

Abstract
Background and Purpose
Orexins (A and B) are hypothalamic peptides that interact with OX1 and OX2 receptors and are involved in the sleep/wake cycle. We previously demonstrated that OX1 receptors are highly expressed in colon cancer tumours and colonic cancer cell lines where orexins induce apoptosis and inhibit tumour growth in preclinical animal models. The present study explored the structure–function relationships of orexin‐B and OX1 receptors.
Experimental Approach
The contribution of all orexin‐B residues in orexin‐B‐induced apoptosis was investigated by alanine scanning. To determine which OX1 receptor domains are involved in orexin‐B binding and apoptosis, a 3D model of OX1 receptor docked to the orexin‐B C‐terminus (AA‐20–28) was developed. Substitution of residues present in OX1 receptor transmembrane (TM) domains by site‐directed mutagenesis was performed.
Key Results
Alanine substitution of orexin‐B residues, L11, L15, A22, G24, I25, L26 and M28, altered orexin‐B's binding affinity. Substitution of these residues and of the Q16, A17, S18, N20 and T27 residues inhibited apoptosis in CHO‐S–OX1 receptor cells. The K120, P123, Y124, N318, K321, F340, T341, H344 and W345 residues localized in TM2, TM3, TM6 and TM7 of OX1 receptors were shown to play a role in orexin‐B recognition and orexin‐B/OX1 receptor‐induced apoptosis.
Conclusions and Implications
The C‐terminus of orexin‐B (i) plays an important role in its pro‐apoptotic effect; and (ii) interacts with some residues localized in the OX1 receptor TM. This study defines the structure–function relationship for orexin‐B recognition by human OX1 receptors and orexin‐B/OX1 receptor‐induced apoptosis, an important step for the future development of new agonist molecules.
Abbreviations
- CHARMM
chemistry at harvard macromolecular mechanics
- CMAP
grid-based energy correction maps
- ECL
extracellular loop
- LINCS
Linear Constraint Solver
- TM
transmembrane
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes b |
| OX1 receptor | Caspase‐3 |
| OX2 receptor | Caspase‐7 |
| LIGANDS | |
|---|---|
| IP3 | Orexin‐B |
| Orexin‐A | Suvorexant |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, 2013b).
Introduction
Orexins A and B (also known as hypocretins 1 and 2) are hypothalamic 33‐aminoacid and 28‐aminoacid neuropeptides, respectively, which originate from prepro‐orexin, a 131‐residue precursor. Orexin‐A contains two intramolecular disulfide bonds between positions 6 to 12 and 7 to 14, while orexin‐B does not have any. These two peptides share the same effects, regulating sleep, wakefulness, feeding, energy homeostasis, obesity, diabetes, breathing, reward system or drug addiction (Laburthe and Voisin, 2012). Orexins trigger biological effects by interacting with two members of the class A GPCR family, that is, orexin receptor‐1 (OX1 receptor) and orexin receptor‐2 (OX2 receptor) (Thompson et al., 2014). Activation of these receptors by orexins classically induces cellular calcium transients through Gq‐dependent and Gq‐independent pathways (Laburthe et al., 2010). Besides these central actions, the orexins/receptor system is also involved in peripheral effects, including modulation of the cardiovascular system and the regulation of neuroendocrines and reproduction (Xu et al., 2013). Recently, our group demonstrated that orexin‐A and orexin‐B, bound to OX1 receptors, can induce massive apoptosis, resulting in the drastic reduction of cell growth in various colonic cancer cell lines, including HT‐29, LoVo, Caco‐2 and others (Voisin et al., 2011). An entirely novel mechanism, not related to Gq‐mediated phospholipase C activation, was shown to trigger orexin‐induced apoptosis (Voisin et al., 2008; El Firar et al., 2009). In fact, orexins induced the tyrosine phosphorylation of two immunoreceptor tyrosine‐based motifs (ITIMs) located at the interface between transmembrane (TM) domain 2 and TM 7 of the OX1 receptor and the cytoplasm (Voisin et al., 2008). The resulting phosphorylated receptor could then recruit and activate the phosphotyrosine phosphatase, SHP‐2, which is responsible for mitochondrial apoptosis, involving cytochrome c release from mitochondria to cytosol and caspase‐3 and caspase‐7 activation (El Firar et al., 2009). The pro‐apoptotic effect of orexins has also been extended to other cancer cell lines derived from human neuroblastoma (SK‐N‐MC cell line) and rat pancreatic cancer (AR42J cell line) (Rouet‐Benzineb et al., 2004; Voisin et al., 2006). Recent data demonstrated that the OX1 receptors is aberrantly expressed in all resected primary colorectal tumours and liver metastases tested but is not present in normal colon tissues (Voisin et al., 2011). Moreover, an injection of exogenous orexins to mice strongly reduced in vivo tumour growth and reversed the development of established tumours in mice xenografted with colon cancer cell lines such as HT‐29 or LoVo, due to robust apoptosis induction (Voisin et al., 2011). Taken together, these observations suggest that the orexins/ OX1 receptor system may represent a new promising target in colorectal cancer therapy, and most probably in other cancers, including pancreatic cancers neuroblastoma, and/or prostate cancer (Alexandre et al., 2014). In this context, structure–function relationship studies of the orexins/ OX1 receptor system are essential for the development of new agonists of OX1 receptors that may represent new therapeutic approaches.
