosophila, clustered in many loci throughout the genome (Genomic tRNA database, http://gtrnadb.ucsc.edu). The transcription of tRNA genes gives rise to pre‐tRNAs, which are processed into mature tRNAs by the action of two highly conserved RNases (Jarrous & Gopalan, 2010; Lai et al, 2010): RNase P, which cleaves the 5′ trailer, and RNase Z, which cleaves the 3′ trailer (Fig 1B). A non‐templated CCA motif added at the 3′ end acts as a substrate for aminoacylation. Around one hundred posttranscriptional modifications have been reported for tRNAs, but the functional significance of very few has been tested (Engelke & Hopper, 2006; Durdevic & Schaefer, 2013). While critical defects in tRNA processing cause cell lethality, more subtle point mutations in the processing machinery can induce surprisingly specific phenotypes. For example, point mutations in the RNA kinase CLP1 disrupt tRNA splicing and cause brain disorders in mice and humans (Hanada et al, 2013; Karaca et al, 2014; Schaffer et al, 2014). Interestingly, this phenotype can be rescued by inactivating the stress sensor protein p53 in clp1 mutant mice (Hanada et al, 2013). This indicates that defects in the central nervous system are not simply consequences of protein synthesis and growth abnormalities. Moreover, additional reports have linked mutations in the tRNA biogenesis pathway with sterility phenotypes in humans, animals, and plants (Wang et al, 2012; Hussain et al, 2013; Lin et al, 2013; Pierce et al, 2013; Xie et al, 2013; Abbott et al, 2014). Less appreciated is the fact that tRNA genes are, together with rRNAs, the most transcribed genes in the genome and tRNAs represent major docking sites for RNA pol III (Dieci et al, 2007). From yeast to mammals, this peculiar feature renders these loci rather challenging to replicate, due to local conflicts between RNA pol III and DNA polymerase during S phase. The replication fork often pauses at tRNA loci, which can consequently become fragile sites for DNA lesions (Clelland & Schultz, 2010; Helmrich et al, 2013). Additionally, it has been shown that RNA pol III binding can define epigenetic boundaries involved in the formation and restriction of heterochromatic domains (Noma et al, 2006; Donze, 2012; Raab et al, 2012). Thus, the emerging view is that tRNA gene loci fulfill multiple roles as chromatin organizers and regulators of gene expression around their genomic localization, beyond their function in the production of tRNAs (Hull et al, 1994; Van Bortle & Corces, 2012; Good et al, 2013; Van Bortle et al, 2014). It thus remains unclear in tRNA‐linked pathologies whether the cause(s) of disease involve abnormal protein synthesis, abnormal tRNA fragments, or indirect defects on the expression of other genes.
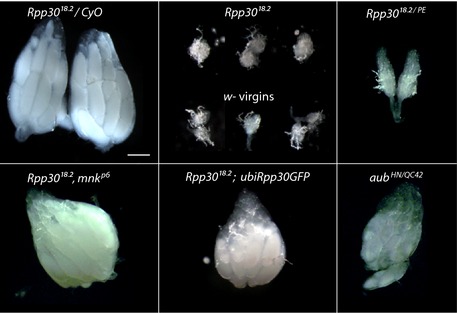
Figure 1. Rpp30 mutations induce premature arrest of oogenesis in Drosophila .
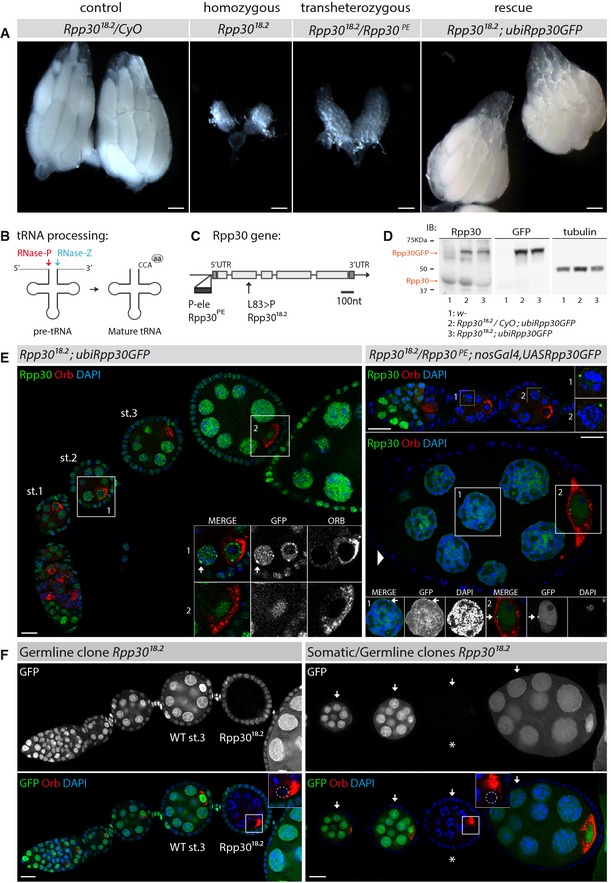
- Ovaries from control heterozygous Rpp30 18.2 flies (left), homozygous Rpp30 18.2 or transheterozygous Rpp30 18.2 /Rpp30 PE flies (middle), and homozygous Rpp30 18.2 carrying a copy of ubiRpp30GFP transgene (right). Scale bar, 100 μm.
- Rpp30 is a subunit of RNase P (red), a ribozyme involved in pre‐tRNA processing by cleaving the 5′ tail. The RNase Z (blue) cuts the 3′ tail of the pre‐tRNA, and CCA motif is added, forming mature tRNA ready to bind an amino acid (aa).
- Gene region corresponding to Rpp30 (RNase P protein 30, adapted from Flybase). The P‐element insertion (Rpp30 PE, dark gray) and the point mutation leucine to proline (Rpp30 18.2, arrow) are shown.
- Ovaries were dissected and lysed and protein extracts analyzed by Western blot using anti‐Rpp30, anti‐GFP, or anti‐tubulin antibodies. Orange arrows indicate overexpressed and endogenous Rpp30 signals. Genotypes are numbered as 1, 2, and 3.
- Rpp30‐dependent early oogenesis arrest is restored by the expression of Rpp30GFP transgene in two different genetic backgrounds. Left: Homozygous Rpp30 18.2 ovariole expressing ubiquitous ubiRpp30GFP (st, stage). Right: Transheterozygous Rpp30 18.2 /Rpp30 PE ovarioles overexpressing germ line‐specific nanosGal4,UASRpp30GFP. Note follicular somatic cells with no Rpp30GFP expression (arrowhead). Magnifications show nuclear Rpp30 localization, Rpp30 perinuclear foci (arrows), and Orb (a specific oocyte marker). Scale bar, 10 μm.
- flp/FRT clones mutant for Rpp30 18.2 specifically in the germ line (star) and/or the follicular cells (arrows) are detected by the absence of GFP. Magnifications show mutant oocytes with Orb mislocalization next to the karyosome (dotted circle) instead of being localized to the posterior, as seen in stage 3 (st.3) wild‐type clones. Scale bar, 10 μm.
A second class of abundant RNAs required for fertility and also for neuronal development are Piwi‐associated RNAs (piRNAs) (Siomi et al, 2011; Perrat et al, 2013; Ross et al, 2014). piRNAs are small non‐coding RNAs of 23–28 nt, which silence transposable elements (TEs) by guiding Argonaute proteins to TE RNAs using sequence complementarity (Malone & Hannon, 2009; Senti & Brennecke, 2010). Transposons represent 15% of the Drosophila genome and almost 50% of the human genome. TE mobilization can cause DNA damage, insertional mutagenesis, and chromosomal rearrangements, therefore constituting a threat to genome integrity. piRNAs silence TEs both at the transcriptional level (TGS, transcriptional gene silencing) and at the posttranscriptional level (PTGS, posttranscriptional gene silencing) (Guzzardo et al, 2013). During PTGS, piRNAs guide Aubergine (Aub) and Argonaute 3 (Ago3) to TE transcripts, hence inducing their cleavage through Aub/Ago3 slicing activity (Brennecke et al, 2007; Li et al, 2009; Malone et al, 2009). During TGS, piRNAs guide Piwi to genomic TE copies, likely through nascent transcript recognition, where it promotes heterochromatinization through the deposition of repressive H3K9 methylation marks (Sienski et al, 2012; Darricarrere et al, 2013; Huang et al, 2013; Le Thomas et al, 2013; Rozhkov et al, 2013). In the absence of piRNAs, TEs become expressed and induce various oogenesis defects (early arrest of egg chamber development, dorsoventral patterning defects, etc.) leading to sterility. Interestingly, these phenotypes are due to the activation of DNA damage checkpoint proteins of the ATR/Chk2 pathway (Chen et al, 2007; Klattenhoff et al, 2007; Pane et al, 2007). Indeed, inactivating chk2 in piRNA mutants such as aubergine (aub) or armitage (armi) rescues most of the morphological defects during oogenesis. Fertility, however, is not restored in these double mutants.
piRNAs are produced by about 140 loci or clusters mostly localized in centromeric and telomeric regions of the Drosophila genome (Brennecke et al, 2007). These clusters are thought to be transcribed into long transcripts made of intermingled TE sequences, which are then processed into small piRNAs (Senti & Brennecke, 2010). Some of these clusters are very similar to regular genes. One such example is the flamenco locus, which produces most of the somatic piRNAs in follicle cells. Its transcription is initiated at a defined starting site and promoter; it is transcribed by the RNA polymerase II in one direction and transcripts are spliced alternatively (Goriaux et al, 2014). In contrast, other piRNA clusters such as the cluster 1 (42AB), which produces most of the germ line piRNAs, have unusual properties (Le Thomas et al, 2014a; Mohn et al, 2014). Firstly, cluster 1/42AB is transcribed by pol II in two opposite directions by a non‐canonical mechanism. Secondly, it requires repressive H3K9me3 marks to be actively transcribed. In support of the latter point, mutations in the H3K9 methyltransferase eggless lead to a major loss of piRNAs, early oogenesis arrest, and sterility (Rangan et al, 2011). What defines a locus as a source of piRNAs is not fully understood (Le Thomas et al, 2014b). Recent genome‐wide analysis, however, suggests that the chromatin environment plays a crucial role (Mohn et al, 2014). This includes the H3K9me3 mark as mentioned above, but also specific heterochromatin proteins such as Rhino, and the Rhino‐binding proteins, Deadlock, and Cutoff. In addition, recent studies have shown that single TE insertions are also significant sources of piRNAs, besides piRNA clusters (Le Thomas et al, 2014a; Mohn et al, 2014; Shpiz et al, 2014).
Drosophila oogenesis has proven to be a crucial model system to study the roles of chromatin modifications and small RNAs‐based mechanisms on germ cell development (Molla‐Herman et al, 2014). The elementary unit of oogenesis is the egg chamber, which is made of 16 germ cells surrounded by an epithelium of somatic follicle cells. Only one of these germ cells will become the oocyte, the future egg (Huynh & St Johnston, 2004). The 15 other cells are nurse cells, which undergo many rounds of DNA replication without mitosis and become highly polyploid. Nurse cells transcribe actively their genome and provide the oocyte with RNA species and nutrients required for its development (Dej & Spradling, 1999; Huynh & St Johnston, 2004). In particular, they produce germ line piRNA precursors, which are processed into mature piRNAs in a structure surrounding each nurse cell nucleus called the “nuage” (Brennecke et al, 2007; Lim & Kai, 2007; Pane et al, 2007; Chambeyron et al, 2008; Zhang et al, 2012). In contrast, the oocyte is arrested in prophase I of meiosis, and its DNA is highly compacted into a karyosome and transcriptionally inactive. The follicle cells encasing the germ line cyst also become polyploid at mid‐oogenesis and are also actively replicating and transcribing their genome (Dej & Spradling, 1999). Moreover, follicle cells also produce many piRNAs, mainly originating from the flamenco locus localized close to the X chromosome centromere (Li et al, 2009; Senti & Brennecke, 2010; Goriaux et al, 2014). Egg chambers are continuously produced throughout the adult life of the Drosophila female from both germ line and somatic stem cells, which are located in the germarium at the anterior tip of each ovary (Huynh & St Johnston, 2004).
In recent years, it has become clear that many important developmental decisions on cell fate, cell polarization, and genome defense are set up during the early stages of oogenesis, from stem cells to stages 3–4 (Huynh & St Johnston, 2004; Jagut et al, 2013; Molla‐Herman et al, 2014; Yan et al, 2014). Here, we report the identification of mutations affecting a subunit of RNase P causing an arrest of oogenesis at early stages. Our study reveals surprising links between tRNA processing, DNA replication, and piRNA transcription.
