

Abstract
Background and Purpose
Posttraumatic stress disorder (PTSD) is a mental disorder with enhanced retention of fear memory and has profound impact on quality of life for millions of people worldwide. The β‐adrenoceptor antagonist propranolol has been used in preclinical and clinical studies for the treatment of PTSD, but the mechanisms underlying its potential efficacy on fear memory retention remain to be elucidated.
Experimental Approach
We investigated the action of propranolol on the retention of conditioned fear memory, the surface expression of glutamate receptor GluA1 subunits of AMPA receptors and synaptic adaptation in the lateral amygdala (LA) of rats.
Key Results
Propranolol attenuated reactivation‐induced strengthening of fear retention while reducing enhanced surface expression of GluA1 subunits and restoring the impaired long‐term depression in LA. These effects of propranolol were mediated by antagonizing reactivation‐induced enhancement of adrenergic signalling, which activates PKA and calcium/calmodulin‐dependent protein kinase II and then regulates the trafficking of AMPA receptors via phosphorylation of GluA1 subunits at the C‐terminus. Both i.p. injection and intra‐amygdala infusion of propranolol attenuated reactivation‐induced enhancement of fear retention.
Conclusions and Implications
Reactivation strengthens fear retention by increasing the level of noradrenaline and promotes the surface expression of GluA1 subunits and the excitatory synaptic transmission in LA. These findings uncover one mechanism underlying the efficiency of propranolol on retention of fear memories and suggest that β‐adrenoceptor antagonists, which act centrally, may be more suitable for the treatment of PTSD.
Abbreviations
- CS
conditioned stimulus
- PTSD
posttraumatic stress disorder
- LA
lateral amygdala
- LFS
low‐frequency stimulation
- LTD
long term depression
- LTP
long‐term potentiation
Tables of Links
| TARGETS |
|---|
| Ligand‐gated ion channels a |
| AMPA receptor |
| GluA1 |
| GluA2 |
| GPCRs b |
| α1‐adrenoceptor |
| β‐adrenoceptor |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a bAlexander et al., 2013a, 2013b).
Introduction
Posttraumatic stress disorder (PTSD) is an anxiety disorder that is characterized by progressively strengthened retention of memory and feeling for emotionally fearful experiences (American Psychiatric Association, 2013). Increased frequency of suicidal behaviours (including suicidal thoughts, plans or acts) and other mood disorders and anxious symptoms have been reported in PTSD (Panagioti et al., 2012; Pietrzak et al., 2014). Although many pharmacological therapies have been used for the treatment of PTSD, a substantial portion of patients fail to show an adequate therapeutic response (Fitzgerald et al., 2014). Although the exact mechanism of PTSD remains elusive, accumulated evidence indicates that disrupted regulation of fear memory and enhanced amygdala activity to stress are intimately associated with the occurrence of PTSD (Bremner et al., 2008; Samuelson, 2011).
Weakening and erasure of fear memory traces are critical for the management of anxiety disorders including PTSD (Mirante et al., 2014). Fear conditioning is a preferred animal model that captures some of the processes involved in PTSD (Debiec and LeDoux, 2006). It has been generally recognized that fear memory formed by acquisition and consolidation can be retrieved (also termed as reactivated) and become unstable, which means it can be either reconsolidated or extinguished by different manipulations or drugs (Mahan and Ressler, 2012). The neural connectivity and synaptic plasticity of the amygdala are closely related to the formation and extinction of fear memory (Kim et al., 2007; Hong et al., 2009; Sierra‐Mercado et al., 2011; Nabavi et al., 2014). Amygdala excitatory synaptic strengthening is thought to contribute to conditioned fear and anxiety. Enhancements of synaptic transmission from the conditioned stimulus (CS) pathways to the lateral amygdala (LA) contribute in the acquisition of fear memory during auditory fear conditioning (McKernan and Shinnick‐Gallagher, 1997; Rumpel et al., 2005). In the amygdala, depotentiation of synaptic transmission may represent an endogenous cellular substrate for fear memory erasure (Kim et al., 2007; Hong et al., 2009; Nabavi et al., 2014). It has been suggested adrenergic activation‐induced long‐term depression (LTD) in amygdala may mediate a form of synaptic meta‐plasticity that recalibrates fear memory processing, especially in the memory storage stage (Clem and Huganir, 2013). Recently, it is also proposed that LA‐LTD with different molecular pathways underlies the erasure of traumatic memory (Mirante et al., 2014). The phosphorylation and trafficking of AMPA receptors containing glutamate receptor GluA1 subunits are essential for the expression of LTD (Hong et al., 2009; Huganir and Nicoll, 2013; Nabavi et al., 2014). Herein, we investigated the relationship of AMPA receptor‐dependent LTD in LA and fear retention.
Emotionally arousing events stimulate the activation of the locus coeruleus neurons, which send adrenergic projections and release noradrenaline to different brain regions (Galvez et al., 1996). Animal and human studies have shown that there are elevations of noradrenaline in the brain during the pathogenesis of PTSD or the occurrence of PTSD‐like event (O'Donnell et al., 2004; Soeter and Kindt, 2011). Noradrenaline is an important neurotransmitter and modulator in the CNS and plays crucial roles in the modulation of glutamatergic and GABAergic neurotransmission in brain regions involved in emotional regulation and expression (Farb et al., 2010; Zhou et al., 2013). Noradrenaline is also believed to be the substantial contributor in the enhanced memory for emotional events, such as fear conditioning and PTSD, compared with emotionally neutral experiences (Debiec et al., 2011; Soeter and Kindt, 2011). There is good recent evidence that noradrenaline enhances the acquisition and retention of conditioned fear memory (Roozendaal et al., 2006; Tully and Bolshakov, 2010). However, the mechanism of action of noradrenaline on reactivation‐dependent fear retention has not been elucidated.
The β‐adrenoceptor antagonist propranolol has been used in preclinical and clinical studies for the prevention and treatment of PTSD, but the molecular mechanism is still unclear (Pitman et al., 2002; Shad et al., 2011). An investigation reported that systemic administration of propranolol has limited efficacy in disrupting fear reconsolidation (Muravieva and Alberini, 2010). Other studies suggest that blockade of β‐adrenoceptors before or after retrieval decreases fear retention in fear conditioning (Pitman et al., 2002; Debiec and LeDoux, 2006; Debiec et al., 2011). Thus, we carried out research to identify the association between AMPA receptors, LTD in the LA and fear memory retention, aiming at a better understanding of the effect of propranolol on event‐based fear memory and providing therapeutic guidelines for the treatment of PTSD with β‐adrenoceptor antagonists.
Methods
Animals
All animal care and experimental procedures were in accordance with the Guide for Care and Use of Laboratory Animals, as adopted and promulgated by the National Institutes of Health of USA, and were approved by the Animal Welfare Committee of Huazhong University of Science & Technology. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 352 animals were used in the experiments described here. Male Sprague‐Dawley rats (4–5 months old) were used for experiments (Laboratory Animal Center of Tongji Medical College, Huazhong University of Science and Technology). Animals were maintained on a controlled 12:12 light cycle at constant temperature (22 ± 2°C) and humidity (55–65%) with food and water provided ad libitum.
Surgical procedure and microinjection
Stereotaxic surgery was carried out as described earlier (Jiang et al., 2013) with alterations needed (see Supporting Information for details).
Adenoviral infection and overexpression
The GluA1 carboxyl terminus (GluA1‐C‐tail) sequence was obtained and used as our previous study (Luo et al., 2015) (see Supporting Information for details).
Fear conditioning tasks
The cue‐fear conditioning experiment was performed as described previously (Wang et al., 2011; Li et al., 2012; Wang et al., 2015). The experiment was conducted over 3 days, conditioning day, reactivation day and testing day. On day 1, after 5 min habituation in the conditioning chamber A, animals were presented with six pairings of a tone for 29 s as the CS (80 dB), co‐terminated with a foot shock as the unconditioned stimulus (1 s). The intertrial interval was 60 s, and the shock intensity for rats was 0.75 mA. Rats were left in the conditioning chamber A for 60 s after termination of the procedure and then returned to their home cages. To assess the effect of propranolol on cue‐fear retention, rats were given i.p. injections of propranolol three times a day (t.i.d), immediately after conditioning, 12 h after conditioning and 30 min before reactivation, after conditioning or intra‐amygdala infusion of propranolol before reactivation. Thirty minutes after the last i.p. injection or 10 min after intra‐amygdala infusion, rats were placed into the consolidation chamber B and observed for 5 min. Cue‐dependent memory was reactivated by three pulses of CSs with 90 s of intertrial interval (day 2). Rats in non‐reactivation groups were placed into the consolidation chamber B for 5 min, but without cues. Still after 24 h (day 3), rats were placed into the consolidation chamber B for testing. After 5 min observation, cue‐fear memory was tested with five pulses of CSs, and the intertrial interval was 60 s. Behavioural indicator freezing was defined and measured during the entire tasking time by software (AniLab 7.0, Yihong Technology Co., Ltd., Wuhan, China). Freezing was defined as the complete absence of activity except for respiratory movement. The data were converted to the percentage of freezing.
Pain threshold measurement and open‐field test
Procedures for pain threshold measurement and open‐field test were processed according to our previous studies with some modifications (Wang et al., 2011; Li et al., 2012; Wang et al., 2015) (see Supporting Information for details).