Until now, little has been known about the structure–function relationship of the orexins/ OX1 receptors system. The determination of the 3D structure of orexin‐B in solution by 2D NMR spectroscopy revealed the presence of two α‐helices encompassing residues Leu7 to Gly19 and residues Ala23 to Met28, connected by a short flexible loop (Lee et al., 1999). In addition, pharmacological tools have been developed to discriminate between OX1 and OX2 receptors (Gotter et al., 2012; Laburthe and Voisin, 2012), including selective peptide agonists for the OX2 receptor such as [Ala11, D‐Leu15] orexin‐B6–28 (Asahi et al., 2003), [Ala27] orexin‐B6–28 (Lang et al., 2004), [Pro11] orexin‐B6–28 (Lang et al., 2004) and other selective non‐peptide antagonists, including TCS‐orexin 2‐29, JNJ‐10397049, EMP4 for OX2 receptors or the non‐peptide molecule antagonists SB‐334867, SB‐408124 and SB‐674042 for OX1 receptors (review by Gotter et al., 2012). Nevertheless, the residues of orexin‐B and the OX1 receptor involved in apoptosis are unknown.
In the earlier context described, the present work aims to investigate the role of each residue of orexin‐B in the induction of apoptosis by the neuropeptide by using an ‘alanine‐scanning’ strategy. Here, we showed that the C‐terminus of orexin‐B is crucial for the ability of the peptide to induce apoptosis. Based on the recent X‐ray structure of the OX2 receptor (Yin et al., 2015), a 3D model of the OX1 receptor was developed and used to design OX1 receptor site‐directed mutagenesis experiments, which demonstrated that seven specific residues are involved in the interaction of orexin‐B with OX1 receptors and in the ability of the peptide to induce a pro‐apoptotic response. This study advances our knowledge on the molecular mechanism of the pro‐apoptotic properties of orexins, which is essential to design new agonists and/or biased agonists as novel therapeutic approaches for digestive and other types of cancers.
Methods
Stable expression of cDNA encoding human OX1 and mutated OX1 receptors in CHO‐S cells
Full‐length OX1R cDNAs were cloned into the expression vector pEYFP‐N2 in fusion at the C‐terminus with the yellow fluorescent protein as described previously (El Firar et al., 2009). The plasmid encoding the human OX1 receptor was stably transfected into CHO‐S cells (ECACC 85050302) using X‐tremeGENE (ROCHE Diagnostics, Meylan, France) according to the manufacturer's protocol. Transfected CHO‐S cells were selected in the presence of 1 mg·mL−1 geneticin (G418, Life Technologies) for 2 weeks and cloned (Ceraudo et al., 2008). OX1 receptor mutants shown in Table 2 were obtained by site‐directed mutagenesis as previously described (Ceraudo et al., 2008). Each mutation was verified by sequencing. The recombinant mutants were stably expressed in CHO‐S cells as described earlier.
Table 2.