Results
Mutations in Rpp30, a subunit of RNase P, cause an early arrest of oogenesis
In an EMS mosaic screen for mutations affecting the early stages of Drosophila oogenesis, we isolated one mutant line, 18.2, which gave rise to viable but completely sterile females and to subfertile males (Jagut et al, 2013). Although viability was reduced (10% of expected progeny at 25°C and 20% at 18°C, see Table 1), homozygous mutant flies were of wild‐type size. In contrast, homozygous mutant ovaries were rudimentary (Fig 1A), resulting from an arrest of oogenesis at early stages of development (stages 3–4, see below). These phenotypes hinted at some specific requirements of the mutated gene for gonad development. The 18.2 mutant line was lethal over the deficiency Df(2L)ED21, and viable but sterile over the P‐element P(lacW)k01901 inserted into the Rpp30 gene (CG11606) (Table 1). The P(lacW)k01901 was lethal over itself and over Df(2L)ED21, indicating that 18.2 was an hypomorphic allele of Rpp30 (Table 1). We renamed the two alleles Rpp30 18.2 and Rpp30 PE for the 18.2 mutation and the P‐element insertion, respectively. Rpp30 is a small subunit of the highly conserved ribozyme RNase P, whose best described function is to cleave the 5′ trail sequence of pre‐tRNAs (Fig 1B) (Xiao et al, 2002; Jarrous & Gopalan, 2010). We sequenced the 18.2 line and found that the Rpp30 18.2 mutation changed the conserved leucine 83 into a proline and that the P(lacW)k01901 was inserted into the 5′ UTR of Rpp30 (Fig 1C). In addition, our genome‐wide RNA‐seq experiments (see below) revealed that levels of Rpp30 mRNA were reduced in Rpp30 18.2 mutant ovaries (Fig EV1A). Moreover, Western blot analysis of ovarian extracts showed that amounts of Rpp30 protein were also diminished in Rpp30 18.2 mutant ovaries (Fig 1D, lane 3). In addition, using the same antibody for immunostaining on ovaries, we found that the nuclear signal of Rpp30 was strongly affected in Rpp30 18.2 mutant flies (Fig EV1B and C). Importantly, all phenotypes induced by Rpp30 18.2 and Rpp30 PE mutations could be fully rescued by the ubiquitous expression of an Rpp30 cDNA tagged with GFP at the C‐terminal end of the protein (Fig 1A and E, left panel). As expected from the known co‐transcriptional maturation of tRNAs, Rpp30‐GFP localization was mainly nuclear (Jarrous & Reiner, 2007; Wichtowska et al, 2013). We also noticed localization at some perinuclear foci. Furthermore, the expression of Rpp30‐GFP only in germ cells, using the nanos‐Gal4 driver, was sufficient to completely rescue oogenesis (Fig 1E, right panel). This result suggested an important requirement for Rpp30 in germ cells. To further test this hypothesis, we induced clones of homozygous cells mutant for Rpp30 18.2 only in somatic or in germ line cells using the Flp/FRT technique (Xu & Rubin, 1993). We observed that egg chambers with germ cells mutant for Rpp30 18.2 were arrested as early as in homozygous mutant ovaries (Fig 1F, left). In contrast, egg chambers with entirely mutant follicle cells developed normally (Fig 1F, right). We also did not observe an additive effect when both germ cells and somatic cells were mutant in the same egg chamber (Fig 1F, right). We concluded that Rpp30 mutations affected oogenesis mainly by disrupting germ cell development.
Table 1.
Viability and fecundity in Rpp30 mutants
| Female genotype | Viabilitya | Fecundity | N |
|---|---|---|---|
| Rpp30 18.2 /Deficiency Df(2L)ED21 | 0 | ||
| Rpp30 PE /Deficiency Df(2L)ED21 | 0 | ||
| Rpp30 PE /Rpp30 PE | 0 | ||
| Rpp30 18.2 /CyO | 1.44 | Fertile | 780/808 |
| Rpp30 18.2 | 0.10 | Sterile | 28/808 |
| Rpp30 18.2 /Rpp30 PE | 1.08 | Sterile | 80/221 |
| FRT40A Rpp30 18.2 /CyO; claspin EP | 1.37 | Fertile | 751/821 |
| FRT40A Rpp30 18.2 ; claspin EP | 0.26 | Sterile | 70/821 |
| FRT40A Rpp30 PE /Rpp30 18.2 ; claspin EP | 1.01 | Sterile | 89/527 |
| FRT40A Rpp30 18.2 /CyO; claspin 45 /TM3 Ser | 1.50 | Fertile | 845 |
| FRT40A Rpp30 18.2 ; claspin 45 | 0 | ||
| Rpp30 18.2 /CyO; p53 11‐1B‐1 | 1.37 | Fertile | 476/518 |
| Rpp30 18.2 ; p53 11‐1B‐1 | 0.24 | Sterile | 42/518 |
| Rpp30 18.2 /CyO; p53 11‐1B‐1 /p53 5A‐1‐4 | 1.40 | Fertile | 826/884 |
| Rpp30 18.2 ; p53 11‐1B‐1 /p53 5A‐1‐4 | 0.20 | Sterile | 58/884 |
The viability (observed/expected ratio) and the fecundity (presence of hatched larvae) and the number of quantified flies observed.
Observed/total.
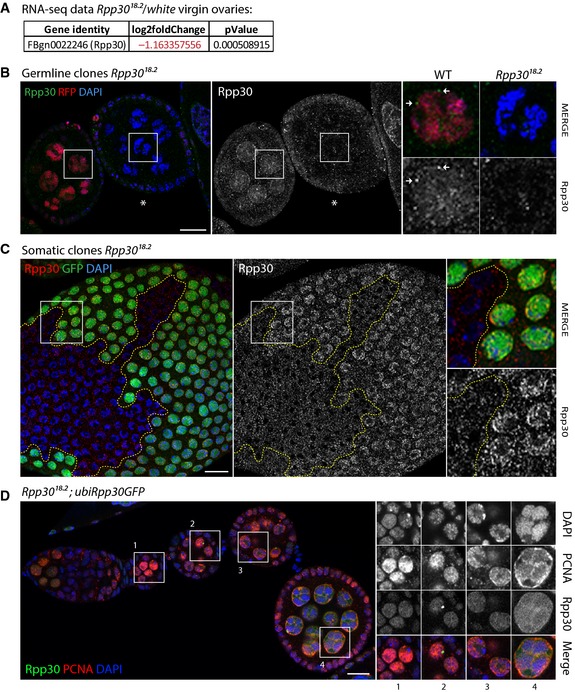
Figure EV1. Rpp30 protein expression is affected in Rpp30 18.2 mutants and PCNA loss in Rpp30 18.2 ovaries is restored when overexpressing ubiRpp30GFP (related to Figs 1 and 3).
-
ANormalized fold change and P‐value between Rpp30 18.2 mutant ovaries and white virgins issue from RNA‐seq transcriptome analysis showing a downregulation of Rpp30 transcripts in Rpp30 18.2 mutant ovaries.
-
B, Cflp/FRT clones mutant for Rpp30 18.2 specifically in the germ line (B) or follicular cells (C) are detected by the absence of RFP or GFP, respectively. Magnifications show Rpp30 absence in the nucleus of mutant cells. Perinuclear Rpp30 foci are indicated by white arrows in a wild‐type nurse cell (B). Scale bar, 10 μm.
-
DRpp30 18.2 homozygous ovaries overexpressing ubiRpp30GFP were dissected, fixed, and stained for PCNA (red). Rpp30GFP is in green and DAPI is in blue. Scale bar, 10 μm.
An early arrest of oogenesis can signal a failure to grow before vitellogenesis. Weak growth can be caused by defects in ribosome biogenesis, such as in Minute mutants, which are viable but sterile (Cramton & Laski, 1994; Fichelson et al, 2009; Zhang et al, 2014). This can also be caused by defects in RNA polymerase III activity, which strongly reduces the bulk of tRNAs (Marshall et al, 2012; Rideout et al, 2012). In both cases, sterility is associated with flies or larvae of much reduced size. We did not observe such a reduction in size in Rpp30 18.2 homozygous or in Rpp30 18.2/Rpp30 PE transheterozygous flies. We thus performed further experiments to determine the cause(s) of sterility in Rpp30 mutant flies.
Rpp30 is required for the correct processing of pre‐tRNA in Drosophila
As Rpp30 is a conserved subunit of RNase P, we first tested whether pre‐tRNA processing was affected in Rpp30 mutant flies. We performed Northern blot analysis of wild‐type and mutant extracts using probes directed against the 5′ and 3′ trail regions of the pre‐tRNAHis and against an internal region retained in the mature tRNAHis (Fig 2) (Xie et al, 2013). Using the 5′ probe, we could detect the pre‐tRNA (~125 nt) and an intermediate form retaining the 5′ trail region (~100 nt) in mutant extracts (Fig 2, lanes 2 and 3), which were not detected in wild‐type extracts (Fig 2, lane 1). Similarly, using the 3′ probe, we detected the pre‐tRNA form and an intermediate form containing the 3′ trail region (~100 nt) in mutant extracts (Fig 2, lanes 2 and 3), but not in wild‐type extracts (Fig 2, lane 1). In the presence of a wild‐type Rpp30‐GFP rescue transgene (Fig 2, lanes 5 and 6), the intermediate form was processed rapidly and the corresponding band was hardly visible. The levels of mature tRNA forms (~75 nt) were not visibly changed in mutant extracts compared to wild‐type extracts, as revealed by the internal probe. We concluded that although Rpp30 mutations affected efficient tRNA processing, the bulk of mature tRNAs was of the right size and quantity.
Figure 2. Rpp30 is required for pre‐tRNA processing.
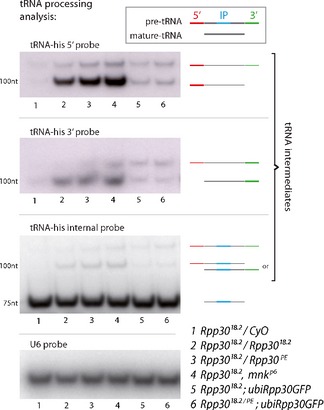
Whole fly RNA extracts were used to study tRNA processing by Northern blot. Top box: the three different probes used are depicted: 5′ and 3′ probes (red and green, respectively) can detect full pre‐tRNAs and tRNAs intermediates, but will not detect mature tRNAs. An internal probe (IP, blue) corresponding to the anticodon region was used to detect all non‐mature and mature tRNAs. The genotypes used in this experiment are numbered at the bottom (1–6). Northern blot panels: tRNA‐his 5′ probe (top), tRNA‐his 3′ probe (middle), and tRNA‐his IP (bottom). U6 probe was used as a loading control.
Rpp30 mutations activate the DNA damage checkpoint
As major tRNA processing events seemed to occur properly, it raised the possibility that the arrest of oogenesis in Rpp30 mutant ovaries was due to the presence of a fraction of misprocessed tRNAs, rather than to a shortage of mature tRNAs. In mice mutant for the RNA kinase CLP1, the appearance of an aberrant form of pre‐tRNATyr is linked to the specific loss of motor neurons, while the steady‐state levels of mature tRNA are normal (Hanada et al, 2013; Karaca et al, 2014). Interestingly, this phenotype can be rescued by inactivating p53 in CLP1 mutant mice, indicating that the defects are caused by a p53‐dependent stress response (Hanada et al, 2013). We thus genetically removed p53 in flies mutant for Rpp30. Strikingly, in double‐mutant flies for Rpp30 18.2; p53 11‐1B‐1, or Rpp30 18.2; p53 11‐1B‐1/p53 5A‐1‐4, we found egg chambers developing to late stages of oogenesis and even forming a few eggs (Fig 3A and Table 1). This rescue was only partial, as the majority of egg chambers were arrested and double‐mutant flies remained sterile (Fig 3F and Table 1). Nonetheless, late stages of oogenesis were never observed in Rpp30 single mutants, indicating that one cause of arrest was the activation of a p53‐dependent checkpoint. In flies, DNA damage is the main cause of p53 activation by the checkpoint effector kinase Chk2. We thus generated double‐mutant flies for Rpp30 and chk2 (also known as mnk/loki in Drosophila) (Oishi et al, 1998; Xu et al, 2001). We found that most egg chambers developed into eggs in Rpp30 18.2; mnk p6, or Rpp30 18.2/Rpp30 PE; mnk p6 mutant flies (Fig 3B). In addition, these eggs were laid and fertilized, indicating that fertility was also restored. We also noticed a better rescue in aged flies than in young adults (Fig 3F). Inactivating chk2 thus led to a more complete rescue of oogenesis than inactivating p53. Importantly, we found that tRNA processing defects remained in Rpp30; chk2 double mutants, although oogenesis and fertility were rescued (Fig 2, lane 4). This result indicated that oogenesis can proceed normally despite the presence of elevated levels of intermediate forms of tRNAs when Chk2 is inactivated. We conclude that it is the activation of DNA damage checkpoints, which arrests oogenesis in Rpp30 mutant flies.
Figure 3. Rpp30 mutation leads to the activation of several checkpoint proteins and to replication stress.
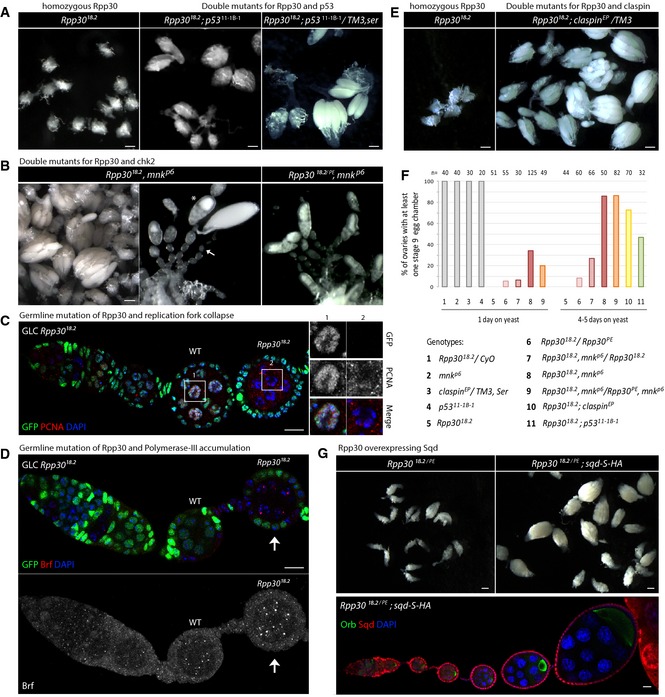
- Rpp30 18.2 homozygous early oogenesis arrest is partially rescued by p53 mutation. Scale bar, 100 μm.
- Early oogenesis arrest found in Rpp30 18.2 homozygous ovaries is rescued by chk2 mutation (Rpp30 18.2, mnk p6 and Rpp30 18.2 /Rpp30 PE , mnk p6). Scale bar, 100 μm. Asterisk: example of one rescued stage 9 egg chamber. Arrow: example of non‐rescued egg chambers.
- Germ line clones mutant for Rpp30 18.2 were immunostained for PCNA (red). DAPI is in blue. Magnifications: PCNA signal in a control or mutant nurse cell. Scale bar, 10 μm.