Electrophysiological recording
Electrophysiological recording for field EPSP (fEPSP) was based on our previous study with some modifications (Li et al., 2012; Luo et al., 2015). For the fear conditioned rats, brain slices for electrophysiological recording were prepared immediately after fear reactivation. The stimulation intensities were adjusted to produce a fEPSP with two‐thirds of the maximal amplitude. After 30 min of baseline recording, LTD was induced by low‐frequency stimulation (LFS; 1 Hz for 15 min). Responses were recorded for 60 min following LFS. Patch‐clamp recordings were also performed according to our previous study (Li et al., 2012). See Supporting Information for more details.
Microdialysis and HPLC
For the measurement of noradrenaline levels in the amygdala, we performed microdialysis and HPLC analysis, and the procedure was similar to our previous study (Luo et al., 2015) (see Supporting Information for details).
Surface protein cross‐linking with BS3
The surface protein cross‐linking was performed according to our previous study with slight modification (Jiang et al., 2013; Lu et al., 2014). The brain slices of rats were dissected, and the LA tissues were isolated under a stereomicroscope (see Supporting Information for details).
Enrichment of synaptic and extrasynaptic membrane fractions
Separation of synaptic and extrasynaptic fractions was performed as described in an earlier protocol (Li et al., 2011) with minor modifications (see Supporting Information for details).
Western blotting
Biological samples of the LA tissues were prepared for Western blot analysis. For the fear conditioned rats, brain slices were cut immediately after fear reactivation, and the LA tissue samples were isolated and made for Western blotting. The performance was based on our previous protocol (Jiang et al., 2013; Lu et al., 2014) (see Supporting Information for details).
Data analysis
All analysis was performed using spss 18.0 software (SPSS Inc., USA), and data are expressed as mean ± SEM. The results from fear conditioning training, reactivation and cue‐dependent fear memory test were statistically evaluated using repeated measures anova. The results from baseline behaviours, microdialysis, Western blotting, patch‐clamp recording and the level of stable LTD were statistically evaluated using one‐way ANOVA. Post hoc tests were performed using least significant difference test. Differences at the P < 0.05 level were considered statistically significant.
Materials
Bicuculline, 6‐cyano‐7‐nitroquinoxaline‐2, 3‐dione, D(−)‐2‐amino‐5‐phosphonovaleric acid (AP5), (±)‐noradrenaline (+)‐bitartrate salt, (−)‐adrenaline, (±)‐propranolol hydrochloride, phentolamine hydrochloride, nadolol, H89 dihydrochloride and KN93 phosphate salt were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Crosslinker bis(sulfosuccinimidal) suberate (BS3) was purchased from Thermo Scientific (Rockford, IL, USA). Other agents were obtained from commercial suppliers. The stock solutions of chemicals were freshly diluted with artificial cerebrospinal fluid (containing in mM: 119 NaCl, 4 KCl, 1.2 MgSO4, 2 CaCl2, 1 NaH2PO4, 26 NaHCO3, 10 glucose, pH7.4) before application. When dimethyl sulfoxide (Sigma‐Aldrich) was used to prepare solutions, its final concentration was 0.1% or less.
Results
Propranolol attenuates reactivation‐induced fear retention and LA‐LTD impairment in rats
Fear reactivation is recognized as a preferred candidate for mimicking the spontaneous recall of fear in human (Agren et al., 2012). We designed fear conditioning experiments with reactivation to examine the effect of endogenous noradrenaline on fear memory retention and LA‐LTD (Figure 1A). Propranolol was given t.i.d. through i.p. injection, after conditioning. We observed that propranolol at both doses significantly reduced the freezing percentage on the reconsolidation testing day following reactivation (F(2, 21) = 10.9, P = 0.001) (Figure 1B). In the consolidation test without reactivation, no difference between saline and propranolol treatments was observed [F(2, 21) = 0.739, no significance (NS)] (Figure 1C). Freezing behaviour of all groups during conditioning training (non‐reactivation: F(2, 21) = 0.66, NS, Figure S1A; reactivation: F(2, 21) = 0.72, NS, Figure S1B) and fear reactivation (F(2, 21) = 0.07, NS, Figure S1B) had no significant differences. Reactivation also induced an increased noradrenaline content in LA (F(1, 14) = 6.54, P = 0.043) (Figure 1D), suggesting reactivation induces the release of noradrenaline in LA and subsequently enhances fear retention.
Figure 1.
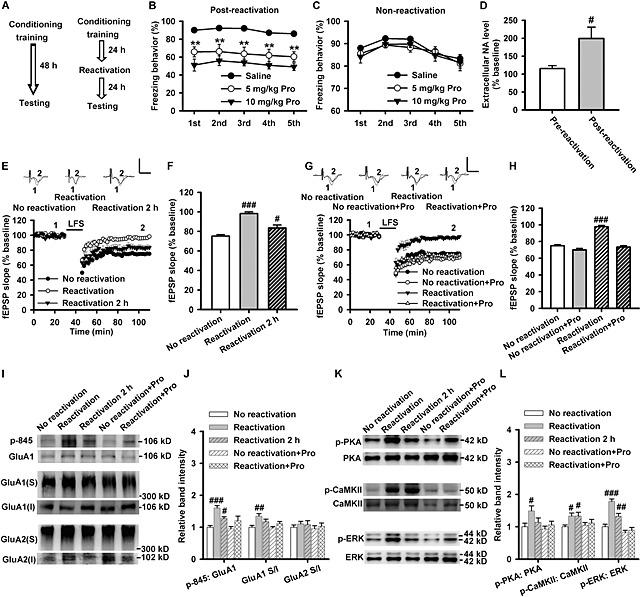
Propranolol attenuates reactivation‐induced fear retention and LA‐LTD impairment. (A) The fear conditioning procedure. (B) The percentage of freezing behaviour at 24 h after reactivation. The freezing behaviour was significantly reduced in 5 (n = 8) and 10 mg·kg−1 (n = 8) propranolol (Pro)‐treated groups compared with saline‐treated rats (n = 8). (C) The percentage of freezing behaviour at 48 h after conditioning (training). The freezing behaviour was not significantly different among saline (n = 8), 5 mg·kg−1 (n = 8) and 10 mg·kg−1 (n = 8) propranolol‐treated groups. (D) Measurement of extracellular noradrenaline (NA) concentration in LA region 20 min before and 20 min after reactivation via microdialysis. The extracellular noradrenaline concentration increased after fear reactivation (n = 8). (E) Time course of the fEPSP evoked by stimulation of Thalamic afferents (T) and recorded in LA slices from non‐reactivation (n = 11 slices from six rats), reactivation (n = 10 slices from six rats) and 2 h after reactivation (n = 8 slices from six rats) groups. Inset: The representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS (1 Hz for 900 s) in rats of non‐reactivation (left), reactivation (middle) and 2 h after reactivation (right). (F) The histogram showing the level of LTD at 60 min after LFS in rats of non‐reactivation, reactivation and 2 h after reactivation. (G) Time course of the fEPSP evoked by stimulation of T and recorded in LA slices from non‐reactivation (n = 11 slices from six rats), non‐reactivation + propranolol (n = 13 slices from six rats), reactivation (n = 11 slices from six rats) and reactivation + propranolol (n = 8 slices from six rats)‐treated rats. Inset: The representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS in rats of non‐reactivation, non‐reactivation + propranolol, reactivation and reactivation + propranolol groups. (H) The histogram showing the level of LTD at 60 min after LFS in rats of non‐reactivation, non‐reactivation + propranolol, reactivation and reactivation + propranolol. (I) Representative images of Western blotting. (J) The histogram showing the p‐845‐GluA1 (n = 6) and surface expression of GluA1 subunits (n = 6) were enhanced after reactivation and this response was prevented by 10 mg·kg−1 propranolol via i.p. injection t.i.d. The surface expression of GluA2 subunits (n = 6) was not affected after reactivation. (K) Representative images of Western blotting. (L) The histogram showing the phosphorylation level of PKA (n = 6), CaMKII (n = 6) and ERK (n = 6) were enhanced after reactivation and this response was prevented by 10 mg·kg−1 propranolol via i.p. injection t.i.d. Calibration: 0.8 mV, 5 ms. **P < 0.01 versus saline. # P < 0.05, ## P < 0.01, ### P < 0.001 versus no reactivation.
Because the differences in locomotive activity, anxiety‐related behaviour and pain response may influence the performances of rats in conditioned fear tasks, we carried out the open‐field and pain threshold tests to detect the potential differences in locomotive activity, anxiety‐related behaviour and pain threshold between the groups. The pain thresholds to foot shock (flinching: F(2, 33) = 1.19, NS, Figure S1C; jumping: F(2, 21) = 2.20, NS, Figure S1D) and locomotive (F(2, 21) = 0.86, NS, Figure S1E) and anxiety‐related (F(2, 21) = 1.45, NS, Figure S1F) behaviours were similar between the groups, which indicated that the differences in fear retention tests were not caused by the potential differentiations in pain threshold, locomotive and anxiety‐related behaviours.
We then performed electrophysiological experiments and found that fear reactivation significantly impaired LA‐LTD, with stable fEPSPs levels 60 min after LFS (1 Hz for 15 min) increasing from 75 ± 1.5% in non‐reactivation group to 98 ± 1.8% in reactivation group and to 84 ± 3.1% at 2 h after reactivation exposure (F(2, 26) = 31.98, P < 0.001) (Figure 1E and F). Moreover, propranolol (10 mg·kg−1) pretreatment restored the impairment of LA‐LTD induced by fear reactivation (F(3, 39) = 42.20, P < 0.001) (Figure 1G and H).