Site‐directed mutagenesis of OX1 receptors (OX1R)
| Mutants | Localization | Binding affinity IC50 (nM) | IC50mut/IC50wt | Apoptosis induction EC50 (nM) | EC50mut/EC50wt |
|---|---|---|---|---|---|
| wt | – | 29.0 ± 5.2 | 1.0 | 28.3 ± 4.8 | 1.0 |
| I98A | TM2 | 41.4 ± 2.1* | 1.4 | 11.4 ± 0.7* | 0.4 |
| C99A | TM2 | 58.6 ± 12.0* | 2.0 | 31.2 ± 5.2 | 1.1 |
| P101A | TM2 | 58.1 ± 2.1* | 2.0 | 111.4 ± 10.8* | 3.9 |
| K120A | TM3 | >10 000 | >400 | >10 000 | >400 |
| V121A | TM3 | 52.1 ± 11.6* | 1.8 | 78.1 ± 12.4* | 2.8 |
| P123A | TM3 | >10 000 | >400 | >10 000 | >400 |
| Y124A | TM3 | 408.2 ± 73.5* | 14.0 | 295.0 ± 68.4* | 10.5 |
| Y211A | TM5 | 20.9 ± 0.9 | 2.3 | 35.5 ± 5.7 | 1.3 |
| F220A | TM5 | 21.4 ± 4.2 | 0.7 | 17.5 ± 3.8 | 0.6 |
| N318A | TM6 | 165.6 ± 44.7* | 5.7 | 151.1 ± 22.3* | 5.3 |
| V319A | TM6 | 24.8 ± 3.1 | 0.9 | 33.5 ± 6.2 | 1.2 |
| L320A | TM6 | 51.4 ± 17.8 | 1.8 | 17.5 ± 4.7 | 0.6 |
| K321A | TM6 | 132.5 ± 23.2* | 15.0 | 71.8 ± 16.2* | 2.5 |
| F340A | TM7 | 700 ± 74.9* | 24.0 | 517.5 ± 42.6* | 18.0 |
| T341A | TM7 | 358.1 ± 120.2* | 12.0 | 261.5 ± 58.2* | 9.0 |
| H344A | TM7 | >10 000 | >400 | >10 000 | >400 |
| W345A | TM7 | 2094.8 ± 88.5* | 72.0 | 1792.6 ± 74.5* | 64.0 |
| Y348A | TM7 | 37.2 ± 5.8 | 1.2 | 151.5 ± 28.9* | 5.4 |
IC50 for inhibition of [125I]‐orexin‐A binding and EC50 for apoptosis induction. All parameters were determined in CHO‐S cells stably expressing human recombinant OX1R. Data are mean ± SEM of at least three experiments performed in triplicate. wt, wild type. * P < 0.05, significantly different from wt.
Cell culture and radioreceptor assays
CHO‐S cells expressing recombinant native or mutated OX1 receptors were grown as previously described (El Firar et al., 2009) and maintained at 37 °C in a humidified 5% CO2/air incubator. [125I]‐orexin‐A (74 TBq·mmol−1) was prepared as previously described (El Firar et al., 2009). Binding of [125I]‐orexin‐A to subconfluent cells was conducted as described previously (El Firar et al., 2009). It should be noted that orexin‐A is used for this assay, as it contains a tyrosine residue that can be iodinated, which is not the case for orexin‐B. Briefly, cells were incubated for 30 min at room temperature in 300 μL of 20 mM Tris/EDTA/saline binding buffer (pH 7.4) containing 0.5% (w v‐1) BSA, 5 mM KCL, 1 mM CaCl2, 1.2 mM MgCl2, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 10 mM glucose, 1 mM probenecid and 0.001% (v v‐1) Tween 20, in the presence of 0.25 nM [125I]‐orexin‐A with or without peptide analogues to be tested. Free peptides were removed by two cell washing cycles with PBS. The non‐specific binding was determined in the presence of 1 or 10 μM unlabelled orexin‐B and represented <5% of the total binding. All of the experiments were performed in triplicate and repeated at least three times.
Apoptosis and inositol phosphate assays
CHO‐S cells expressing recombinant native or mutated OX1 receptors were seeded and grown as described earlier. After 24 h of culture, cells were treated with or without peptide analogues to be tested at the concentration indicated in the figure legends. After 48 h of treatment, adherent cells were harvested by TryplE (Life Technologies). Apoptosis was determined using the Guava personal cell analysis system and the Guava nexin kit as previously described (Voisin et al., 2008). Results are expressed as % of apoptotic annexin V‐phycoerythrin‐positive cells.