- Germ line clones mutant for Rpp30 18.2 were immunostained for Brf (pol III) (red). DAPI is in blue. A Z‐projection of Brf staining is shown. The arrow points to Brf aggregates in a mutant chamber. Scale bar, 10 μm.
- Early oogenesis arrest found in Rpp30 18.2 homozygous ovaries is partially rescued by claspin mutation (Rpp30 18.2 ; claspin EP /TM3). Scale bar, 100 μm.
- Quantification of rescued ovaries harboring at least one stage 9 egg chamber in genotypes numbered from 1 to 11 after 1 day or several days on yeast. Gray bars (1–4) are control flies. n = number of ovaries.
- Upper panel: Early oogenesis arrest found in Rpp30 182./PE transheterozygous ovaries is rescued by sqd‐S‐HA overexpression. Scale bars: 100 μm. Lower panel: Rpp30 18.2/PE; sqd‐S‐HA/sqd‐S‐HA ovaries were dissected and stained for Orb (green), HA (squid, red), and DAPI (blue). Scale bar, 10 μm.
Source data are available online for this figure.
Rpp30 mutations increase DNA replication stress
What could be the source(s) of DNA damage in Rpp30 mutant ovaries? It is well‐established in yeast and vertebrate cells that tRNA transcription creates stress during replication at specific loci in the genome (Deshpande & Newlon, 1996; Helmrich et al, 2013). Even under normal conditions, the high occupancy of RNA polymerase III (pol III) at tRNA gene loci is a block for the DNA polymerase (DNA pol) when replicating DNA. These pol III/DNA pol conflicts can destabilize the replication fork, lead to DNA lesions, and activate checkpoint proteins (Clelland & Schultz, 2010; Nguyen et al, 2010). Thus, given the known roles of tRNA loci as replication barriers and chromatin organizers, we hypothesized that replication stress could be a possible source of DNA damage in Rpp30 mutant ovaries, knowing that nurse cells undergo high rates of endoreplication (Noma et al, 2006; Donze, 2012; Raab et al, 2012). To test this hypothesis, we analyzed the localization of DNA pol and RNA pol III components in mutant and wild‐type conditions. We found that PCNA, a core component of the replication machinery, was not associated with DNA in Rpp30 18.2 germ line clones, suggesting a collapse of replication forks (Fig 3C) (Mailand et al, 2013). This PCNA loss was restored in homozygous Rpp30 18.2 flies expressing ubiRpp30::GFP transgene (Fig EV1D). Furthermore, we found that Brf (a subunit of RNA pol III) accumulated as aggregates specifically in Rpp30 mutant germ cells (Fig 3D). It was thus plausible that high accumulation of Pol III could increase replication fork collapses and replication stress. To investigate whether replication stress could block oogenesis, we generated flies mutant for both Rpp30 and a component of the replication stress checkpoint. Depending on the species, replication stress activates the ATR/Chk1/Claspin pathway, the ATM/Chk2 pathway, or both. In Drosophila, Claspin appears most specific to replication stress (Lee et al, 2012), as Chk1 and Chk2 respond to a wider range of DNA damages (Reinhardt & Yaffe, 2009). Furthermore, Claspin is part of the replication fork machinery and is required for efficient replication (Lou et al, 2008; Scorah & McGowan, 2009). We could not obtain flies double mutant for Rpp30 and a null allele of claspin 45, even when heterozygous, indicating synthetic lethality between the two genes (Table 1). We used instead a hypomorphic allele of claspin and found that Rpp30 18.2; claspin EP/+ and Rpp30 18.2; claspin EP mutant ovaries were partially rescued (Fig 3E and F, and Table 1), to a similar extent as with the inactivation of p53. We also observed an increase in viability of double‐mutant flies (Table 1). We confirmed these results by using three additional hypomorphic alleles of claspin, claspin 279, claspin aq4 , and claspin aq5 , which could all partially rescue Rpp30 mutant phenotypes (Table 2) (Lee et al, 2012). We further noticed an inverse correlation between the strength of the allele and the degree of rescue, with the weakest alleles rescuing the best (claspin aq4 < claspin 279 < claspin aq5). We further tested this model by introducing two additional copies of Squid in Rpp30 mutant ovaries. Squid is an abundant shuttling hnRNP similar to yeast Npl3 and is required for fertility in flies (Norvell et al, 1999). The helicase Rrm3 and the hnRNP Npl3 were shown to promote DNA replication at difficult‐to‐replicate loci such as tRNA genes and rDNA in S. cerevisiae (Kelley, 1993; Ivessa et al, 2003; Azvolinsky et al, 2009; Santos‐Pereira et al, 2013; Herrera‐Moyano et al, 2014). We overexpressed the nuclear Squid‐S isoform, which can rescue squid mutant ovaries (Norvell et al, 1999). We found that ovaries of Rpp30 18.2/Rpp30 PE; squid‐S/squid‐S flies produced egg chambers developing into eggs, and fertility was also partially restored (Fig 3G). We concluded that DNA replication stress was increased in Rpp30 mutant ovaries. This increase could partially explain the arrest of oogenesis in Rpp30 mutant ovaries, as both reducing the response to DNA replication damage and facilitating DNA replication at tRNA gene loci could partially rescue oogenesis and fertility. However, the partial, and not full, rescue of oogenesis indicated the presence of other sources of DNA damage in addition to replication stress.
Table 2.
Rpp30 oogenesis arrest is rescued by claspin mutations
| Female genotype | Ovaries with stage 9 chambers | Total observed | % Rescue |
|---|---|---|---|
| Rpp30 18,2 /CyO | 40 | 40 | 100 |
| Rpp30 18,2 /Rpp30 PE | 2 | 106 | 1.9 |
| Rpp30 18,2 /Rpp30 PE ; claspin aq5 /TM3 | 5 | 46 | 11 |
| Rpp30 18,2 /Rpp30 PE ; claspin aq5 | 13 | 64 | 20 |
| Rpp30 18,2 /Rpp30 PE ; claspin 279 /TM3 | 10 | 39 | 26 |
| Rpp30 18,2 /Rpp30 PE ; claspin 279 | 17 | 62 | 27 |
| Rpp30 18,2 /Rpp30 PE ; claspin aq4 /TM3 | 20 | 29 | 69 |
| Rpp30 18,2 /Rpp30 PE ; claspin aq4 | 14 | 39 | 36 |
Quantification of rescued ovaries harboring at least one stage 9 egg chamber in the indicated genotypes after several days on yeast.
Mutations in Rpp30 block transcription at major piRNA‐producing clusters
In addition to creating DNA damage, the collapse of replication forks can disrupt locally the organization of chromatin and the expression of nearby genes (Lambert & Carr, 2013). We thus performed genome‐wide RNA‐seq of Rpp30 mutant ovaries using, as control, ovaries of newborn wild‐type females with similar developmental stages. We focused on genes that were linked to fertility, downregulated in mutant ovaries, and localized close to clusters of tRNA genes. A family of genes producing piRNAs (also known as piRNA clusters) attracted our attention, because they fitted all three criteria: (i) piRNAs are required for fertility as they silence transposable elements (TEs) in the fly reproductive organs. Flies mutant for components of the piRNA biogenesis pathway are sterile. (ii) We found that transcription of at least 30 clusters was strongly downregulated in Rpp30 mutant ovaries. In particular, transcription of piRNA cluster 1/42AB was almost completely absent and transcription of cluster 2 was significantly reduced in mutant ovaries (Fig 4A and B, and Table 3). These two clusters produce the vast majority of germ line piRNAs. (iii) Cluster 1/42AB is localized close to the main cluster of tRNA genes at the centromere of chromosome II (~50 Kb) (Fig 4A). Cluster 2 is also localized close to several tRNA genes at the centromere of the X chromosome.
Figure 4. Rpp30 mutation leads to piRNA transcription defects.
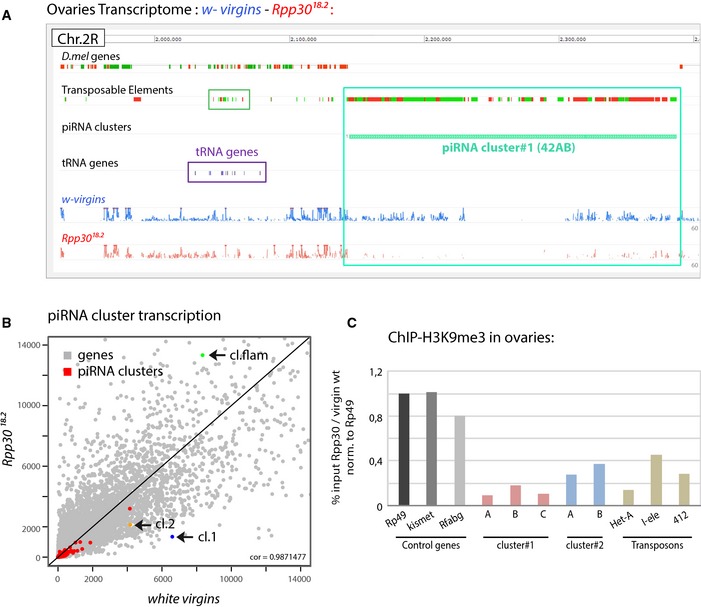
- RNA extracts from about 1,000 white virgin and Rpp30 18.2 ovaries were sequenced and the corresponding reads from chromosome 2R peri‐centromeric region are shown using Galaxy10 Mississippi Genome Browser (drosophile.org). Flybase D.mel genes, transposable elements (TEs), piRNA clusters, and tRNA gene clusters are indicated. Highlighted squares correspond to TEs, green; tRNA genes, violet; and piRNA clusters, turquoise.
- Scatterplot comparing the expression of piRNA clusters issue from RNA‐seq data in white virgins versus Rpp30 18.2 mutant ovaries. Gray dots correspond to genes and red dots to the 141 Drosophila piRNA clusters. Cluster 1 (42AB) is in blue, cluster 2 is in orange, and cluster 8 flamenco is in green.
- About 1,400 ovaries from white virgins and Rpp30 18.2 were used for ChIP experiments with H3K9me3 antibody. The corrected percentage of input normalized to control genes is shown as the ratio between the mutant and the control, reflecting a decrease in methylation profiles in different regions of the two principal piRNA clusters (1 and 2) and in some TEs (Het‐A, I‐ele, and 412).
Table 3.
piRNA clusters expression in Rpp30 18.2 mutants
| Fold change Rpp30 versus white virgins from RNA‐seq data | |||
|---|---|---|---|
| piRNA cluster number | Log2 fold change | P‐value | Chromosome |
| 19 | 1.75 | 6.54E‐05 | X |
| 8 | 0.66 | 0.0099094 | X |
| 2 | −0.95 | 1.70E‐05 | X |
| 62 | −1.5 | 0.0017961 | 2L |
| 5 | −0.8 | 0.0098801 | 2L |
| 35 | −0.89 | 0.0036137 | 2LHet |
| 1 | −2.26 | 3.02E‐41 | 2R |
| 56 | −1.49 | 0.0003018 | 2R |
| 142 | −0.86 | 0.0004981 | 2R |
| 97 | −2.09 | 0.0034424 | 2R |
| 6 | −1.16 | 4.25E‐06 | 3L |
| 32 | −1.52 | 0.0026484 | 3L |
| 26 | −1.34 | 4.02E‐09 | 3LHet |
| 15 | −1.36 | 1.45E‐06 | 3LHet |
| 44 | −1.78 | 2.67E‐05 | 3LHet |
| 34 | −1.53 | 0.000248 | 3LHet |
| 12 | −1.14 | 0.0003177 | 3LHet |
| 24 | −1.2 | 0.0011207 | 3LHet |
| 57 | −0.89 | 0.0035816 | 3LHet |
| 72 | −0.99 | 0.0036892 | 3LHet |
| 59 | −2.11 | 1.53E‐09 | 3RHet |
| 37 | −2.06 | 8.50E‐07 | 3RHet |
| 16 | −1.47 | 0.0004072 | 3RHet |
| 29 | −1.28 | 0.0001272 | 4 |
| 43 | −2.72 | 2.12E‐09 | U |
| 10 | −0.93 | 0.0009059 | U |
| 7 | −1.69 | 0.0020243 | U |
| 94 | −1.66 | 0.0021603 | U |
| 68 | −1.38 | 0.0024001 | U |
| 77 | −1.16 | 0.0034278 | U |
| 74 | −1.8 | 0.0040003 | U |
| 14 | −1.08 | 0.0066757 | U |
piRNA clusters highlighted in Figure 4B are shown in bold.
The normalized fold change, the P‐value, and the chromosome localization of the piRNA clusters that are affected in Rpp30 18.2 mutants ovaries over white virgin transcriptome RNA‐seq analysis.
How could the transcription of piRNA clusters be affected? The regulation of piRNAs transcription is not well understood. However, it has recently been proposed that the deposition of the heterochromatin mark H3K9me3 may be required for the transcription of dual‐strand clusters in the germ line (Rangan et al, 2011; Mohn et al, 2014). We thus tested whether a loss of this mark could play a part in the loss of piRNA precursor transcription. We carried out two independent ChIP experiments for H3K9me3 marks using ovaries of Rpp30 18.2 females and ovaries of similar size from newborn wild‐type females, as developmental control (Figs 4C and EV2). We found that H3K9me3 occupancy was diminished to about 10% of wild‐type levels in Rpp30 mutant ovaries for cluster 1 and to 30% for cluster 2. Importantly, this reduction in heterochromatin marks was not a general feature, as a euchromatic gene (kismet) and a heterochromatic gene (rfabg) showed similar levels of H3K9 trimethylation in wild‐type and mutant conditions. The RNA‐seq data also revealed that expression of flamenco/cluster 8, the main piRNA‐producing cluster in somatic cells was not reduced and appeared even slightly increased in mutant ovaries (Fig 4B and Table 3). Accordingly, flamenco transcription does not depend on H3K9me3, and somatic Rpp30 mutant clones were not associated with visible oogenesis defects (Fig 1F, right panel). Altogether, these data strongly suggest that Rpp30 mutations disrupt the chromatin surrounding the main tRNA genes locus and lead to a strong reduction in the transcription of the nearby major piRNAs clusters in germ cells.