Fear reactivation promoted the surface delivery of GluA1 subunits (F(4, 25) = 5.075, P = 0.004), but did not influence the surface expression of GluA2 subunits (F(4, 25) = 0.398, P = 0.808), and propranolol (10 mg·kg−1) prevented the reactivation‐induced surface delivery of GluA1 (P = 0.172, reactivation + Pro vs. non‐reactivation + Pro) (Figure 1I and J). Because phosphorylation of GluA1 subunits at Ser845 within the C‐terminus by PKA is an important factor in the maintenance of AMPA receptor function and dephosphorylation of Ser845 is responsible for the endocytosis of AMPA receptors and the formation of LTD in an activity‐dependent manner (Wang et al., 2005), we asked whether fear reactivation influences the phosphorylation of Ser845 in GluA1 subunitsand the activation of PKA signals. We found that after reactivation, the phosphorylation level of Ser845 (F(4, 25) = 8.482, P < 0.001; reactivation: P < 0.001; reactivation 2 h: P = 0.044) (Figure 1I and J), PKA (F(4, 25) = 3.077, P = 0.034; reactivation: P = 0.010), calcium/calmodulin‐dependent protein kinase II (CaMKII) (F(4, 25) = 2.524, P = 0.066; reactivation: P = 0.031; reactivation 2 h: P = 0.031) and ERK (F(4, 25) = 24.887, P < 0.001; reactivation: P < 0.001; reactivation 2 h: P = 0.008) were all significantly increased when compared with the non‐reactivation group. We found propranolol attenuated these effects induced by reactivation (Ser845: P = 0.078, PKA: P = 0.471, CaMKII: P = 0.601, ERK: P = 0.533, reactivation + Pro vs. non‐reactivation + Pro) (Figure 1K and L).
These results suggest that fear reactivation promoted the release of noradrenaline in LA, facilitated the surface expression of GluA1 subunits and enhanced fear retention. Activation of β‐adrenoceptors and PKA/CaMKII signals may mediate the reactivation‐induced effects via strengthening the surface delivery of GluA1‐containing AMPA receptors.
Exogenous noradrenaline impairs LA‐LTD and increases the surface expression of GluA1 subunits
Endogenous and exogenous (10 μM) noradrenaline increases the phosphorylation level of GluA1 subunits at the C‐terminal domain in the hippocampus of mice (Hu et al., 2007). As we detected an enhanced release of noradrenaline in LA and a series of alterations in molecular, cellular and behavioural levels induced by reactivation, we further examined whether exogenous noradrenaline could impair LTD and enhance surface expression and function of AMPA receptors in LA.
Bath incubation with noradrenaline (2.5, 5, 10 and 20 μM) significantly impaired LA‐LTD when compared with that of the control group (control: 64.62 ± 2.56%, 2.5 μM: 89 ± 1.4%, 5 μM: 96 ± 2.4%, 10 μM: 98 ± 3.0%, 20 μM: 101 ± 1.6%; F(4, 48) = 49.1, P < 0.001) (Figure 2A and B). We also tested the basal fEPSPs (Figure S2A) and paired‐pulse ratio (Figure S2B) in LA and found that noradrenaline (10 μM) did not affect these indicators, suggesting that noradrenaline impaired LFS‐induced LA‐LTD without influencing the basal excitatory synaptic transmission or presynaptic release.
Figure 2.
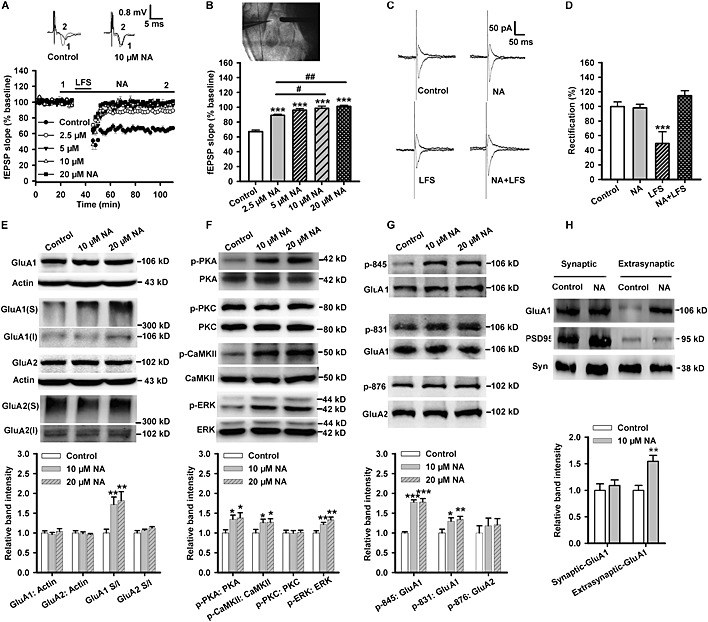
Exogenous noradrenaline (NA) impairs LA‐LTD and promotes the surface expression of GluA1 subunits. (A) Time course of the fEPSP from control (n = 17 slices from eight rats), 2.5 (n = 9 slices from five rats), 5 (n = 9 slices from five rats), 10 (n = 7 slices from four rats) and 20 μM noradrenaline‐treated slices (n = 11 slices from six rats). Inset: The representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS in either control (left) or 10 μM noradrenaline‐treated (right) slices. (B) The figure showing the location of recording (left) and stimulating (right) electrodes in the T‐LA pathway in brain slice (upper). Histogram showing the level of LTD at 60 min after LFS in control or noradrenaline‐treated slices (lower). (C) Example of evoked AMPA receptor‐mediated synaptic responses recorded at −70 mV and + 40 mV from control, noradrenaline, LFS or noradrenaline + LFS‐treated slices. (D) Average rectification values (I‐70 mV/I + 40 mV) for control (n = 6), noradrenaline (n = 6), LFS (n = 9) or noradrenaline + LFS (n = 7)‐treated slices. Values are normalized to control cells. (E) Inset: Representative images of Western blotting. The histogram showing that the surface expression of GluA1 subunits (n = 8) was enhanced, while the level of total GluA1 (n = 7), GluA2 (n = 8) or the surface expression of GluA2 subunits (n = 8) was not affected after noradrenaline treatment. (F) Inset: Representative images of Western blotting. The histogram showing that the phosphorylation level of PKA (n = 7), CaMKII (n = 8) or ERK (n = 8) was enhanced, while PKC (n = 8) was not affected after noradrenaline treatment. (G) Inset: Representative images of Western blotting. The histogram showing the phosphorylation levels of GluA1‐845 (n = 8) and GluA1‐831 (n = 8) were enhanced, while phosphorylation of GluA2‐876 (n = 8) was not affected after noradrenaline treatment. (H) Inset: Representative images of Western blotting (Syn = synaptophysin). The histogram showing the extrasynaptic expression of GluA1 was enhanced, while the synaptic GluA1 subunits were not affected by noradrenaline (10 μM for 10 min) (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 versus control. # P < 0.05, ## P < 0.01 versus 2.5 μM noradrenaline‐treated slices.
Noradrenaline alone influenced neither the basal amplitude of NMDA (F(1, 13) = 1.11) (Figure S2C and D) and AMPA receptor‐mediated currents (F(1, 14) = 0.46) (Figure S2E and F) nor the rectification values (I−70 mV/I+40 mV) of AMPA receptor currents (F(3, 24) = 22.13, P < 0.001; P = 0.86, noradrenaline vs. control). However, it restored the LFS‐induced reduction in the rectification values of AMPA receptors (noradrenaline + LFS: P < 0.001 vs. control) (Figure 2C and D).
Noradrenaline at both concentrations (10 and 20 μM) did not influence the total expression of GluA1 (F(2, 18) = 0.465) or GluA2 subunits (F(2, 18) = 0.205) after acute treatment for 10 min but increased the surface expression of GluA1 subunits (F(2, 21) = 6.454, P = 0.007) in amygdala slices (Figure 2E). Noradrenaline significantly activated PKA (F(2, 18) = 3.969, P = 0.037), CaMKII (F(2, 21) = 3.512, P = 0.048) and ERK (F(2, 21) = 11.023, P = 0.001), but not PKC (F(2, 21) = 0.044) (Figure 2F). Noradrenaline also increased the phosphorylation of GluA1‐ Ser845 (F(2, 21) = 18.320, P < 0.001) and Ser831 (F(2, 21) = 5.781, P = 0.010) (Figure 2G).
Because noradrenaline did not affect the excitatory synaptic transmission (Figure S2A) and AMPA receptor currents (Figure S2E and F) in baseline conditions, we further examined the extrasynaptic and synaptic distribution of GluA1 subunitsfollowing noradrenaline‐induced surface trafficking. Our results indicated that noradrenaline significantly enhanced the content of GluA1 subunits in the extrasynaptic fraction (F(1, 14) = 13.671, P = 0.002), but had no significant influence on GluA1 subunits trafficking into the synaptic fraction (F(1, 14) = 0.294) (Figure 2H).
These findings suggested that noradrenaline was an excitatory modulator in the amygdala neural circuit, and it may have facilitated the insertion of GluA1 subunits close to, but not at, the plasma membrane of synapses. During the induction of LTD, AMPA receptors translocate into the intracellular space to induce the expression of LTD. GluA1 subunits expressed at extrasynaptic membrane induced by noradrenaline then can laterally translocate into the synapses to prevent the expression of LTD induced by LFS in an activity‐dependent manner.