Subconfluent cells were labelled with [myo‐3H]‐inositol (3.15 TBq·mmol−1) (TRK883; GE Heathcare, Les Ullis, France) for 24 h in standard culture medium. Labelled cells were then incubated for 30 min at 37 °C in Tris/EDTA/saline binding buffer (pH 7.4) containing 20 mM lithium chloride with or without increasing concentrations of the peptide analogues to be tested. Cells were then treated with ice‐cold formic acid, and total IP3 was separated from free [myo‐3H]‐inositol using column chromatography as previously reported (Rouet‐Benzineb et al., 2004).
Statistical analysis
Results are expressed as mean ± SEM. All statistical analyses were performed with GraphPad 4 (http://www.graphpad.com) The Mann–Whitney U test was used for comparison of mean values between two groups. Differences were considered statistically significant at P < 0.05.
Molecular modelling and molecular dynamics simulations
The model of the human OX1 receptor (42–375) was built based on the 3D crystal structure of the human OX2 receptor fusion protein to Pyrococcus abysii glycogen synthase bound to the insomnia drug suvorexant (Protein Data Bank ID code 4S0V) as template structure due to high sequence identity (64%) between the two types of receptor and using SWISS‐MODEL (Arnold et al., 2006; Benkert et al., 2011; Biasini et al., 2014). Protonation states of the titrable residues were assigned based on the pKa values calculated with the Propka server (www.poissonboltzmann.org). This model was embedded in a pre‐equilibrated 1‐palmitoyl‐2‐oleoyl‐phosphatidylcholine (POPC) membrane model using CHARMM‐GUI (Jo et al., 2008; Wu et al., 2014). Potassium and chloride ions were added to the system to a concentration of 150 mM. The final system contained 15 212 water molecules, 96 POPC lipid molecules, the OX1 receptor model and ions with a simulation cell size of 66 × 66 × 152 Å. The system was first relaxed through energy minimization using the steepest descents algorithm. Then, an equilibration and production protocol included the following stages: (i) harmonic positional restrictions on all the heavy atoms of the receptor (five steps of 1 ns each, decreasing gradually the force constant from 1000 to 200 kJ·mol−1·Å−2 in each step); (ii) harmonic positional restrictions on all backbone atoms of the receptor (4 ns); (iii) harmonic positional restrictions on all Cα atoms of the receptor (5 ns); and (iv) fully unrestrained simulations (100 ns). Docking simulations were performed using the NMR structure of the orexin‐B C‐terminus (pdb: 1CQ0). Briefly, the binding pocket of the structure of the OX1 receptor at the end of the molecular dynamic (MD) simulation was determined using Fpocket webserver (Le Guilloux et al., 2009). The largest pocket, localized at the top of the TM domains of the OX1 receptor, was selected. The docking of the orexin‐B C‐terminus (sequence 20–28) into the OX1 receptor was performed with Haddock webserver (http://haddocking.org). This model of the complex OX1 receptor–orexin‐B20–28 was then embedded in a pre‐equilibrated POPC membrane model using CHARMM‐GUI (Jo et al., 2008; Wu et al., 2014). Potassium and chloride ions were added to the system to a concentration of 150 mM. The final system contained 15 018 water molecules, 128 POPC lipid molecules, the model of the complex and ions with a simulation cell size of 75 × 75 × 130 Å. The system was first relaxed through energy minimization using the steepest descents algorithm. Then, an equilibration and production protocol included the following stages: (i) harmonic positional restrictions on all the backbone and side chains heavy atoms of the receptor and peptide (six steps, decreasing gradually the force constant from 4000 to 50 kJ·mol−1·Å−2 for the backbone heavy atoms and from 2000 to 0 kJ·mol−1·Å−2 for the side chain heavy atoms in each step for a total of 15 ns simulation time); and (ii) fully unrestrained simulations (100 ns).
Molecular dynamic simulations were performed with GROMACS 5.0 using the CHARMM22 protein force field with CMAP corrections (MacKerell et al., 1998, 2004), the CHARMM36 lipid force field (Klauda et al., 2010) and TIP3P water molecules. A semi‐isotropic Parrinello–Rahman barostat was employed to maintain the pressure independently in the x‐y plane and in the z direction at 100 kPa with a coupling constant of 5 ps. The isothermal compressibility constant was 4.5 × 10−5·Pa−1. The temperature was kept constant at 36.9 °C with the Nose–Hoover method and by coupling independently the lipid molecules, the protein and the water‐ion groups with a common period of 1 ps. A cutoff of 1.2 nm was employed for the Lennard–Jones potential. The long‐range interactions were calculated using the particle mesh Ewald method with a 1.2 nm real space cutoff, a 0.12 nm space grid and a fourth‐order B‐spline interpolation scheme to optimize the computational performance. The equations of motion were integrated using the leapfrog method with a 2 fs time step. The LINCS algorithm was used to constrain bond lengths in the protein and lipid molecules.