Figure EV2. Size of ovaries used in this work (related to Figs 4, 5, 6).
Ovaries of different genotypes used for small RNA sequencing, RNA sequencing, and ChIP analysis. The left panel of heterozygous Rpp30 18.2 /CyO ovaries has been included again in this figure (also present in Fig 1A) in order to better compare the different ovaries used altogether. Scale bar, 100 μm.
Mutations in Rpp30 cause a dramatic depletion of the piRNA population
Endo‐siRNAs and piRNAs are the two main classes of small RNAs required to silence TEs in flies (Senti & Brennecke, 2010). To analyze these small RNA populations, we performed small RNA‐seq in Rpp30 18.2 and Rpp30 18.2/Rpp30 PE mutant ovaries, and used as controls Rpp30 18.2/CyO adults, newborn wild‐type females (developmental control) and Rpp30 18.2 ; ubi‐Rpp30‐GFP ovaries (genetic background control), as well as aub mutant ovaries (positive control) (see Fig EV2 for size of ovaries). By investigating the size distribution of small RNAs matching TEs sequences, we found that the 23–28 nt population corresponding to piRNAs was dramatically decreased in Rpp30 mutant ovaries, almost as strongly as in aub (Fig 5A). In contrast, the population of endo‐siRNAs (centered on 21 nt) was globally not affected, indicating that Rpp30 mutations disrupted piRNA production specifically. We focused on piRNAs mapping to unique piRNA‐producing loci in the genome (Brennecke et al, 2007). We found that the main piRNA clusters were dramatically affected in Rpp30 mutants, such as cluster 1/42AB, cluster 2, and cluster 3, which account for the majority of germ line piRNAs (Fig 5B). Next, we investigated whether the processing of the remaining piRNAs in mutant germ cells was affected by analyzing the “ping‐pong” signal, which is the probability of complementary sense and antisense piRNAs to overlap by 10 nt (Brennecke et al, 2007; Gunawardane et al, 2007). It is a signature of piRNA biogenesis happening in the “nuage” of nurse cells by the slicing activity of Ago3 and Aub. We found that the overall percentage of ping‐pong signal was strongly reduced in mutant germ cells (Fig 5C), but not completely abolished as in aub mutant ovaries (Brennecke et al, 2007; Gunawardane et al, 2007). This strong reduction in the ping‐pong signal could reflect a strong reduction in the total amount of piRNAs. We thus calculated the z‐score of the ping‐pong signal and found that in Rpp30 18.2 mutant ovaries, it was close to wild‐type levels, indicating that the processing of the remaining piRNAs in the germ line was not strongly affected by Rpp30 mutations. We then explored the cellular consequences of these defects by analyzing the morphology and composition of the nuage surrounding nurse cell nuclei, where the ping‐pong processing of piRNAs takes place. The localization of Aubergine was dramatically altered: While Aub is normally organized as rings around each nurse cell nucleus, it was dispersed throughout the cytoplasm of mutant germ cells (Fig 5D). Ago3 and Maelstrom localizations were also affected forming clumps instead of adopting an even distribution around each nucleus as in the wild‐type situation (Fig 5D). However, the structure of the nuage itself was globally preserved as revealed by the normal pattern of Krimper and Vasa (Fig EV3A). We concluded that Rpp30 mutations induced a dramatic depletion of germ line piRNAs. However, Rpp30 did not seem to play a critical role in processing piRNA precursors through the ping‐pong cycle.
Figure 5. Rpp30 mutation leads to a collapse of piRNAs.
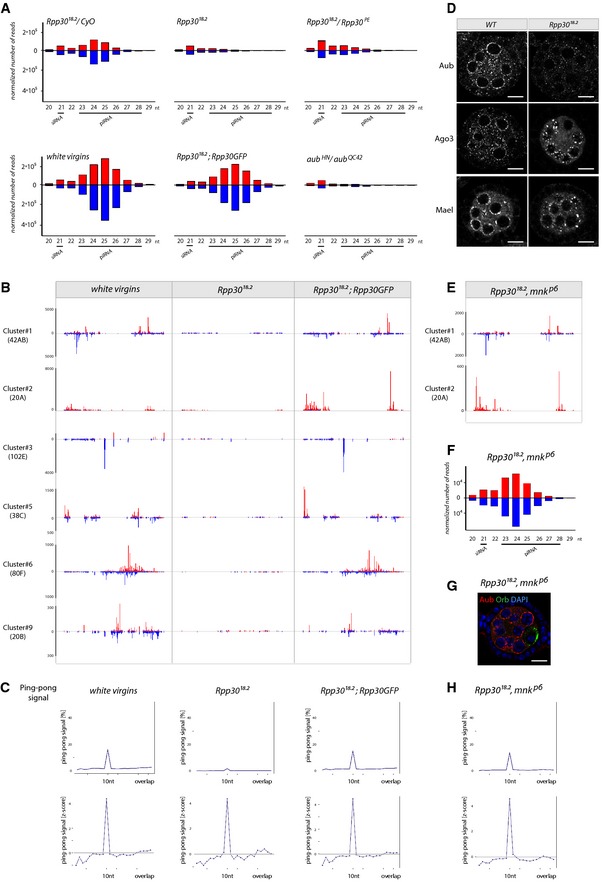
-
ASmall RNAs extracted from ovaries of different genotypes were sequenced. Size histograms of normalized RNA populations matching transposable elements (TE) are shown. Red: sense. Blue: antisense. Endo‐siRNA (21 nt) and piRNAs (23–28 nt) are indicated. Rpp30 mutations in two different genetic backgrounds (Rpp30 18.2 and Rpp30 18.2 /Rpp30 PE) specifically alters piRNA populations, compared to heterozygous control ovaries. The white virgin ovaries were used as a developmental control. Rpp30 18.2 carrying the ubiRpp30GFP transgene (genetic background control) rescues general piRNA populations. The aub HN /aub QC42 was used as a positive control.
-
BSpecific normalized unique mapper piRNA populations (23–28 nt) from the major piRNA clusters are shown for the indicated genotypes. The scale of reads is indicated on the left.
-
CThe ping‐pong signal calculated on multimappers piRNA populations is shown for the different genotypes.
-
DNuage‐specific markers (Aub, Ago3, and Mael) are shown in stage 3 wild‐type and germ line mutant clones for Rpp30 18.2. Scale bar, 10 μm.
-
E–HDouble mutant ovaries for Rpp30 and chk2 (Rpp30 18.2 , mnk p6 ) rescued the specific and general piRNA populations (E, F), nuage morphology (G), and the characteristic ping‐pong signal (H). Scale bar, 10 μm.
Figure EV3. piRNAs are rescued in double mutants Rpp30, chk2 (related to Fig 5).
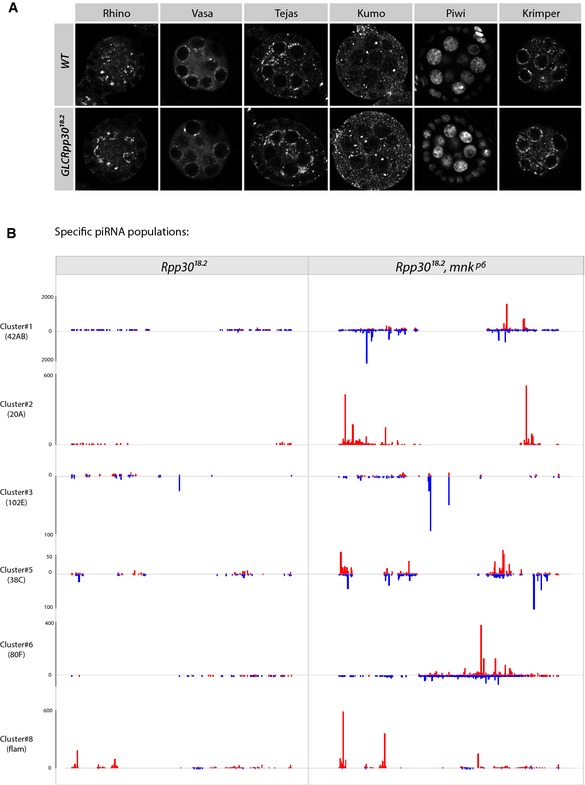
- Nuage‐specific factors (Vasa, Tejas, Kumo, and Krimper) and Rhino and Piwi protein localizations are shown in wild‐type and germ line mutant clones for Rpp30 18.2.
- piRNA‐specific unique mappers from different piRNA clusters are shown in Rpp30 18.2 homozygous ovaries versus Rpp30 18.2 , mnk p6 double‐mutant ovaries. The scales are indicated on the left. Red, sense. Blue, antisense.
Source data are available online for this figure.
One striking feature of Rpp30; chk2 double mutant was a partial rescue of fertility. It contrasted with the inactivation of the same ATM/Chk2 checkpoint in aub, armi, or zucchini mutants, which alleviates oogenesis defects, but does not restore fertility (Klattenhoff et al, 2007; Pane et al, 2007). We thus analyzed the piRNA population in Rpp30; chk2 double‐mutant ovaries. We found that the global population of piRNAs and unique mappers were significantly restored (Figs 5E and F, and EV3B). Accordingly, we found by RT–qPCR that transcription of the main piRNA clusters 1 and 2 was significantly increased in Rpp30, chk2 double mutant compared to Rpp30 single mutant (Fig EV4A). In addition, we observed that this rescue of piRNA clusters transcription correlated well with a rescue of the H3K9me3 marks in Rpp30, chk2 double‐mutant ovaries (Fig EV4C). We performed ChIP experiments for H3K9me3 marks and found for cluster 1 that in Rpp30, chk2 double‐mutant ovaries, H3K9me3 levels were around 60% of wild‐type as compared to 10% in Rpp30 single mutant; and for cluster 2, H3K9me3 levels were back to around 90% of wild‐type as compared to 30% in Rpp30 single mutant (Fig EV4C). We also found that the localization of Aub to the nuage, Orb to the posterior cortex, and PCNA to nurse cell DNA, as well as the ping‐pong signal, was all mostly rescued (Figs 5E–H and EV4D). These results demonstrated that Rpp30 cannot have a crucial role in processing most piRNA precursors, despite the known endonuclease activity of RNase P. We concluded that Rpp30 mutations most likely affected the transcription of piRNAs but not their processing.
Figure EV4. Rpp30, chk2 double mutant characterization (related to Figs 3, 4, and 6).
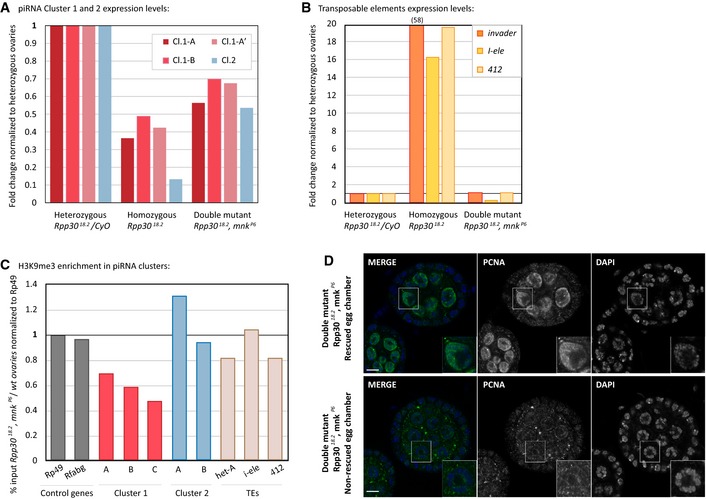
-
A, BRNA extracted from heterozygous Rpp30 18.2 /CyO, homozygous Rpp30 18.2 and double mutant Rpp30 18.2 ,mnk p6 ovaries was used to study piRNA clusters 1 and 2 expression levels (A) and transposable element expression (B) by RT–qPCR.
-
CAbout 100 ovaries from white and Rpp30 18.2 , mnk p6 were used for ChIP experiments with H3K9me3 antibody. The corrected percentage of input normalized to control genes is shown as the ratio between the double mutant and the control, reflecting a rescue of methylation profiles in different regions of the two principal piRNA clusters (1 and 2) and in some TEs (Het‐A, I‐ele, and 412) in double‐mutant ovaries.
-
DDouble‐mutant ovaries Rpp30 18.2 , mnk p6 were fixed and stained for PCNA. Example of one rescued and one non‐rescued egg chamber are shown. Scale bar, 10 μm.
Transposable elements are de‐repressed in Rpp30 mutant ovaries
We then analyzed the consequences of this reduction in piRNA production on transposable elements (TEs) regulation. We measured the steady‐state mRNA levels of different TE families by RT–qPCR (Fig 6A). Most TEs showed a modest upregulation in Rpp30 18.2 and Rpp30 18.2/Rpp30 PE mutant ovaries compared to Rpp30 18.2 /CyO and Rpp30 18.2/Rpp30 PE ; ubi‐Rpp30‐GFP (meaning in the same genetic background). In contrast, specific TEs, such as I‐element or 412, exhibited strong de‐repression in Rpp30 mutant when compared to heterozygous flies (20‐ and 15‐fold increase, respectively). These levels were comparable to the increases observed in aub mutants in which germ line piRNAs are almost completely absent. To obtain a genome‐wide quantitative and qualitative assessment of TE deregulation, we used our RNA‐seq datasets of Rpp30 18.2 mutant ovaries (Fig 6B and C). We found that on average TEs were significantly de‐repressed, such as 412, and that some of them were highly de‐repressed, such as I‐element, as we previously showed (Fig 6A). P‐element expression was also very high. Finally, we tested the genomic copies of TE families that were highly expressed in the Rpp30 mutant background and found that their levels of H3K9 trimethylation were also greatly diminished (Fig 4C). Overall, these results demonstrate that transposable elements were de‐repressed in Rpp30 18.2 mutant ovaries. In contrast, we found that in Rpp30; chk2 double‐mutant ovaries, representative TEs were repressed, and that their levels of H3K9me3 were close to those of wild‐type (Fig EV4B and C).