Propranolol prevents noradrenaline‐induced carboxyl terminal phosphorylation and surface expression of GluA1 subunits in LA
Our in vitro experiments showed that noradrenaline activated PKA but not PKC, which indicates that it is the β‐adrenoceptors rather than α1‐adrenoceptors that are mainly activated in LA by noradrenaline. So we further tested whether propranolol could prevent noradrenaline from inducing alterations in vitro.
Pretreatment with propranolol (10 μM) (F(3, 33) = 27.33, P < 0.001) (Figure 3A and B), but not phentolamine (50 μM) (Figure S3A and B), effectively attenuated noradrenaline‐induced impairment of LA‐LTD. Data from patch‐clamp recording suggested that propranolol (F(1, 9) = 0.161) (Figure S3C), not phentolamine (Figure S3D), significantly prevented the effect of noradrenaline on LFS‐induced rectification of AMPA receptors. Western blot analysis showed that propranolol (10 μM) attenuated noradrenaline‐induced activation of PKA (F(5, 30) = 3.076, P = 0.023; Pro + NA: P = 0.586 vs. control; Phen + NA: P = 0.008 vs. control), CaMKII (F(5, 30) = 3.521, P = 0.013; Pro + NA: P = 0.561 vs. control; Phen + NA: P = 0.015 vs. control) and ERK (F(5, 30) = 20.143, P < 0.001; Pro + NA: P = 0.089 vs. control; Phen + NA: P < 0.001 vs. control) (Figure 3C and D) and phosphorylation of GluA1‐Ser845 (F(5, 30) = 6.686, P < 0.001; Pro + NA: P = 0.282 vs. control; Phen + NA: P = 0.001 vs. control) and surface expression of GluA1 subunits (F(5, 36) = 5.765, P = 0.001; Pro + NA: P = 0.969 vs. control; Phen + NA: P = 0.034 vs. control) (Figure 3E and F). These findings suggest that blocking β‐adrenoceptors with propranolol attenuated the noradrenaline‐induced surface delivery of GluA1 subunits and the impairment of LA‐LTD.
Figure 3.
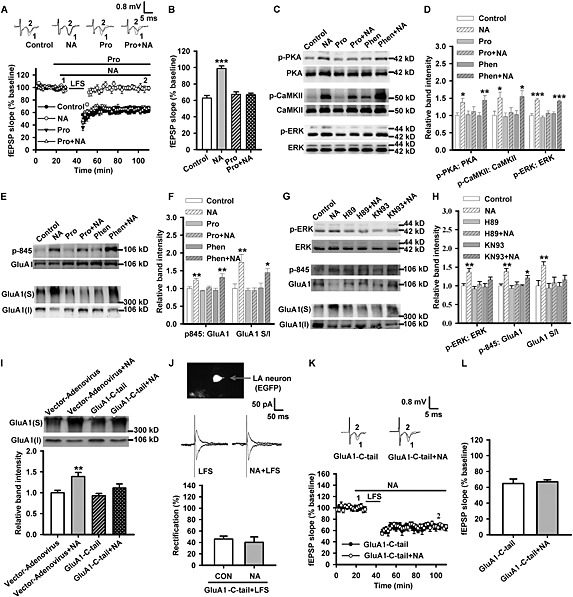
Propranolol prevents noradrenaline‐induced carboxyl terminal phosphorylation and surface expression of GluA1 subunits in LA. (A) Time course of the fEPSP from control (n = 17 slices from eight rats), noradrenaline (NA; n = 9 slices from five rats), propranolol (Pro; n = 9 slices from five rats) and propranolol + noradrenaline‐treated slices (n = 11 slices from six rats). Inset: The representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS in control, noradrenaline, propranolol or propranolol + noradrenaline‐treated slices. (B) The histogram showing the level of LTD at 60 min after LFS in control, noradrenaline, propranolol or propranolol + noradrenaline‐treated slices. (C) Representative images of western blotting. (D) The histogram showing the phosphorylation level of PKA (n = 8), CaMKII (n = 8) or ERK (n = 8) was enhanced by noradrenaline and this effect was inhibited by pretreatment with propranolol but not phentolamine (Phen). (E) Representative images of western blotting. (F) The histogram showing the phosphorylation level of GluA1‐845 (n = 6) or the surface expression of GluA1 (n = 7) was enhanced by noradrenaline, and this effect was inhibited by pretreatment with propranolol but not phentolamine. (G) Representative images of western blotting. (H) The histogram showing the phosphorylation level of ERK (n = 6), GluA1‐845 (n = 4) or the surface expression of GluA1 (n = 6) was enhanced by noradrenaline, and this effect was inhibited by pretreatment with H89 or KN93. (I) Representative images of western blotting (upper). The histogram showing the surface expression of GluA1 was enhanced by treatment with 10 μM noradrenaline for 10 min, and this effect was prevented by overexpression of GluA1‐C‐tail, but not vector‐adenovirus (n = 6). (J) Top: Principal LA neuron infected with the overexpression‐adenovirus (indicated by arrow) visualized under epifluorescent illumination with GFP filter. Middle: Example of evoked AMPA receptor‐mediated synaptic responses recorded at −70 mV and +40 mV from GluA1‐C‐tail adenovirus‐infected and LFS or noradrenaline + LFS‐treated slices. Lower: Average rectification values (I−70 mV/I+40 mV) for LFS (n = 5 from five rats) or noradrenaline + LFS (n = 5 from five rats)‐treated slices. Values are normalized to control cells. (K) Time course of the fEPSP from overexpression of GluA1‐C‐tail (n = 8 slices from six rats) and GluA1‐C‐tail + noradrenaline‐treated rats (n = 7 slices from six rats). Inset: Representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS. (L) Histogram showing the level of LTD at 60 min after LFS in GluA1‐C‐tail or GluA1‐C‐tail + noradrenaline‐treated rats. *P < 0.05, **P < 0.01, ***P < 0.001 versus control or vector‐adenovirus.
We then asked whether activation of PKA and CaMKII is involved in noradrenaline‐induced phosphorylation and surface trafficking of GluA1 subunits. H89 (10 μM), a selective inhibitor of PKA, prevented noradrenaline‐induced activation of ERK (F(5, 30) = 4.401, P = 0.004; H89: P = 0.482 vs. control; H89 + NA: P = 0.307 vs. control), phosphorylation (F(5, 18) = 7.189, P = 0.001; H89: P = 0.489 vs. control; H89 + NA: P = 0.251 vs. control) and surface expression of GluA1 subunits (F(5, 30) = 3.356, P = 0.016; H89: P = 0.745 vs. control; H89 + NA: P = 0.416 vs. control) (Figure 3G and H). KN93 (20 μM), a selective inhibitor of CaMKII, also attenuated noradrenaline‐induced activation of ERK (KN93: P = 0.814 vs. control; KN93 + NA: P = 0.099 vs. control) and surface expression of GluA1 subunits (KN93: P = 0.968 vs. control; KN93 + NA: P = 0.328 vs. control) and, to a less extent, the phosphorylation of GluA1‐Ser845 (KN93: P = 0.943 vs. control; KN93 + NA: P = 0.023 vs. control) (Figure 3G and H).
To test the assumption that phosphorylation of the carboxyl terminal domain of GluA1 subunits mediates the function of noradrenaline in LA, we blocked the noradrenaline‐induced phosphorylation of GluA1‐C‐terminus by overexpression of GluA1‐C‐tail in LA region. Efficiency of adenovirus infection was assessed by the intensity and location of fluorescence (Figure S3E). Overexpression of GluA1‐C‐tail did not influence the surface expression of GluA1 subunits and the LFS‐induced LA‐LTD in basal condition but largely suppressed the effects of noradrenaline on the surface delivery of GluA1 subunits (F(3, 20) = 6.477, P = 0.003; P = 0.003, vector + NA vs. vector; P = 0.527, GluA1‐C‐tail vs. vector; P = 0.115, GluA1‐C‐tail + NA vs. GluA1‐C‐tail) (Figure 3I), the rectification of AMPA receptor‐mediated currents (GluA1‐C‐tail + LFS: F(1, 8) = 0.298, NA vs. control; Figure 3J) (vector‐adenovirus + LFS: F(1, 8) = 8.367, P = 0.02, NA vs. control; Figure S3F) and the LA‐LTD (F(1, 13) = 15.63, P = 0.002; Figure S3G and H) (F(1, 16) = 0.43, P = 0.521; Figure 3K and L). These results indicate that the phosphorylation of GluA1‐C‐terminal domain mediated the noradrenaline‐induced changes of AMPA receptors in LA.
Propranolol suppresses adrenaline‐induced LA‐LTD impairment and GluA1 subunit surface trafficking via a central action
Furthermore, we tested whether adrenaline (i.p. injection)‐induced noradrenaline release in the brain (Frankland et al., 2004; Luo et al., 2015) can mimic the effects of fear reactivation and exogenous noradrenaline. Adrenaline (0.5 mg·kg−1, 15 min) induced the impairment of LA‐LTD (F(2, 24) = 24.56, P < 0.001) (Figure 4A), activation of PKA (F(6, 21) = 5.150, P = 0.002) (Figure 4D) and CaMKII (F(6, 21) = 2.957, P = 0.030) (Figure 4D) and the phosphorylation of GluA1‐Ser845 (F(6, 35) = 6.923, P < 0.001) (Figure 4E), only in a relatively short period of time. However, the promoting effect of adrenaline on the activation of ERK (F(6, 21) = 13.792, P < 0.001; Adr: P = 0.002; Adr 2 h: P = 0.020) (Figure 4D) and surface expression of GluA1 subunits (F(6, 35) = 4.783, P = 0.001; Adr: P = 0.008; Adr 2 h: P = 0.046) (Figure 4E) lasted more than 2 h after its injection.