Molecular dynamic trajectories analyses were conducted using GROMACS utilities. All the calculations were performed with the supercomputing resources available at the Centre de Calcul Recherche et Technologie (CEA, France).Three‐dimensional representations of the interaction between the C‐terminal region of orexin‐B and the 3D model of the OX1 receptor were drawn using Chimera software (http://www.cgl.ucsf.edu/chimera/).
Materials
Enzymes, vectors and culture medium were obtained from Life Technologies (Saint Aubin, France). All orexin‐B analogues were obtained by custom solid‐phase synthesis from GL Biochem (Shanghai, China), and all were at least 95% pure. [125I]‐orexin‐A was prepared and purified as previously described (Voisin et al., 2008). All other highly purified chemicals were from Sigma‐Aldrich (Saint Quentin‐Fallavier, France).
Results
Structure–function relationship of orexin‐B
Previous reports have clearly demonstrated that orexin‐A and orexin‐B induce mitochondrial apoptosis in colonic cancer cell lines as HT‐29, LoVo and other cell lines, which express OX1 but not OX receptors (Voisin et al., 2011). In order to identify the pharmacophore of orexin‐B responsible for this effect, we used the alanine scanning technique (Nicole et al., 2000). Twenty‐five mutants of orexin‐B were synthesized in which the 25 residue side chains were individually replaced with alanine. As the residues in position 17, 22 or 23 of the native peptide are alanine, three additional mutants were synthesized, substituting the alanine for a leucine residue. All mutants were tested for their interaction with human recombinant OX1 receptor, stably expressed in CHO‐S cells, by competitive inhibition of [125I]‐orexin‐A binding. The slopes of the mutant dose–response curves for inhibiting [125I]‐orexin‐A binding were identical for all mutants (not shown). All competitor curves analysed with the GraphPad software fitted to a monophasic binding pattern, indicating the presence of only one binding site (Supporting Information Figures S1B and S2B). When the IC50 of all mutants was analysed, the largest decrease in affinity for the OX1 receptor occurred when A22, G24, I25, L26 and M28 were substituted for alanine or leucine, resulting in a 40‐fold to 200‐fold decrease (Table 1, Figure 1 and Supporting Information Figure S2B). Alanine substitution of L11 and L15 resulted in significant decrease in IC50 but by only 10‐fold (Table 1, Figure 1 and Supporting Information Fig. S2A). It should be noted that alanine substitutions at positions 20 and 27 induced a significant weak decrease in affinity, that was, lesser than fivefold. Substitution at other positions, including S18, N20 and T27, did not change the affinity or resulted only in decreases of less than one log (Table 1 and Figure 1).
Table 1.