Figure 6. Rpp30 mutation leads to transposable elements overexpression.
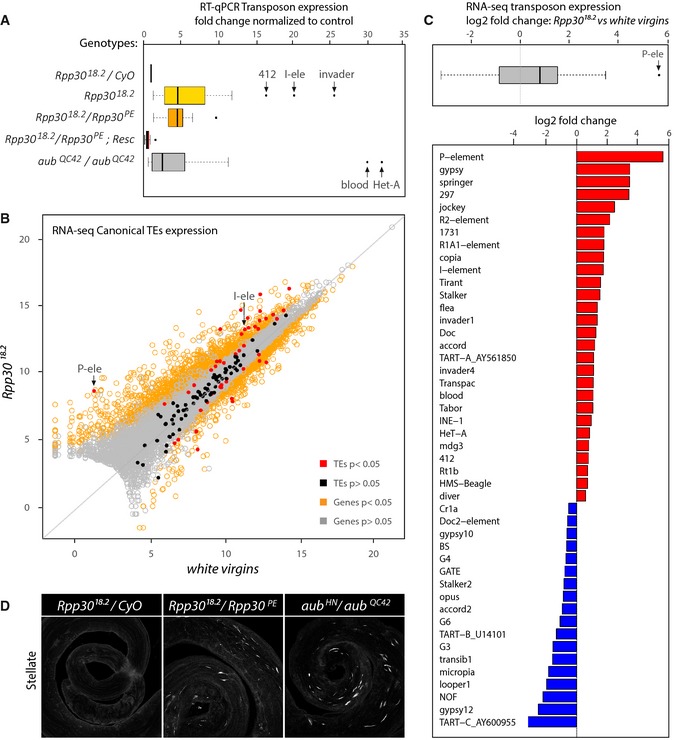
- Ovaries of different genotypes were dissected for RNA extraction. RT–qPCR was performed using transposable elements‐specific oligonucleotides. The boxplots show several pooled transposon fold changes obtained for different genotypes after normalization to control heterozygous Rpp30 18.2/CyO. Homozygous and transheterozygous Rpp30 flies (yellow and orange, respectively) show transposon overexpression, whereas Rpp30 18.2 /Rpp30 PE ovaries carrying the ubiRpp30GFP transgene are similar to control. aub HN/QC42 ovaries (gray) were used as a positive control. Arrows point to some highly expressed transposons.
- Scatterplot showing the expression of canonical transposable elements from RNA‐sequencing data obtained from white virgins versus Rpp30 18.2 ovaries. Arrows point to P‐element (P‐ele) and I‐element (I‐ele). Transposons are indicated as red or black dots (significant or not significant fold change, respectively). Genes are indicated as yellow or gray dots (significant or not significant fold change, respectively).
- Significant transposons fold changes (P < 0.05) were pooled and shown in a boxplot (gray). Note that the median value (black bar) is above 0. Arrow indicates the P‐element (P‐ele). Fold changes of individual transposons are shown in a barplot. Red bars, upregulated transposons. Blue bars, downregulated transposons.
- Testes from control heterozygous, transheterozygous Rpp30 18.2 /Rpp30 PE or aub HN/QC42 were fixed and stained for Stellate crystal formation. Z‐projections are shown.
To test whether Chk2 activation could directly inhibit piRNA transcription and lead to TEs transcription, we used spnA mutant ovaries. SpnA is a Rad51 protein required to repair meiotic DSBs in Drosophila germ cells (Staeva‐Vieira et al, 2003). spnA mutant ovaries show meiotic defects, ventralized eggs, and sterility. These phenotypes are due to the activation of Chk2, as these defects (except sterility) are rescued in spnA, chk2 double‐mutant ovaries (Staeva‐Vieira et al, 2003). As a control, we used spnE mutant flies, which show well‐characterized defects in piRNA biogenesis (Malone et al, 2009). We performed RT–qPCR on representative TEs in spnA and spnE mutant ovaries and found that TEs were not upregulated in spnA mutant ovaries, whereas TEs were de‐repressed in spnE ovaries as published (Malone et al, 2009) (Fig EV5A). Aubergine staining in the nuage was also similar to wild‐type in spnA ovaries, while it was much reduced in spnE nurse cells (Fig EV5B, left panel). Finally, we found that PCNA staining was identical to wild‐type in both spnA and spnE mutant ovaries (Fig EV5B, right panel).We concluded that activating Chk2 by other means (here unrepaired meiotic DSBs) is not sufficient to disrupt the nuage and the silencing of TEs.
Figure EV5. Chk2 activation by spnA mutation does not recapitulate Rpp30 phenotype (related to Figs 3, 5, and 6).
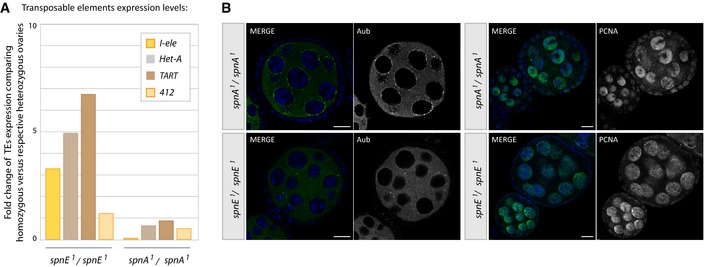
- RNA extracted from heterozygous or homozygous spnA 1 and spnE 1 flies was used to study transposable element expression by RT–qPCR. The fold change of TEs expression comparing homozygous versus respective heterozygous ovaries is shown.
- Ovaries from homozygous spnA 1 and spnE 1 flies were fixed and stained for Aub (left) or PCNA (right). Scale bar, 10 μm.
Because fertility defects were also observed in males carrying the Rpp30 18.2 mutation, we tested other genetic elements than TEs, which are also regulated in a piRNA‐dependent manner. The Stellate (Ste) locus is made of about 200 tandem repeats of the Ste gene, which are specifically silenced in males by piRNA species produced from the Su(Ste) locus located on the Y chromosome (Tulin et al, 1997; Aravin et al, 2004). In aubergine mutant testes, Ste is overexpressed and forms crystals, as a result of defective piRNA production (Aravin et al, 2004; Klattenhoff et al, 2007). In Rpp30 18.2/Rpp30 PE mutant testes, we found crystals of Ste. Although the amount of crystals was lower than that in aub mutant, these crystals were never seen in wild‐type testes (Fig 6D).
We concluded that transposons were de‐repressed in Rpp30 mutant germ cells. De‐repression of TEs can induce their mobilization and the accumulation of DNA lesions throughout the genome. We propose that both TEs and replication stress contribute to the activation of DNA damage checkpoint proteins and the arrest of oogenesis in Rpp30 mutant ovaries.
Discussion
This study started by the surprising observation that hypomorphic mutations in a tRNA processing enzyme could specifically induce sterility. Our results reveal that tRNA defects per se are not causing a failure to produce mature eggs. Instead, this defect is linked to the activation of several DNA damage checkpoint proteins leading to oogenesis arrest. We find that replication stress and transposon de‐repression are the likely causes of DNA damage in Rpp30 mutants, leading to the activation of the checkpoint proteins p53, Claspin, and Chk2, and premature interruption of oogenesis. tRNA‐related pathologies in humans are associated with complex clinical phenotypes, including neuropathologies and sterility (Abbott et al, 2014). By revealing the chain of events leading to sterility in Drosophila, we hope that our study will shed light onto the etiology of these diseases. Here, we propose a model by which tRNA defects impact on chromatin organization in cis of tRNA‐producing loci, inducing replication stress and collapse of piRNA transcription (Fig 7).
Figure 7. Rpp30 mutation leads to tRNA processing and piRNA transcription defects.
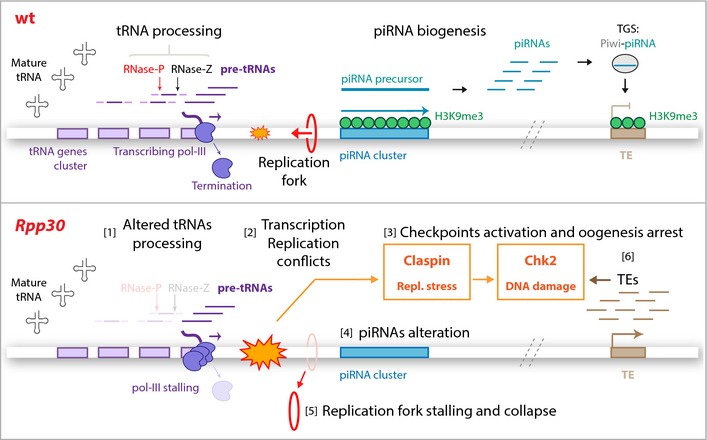
Top: In wild‐type ovaries, tRNA genes (violet boxes) are transcribed by pol III. tRNAs are then processed by RNase P and Z ribozymes that cleave tRNAs precursors (pre‐tRNAs, dark violet). tRNA gene clusters can be localized near piRNA clusters (turquoise), which are characterized by H3K9me3 epigenetic mark (green) required for piRNA transcription. piRNAs arising from piRNA precursors guide Piwi proteins to transcriptionally silence transposable element expression (TGS), protecting genome integrity. Bottom: In Rpp30 ovaries, tRNA processing defects [1] lead to transcription–replication conflicts (orange) [2]. These conflicts activate both replication stress and DNA damage checkpoint proteins, Claspin and Chk2 [3], which induce piRNA transcription defects [4] and replication fork stalling and PCNA collapse [5]. Downregulation of piRNAs results in TEs overexpression [6], which also activate DNA damage checkpoint [3].
A model linking tRNA defects, replication stress, and piRNAs transcription
We found that Rpp30 mutations exacerbate replication conflicts, as shown by the accumulation of the pol III subunit Brf, the collapse of PCNA, the activation of Claspin, and the rescue by overexpression of Squid. In turn, these defects could disrupt the deposition of H3K9me3 heterochromatin marks in the surrounding region, as observed in our H3K9me3 ChIP experiments. In Drosophila, loss of H3K9me3 near tRNA genes would affect the transcription of neighboring loci sensitive to H3K9me3 levels. As demonstrated recently, loci producing piRNAs such as single TE insertions and dual‐strand clusters are particularly sensitive to H3K9me3 levels (Mohn et al, 2014). Our model could thus explain why transcription of such loci is strongly affected if they are in close proximity to tRNA genes. Consequently, the strong reduction in piRNAs production in Rpp30 mutant ovaries leads to the de‐repression of TEs, an increase in DNA damage, and an early arrest of oogenesis.
There are about 300 tRNA genes in Drosophila and about 140 piRNA clusters as defined in Brennecke et al (2007). Although we did not find a perfect correlation between tRNA genes and piRNA cluster proximity for every single locus, the major piRNA clusters, accounting for the majority of germ line piRNAs, are all close to tRNA genes. For example, cluster 1 localized at 42AB and producing more than 30% of germ line piRNAs, is less than 40 kb away from the main cluster of tRNA genes containing more than 15 tRNA genes (Fig 4A and Flybase 2R:2015985‐2163232). Cluster 2 and cluster 6, which are also major producers of germ line piRNAs, are found close to several tRNA genes and are strongly affected in Rpp30 mutants. In contrast, transcription of flamenco, which has only low levels of H3K9me3, is slightly increased in Rpp30 mutant ovaries. In addition to piRNA clusters, we found that tRNA genes were very often intertwined with transposable elements (Fig 4A). Recently, these stand‐alone TE insertions were shown to be major sources of piRNAs in addition to piRNA clusters (Mohn et al, 2014; Shpiz et al, 2014). Loss of H3K9me3 marks around tRNA genes would also affect these sources of piRNAs and further decrease the overall population of piRNAs in Rpp30 mutant germ cells.
How could mutations in the RNase P enzyme affect tRNA transcription and increase replication stress? We can envision at least three mutually non‐exclusive hypotheses. Firstly, in human cells and in yeast, RNase P has been shown to bind chromatin at tRNA gene loci and to enhance their transcription by pol III (Reiner et al, 2006; Jarrous & Reiner, 2007). This function of RNase P directly couples tRNA transcription and early processing. Interestingly, in an RNase P mutant, pol III complexes are still normally recruited to tRNA genes, but their transcription levels are very low, indicative of pol III immobilization on tRNA promoters (Reiner et al, 2006). According to our model, higher pol III occupancy would significantly increase replication stress. Secondly, we can speculate that the production of misprocessed tRNAs could activate a feedback response prompting the cell to produce more tRNAs in order to compensate. Increased occupancy of pol III on tRNA gene promoters would also ensue here. Finally, it has been recently shown that correct tRNA folding is required for releasing pol III from tRNA loci (Nielsen et al, 2013). If tRNAs are not properly folded, RNA pol III does not terminate transcription and remains on the locus. Regarding the function of Rpp30, the aberrant presence of the 5′ tail of pre‐tRNA in Rpp30 mutants could induce improper folding of the transcript, inducing pol III stalling. A common event in these three hypotheses is the increased binding time of pol III (or associated factors) on tRNA genes, with conflicting effects on the progression of the replication machinery (Nguyen et al, 2010). These defects would be especially pronounced in nurse cells, which are actively endoreplicating their DNA. Accordingly, we found an accumulation of the pol III subunit Brf in Rpp30 mutant nurse cells. It should be noted that the overall amount of mature tRNAs seems normal in Rpp30 mutants, and thus, only a subset of tRNA genes is likely to be affected at a given time and/or in a given cell. Nonetheless, our data show that defects even restricted to a subset of tRNA loci can induce replication stress at a threshold sufficient to activate dedicated checkpoints and arrest oogenesis.