Figure 4.
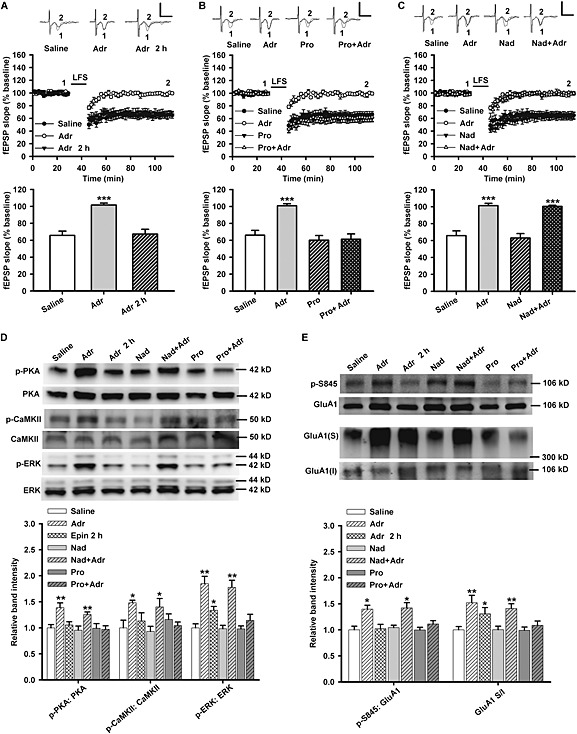
Propranolol not nadolol suppresses adrenaline‐induced LA‐LTD impairment and surface expression of GluA1 subunits. (A) Time course of LTD in slices from rats after treatment with saline (n = 10 from five rats), immediate adrenaline (Adr; n = 11 from six rats) and adrenaline 2 h earlier (n = 6 from four rats). Bar graphs showing the mean decrease in fEPSPs slope after LFS. (B) Time course of LTD in slices from rats treated with saline (n = 9 from five rats), adrenaline (n = 10 from five rats), propranolol (Pro; n = 10 from five rats) and propranolol + adrenaline (n = 10 from five rats). Bar graphs showing the mean decrease in fEPSPs slope after LFS. (C) Time course of LTD in slices from rats treated with saline (n = 9 from five rats), adrenaline (n = 10 from five rats), nadolol (Nad; n = 11 from five rats) and nadolol + adrenaline (n = 7 from four rats). Bar graphs showing the mean decrease in fEPSPs slope after LFS. (D) Inset: Representative Western blotting images (upper). The histogram showing propranolol not nadolol prevents the increase in p‐PKA, p‐CaMKII and p‐ERK in amygdala induced by adrenaline (n = 4 for each group). (E) Inset: Representative Western blotting images (upper). The histogram showing propranolol but not nadolol prevents the increase in p‐S845 and surface expression of GluA1 subunits in amygdala induced by adrenaline (n = 6 for each group). Calibration: 0.8 mV, 5 ms. *P < 0.05, **P < 0.01, ***P < 0.001 versus saline.
Unlike propranolol, nadolol is a selective β‐adrenoceptor antagonist with poor penetration of the blood–brain barrier (Johnson et al., 2008). The rats were pretreated with propranolol (10 mg·kg−1) or nadolol (10 mg·kg−1) via i.p. injection 15 min before the administration of adrenaline. Only propranolol and not nadolol attenuated adrenaline‐induced impairment of LA‐LTD (P = 0.528, Pro + Adr vs. Saline; Figure 4B) (P < 0.001, Nad + Adr vs. Saline; Figure 4C), activation of PKA (P = 0.835, Pro + Adr vs. Saline; P = 0.009, Nad + Adr vs. Saline) (Figure 4D), CaMKII (P = 0.498, Pro + Adr vs. Saline; P = 0.012, Nad + Adr vs. Saline) (Figure 4D) and ERK (P = 0.252, Pro + Adr vs. Saline; P = 0.002, Nad + Adr vs. Saline) (Figure 4D) and phosphorylation of GluA1‐Ser845 (P = 0.181, Pro + Adr vs. Saline; P = 0.002, Nad + Adr vs. Saline) (Figure 4E) and surface delivery of GluA1 subunits (P = 0.379, Pro + Adr vs. Saline; P = 0.005, Nad + Adr vs. Saline) (Figure 4E). Therefore, blocking β‐adrenoceptors in the CNS rather than in the periphery, by systemically administrated propranolol, diminishes the effects of noradrenaline in LA.
Propranolol and overexpression of GluA1‐C‐tail both attenuate reactivation‐induced LA‐LTD impairment and fear retention
Propranolol (1 µg per site) or saline was given via intra‐amygdala (LA or 2 mm above LA) infusion before fear reactivation, and the rats in non‐reactivation groups were also given the same treatments (Figure 5A).
Figure 5.
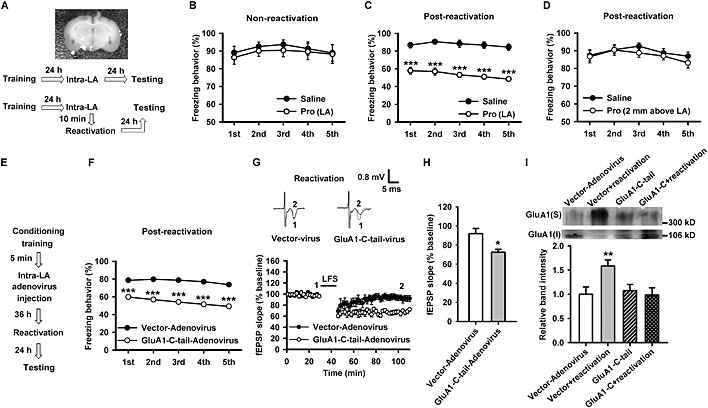
Propranolol and overexpression of GluA1‐C‐tail in LA both attenuate reactivation‐induced LTD impairment and fear retention. (A) Image showing cannulae placements in LA of rats (upper). The behavioural procedure used in the experiment (lower). (B) The percentage of freezing behaviour at 48 h after conditioning (training). The freezing behaviour was not significantly different between saline (n = 7) and propranolol (Pro)‐treated groups (n = 9). (C) The percentage of freezing behaviour at 24 h after reactivation. The freezing behaviour was significantly reduced in the propranolol‐treated group (n = 7) compared with saline‐treated rats (n = 7). (D) The percentage of freezing behaviour at 24 h after reactivation. The freezing behaviour was not significantly different between saline (n = 7) and propranolol‐treated groups (n = 8). (E) The behavioural procedure used in the experiment. (F) The freezing behaviour was significantly reduced in overexpression of GluA1‐C‐tail‐treated rats (n = 9) compared with vector‐adenovirus‐treated rats (n = 8). (G) Time course of the fEPSP from vector‐adenovirus (n = 7 slices from four rats) or overexpression of GluA1‐C‐tail (n = 8 slices from four rats)‐treated rats after reactivation. Inset: The representative fEPSPs recorded in individual slices before (1) and 60 min after (2) LFS. (H) The histogram showing the level of LTD at 60 min after LFS in vector‐adenovirus or overexpression of GluA1‐C‐tail‐treated rats after fear reactivation. (I) Representative images of western blotting (upper). The histogram showing that surface expression of GluA1 subunits was enhanced after reactivation and this effect was suppressed by overexpression of GluA1‐C‐tail, but not vector‐adenovirus (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001 versus saline or vector‐adenovirus‐treated rats.
Intra‐LA infusion of propranolol significantly attenuated fear memory retention in a reactivation‐dependent manner (F(1, 12) = 0.169, NS; Figure 5B) (F(1, 14) = 78.10, P < 0.001; Figure 5C), and infusion of propranolol at 2 mm above LA did not affect reactivation‐induced fear retention (F(1,13) = 0.937, NS; Figure 5D). Administration of propranolol via intra‐amygdala infusion had no distinguishable effect on the freezing behaviour of rats in habituation, conditioning training and reactivation (Figure S4A–C). These results further support our hypothesis that β‐adrenoceptor activation in LA mediated the effects of fear reactivation.
In behavioural experiments with adenovirus infection and overexpression of GluA1‐C‐tail in LA region (Figure 5E), we found that blocking the phosphorylation of GluA1‐C‐terminal domain also significantly suppressed reactivation‐induced fear retention (F(1, 15) = 95.72, P < 0.001; Figure 5F). Overexpression of GluA1‐C‐tail in LA did not influence the freezing behaviour in habituation, conditioning training and fear reactivation (Figure S4D).
Overexpression of GluA1‐C‐tail also restored reactivation‐induced impairment of LA‐LTD from 92 ± 5.5% in vector to 73 ± 3.0% in the overexpression group (F(1, 13) = 10.33, P = 0.007, GluA1‐C‐tail vs. vector; Figure 5G and H). Meanwhile, Western blot analysis showed that overexpression of GluA1‐C‐tail (F(3, 20) = 4.203, P = 0.019; P = 0.653, GluA1‐C‐tail + reactivation vs. GluA1‐C‐tail), but not vector‐adenovirus, eliminated reactivation‐induced surface expression of GluA1 subunits (Figure 5I).