Biological activity of orexin‐B mutants in CHO‐S cells stably expressing human recombinant OX1 receptors (OX1R)
| Orexin‐B residues | Substitution | Binding affinity IC50 (nM) | IP3 production EC50 (nM) | Apoptosis induction EC50 (nM) |
|---|---|---|---|---|
| wt | – | 31.2 ± 1.4 | 28.0 ± 5.8 | 19.6 ± 2.1 |
| R1 | Ala | 34.4 ± 2.7 | 51.0 ± 6.7* | 10.9 ± 0.9* |
| S2 | Ala | 51.0 ± 6.7* | 63.0 ± 49.6 | 30.3 ± 10.2 |
| G3 | Ala | 27.3 ± 2.2 | 14.0 ± 4.9 | 24.7 ± 4.1 |
| P4 | Ala | 34.3 ± 2.7 | 8.8 ± 5.6 | 21.1 ± 2.8 |
| P5 | Ala | 5.5 ± 0.8* | 10.8 ± 3.5 | 29.1 ± 7.0 |
| G6 | Ala | 46.7 ± 3.4* | 96.3 ± 18.2* | 33.9 ± 5.9 |
| L7 | Ala | 33.4 ± 8.3 | 32.2 ± 4.3 | 24.2 ± 4.6 |
| Q8 | Ala | 9.3 ± 0.7* | 8.1 ± 1.1* | 24.7 ±4.1 |
| G9 | Ala | 46.1 ± 11.2 | 38.7 ± 11.4 | 50.2 ± 10.8 |
| R10 | Ala | 34.3 ± 2.7 | 14.2 ± 3.0 | 27.3 ± 2.2 |
| L11 | Ala | 300.1 ± 48.9* | 360.6 ± 136.0* | 1109.5 ± 475.5* |
| Q12 | Ala | 23.4 ± 1.7* | 6.7 ± 3.0* | 26.0 ± 7.1 |
| R13 | Ala | 32.2 ± 4.3 | 13.2 ± 2.6 | 20.3 ± 2.7 |
| L14 | Ala | 21.4 ± 5.5 | 14.0 ± 4.1 | 21.5 ± 2.4 |
| L15 | Ala | 375.1 ± 63.1* | 105.8 ± 26.4* | 3223.5 ± 1763.9* |
| Q16 | Ala | 29.4 ± 2.2 | 30.0 ± 4.9 | 368.6 ± 138.9* |
| A17 | Leu | 34.3 ± 2.7 | 31.6 ± 2.3 | 491.6 ± 258.3* |
| S18 | Ala | 20.7 ± 0.8* | 43.9 ± 6.2 | 5987.3 ± 2006.3* |
| G19 | Ala | 29.4 ± 2.2 | 12.5 ±4.3 | 26.9 ± 7.0 |
| N20 | Ala | 157 ± 39* | 238.4 ±79.9* | >10 000 |
| H21 | Ala | 30.5 ± 1.1 | 23.9 ± 3.9 | 106.0 ± 4.9* |
| A22 | Leu | 3491 ± 490* | >10 000 | 7504 ± 111.4* |
| A23 | Leu | 10.9 ± 0.9* | 18.4 ± 0.9 | 64.8 ± 12.0* |
| G24 | Ala | 3982.3 ± 820.3* | >10 000* | 7733.3 ± 2266.7* |
| I25 | Ala | 6674 ± 1663* | >10 000* | 3799.3 ± 1133.5* |
| L26 | Ala | 3563.7 ± 236.6* | >10 000* | >10 000* |
| T27 | Ala | 215.6 ± 70.4* | 142.4 ± 58.6* | >10 000* |
| M28 | Ala | 1263 ± 370.8* | 317.3 ± 93.2* | 7333.3 ± 2666.7* |
| OxB6–28 | – | 47.4 ± 8.9 | 81.7 ± 24.9* | 75.1 ± 20.4* |
| OxB7–28 | – | 388.0 ± 112.5* | 1513.7 ± 255.4* | 2183.0 ± 1133.6* |
| S18, N20 | Ala, Ala | 200.2 ± 58.8* | >10 000* | >10 000* |
| S18, T27 | Ala, Ala | 175.0 ± 24.5* | >10 000* | >10 000* |
IC50 for inhibition of [125I]‐orexin‐A binding and EC50 for stimulation of IP3 production or apoptosis induction. All parameters were determined in CHO‐S–OX1R cells. Data are mean ± SEM of at least three experiments performed in triplicate. wt, wild type. * P < 0.05, significantly different from wt.
Figure 1.
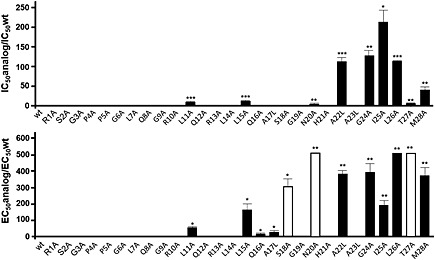
Binding characteristics (top panel) and pro‐apoptotic properties (bottom panel) of wt human orexin‐B and peptide analogues. Single amino acids in the wt orexin‐B sequence were replaced by l‐alanine or l‐leucine as shown ont the x‐axis. Top panel: wt OX1 receptors were stably expressed in CHO‐S cells, and competitive inhibition of [125I]‐orexin‐A binding by unlabelled mutants was analysed. The concentration of each mutant that half maximally inhibited specific [125I]‐orexin‐A binding (IC50) was determined and the ratio IC50mutant/IC50wt was calculated; Bottom panel: pro‐apoptotic activity of wt orexin‐B and peptide analogues determined in CHO‐S–OX1 receptor cells. The concentration of each mutant that half maximally induced cellular apoptosis (EC50) was determined and the ratio EC50mutant/EC50wt was calculated. * P < 0.05, ** P < 0.001, *** P < 0.0001.