It has been previously proposed that stalling of the replication fork can be rescued by homologous recombination (Li & Heyer, 2008; O'Donnell et al, 2010; Alabert & Groth, 2012). As a result, epigenetic marks such as histone modifications lack from both DNA strands in the neighboring region, as we observed with the H3K9me3 mark. Our model is thus consistent with what has been proposed for RNAi mutants in S. pombe, where increased transcription–replication conflicts compromise H3K9me2 enrichment at centromeres (Kato et al, 2005; Zaratiegui et al, 2011). Recently, these results were extended to genome‐wide level in a Dicer 1 mutant yeast where tRNA genes loci were shown to be prominent sites of transcription–replication conflicts between RNA pol II and DNA pol (Castel et al, 2014). Our model could thus be generally applicable in different species.
tRNA genes and transposable element insertion
Our model is based on the local effect of replication stress on chromatin and thus relies on the genomic proximity of tRNA genes and piRNAs producing sources. These sources can be piRNA clusters but also single TE insertions as demonstrated recently (Mohn et al, 2014; Shpiz et al, 2014). We would like to speculate that this proximity is not random, as tRNA genes are often associated with single transposons or transposon remnants, such as piRNA clusters. In Saccharomyces cerevisiae and Dictyostelium discoideum, retrotransposons of the Ty family show a strong bias of insertions upstream of pol III transcripts (Qi et al, 2012). In particular, the main Ty1 and Ty3 retrotransposons are inserted almost exclusively upstream of tRNA genes. This insertion bias is driven by direct interactions between TE integration proteins and TFIIIB, an essential component of RNA pol III. This proximity may help amplification of LTR‐containing TEs, as they require tRNAs to prime their retro‐transcription (Mak & Kleiman, 1997). Such a genome‐wide analysis has not been done in other organisms. Interestingly, it was recently reported in C. elegans that tRNA genes are also linked to the production of piRNAs (also known as 21U‐RNAs) in germ cells (Kasper et al, 2014). The SNPC‐4 protein was shown to bind to tRNA genes located within the two main piRNA‐producing loci and to be required for the transcription of piRNAs in germ cells (Kasper et al, 2014). The authors also proposed that tRNA genes may create a chromatin environment facilitating piRNAs transcription. The genomic and functional proximity of tRNA genes and piRNAs sources could thus be conserved across many species.
The single most de‐repressed TE in our study is the P‐element, a DNA transposon. In contrast to retrotransposons, DNA transposons cannot expand by retro‐transposition (“copy‐and‐paste”), but move by a “cut‐and‐paste” mechanism, which cannot explain an increase in copy number (Siomi et al, 2011). An elegant study has shown that P‐elements transpose preferentially to origins of replication (Spradling et al, 2011). This mechanism proposes that coordinating transposition with replication could expand the number of P‐element insertions, not only by sister‐strand mediated repair (due to the excision), but it would also allow one P‐element to translocate to several replication forks in a single S phase, thus multiplying the number of integrated copies. In light of our results and others, tRNA genes are strong replication blocks on which replication forks are paused for extended periods of time. This would increase the possibilities of expansion for P‐elements and explain the preferential insertions of these TEs in hard‐to‐replicate regions such as centromeres, telomeres, and tRNA gene loci. The relationship between tRNA genes and transposable elements could thus be one of the drivers that shaped the Drosophila genome during evolution.
Materials and Methods
Fly stocks
All flies were raised at 25°C with some exceptions (see below). The following fly stocks were used: w1118; Df(2L)ED21, P{3′.RS5 + 3.3′}ED21/SM6a (Rpp30 deficiency, Bloomington 9177); y,w,hs‐FLP;nls‐GFP,FRT40A, Bloomington (Bl.); hs‐FLP;His2B‐RFP,FRT40A was kindly provided by Yohanns Bellaïche (Institut Curie, Paris); y,w, FRT40A‐Rpp30 18.2 stock was generated in a EMS screen in the laboratory (Rpp30 18.2, Jagut et al, 2009); y, w; P{lacW}Rpp30 k01901/CyO (Rpp30 PE, Bl.10507); mnk p6 was kindly provided by Uri Abdu (Ben Gurion University, Israel); p53 11‐1B‐1 (Bl. 6816); p53 5A‐1‐4 (Bl.6815); aub HN2 , cn 1 ,bw 1/CyO (Bl. 8517); aub QC42 ,cn 1 ,bw 1/CyO (Bl. 4968); w 1118 (Bl. 3605); y,w; claspin EY11302 (Bl. 20287); claspin 45, claspin 279, claspin aq4 , and claspin aq5 were kindly provided by Young‐Han Song (Hallym University, Republic of Korea). Rpp30; claspin double mutants and FRT40A‐Rpp30 18.2; ubiRpp30GFP/TM3, Ser stocks were raised at 22°C. Rpp30 18.2 , mnk p6 double mutant was obtained by standard recombination techniques, and mnk p6 mutation was verified by PCR. Sqd‐S‐HA transgenic flies were kindly provided by Trudi Schupbach (Princeton University) (spnA 1 (Bl. 3322); spnE 1 (Bl. 3327)).
Rpp30 18.2 allele sequencing
DNA was extracted from single adult females homozygous for Rpp30 18.2 by squeezing one fly in 50 μl of squeezing buffer (10 mM Tris–Cl pH 8.2; 1 mM EDTA; 25 mM NaCl; 200 μg/ml of proteinase K). After 30‐min incubation at 37°C and 95°C for 1–2 min, the solution was centrifuged for 5 min at maximum speed. The extracted DNA was sequenced (Cogenics‐Beckman). The pairs of oligonucleotides (Sigma Aldrich) spanning the whole gene region are detailed below.
The Geneious software was used to align the sequenced products of Rpp30 (Flybase CG11606).
Rpp30GFP transgene constructs
cDNA corresponding to Rpp30 (DGRC, GenBank AY075315) was used to do a PCR (5′‐CACCATGGAGCAAACAAGGCC‐3′ and 5′‐CGCCACTTTAAGTCGTTTAATAGCG‐3′). The PCR product was purified and cloned into the pENTR/D‐TOPO Gateway vector (Invitrogen) and subcloned into the pUbi (ubiquitous expression) and pUASp (germ line‐specific expression) vectors, with a C‐terminal GFP tag (Murphy and Huynh laboratory, DGRC, the Gateway vector done by Clara Moch). Transgenic lines were generated by standard methods (BestGene).
Flp/FRT clone generation
y,w, flp; FRT40AGFP or FRT40ARFP were crossed with FRT40A Rpp30 18.2/CyO flies. Resulting 3rd instar larvae were heat‐shocked two times at 37°C for 2 h (morning and then afternoon). Ovaries from 4 to 5 days adult flies were dissected (see below). Mutant homozygous clones were recognized by the absence of GFP or RFP.
Immunofluorescence experiments in ovaries and testes
Ovaries were dissected in PBS and then fixed in PFA 4% for 15 min (or 5 min for nuage components). After 3 washes in PBS, ovaries were permeabilized for 30 min with PBT (PBS Triton 0.2%). Ovaries were incubated with primary antibody overnight (O/N) at 4°C, then washed and incubated with secondary antibodies for 2 h at room temperature. After several washes, ovaries were incubated with Hoechst for 5 min, PBS was removed, and mounting solution was added (cityfluor Biovalley). The ovarioles were mounted on a microscope slide for observation removing late stages and eggs to better observe early stages. Testes were dissected in PBS and then fixed in 4% PFA in PBT 0.3% plus 3 volumes of heptane and incubated for 20 min at room temperature. After two washes in PBT 0.2% and 1 h in PBT 0.3% and BSA 3%, testes were incubated with primary antibody O/N in PBT 0.2%. After several washes, secondary antibody was added for 2 h in PBT 0.2%, testes were washed, incubated with Hoechst for 15 min in PBS, and incubated in cityfluor at 4°C until mounting. The following antibodies were used: mouse anti‐Orb used at 1/250 [clones 6H8 and 4H8 Developmental Studies Hybridoma Bank]; rabbit anti‐Aubergine (kindly provided by Paul Lasko, McGill University, Quebec) used at 1/10,000; rabbit anti‐Argonaute3, anti‐Piwi, anti‐Krimper, anti‐Stellate, anti‐Vasa, anti‐Rhino, and guinea pig anti‐Maelstrom were kindly provided by Toshie Kai (Temasek, Singapore) and/or William Theurkauf (University of Massachusetts), and were used at 1/500 or 1/1,000. Mouse anti‐PCNA, used at 1/5,000 (clone PC10, Dako), was kindly provided by Nathalie Dostatni (Institut Curie, Paris). Rabbit anti‐Brf, used at 1/500 was kindly provided by W.E. Stumph (San Diego University). Ovaries and testes were visualized with a confocal microscope Zeiss LSM 780. Images were acquired with Zen image software. Images were processed and mounted with Adobe Photoshop and Illustrator CS4. For testes, Z‐stacks are shown as a Z‐projection with the same acquisition settings in control and mutant conditions.
Quantification of Rpp30 18.2 oogenesis arrest rescued by chk2, p53, and claspin mutations
Flies were kept for up to 1 week with yeast, ovaries were dissected in PBS and the percentage of ovaries with at least one stage 9 egg chamber were quantified as rescued.
RNA extraction from ovaries
Dissected ovaries were collected in cold PBS. Ovaries were homogenized with a pestle in TRIzol (Invitrogen) and were snap‐frozen and stored at −80°C until RNA extraction. RNA was precipitated by standard methods with chloroform and isopropanol, washed in EtOH 70%, and resuspended in RNase‐free H2O (Sigma). DNA was digested with DNase following the indicated procedure (Ambion). RNA was precipitated with phenol–chloroform, chloroform, NaAc (0.3 M, pH 5.5), and isopropanol treatments. RNAs were washed with EtOH 100% and resuspended in RNase‐free H20. RNA quality was controlled by Bioanalyzer 2100 (Agilent technologies). RNA concentration was measured with a Nanodrop (Thermo Scientific) or a Qbit (Invitrogen). For RT–qPCR or small RNAs deep sequencing (Fasteris), ovaries from 50 control flies, 500 Rpp30 mutant flies, and about 50 Rpp30 18.2 , mnk p6 double‐mutant flies were used. For RNA‐sequencing transcriptome analysis (Genomic Paris Centre), 900 ovaries of white virgins and 900 ovaries of Rpp30 18.2 were used.
Small RNA sequencing
RNA samples of 5 μg were prepared from ovaries' extractions (see above). High‐throughput sequencing was done with Illumina HiSeq, 10% single‐reads lane 1× 50 bp. (Fasteris). 15–29 nt RNAs sequences excluding rRNA (riboZero) were sequenced. For genomic and canonical transposons annotations, we used the D. melanogaster release 5.49 gene or all‐transposon annotation files from FlyBase (http://flybase.org). For piRNA cluster positions, we used the coordinates given in Brennecke et al (2007). All the analyses were performed with Galaxy tools https://mississippi.snv.jussieu.fr. Reads were normalized first by obtaining bank size factors by DESeq geometrical normalization (version 1.0.0) from miRNA count lists (Bowtie tool sRbowtie, version 1.1.0, Dmel_miR_r20, 1 mismatch allowed) and then by normalizing the different banks to the smallest one (Rpp30 18.2/CyO). General piRNA populations were identified by matching the reads to transposable elements (Bowtie tool sRbowtie, version 1.1.0, Dmel_all‐transposon, 1 mismatch allowed) and then by analyzing the size of the reads: the piRNAs being from 23 to 28 nt. To study the specific piRNA sequences, unique mappers were obtained (Bowtie tool, sRbowtie, version 1.1.0, Dmel_r5.49, 1 mismatch allowed) and then matched on specific positions corresponding to piRNA clusters, as determined by Brennecke et al (2007). Ping‐pong signal (23‐ to 28‐nt RNA reads whose 5′ ends overlapped with another 23‐ to 28‐nt RNA read on the opposite strand) was calculated with the multimapper piRNA populations (Bowtie tool, sRbowtie, version 1.1.0, Dmel_all‐transposon, 1 mismatch allowed) and shown as a percentage or as a z‐score, as previously done (de Vanssay et al, 2012; Antoniewski, 2014).
RNA sequencing
RNA samples of 1 μg were prepared from ovaries' extractions from white virgins or Rpp30 18.2 stocks raised at 18°C. RNAs were used for directional RNA sequencing in Genomic Paris Centre. ScriptSeq™ mRNA‐Seq Library Preparation Kit (epicenter) and Ribosomal RNAs Ribo‐Zero™ rRNA Removal Kit (epicenter) were used. After quality control analysis, reads were matched to the genome with the help of Tophat2 tools. Bank size differences were solved with DESeq geometrical tool, and reads were normalized to the smallest bank. DESeq2 tool was used to infer significant differences between control and mutant situations. The base mean of biological duplicates, the fold change, and the P‐value were calculated. The expression profiles of genes, transposons, or piRNAs clusters are shown with boxplots, scatterplots, or barplots obtained using R software (http://www.r-project.org/). The visualization of reads was done in Galaxy (https://mississippi.snv.jussieu.fr).
Datasets deposition
Small RNA sequencing and RNA‐sequencing data have been deposited in the European Nucleotide Archive (ENA) of the EMBL‐EBI (http://www.ebi.ac.uk/ena), accession number: PRJEB10569. Galaxy analyses histories are available upon request.
RT–qPCR
cDNA was prepared from 5 μg of RNA using standard methods: random primers, 10 μM of dNTP mix, 5× first strand buffer, 0.1 M DTT, RNase out inactivator, and Superscript III RT 200 U/μl (Invitrogen). For qPCR, serial dilutions of cDNA were used to analyze oligonucleotides efficiency and to estimate the optimal cDNA quantity to use. The 384‐well plates (Thermo Scientific) were used, each containing 2 μl of cDNA mix and 8 μl of Primer mix with Syber Green (Roche). Each point was tested in triplicate with 2 different biological samples. The real‐time qPCR was done with Applied Biosystems® ViiA™ 7. The results were normalized to control conditions (heterozygous Rpp30 18.2/CyO), and the fold change was calculated. The fold changes found for several transposons were pooled and are shown in boxplots obtained with the R software for different genotypes. The oligonucleotides used are detailed below.