Taken together, these findings further demonstrated that inhibition of β‐adrenoceptors or GluA1‐C‐terminal phosphorylation, which leads to the surface trafficking of GluA1‐containing AMPA receptors in LA, attenuated reactivation‐induced fear memory retention. This proposal is presented in Figure 6, delineating the interfering stages for propranolol and GluA1‐C‐tail overexpression in fear retention.
Figure 6.
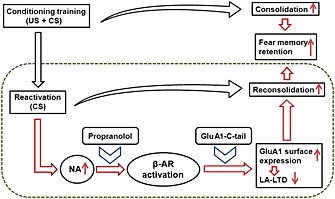
Scheme of the stages at which propranolol and GluA1‐C‐tail can interfere with fear reconsolidation. After acquisition of the associative fear memory (US + CS), drugs that strengthens the consolidation process lead to enhancement of fear retention. After fear reactivation (CS for one time), endogenous noadrenaline strengthens the process of reconsolidation, resulting in the enhancement of fear memory retention. Blocking β‐adrenoceptors by propranolol, or preventing the phosphorylation and surface expression of GluA1 subunitsby GluA1‐C‐tail overexpression in LA, attenuates reactivation‐induced fear memory retention. US, unconditioned stimulus (foot shock); CS, conditioned stimulus (tone).
Discussion
The present study demonstrated for the first time that fear reactivation‐induced fear memory retention is mediated by increased release of noradrenaline, activation of β‐adrenoceptors and enhanced surface expression of GluA1‐containing AMPA receptors, especially the surface expression of extrasynaptic GluA1 subunits in LA. Most importantly, we found that propranolol prevented the alterations induced by noradrenaline and reactivation, via modulating the stability of GluA1 subunits in LA. Our results provide a better understanding for the occurrence of PTSD‐like events and the efficacy of propranolol in the treatment of PTSD.
Fear reconsolidation following reactivation contains a process of traumatic memory recall (Agren et al., 2012), which is parallel with the surge of noradrenaline release in the brain. The level of noradrenaline in the brain is unduly increased in PTSD patients (O'Donnell et al., 2004; Soeter and Kindt, 2011). Similarly, previous studies have found that both fear conditioning and reactivation increase noradrenaline levels in the CNS (McIntyre et al., 2002; Morilak et al., 2005; Luo et al., 2015). However, others and our results showed that propranolol was effective only in reactivation‐dependent fear retention (Debiec and Ledoux, 2004; Bush et al., 2010), so we detected noradrenaline level in LA before and after reactivation and observed an increased noradrenaline level after fear reactivation. Elevation of noradrenaline content during fear memory recall may explain the strengthening of emotional memories compared with neutral memories (LaLumiere et al., 2003; O'Donnell et al., 2004; Soeter and Kindt, 2011). This is probably the main reason why propranolol may have beneficial effect for the treatment of PTSD. However, it has been reported that propranolol is effective against fear extinction only with the exposure of fear memory recall (Ouyang and Thomas, 2005; Mueller and Cahill, 2010; Fitzgerald et al., 2014). Other reports suggested that propranolol is not effective in conditioned fear consolidation (Debiec and Ledoux, 2004; Bush et al., 2010) and only has limited efficacy on reconsolidation of fear memory when given systemically (Muravieva and Alberini, 2010). We suppose these contradictions may be due to differences in trial protocols, the releasing pattern of noradrenaline and even hereditary factors.
When compared with single brain regions, the extended nervous network integrity is also very important for efficient behavioural regulations (Vertes, 2006; Bonnelle et al., 2012). According to neuroimaging studies, the amygdala is part of an extended nervous network, which is named ‘visceromotor’ network and modulates emotional behaviour (Nugent et al., 2006). Because the basal and LA receives several nervous projections from other brain regions, including the prefrontal cortex, the hippocampus and the thalamus, it has been accepted that plasticity of the amygdala may be a result of the plasticity at an extended network level (Vertes, 2006; Bonnelle et al., 2012). And in the other way around, amygdala innervates nervous projections to downstream targets such as the bed nucleus of the stria terminalis and the brainstem nuclei; thus, the plasticity of amygdala also influences the plasticity and behavioural processes at the associated network level (Stamatakis et al., 2014). Because the neurotransmission from the thalamus to the LA plays a key role in auditory fear memory regulation (McKernan and Shinnick‐Gallagher, 1997; Rumpel et al., 2005), this study focused on the LA to investigate the LA‐LTD and fear memory retention in rats.
Our behavioural experiments with reactivation were carried out to clarify the intrinsic relationship of noradrenaline and amygdala‐dependent fear retention and the interfering property of propranolol on PTSD‐like event. Although most previous research found that propranolol given after reactivation was effective in attenuating fear retention (Debiec and Ledoux, 2004; Debiec and LeDoux, 2006; Brunet et al., 2008), it has been recently demonstrated that pre‐reactivational administration of propranolol did not modify PTSD symptoms (Wood et al., 2015). However, other studies reported that propranolol given before reactivation impaired the return of fear memory (Kindt et al., 2009; Lonergan et al., 2012; Schwabe et al., 2012). Because therapeutic intervention for PTSD was usually applied after the exposure of traumatic stress, rather than fear reactivation (Vaiva et al., 2003), treatments with propranolol were started before the reactivation event and the drug was given three times 1 day (Vaiva et al., 2003). We found that propranolol did not influence freezing behaviour in the reactivation process but significantly weakened fear retention in the reconsolidation test. These results indicated propranolol given before reactivation was effective in preventing reconsolidation‐related fear retention. Meanwhile, we found no significant effect of propranolol on fear consolidation. We suppose that reactivation on day 2 elevates the release of noradrenaline and it promotes fear reconsolidation via β‐adrenoceptors in LA. A confusing phenomenon is that the reactivation event itself does not increase fear memory retention tested on day 3. One explanation that needs further evidence is that the reactivation stimulus contains only the CS (three pulses of tone), which itself has memory unstabilizing and transforming action, at the same time as offering fear reactivation.
Noradrenaline affects synaptic plasticity in different brain areas, and the α1, α2 and β‐adrenoceptors have distinctive patterns of expression in the CNS, which determines the function of noradrenaline in each neuroanatomical region (Hein, 2006; Tully and Bolshakov, 2010). In the hippocampal CA1 region and the bed nucleus of the stria terminalis, noradrenergic activation influences long‐term potentiation (LTP) and LTD via α1 and β‐adrenoceptors (Wang et al., 2005; McElligott et al., 2010; Nobis et al., 2011). It has been suggested that adrenergic activation strengthens the amygdala LTP in neural circuits involving glutamatergic and GABAergic transmission (Tully et al., 2007; Silberman et al., 2010). While the inhibition of GABAergic neurotransmission by noradrenaline (Tully et al., 2007) may be the result of activation of α‐adrenoceptors, we focused on AMPA receptors to explore the long‐term changes of glutamatergic transmission. LTD of the amygdala synaptic transmission is proposed as a cellular mechanism determining fear memory retention or erasing (Maroun, 2006; Kim et al., 2007; Hong et al., 2009; Danielewicz and Hess, 2014). A recent study has demonstrated that optogenetic delivery of amygdala‐LTD leads to fear memory erasure (Nabavi et al., 2014). It has been recognized that fear memory can be reactivated and become unstable, suggesting that it can be either reconsolidated or extinguished (Mahan and Ressler, 2012). We found that fear reactivation induced an increased release of noradrenaline and caused an impairment of LA‐LTD by activating β‐adrenoceptors. So, reactivation‐induced impairment of LA‐LTD may be the critical cellular mechanism for the destabilization and reconsolidation of fear memory. Noradrenaline can enhance fear memory by promoting acquisition and consolidation (Roozendaal et al., 2006; Tully and Bolshakov, 2010). While fear responses may represent a biological mechanism for memory enhancement, it may also be a fundament for PTSD‐like events in animal models. The noradrenaline‐based mechanism may also be involved in some other emotion‐aroused learning experiences, such as appetitive conditioning (Salinas et al., 1997). But the mechanism for noradrenergic activation‐induced fear memory extinction (Berlau and McGaugh, 2006) should be different, which may include an LTP‐enhancing and new memory (for extinction events) acquisition and consolidation processes. In this research, we further explored the mechanism of action of noradrenaline and the preventing efficacy of propranolol on reactivation‐related fear retention.
Activation of β‐adrenoceptors by noradrenaline results in facilitation of excitatory synaptic transmission via the activation of PKA signalling, contributing to the strengthening of emotional memory (Evans et al., 2010). Our results indicate only the β‐adrenoceptor antagonist was effective on noradrenaline‐induced responses in LA and reactivation‐induced fear retention. Interestingly, we also found only the central action of propranolol is involved in attenuating the effect of noradrenaline. We used a dose of adrenaline (0.5 mg·kg−1) that was previously shown elevating the contextual fear memory performance in mice (Frankland et al., 2004; Hu et al., 2007). It was propranolol, rather than nadolol, that is a peripherally selective β‐adrenoceptor antagonist that scarcely crosses the blood–brain barrier (Johnson et al., 2008), which blocked the effects of adrenaline. Thus, selectively blocking β‐adrenoceptors in the CNS may exert much better efficacy than systemic administration. Therefore, we gave propranolol by intra‐amygdala infusion. The results suggested that the action of propranolol in LA was specifically essential in its attenuating efficiency on reactivation‐induced fear retention.