All orexin‐B mutants were tested for their ability to stimulate IP3 production in CHO‐S–OX1 receptor cells (Table 1). The dose–response curves of IP3 production for all mutants were identical to those of native orexin‐B (Supporting Information Fig. S1A and B). There was a good correlation between the EC50 for intracellular IP3 production and the IC50 for the binding inhibition of [125I]‐orexin‐A to OX1 receptors (Table 1). A straight line (r = 0.92) was obtained when plotting logEC50 versus logIC50, indicating that all mutants behaved as OX1 receptor agonists, with identical or lower potencies than native orexin‐B (Figure 2). Taken together, these data demonstrate the importance of five crucial residues in the C‐terminal sequence of orexin‐B, that is, A22, G24, I25, L26 and M28 (Figure 1, Supporting Information Fig. S1A and B). Nevertheless, a minor contribution of the central part of the peptide is also to be noted, specifically the L11 and L15 residues (Table 1, Figure 1 and Supporting Information Fig. S1A). All mutants were then tested for their ability to trigger apoptosis in CHO‐S–OX1 receptor cells. As expected, alanine substitution of L11, L15, A22, G24, I25, L26 and M28, which all played a role in orexin‐B affinity and cellular IP3 production (see preceding text), decreased peptide‐induced apoptosis by 50‐fold to 500‐fold (Table 1, Figure 1, Supporting Information Figs S1A and B).
Figure 2.
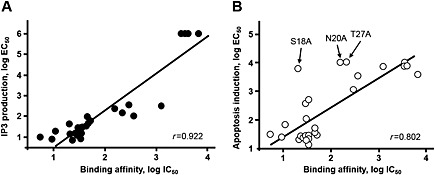
Correlation between the EC50 values and IC50 values determined for native orexin‐B and all singly substituted mutants of orexin‐B. (A) Correlation between the EC50 values and K i values determined for the IP3 and binding assays in CHO‐S cells stably expressing recombinant OX1 receptors (CHO‐S–OX1R). (B) Correlation between the EC50 values and IC50 values determined for the apoptosis and binding assays in CHO‐S–OX1R. Note the S18A, N20A and T27A mutants for which the EC50 for inducing cellular apoptosis was much higher than their IC50 for binding. See Table 1 for details.
Interestingly, a strong decrease in the ability to induce apoptosis occurred when S18, N20 and T27 were substituted for alanine (Table 1 and Figure 1). Indeed, these substitutions produced at least a 100‐fold drop in orexin‐B‐induced apoptosis (Table 1 and Figure 1), even though they were able to bind to OX1 receptors with relatively good affinity (Table 1 and Figure 1). Similarly, although to a lesser extent, mutants Q16A and A17L, which bind to the OX1 receptor with a similar affinity as wild‐type (wt) orexin‐B, exhibited a decrease in pro‐apoptotic activity by about 20‐fold (Table 1). Globally, a good correlation (r = 0.80) between the EC50 for induction of apoptosis and IC50 for inhibiting [125I]‐orexin‐A binding was observed for all mutants, except for the S18A, N20A and T27A mutants (Figure 2), suggesting that these peptides behaved as partial OX1 receptor agonists. When they were omitted from the regression curve, the correlation coefficient improved (r = 0.90), indicating a lack of correlation between OX1 receptor affinity and apoptosis induction for the S18A, N20A and T27A mutants.