Small RNAs enrichment and Northern blot
RNA was extracted from 100 whole adult females for wild‐type or mutant conditions. To obtain a fraction enriched with low molecular weight (LMW) RNAs (< 200 nt), high molecular weight (HMW) RNAs were precipitated by incubating 100 μg of total RNA with NaCl (5 M) and 20% PEG 8000. After 30 min at 4°C, the mixture was spin for 10 min at 4°C at maximum speed and the supernatant was transferred with the LMW‐RNAs into a new tube. Small RNAs were precipitated by adding 3 volumes of EtOH 100%, mixed, incubated O/N at −80°C, and centrifuged for 15 min at 4°C at maximum speed. Small RNAs were dissolved in 50% of deionized formamide (Sigma). RNA samples (2.5 μg) were incubated for 5 min at 95°C for linearization, then immediately placed on ice. Samples were migrated in a 12% SDS gel using acrylamide/bisacrylamide 19:1 (Sigma). After migration, RNAs were transferred onto a hybond membrane N+ (GE Healthcare) by semi‐dry transfer using Trans‐Blot Turbo system (Biorad). RNAs were cross‐linked by UV treatment (Stratalinker 2400, Stratagene). Probes to detect tRNAs were radiolabeled with the PNK kit (Fermentas) with 25 μCi of γ‐ATP P32 for 1 h at 37°C, and purified with G25 columns (GE Healthcare). Membranes were prehybridized (Sigma buffer) at 42°C and then hybridized with the radiolabeled probes O/N at 42°C, at slow velocity. After several washes, the membrane signal was analyzed with a Thyphoon phosphorimager (Amersham/GE Healthcare). The sequences of probes used are detailed below. RNA sizes were analyzed with Dynamarker (B2 Scientific).
ChIP experiments
For ChIP experiments, we used 25 adult white flies for control ovaries, 900 flies for white virgins or Rpp30 18.2 mutants (stocks raised at 18°C), and 50 flies for double mutants Rpp30 18.2 , mnk p6. Ovaries were dissected in cold PBS then fixed with 1.8% formaldehyde for 10 min at room temperature (RT) and then quenched with glycine 125 mM for 5 min at RT. After 3 washings in cold PBS, liquid‐free ovaries were snap‐frozen and stored at −80°C until used. For cell lysis and DNA fragmentation, ovaries were sequentially incubated for 10 min on ice with the following ex‐tempo prepared buffers in the presence of protease inhibitors: (1) HEPES–KOH 50 mM, pH 7,5; NaCl 140 mM; EDTA 1 mM, pH 8; glycerol 10%; NP‐40 0.5%; Triton X‐100 0.25%; (2) NaCl 200 mM; EDTA 1 mM, pH 8; EGTA 0.5 mM, pH 8; Tris 10 mM, pH 8; (3) EDTA 1 mM, pH 8; EGTA 0.5 mM, pH 8; Tris 10 mM, pH 8; sarkosyl 0.5%. After incubation with 1 ml of buffer 3, ovaries were lysed with a Bioruptor (Diagenode) for 10 min in high position, making pulses of 30 s on/off, followed by centrifugation at maximum speed for 10 min at 4°C. The supernatant was kept and an aliquot of 20–30 μl was set aside. To test the quality and quantity of the DNA extract, this aliquot was incubated with TE‐SDS 1% (TES) buffer, and the cross‐link was reversed in a water bath O/N at 65°C. After dilution to decrease the SDS concentration to 0.5%, RNase‐A was added (200 μg/ml) and incubated for 2 h at 37°C and, then, proteinase K (200 μg/ml) and CaCl2 5 mM were added and incubated for 30 min at 55°C. DNA was obtained by standard phenol/chloroform/isoamylic acid purification protocols and extracted using Phase Lock Gel Light (5prime). After centrifugation, the aqueous phase with DNA was precipitated using EtOH 100%, NaAc 0.3 M, and glycogen 20 μg/ml. The DNA concentration was measured with QuBit (Invitrogen) and the quality of DNA analyzed by agarose gel electrophoresis 1%, observing the typical smear of fragmented DNA around 500 bp. For immunoprecipitation, 50 μl of Dynabeads‐A (Invitrogen) were incubated with 500 μl of ChIP blocking buffer (CBB) (Tween‐20 0.5%; BSA 5 mg/ml in PBS) for 30 min at 4°C in movement. After 2 washes with CBB at 4°C, beads were resuspended in 195 μl of CBB, and 5 μl of antibody H3K9me3 (Actif motif Ref.39161, batch number 13509002) was added and incubated O/N at 4°C. Beads were then washed 3 times with CBB at 4°C and resuspended in 100 μl of CBB. In parallel, chromatin was treated with incubation buffer prepared ex‐tempo (Triton X‐100 0.3%, NaDOC 0.3%, EDTA 15 mM, pH 8, PMSF 3 mM, protease inhibitors 1×), keeping 10% of the volume for the input control. The chromatin was mixed with beads and incubated O/N at 4°C in movement. After 7 washes with ChIP‐RIPA buffer at 4°C and one wash in TE/NaCl 50 mM, the mixture was resuspended in 200 μl of TES buffer and incubated for 30 min at 65°C under shaking conditions. After centrifugation at maximum speed, the supernatant containing the methylated chromatin was incubated O/N at 65°C to reverse the cross‐link and then treated with RNase‐A and proteinase K as described above, to purify the DNA and to do qPCR analysis. To estimate the methylation level of different piRNA clusters, transposon elements, and genes, the PCR results were analyzed as following: the percentage of input was calculated: % input = 2(corrected CT input − CT) × 100. These values were normalized to a control gene (actin‐5C or Rp49). The oligonucleotides used are detailed below.
Western blots and Rpp30 antibody
Ovaries were dissected in cold PBS, incubated in lysis buffer for 20 min at 4°C (Chromoteck), and centrifuged for 10 min at maximum speed at 4°C to exclude cell debris and yolk. Denatured ovary protein extracts were separated on 4–15% Mini PROTEAN TGX gel (Biorad) and electrotransferred to nitrocellulose membranes with Trans‐Blot Turbo Mini Nitrocellulose Transfer Packs (Biorad). Membranes were blocked O/N at 4°C in blocking buffer (PBS, 0.1% Tween‐20, 5% milk) and incubated O/N in the same buffer containing primary antibodies at the following dilutions: rabbit anti‐GFP at 1/10,000 (Ahmed El Marjou, Institut Curie, proteomic platform), mouse anti‐α‐tubulin (clone DM1A, Sigma) at 1/5,000, and purified rabbit anti‐Rpp30 at 1/500 (Proteogenix). After three washes in PBS‐T (PBS, 0.1% Tween‐20), membranes were incubated for 2 h with secondary antibody in blocking buffer containing HRP‐Goat anti‐rabbit or anti‐mouse (Jackson Immunoresearch) diluted at 1/10,000. After three washes in PBS‐T, bands were detected with the SuperSignal West Pico Chemiluminescent Substrate (Pierce) and visualized using a mini‐LAS‐4000 Imaging System (Fujifilm). Protein sizes were analyzed using Precision Plus Protein Dual Color Standards (Biorad). The purified rabbit anti‐Rpp30 was made by Proteogenix with the following Rpp30 C‐terminal peptide: ADAFEVKDGTEHAIKRLKVA (Table EV1).
Author contributions
AMH conceived, designed, performed, and analyzed experiments; and wrote the manuscript. AMV and CGE performed and analyzed experiments. CA designed and performed bioinformatic analyses. JRH conceived, performed, and analyzed experiments; and wrote the manuscript. All authors helped writing and discussing the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 3A
Source Data for Expanded View
Acknowledgements
We are grateful to D. Bourc'his, S. Castel, E. Heard, P. Léopold, A. Le Thomas, A. Lengronne, M. Walter, M. Wassef, N. Zamudio, and S. Jensen for helpful discussions and advices. We thank Déborah Bourc'his, Edith Heard, and Clément Carré for critical reading of the manuscript; DSHB (Iowa University) for antibodies; and Bloomington Drosophila Stock center for fly stocks. We thank W.E. Stumph (San Diego State University) for Brf antibody. We thank the imaging facility (PICT@BDD). This work was supported by the CNRS, ANR (PlasTiSiPi to C.A. and J.R.H.), DEEP LabEx, FSER (Schlumberger), Ville de Paris, and Fondation BNP‐Paribas to JRH; DIM Biotherapies; and ARC Postdoctoral Fellowship (A.M.H.).
The EMBO Journal (2015) 34: 3009–3027
See also: S Yamanaka & H Siomi (December 2015)
References
- Abbott JA, Francklyn CS, Robey‐Bond SM (2014) Transfer RNA and human disease. Front Genet 5: 158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C, Groth A (2012) Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13: 153–167 [DOI] [PubMed] [Google Scholar]
- Antoniewski C (2014) Computing siRNA and piRNA overlap signatures. Methods Mol Biol 1173: 135–146 [DOI] [PubMed] [Google Scholar]
- Aravin AA, Klenov MS, Vagin VV, Bantignies F, Cavalli G, Gvozdev VA (2004) Dissection of a natural RNA silencing process in the Drosophila melanogaster germ line. Mol Cell Biol 24: 6742–6750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azvolinsky A, Giresi PG, Lieb JD, Zakian VA (2009) Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol Cell 34: 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ (2007) Discrete small RNA‐generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103 [DOI] [PubMed] [Google Scholar]
- Castel SE, Ren J, Bhattacharjee S, Chang AY, Sanchez M, Valbuena A, Antequera F, Martienssen R (2014) Dicer promotes transcription termination at sites of replication stress to maintain genome stability. Cell 159: 572–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambeyron S, Popkova A, Payen‐Groschene G, Brun C, Laouini D, Pelisson A, Bucheton A (2008) piRNA‐mediated nuclear accumulation of retrotransposon transcripts in the Drosophila female germline. Proc Natl Acad Sci USA 105: 14964–14969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Pane A, Schupbach T (2007) Cutoff and aubergine mutations result in retrotransposon upregulation and checkpoint activation in Drosophila. Curr Biol 17: 637–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland BW, Schultz MC (2010) Genome stability control by checkpoint regulation of tRNA gene transcription. Transcription 1: 115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramton S, Laski F (1994) String of pearls encodes Drosophila ribosomal protein S2, has Minute‐like characteristics, and is required during oogenesis. Genetics 137: 1039–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darricarrere N, Liu N, Watanabe T, Lin H (2013) Function of Piwi, a nuclear Piwi/Argonaute protein, is independent of its slicer activity. Proc Natl Acad Sci USA 110: 1297–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dej KJ, Spradling AC (1999) The endocycle controls nurse cell polytene chromosome structure during Drosophila oogenesis. Development 126: 293–303 [DOI] [PubMed] [Google Scholar]
- Deshpande AM, Newlon CS (1996) DNA replication fork pause sites dependent on transcription. Science 272: 1030–1033 [DOI] [PubMed] [Google Scholar]
- Dieci G, Fiorino G, Castelnuovo M, Teichmann M, Pagano A (2007) The expanding RNA polymerase III transcriptome. Trends Genet 23: 614–622 [DOI] [PubMed] [Google Scholar]
- Donze D (2012) Extra‐transcriptional functions of RNA Polymerase III complexes: TFIIIC as a potential global chromatin bookmark. Gene 493: 169–175 [DOI] [PubMed] [Google Scholar]
- Durdevic Z, Schaefer M (2013) tRNA modifications: necessary for correct tRNA‐derived fragments during the recovery from stress? BioEssays 35: 323–327 [DOI] [PubMed] [Google Scholar]
- Engelke DR, Hopper AK (2006) Modified view of tRNA: stability amid sequence diversity. Mol Cell 21: 144–145 [DOI] [PubMed] [Google Scholar]
- Fichelson P, Moch C, Ivanovitch K, Martin C, Sidor CM, Lepesant JA, Bellaiche Y, Huynh JR (2009) Live‐imaging of single stem cells within their niche reveals that a U3snoRNP component segregates asymmetrically and is required for self‐renewal in Drosophila. Nat Cell Biol 11: 685–693 [DOI] [PubMed] [Google Scholar]
- Good PD, Kendall A, Ignatz‐Hoover J, Miller EL, Pai DA, Rivera SR, Carrick B, Engelke DR (2013) Silencing near tRNA genes is nucleosome‐mediated and distinct from boundary element function. Gene 526: 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriaux C, Desset S, Renaud Y, Vaury C, Brasset E (2014) Transcriptional properties and splicing of the flamenco piRNA cluster. EMBO Rep 15: 411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardane LS, Saito K, Nishida KM, Miyoshi K, Kawamura Y, Nagami T, Siomi H, Siomi MC (2007) A slicer‐mediated mechanism for repeat‐associated siRNA 5' end formation in Drosophila. Science 315: 1587–1590 [DOI] [PubMed] [Google Scholar]
- Guzzardo PM, Muerdter F, Hannon GJ (2013) The piRNA pathway in flies: highlights and future directions. Curr Opin Genet Dev 23: 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, Minis A, Sato F, Mimata H, Yoshimura A, Tamir I, Rainer J, Kofler R, Yaron A, Eggan KC, Woolf CJ et al (2013) CLP1 links tRNA metabolism to progressive motor‐neuron loss. Nature 495: 474–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Nudler E, Tora L (2013) Transcription‐replication encounters, consequences and genomic instability. Nat Struct Mol Biol 20: 412–418 [DOI] [PubMed] [Google Scholar]
- Herrera‐Moyano E, Mergui X, Garcia‐Rubio ML, Barroso S, Aguilera A (2014) The yeast and human FACT chromatin‐reorganizing complexes solve R‐loop‐mediated transcription‐replication conflicts. Genes Dev 28: 735–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24: 502–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull MW, Erickson J, Johnston M, Engelke DR (1994) tRNA genes as transcriptional repressor elements. Mol Cell Biol 14: 1266–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain S, Tuorto F, Menon S, Blanco S, Cox C, Flores JV, Watt S, Kudo NR, Lyko F, Frye M (2013) The mouse cytosine‐5 RNA methyltransferase NSun2 is a component of the chromatoid body and required for testis differentiation. Mol Cell Biol 33: 1561–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh JR, St Johnston D (2004) The origin of asymmetry: early polarisation of the Drosophila germline cyst and oocyte. Curr Biol 14: R438–R449 [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, Zakian VA (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein‐DNA complexes. Mol Cell 12: 1525–1536 [DOI] [PubMed] [Google Scholar]