There are critical phosphorylation sites within the C‐terminus of GluA1 and GluA2 subunits of AMPA receptors (Wang et al., 2005). The Ser831 and Ser845 residues within the C‐terminal domain are crucial for the synaptic trafficking of GluA1 subunits. The phosphorylation of GluA2 subunitsat Ser880 and Tyr876 residues is important for the synaptic endocytosis of GluA2‐containing AMPA receptors and the formation of LTD (Seidenman et al., 2003; Hayashi and Huganir, 2004). Phosphorylation of GluA1‐Ser845/831 by noradrenaline in the hippocampus is required for the enhancement of emotional memory (Hu et al., 2007). A recent study demonstrated phosphorylation of GluA1‐Ser831 in LA is important for fear renewal in rats (Lee et al., 2013). We found that acute treatment with noradrenaline increased Ser845/831 phosphorylation and surface expression of GluA1 subunits, suggesting phosphorylated modification of GluA1 subunits by PKA signals may be an integral component in adrenergic activation‐induced surface trafficking of these subunits. Our study also showed that noradrenaline‐induced surface expression of GluA1 subunits was mainly located at extrasynaptic sites, which indicates that further stimuli may be necessary to activate the lateral trafficking of GluA1‐containing AMPA receptors into the synapses, resulting in an increased number of AMPA receptors at the postsynaptic density and an enhanced potential for synaptic transmission.
However, it is unclear and deserves further research to identify which subtypes of β‐adrenoceptors are involved in adrenergic activation in the LA. What is more, it needs further exploration to answer whether other cellular signalling events, as addressed in a mechanistic study of fear reconsolidation (Li et al., 2013), and modulation of AMPA receptors in other manners may participate in reactivation‐induced amygdala and behavioural responses.
In summary, the present results suggest that reactivation enhances fear retention by increasing the release of noradrenaline, which promotes the surface expression of GluA1 and strengthens the excitatory synaptic transmission in LA. This work provides new mechanistic insights for the beneficial effect of propranolol on event‐based fear retention and indicates that propranolol combined with exposure therapy (moderate reactivation) may be more effective than propranolol or exposure therapy alone in the treatment of PTSD. Moreover, other β‐adrenoceptor antagonists with CNS selective action may also be targeted for the intervention of PTSD.
Author contributions
J. Z. and Y. L. designed and substantially performed the experiments, analysed the data and wrote the paper. J‐T. Z. helped in the separation of synaptic and extrasynaptic fractions. M‐X. L. helped in microdialysis experiment. C‐M. W. and X‐L. G. helped in animal experiments. P‐F. W., Z‐L. H., Y. J. and L. N. collaborated by providing expert advice. F. W. collaborated by analysing and interpreting the data, developing and editing the manuscript and co‐writing the manuscript. J‐G. C. oversaw the experiments, supervised the project and wrote and revised the manuscript.
Conflict of interest
None declared.
Supporting information
Figure S1 There are no differences in baseline behaviors between saline and propranolol‐treated rats.
Figure S2 Negative effects of NE on glutamatergic synapse transmission.
Figure S3 Activation of β‐AR and phosphorylation GluA1‐C terminus mediate NE‐induced upregulation of AMPAR function in LA.
Figure S4 Neither propranolol nor overexpression of GluA1‐C‐tail in LA influences fear acquisition, consolidation or reactivation of rats.
Supporting info item
Acknowledgements
We acknowledge the support from the National Basic Research Program, the International Science & Technology Cooperation Program of China and the National Natural Scientific Foundation of China.
This work was supported by grants from the National Basic Research Program of China (973 program no. 2013CB531303 to J‐G. C. and no. 2014CB744601 to F. W.), the International Science & Technology Cooperation Program of China (no. 2011DFA32670) and PCSIRT (no. IRT13016) to J‐G. C. This work was also supported by the National Natural Scientific Foundation of China to F. W. (no. 81222048).
Zhou, J. , Luo, Y. , Zhang, J.‐T. , Li, M.‐X. , Wang, C.‐M. , Guan, X.‐L. , Wu, P.‐F. , Hu, Z.‐L. , Jin, Y. , Ni, L. , Wang, F. , and Chen, J.‐G. (2015) Propranolol decreases retention of fear memory by modulating the stability of surface glutamate receptor GluA1 subunits in the lateral amygdala. British Journal of Pharmacology, 172: 5068–5082. doi: 10.1111/bph.13272.
References
- Agren T, Engman J, Frick A, Björkstrand J, Larsson EM, Furmark T et al. (2012). Disruption of reconsolidation erases a fear memory trace in the human amygdala. Science 337: 1550–1552. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Ligand‐Gated Ion Channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th edn. American Psychiatric Publishing: Arlington, pp. 189–195. [Google Scholar]
- Berlau DJ, McGaugh JL (2006). Enhancement of extinction memory consolidation: the role of the noradrenergic and GABAergic systems within the basolateral amygdala. Neurobiol Learn Mem 86: 123–132. [DOI] [PubMed] [Google Scholar]
- Bonnelle V, Ham TE, Leech R, Kinnunen KM, Mehta MA, Greenwood RJ et al. (2012). Salience network integrity predicts default mode network function after traumatic brain injury. Proc Natl Acad Sci U S A 109: 4690–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Elzinga B, Schmahl C, Vermetten E (2008). Structural and functional plasticity of the human brain in posttraumatic stress disorder. Prog Brain Res 167: 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Orr SP, Tremblay J, Robertson K, Nader K, Pitman RK (2008). Effect of post‐retrieval propranolol on psychophysiologic responding during subsequent script‐driven traumatic imagery in post‐traumatic stress disorder. J Psychiatric Res 42: 503–506. [DOI] [PubMed] [Google Scholar]
- Bush DE, Caparosa EM, Gekker A, Ledoux J (2010). Beta‐adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front Behav Neurosci 4: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL (2013). Norepinephrine enhances a discrete form of long‐term depression during fear memory storage. J Neurosci 33: 11825–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielewicz J, Hess G (2014). Early life stress alters synaptic modification range in the rat lateral amygdala. Behav Brain Res 265: 32–37. [DOI] [PubMed] [Google Scholar]
- Debiec J, Bush DE, Ledoux J (2011). Noradrenergic enhancement of reconsolidation in the amygdala impairs extinction of conditioned fear in rats – a possible mechanism for the persistence of traumatic memories in PTSD. Depress Anxiety 28: 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debiec J, Ledoux J (2004). Disruption of reconsolidation but not consolidation of auditory fear conditioning by noradrenergic blockade in the amygdala. Neuroscience 129: 267–272. [DOI] [PubMed] [Google Scholar]
- Debiec J, LeDoux J (2006). Noradrenergic signaling in the amygdala contributes to the reconsolidation of fear memory: treatment implications for PTSD. Ann N Y Acad Sci 1071: 521–524. [DOI] [PubMed] [Google Scholar]
- Evans BA, Sato M, Sarwar M, Hutchinson DS, Summers RJ (2010). Ligand‐directed signalling at beta‐adrenoceptors. Br J Pharmacol 159: 1022–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farb CR, Chang W, Ledoux JE (2010). Ultrastructural characterization of noradrenergic axons and beta‐adrenergic receptors in the lateral nucleus of the amygdala. Front Behav Neurosci 4: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ, Seemann JR, Maren S (2014). Can fear extinction be enhanced? A review of pharmacological and behavioral findings. Brain Res Bull 105: 46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankland PW, Josselyn SA, Anagnostaras SG, Kogan JH, Takahashi E, Silva AJ (2004). Consolidation of CS and US representations in associative fear conditioning. Hippocampus 14: 557–569. [DOI] [PubMed] [Google Scholar]
- Galvez R, Mesches MH, McGaugh JL (1996). Norepinephrine release in the amygdala in response to footshock stimulation. Neurobiol Learn Mem 66: 253–257. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Huganir RL (2004). Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci 24: 6152–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein L (2006). Adrenoceptors and signal transduction in neurons. Cell Tissue Res 326: 541–551. [DOI] [PubMed] [Google Scholar]
- Hong I, Song B, Lee S, Kim J, Choi S (2009). Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur J Neurosci 30: 2089–2099. [DOI] [PubMed] [Google Scholar]
- Hu H, Real E, Takamiya K, Kang MG, Ledoux J, Huganir RL et al. (2007). Emotion enhances learning via norepinephrine regulation of AMPA‐receptor trafficking. Cell 131: 160–173. [DOI] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA (2013). AMPARs and synaptic plasticity: the last 25 years. Neuron 80: 704–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Wang W, Wang F, Hu ZL, Xiao JL, Yang S et al. (2013). The stability of NR2B in the nucleus accumbens controls behavioral and synaptic adaptations to chronic stress. Biol Psychiatry 74: 145–155. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Cortez V, Kennedy SL, Foley TE, Hanson H 3rd, Fleshner M (2008). Role of central beta‐adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav Immun 22: 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G et al. (2007). Amygdala depotentiation and fear extinction. Proc Natl Acad Sci U S A 104: 20955–20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). NC3Rs Reporting Guidelines Working Group. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindt M, Soeter M, Vervliet B (2009). Beyond extinction: erasing human fear responses and preventing the return of fear. Nat Neurosci 12: 256–258. [DOI] [PubMed] [Google Scholar]