- Jagut M, Mihaila‐Bodart L, Molla‐Herman A, Alin MF, Lepesant JA, Huynh JR (2013) A mosaic genetic screen for genes involved in the early steps of Drosophila oogenesis. G3 (Bethesda) 3: 409–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrous N, Reiner R (2007) Human RNase P: a tRNA‐processing enzyme and transcription factor. Nucleic Acids Res 35: 3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrous N, Gopalan V (2010) Archaeal/eukaryal RNase P: subunits, functions and RNA diversification. Nucleic Acids Res 38: 7885–7894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, Erdin S, Isikay S, Franco LM, Gonzaga‐Jauregui C, Gambin T, Gelowani V, Hunter JV, Yesil G, Koparir E, Yilmaz S et al (2014) Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157: 636–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper DM, Wang G, Gardner KE, Johnstone TG, Reinke V (2014) The C. elegans SNAPc Component SNPC‐4 Coats piRNA Domains and Is Globally Required for piRNA Abundance. Dev Cell 31: 145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y (2005) RNA polymerase II is required for RNAi‐dependent heterochromatin assembly. Science 309: 467–469 [DOI] [PubMed] [Google Scholar]
- Kelley RL (1993) Initial organization of the Drosophila dorsoventral axis depends on an RNA‐binding protein encoded by the squid gene. Genes Dev 7: 948–960 [DOI] [PubMed] [Google Scholar]
- Klattenhoff C, Bratu DP, McGinnis‐Schultz N, Koppetsch BS, Cook HA, Theurkauf WE (2007) Drosophila rasiRNA pathway mutations disrupt embryonic axis specification through activation of an ATR/Chk2 DNA damage response. Dev Cell 12: 45–55 [DOI] [PubMed] [Google Scholar]
- Lai LB, Vioque A, Kirsebom LA, Gopalan V (2010) Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett 584: 287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S, Carr AM (2013) Replication stress and genome rearrangements: lessons from yeast models. Curr Opin Genet Dev 23: 132–139 [DOI] [PubMed] [Google Scholar]
- Le Thomas A, Rogers AK, Webster A, Marinov GK, Liao SE, Perkins EM, Hur JK, Aravin AA, Toth KF (2013) Piwi induces piRNA‐guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev 27: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Stuwe E, Li S, Du J, Marinov GK, Rozhkov N, Ariel Chen YC, Luo Y, Sachidanandam R, Fejes Toth K, Patel D, Aravin A (2014a) Transgenerationally inherited piRNAs trigger piRNA biogenesis by changing the chromatin of piRNA clusters and inducing precursor processing. Genes Dev 28: 1667–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Thomas A, Toth KF, Aravin AA (2014b) To be or not to be a piRNA: genomic origin and processing of piRNAs. Genome Biol 15: 204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EM, Trinh TT, Shim HJ, Park SY, Nguyen TT, Kim MJ, Song YH (2012) Drosophila Claspin is required for the G2 arrest that is induced by DNA replication stress but not by DNA double‐strand breaks. DNA Repair (Amst) 11: 741–752 [DOI] [PubMed] [Google Scholar]
- Li X, Heyer WD (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18: 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Vagin VV, Lee S, Xu J, Ma S, Xi H, Seitz H, Horwich MD, Syrzycka M, Honda BM, Kittler EL, Zapp ML, Klattenhoff C, Schulz N, Theurkauf WE, Weng Z, Zamore PD (2009) Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137: 509–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AK, Kai T (2007) Unique germ‐line organelle, nuage, functions to repress selfish genetic elements in Drosophila melanogaster. Proc Natl Acad Sci USA 104: 6714–6719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FJ, Shen L, Jang CW, Falnes PO, Zhang Y (2013) Ikbkap/Elp1 deficiency causes male infertility by disrupting meiotic progression. PLoS Genet 9: e1003516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL (2008) Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell 32: 106–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Gibbs‐Seymour I, Bekker‐Jensen S (2013) Regulation of PCNA‐protein interactions for genome stability. Nat Rev Mol Cell Biol 14: 269–282 [DOI] [PubMed] [Google Scholar]
- Mak J, Kleiman L (1997) Primer tRNAs for reverse transcription. J Virol 71: 8087–8095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Brennecke J, Dus M, Stark A, McCombie WR, Sachidanandam R, Hannon GJ (2009) Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137: 522–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone CD, Hannon GJ (2009) Small RNAs as guardians of the genome. Cell 136: 656–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall L, Rideout EJ, Grewal SS (2012) Nutrient/TOR‐dependent regulation of RNA polymerase III controls tissue and organismal growth in Drosophila. EMBO J 31: 1916–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F, Sienski G, Handler D, Brennecke J (2014) The rhino‐deadlock‐cutoff complex licenses noncanonical transcription of dual‐strand piRNA clusters in drosophila. Cell 157: 1364–1379 [DOI] [PubMed] [Google Scholar]
- Molla‐Herman A, Matias RN, Huynh JR (2014) Chromatin modifications regulate germ cell development and transgenerational information relay. Curr Opin Insect Sci 1: 10–18 [DOI] [PubMed] [Google Scholar]
- Nguyen VC, Clelland BW, Hockman DJ, Kujat‐Choy SL, Mewhort HE, Schultz MC (2010) Replication stress checkpoint signaling controls tRNA gene transcription. Nat Struct Mol Biol 17: 976–981 [DOI] [PubMed] [Google Scholar]
- Nielsen S, Yuzenkova Y, Zenkin N (2013) Mechanism of eukaryotic RNA polymerase III transcription termination. Science 340: 1577–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma K, Cam HP, Maraia RJ, Grewal SI (2006) A role for TFIIIC transcription factor complex in genome organization. Cell 125: 859–872 [DOI] [PubMed] [Google Scholar]
- Norvell A, Kelley RL, Wehr K, Schupbach T (1999) Specific isoforms of squid, a Drosophila hnRNP, perform distinct roles in Gurken localization during oogenesis. Genes Dev 13: 864–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell L, Panier S, Wildenhain J, Tkach JM, Al‐Hakim A, Landry MC, Escribano‐Diaz C, Szilard RK, Young JT, Munro M, Canny MD, Kolas NK, Zhang W, Harding SM, Ylanko J, Mendez M, Mullin M, Sun T, Habermann B, Datti A et al (2010) The MMS22L‐TONSL complex mediates recovery from replication stress and homologous recombination. Mol Cell 40: 619–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi I, Sugiyama S, Otani H, Yamamura H, Nishida Y, Minami Y (1998) A novel Drosophila nuclear protein serine/threonine kinase expressed in the germline during its establishment. Mech Dev 71: 49–63 [DOI] [PubMed] [Google Scholar]
- Pane A, Wehr K, Schupbach T (2007) Zucchini and squash encode two putative nucleases required for rasiRNA production in the Drosophila germline. Dev Cell 12: 851–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrat PN, DasGupta S, Wang J, Theurkauf W, Weng Z, Rosbash M, Waddell S (2013) Transposition‐driven genomic heterogeneity in the Drosophila brain. Science 340: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SB, Gersak K, Michaelson‐Cohen R, Walsh T, Lee MK, Malach D, Klevit RE, King MC, Levy‐Lahad E (2013) Mutations in LARS2, encoding mitochondrial leucyl‐tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet 92: 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X, Daily K, Nguyen K, Wang H, Mayhew D, Rigor P, Forouzan S, Johnston M, Mitra RD, Baldi P, Sandmeyer S (2012) Retrotransposon profiling of RNA polymerase III initiation sites. Genome Res 22: 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab JR, Chiu J, Zhu J, Katzman S, Kurukuti S, Wade PA, Haussler D, Kamakaka RT (2012) Human tRNA genes function as chromatin insulators. EMBO J 31: 330–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangan P, Malone CD, Navarro C, Newbold SP, Hayes PS, Sachidanandam R, Hannon GJ, Lehmann R (2011) piRNA production requires heterochromatin formation in Drosophila. Curr Biol 21: 1373–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner R, Ben‐Asouli Y, Krilovetzky I, Jarrous N (2006) A role for the catalytic ribonucleoprotein RNase P in RNA polymerase III transcription. Genes Dev 20: 1621–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, Yaffe MB (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol 21: 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout EJ, Marshall L, Grewal SS (2012) Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci USA 109: 1139–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RJ, Weiner MM, Lin H (2014) PIWI proteins and PIWI‐interacting RNAs in the soma. Nature 505: 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, Hannon GJ (2013) Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27: 400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Pereira JM, Herrero AB, Garcia‐Rubio ML, Marin A, Moreno S, Aguilera A (2013) The Npl3 hnRNP prevents R‐loop‐mediated transcription‐replication conflicts and genome instability. Genes Dev 27: 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, Rosti RO, Abdellateef M, Caglar C, Kasher PR, Cazemier JL, Weterman MA, Cantagrel V, Cai N, Zweier C, Altunoglu U et al (2014) CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157: 651–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scorah J, McGowan CH (2009) Claspin and Chk1 regulate replication fork stability by different mechanisms. Cell Cycle 8: 1036–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senti KA, Brennecke J (2010) The piRNA pathway: a fly's perspective on the guardian of the genome. Trends Genet 26: 499–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpiz S, Ryazansky S, Olovnikov I, Abramov Y, Kalmykova A (2014) Euchromatic transposon insertions trigger production of novel Pi‐ and endo‐siRNAs at the target sites in the drosophila germline. PLoS Genet 10: e1004138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Donertas D, Brennecke J (2012) Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell 151: 964–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi MC, Sato K, Pezic D, Aravin AA (2011) PIWI‐interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12: 246–258 [DOI] [PubMed] [Google Scholar]
- Spradling AC, Bellen HJ, Hoskins RA (2011) Drosophila P elements preferentially transpose to replication origins. Proc Natl Acad Sci USA 108: 15948–15953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staeva‐Vieira E, Yoo S, Lehmann R (2003) An essential role of DmRad51/SpnA in DNA repair and meiotic checkpoint control. EMBO J 22: 5863–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin AV, Kogan GL, Filipp D, Balakireva MD, Gvozdev VA (1997) Heterochromatic Stellate gene cluster in Drosophila melanogaster: structure and molecular evolution. Genetics 146: 253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, Corces VG (2012) tDNA insulators and the emerging role of TFIIIC in genome organization. Transcription 3: 277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bortle K, Nichols MH, Li L, Ong CT, Takenaka N, Qin ZS, Corces VG (2014) Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol 15: R82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vanssay A, Bougé AL, Boivin A, Hermant C, Teysset L, Delmarre V, Antoniewski C, Ronsseray S (2012) Paramutation in Dro sophila linked to emergence of a piRNA‐producing locus. Nature 490: 112–115 [DOI] [PubMed] [Google Scholar]
- Wang SQ, Shi DQ, Long YP, Liu J, Yang WC (2012) GAMETOPHYTE DEFECTIVE 1, a putative subunit of RNases P/MRP, is essential for female gametogenesis and male competence in Arabidopsis. PLoS ONE 7: e33595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichtowska D, Turowski TW, Boguta M (2013) An interplay between transcription, processing, and degradation determines tRNA levels in yeast. Wiley Interdiscip Rev RNA 4: 709–722 [DOI] [PubMed] [Google Scholar]
- Xiao S, Scott F, Fierke CA, Engelke DR (2002) Eukaryotic ribonuclease P: a plurality of ribonucleoprotein enzymes. Annu Rev Biochem 71: 165–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Dubrovskaya V, Yacoub N, Walska J, Gleason T, Reid K, Dubrovsky EB (2013) Developmental roles of Drosophila tRNA processing endonuclease RNase ZL as revealed with a conditional rescue system. Dev Biol 381: 324–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Rubin G (1993) Analysis of genetic mosaics in developing an adult Drosphila tissues. Development 117: 1223–1237 [DOI] [PubMed] [Google Scholar]
- Xu J, Xin S, Du W (2001) Drosophila Chk2 is required for DNA damage‐mediated cell cycle arrest and apoptosis. FEBS Lett 508: 394–398 [DOI] [PubMed] [Google Scholar]
- Yan D, Neumuller RA, Buckner M, Ayers K, Li H, Hu Y, Yang‐Zhou D, Pan L, Wang X, Kelley C, Vinayagam A, Binari R, Randklev S, Perkins LA, Xie T, Cooley L, Perrimon N (2014) A regulatory network of Drosophila germline stem cell self‐renewal. Dev Cell 28: 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, de Castro E, Marin L, Chang AY, Goto D, Cande WZ, Antequera F, Arcangioli B, Martienssen RA (2011) RNAi promotes heterochromatic silencing through replication‐coupled release of RNA Pol II. Nature 479: 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang J, Xu J, Zhang Z, Koppetsch BS, Schultz N, Vreven T, Meignin C, Davis I, Zamore PD, Weng Z, Theurkauf WE (2012) UAP56 couples piRNA clusters to the perinuclear transposon silencing machinery. Cell 151: 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Shalaby NA, Buszczak M (2014) Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 343: 298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 3A
Source Data for Expanded View