- LaLumiere RT, Buen TV, McGaugh JL (2003). Post‐training intra‐basolateral amygdala infusions of norepinephrine enhance consolidation of memory for contextual fear conditioning. J Neurosci 23: 6754–6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Song B, Kim J, Park K, Hong I, An B et al. (2013). GluA1 phosphorylation at serine 831 in the lateral amygdala is required for fear renewal. Nat Neurosci 16: 1436–1444. [DOI] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011). Soluble Abeta oligomers inhibit long‐term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci 31: 6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Meloni EG, Carlezon WA Jr, Milad MR, Pitman RK, Nader K et al. (2013). Learning and reconsolidation implicate different synaptic mechanisms. Proc Natl Acad Sci U S A 110: 4798–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YK, Wang F, Wang W, Luo Y, Wu PF, Xiao JL et al. (2012). Aquaporin‐4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: involvement of downregulation of glutamate transporter‐1 expression. Neuropsychopharmacology 37: 1867–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan MH, Olivera‐Figueroa LA, Pitman RK, Brunet A (2012). Propranolol's effects on the consolidation and reconsolidation of long‐term emotional memory in healthy participants: a meta‐analysis. J Psychiatry Neurosci27 Nov 2012 . doi:10.1503/jpn.120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HF, Wu PF, Yang YJ, Xiao W, Fan J, Liu J et al. (2014). Interactions between N‐ethylmaleimide‐sensitive factor and GluR2 in the nucleus accumbens contribute to the expression of locomotor sensitization to cocaine. J Neurosci 34: 3493–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Zhou J, Li MX, Wu PF, Hu ZL, Ni L et al. (2015). Reversal of aging‐related emotional memory deficits by norepinephrine via regulating the stability of surface AMPA receptors. Aging Cell 14: 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahan AL, Ressler KJ (2012). Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci 35: 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroun M (2006). Stress reverses plasticity in the pathway projecting from the ventromedial prefrontal cortex to the basolateral amygdala. Eur J Neurosci 24: 2917–2922. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Klug JR, Nobis WP, Patel S, Grueter BA, Kash TL et al. (2010). Distinct forms of Gq‐receptor‐dependent plasticity of excitatory transmission in the BNST are differentially affected by stress. Proc Natl Acad Sci U S A 107: 2271–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre CK, Hatfield T, McGaugh JL (2002). Amygdala norepinephrine levels after training predict inhibitory avoidance retention performance in rats. Eur J Neurosci 16: 1223–1226. [DOI] [PubMed] [Google Scholar]
- McKernan MG, Shinnick‐Gallagher P (1997). Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature 390: 607–611. [DOI] [PubMed] [Google Scholar]
- Mirante O, Brandalise F, Bohacek J, Mansuy IM (2014). Distinct molecular components for thalamic‐ and cortical‐dependent plasticity in the lateral amygdala. Front Mol Neurosci 7: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S et al. (2005). Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry 29: 1214–1224. [DOI] [PubMed] [Google Scholar]
- Mueller D, Cahill SP (2010). Noradrenergic modulation of extinction learning and exposure therapy. Behav Brain Res 208: 1–11. [DOI] [PubMed] [Google Scholar]
- Muravieva EV, Alberini CM (2010). Limited efficacy of propranolol on the reconsolidation of fear memories. Learn Mem 17: 306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R (2014). Engineering a memory with LTD and LTP. Nature 511: 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobis WP, Kash TL, Silberman Y, Winder DG (2011). β‐Adrenergic receptors enhance excitatory transmission in the bed nucleus of the stria terminalis through a corticotrophin‐releasing factor receptor‐dependent and cocaine‐regulated mechanism. Biol Psychiatry 69: 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AC, Milham MP, Bain EE, Mah L, Cannon DM, Marrett S et al. (2006). Cortical abnormalities in bipolar disorder investigated with MRI and voxel‐based morphometry. Neuroimage 30: 485–497. [DOI] [PubMed] [Google Scholar]
- O'Donnell T, Hegadoren KM, Coupland NC (2004). Noradrenergic mechanisms in the pathophysiology of post‐traumatic stress disorder. Neuropsychobiology 50: 273–283. [DOI] [PubMed] [Google Scholar]
- Ouyang M, Thomas SA (2005). A requirement for memory retrieval during and after long‐term extinction learning. Proc Natl Acad Sci U S A 102: 9347–9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagioti M, Gooding PA, Tarrier N (2012). A meta‐analysis of the association between posttraumatic stress disorder and suicidality: the role of comorbid depression. Compr Psychiatry 53: 915–930. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzak RH, el‐Gabalawy R, Tsai J, Sareen J, Neumeister A, Southwick SM (2014). Typologies of posttraumatic stress disorder in the U.S. adult population. J Affect Disorders 162: 102–106. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB et al. (2002). Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biol Psychiatry 51: 189–192. [DOI] [PubMed] [Google Scholar]
- Roozendaal B, Hui GK, Hui IR, Berlau DJ, McGaugh JL, Weinberger NM (2006). Basolateral amygdala noradrenergic activity mediates corticosterone‐induced enhancement of auditory fear conditioning. Neurobiol Learn Mem 86: 249–255. [DOI] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R (2005). Postsynaptic receptor trafficking underlying a form of associative learning. Science 308: 83–88. [DOI] [PubMed] [Google Scholar]
- Salinas JA, Introini‐Collison IB, Dalmaz C, McGaugh JL (1997). Posttraining intraamygdala infusions of oxotremorine and propranolol modulate storage of memory for reductions in reward magnitude. Neurobiol Learn Mem 68: 51–59. [DOI] [PubMed] [Google Scholar]
- Samuelson KW (2011). Post‐traumatic stress disorder and declarative memory functioning: a review. Dialogues Clin Neurosci 13: 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe L, Nader K, Wolf OT, Beaudry T, Pruessner JC (2012). Neural signature of reconsolidation impairments by propranolol in humans. Biol Psychiatry 71: 380–386. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R (2003). Glutamate receptor subunit 2 serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci 23: 9220–9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shad MU, Suris AM, North CS (2011). Novel combination strategy to optimize treatment for PTSD. Hum Psychopharmacol 26: 4–11. [DOI] [PubMed] [Google Scholar]
- Sierra‐Mercado D, Padilla‐Coreano N, Quirk GJ (2011). Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology 36: 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Ariwodola OJ, Chappell AM, Yorgason JT, Weiner JL (2010). Lateral paracapsular GABAergic synapses in the basolateral amygdala contribute to the anxiolytic effects of beta 3 adrenoceptor activation. Neuropsychopharmacology 35: 1886–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeter M, Kindt M (2011). Noradrenergic enhancement of associative fear memory in humans. Neurobiol Learn Mem 96: 263–271. [DOI] [PubMed] [Google Scholar]
- Stamatakis AM, Sparta DR, Jennings JH, McElligott ZA, Decot H, Stuber GD (2014). Amygdala and bed nucleus of the stria terminalis circuitry: implications for addiction‐related behaviors. Neuropharmacology 76PtB: 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Bolshakov VY (2010). Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY (2007). Norepinephrine enables the induction of associative long‐term potentiation at thalamo‐amygdala synapses. Proc Natl Acad Sci U S A 104: 14146–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A et al. (2003). Immediate treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biol Psychiatry 54: 947–949. [DOI] [PubMed] [Google Scholar]
- Vertes RP (2006). Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience 142 (1): 1–20. [DOI] [PubMed] [Google Scholar]
- Wang CM, Yang YJ, Zhang JT, Liu J, Guan XL, Li MX et al. (2015). Regulation of emotional memory by hydrogen sulfide: role of GluN2B‐containing NMDA receptor in the amygdala. J Neurochem 132: 124–134. [DOI] [PubMed] [Google Scholar]
- Wang JQ, Arora A, Yang L, Parelkar NK, Zhang G, Liu X et al. (2005). Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol 32: 237–249. [DOI] [PubMed] [Google Scholar]
- Wang W, Wang F, Yang YJ, Hu ZL, Long LH, Fu H et al. (2011). The flavonoid baicalein promotes NMDA receptor‐dependent long‐term potentiation and enhances memory. Br J Pharmacol 162: 1364–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood NE, Rosasco ML, Suris AM, Spring JD, Marin MF, Lasko NB et al. (2015). Pharmacological blockade of memory reconsolidation in posttraumatic stress disorder: three negative psychophysiological studies. Psychiatry Res 225: 31–39. [DOI] [PubMed] [Google Scholar]
- Zhou HC, Sun YY, Cai W, He XT, Yi F, Li BM et al. (2013). Activation of beta2‐adrenoceptor enhances synaptic potentiation and behavioral memory via cAMP‐PKA signaling in the medial prefrontal cortex of rats. Learn Mem 20: 274–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 There are no differences in baseline behaviors between saline and propranolol‐treated rats.
Figure S2 Negative effects of NE on glutamatergic synapse transmission.
Figure S3 Activation of β‐AR and phosphorylation GluA1‐C terminus mediate NE‐induced upregulation of AMPAR function in LA.
Figure S4 Neither propranolol nor overexpression of GluA1‐C‐tail in LA influences fear acquisition, consolidation or reactivation of rats.
Supporting info item