To confirm these observations, two new orexin‐B mutants were synthesized with double substitution by alanine of the S18 and N20 or S18 and T27 residues. As shown in Table 1 and Figure 3A, both double mutants slightly altered the ability of the peptide to interact with the OX1 receptor (IC50 = 31.2 nM for wt versus 200.2 nM for S18A/N20A and 175 nM for S18A/T27A). In contrast, both strongly abolished the production of IP3 and the induction of apoptosis (Table 1). Thus, these data suggest that the S18A/N20A and S18A/T27A mutants might be partial agonists. To confirm this, the ability of orexin‐B to inhibit cell growth was measured in the presence or absence of either the S18A/N20A or S18A/T27A peptides. As shown in Figure 3, orexin‐B induced a strong inhibition of cell growth in a dose‐dependent manner, with an EC50 of about 25 nM. Addition of various concentrations (0.1 to 10 μM) of mutants partially inhibited the orexin‐B‐induced inhibition of cell growth. Indeed, addition of 10 μM of S18A/N20A or S18A/T27A peptides totally abolished the response induced by 0.1 nM orexin‐B (Figure 3C and D), whereas both mutants partially antagonized the effect of 1 to 10 μM orexin‐B. Schild plots (Arunlakshana and Schild, 1959) derived from these experiments indicated that the regression line slope was about 0.85 for both analogues (Supporting Information Fig. S2), which is not significantly different from 1, suggesting that the mutants are competitive antagonists. These results indicate that the double mutant S18A/N20A and S18A/T27A peptides are partial agonists for apoptosis induction.
Figure 3.
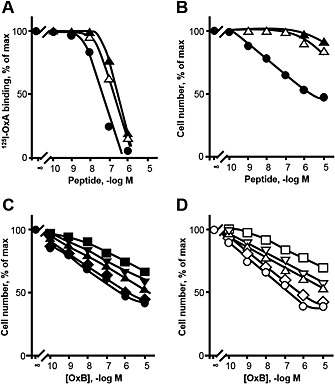
Specific [125I]‐orexin‐A (OxA) binding to CHO‐S–OX1 receptor (OX1R) cells in the presence of increasing concentrations of unlabelled orexin‐B, S18A/N20A and S18A/T27A peptides (A) and determination of the inhibition of cellular growth induced by increasing concentrations of orexin‐B, S18A/N20A (B, C) and S18A/T27A (B, D) peptides in CHO‐S–OX1R cells. (A) CHO‐S–OX1R cells were incubated with the indicated concentrations of orexin‐B, S18A/N20A and S18A/T27A. Results are expressed as % of radioactivity specifically bound in the absence of added unlabelled peptide. (B) CHO‐S–OX1R cells were incubated with the indicated concentrations of orexin‐B, S18A/N20A and S18A/T27A, and cells were counted after 48 h incubation. Results are expressed as % of total viable cells. (C, D) Increasing concentrations of orexin‐B were incubated together with the following concentrations of S18A/N20A mutant (C): 0, 10 nM, 100 nM, 1 μM, 10 μM, or S18A/T27A mutant (D). Each point is the mean of three separate experiments. For clarity, SEM are not indicated; they were always below 15% of mean values.
In order to determine the minimal sequence of orexin‐B having a full biological activity, two deletion mutants were also synthesized (Table 1). Deletion of the first five residues of orexin‐B (orexin‐B6–28) did not alter the peptide's ability to inhibit the binding of [125I]‐orexin‐A to the OX1 receptor as compared with native orexin‐B (Table 1). In contrast, deletion of the first six residues (orexin‐B7–28) induced a decrease in affinity for OX1 receptors (Table 1) by more than one log. Orexin‐B6–28 induced apoptosis and IP3 production similarly to wt orexin‐B (Table 1), while orexin‐B7–28 strongly altered peptide‐induced apoptosis and IP3 production (Table 1). Taken together, these data suggest that the peptide must be at least 22‐aminoacids long to induce these responses.
A 3D model of the OX1 receptor and docking of the orexin‐B C‐terminus
Based on the high degree of sequence identity between human OX1 and OX2 receptor (64%), the crystallographic structure of the OX2 receptor was employed as a template to generate a homology model of the OX1 receptor. This model was employed as a starting conformation for an all‐atom MD simulation in a lipid bilayer. After 100 ns MD production, the model was comparable with OX2 receptor structure with the backbone root mean square deviation (RMSD) value of 1.02 Å between the OX1 receptor model and OX2 receptor structure. The OX1 receptor 3D model exhibited one short helix in the intracellular C‐terminal tail (G363 to S374, Figure 4), as observed in the structure of the OX2 receptor (Yin et al., 2015). The orexin‐B C‐terminus (sequence 20–28) was then docked into the structure of OX1 recepto obtained at the end of the 100 ns MD production. Among the various docking poses obtained, one 3D model was selected on the basis of best score (−112.7 ± 6.8), maximal buried surface area (1492 Å ± 58.8) and spatial orientation of the orexin‐B20–28 C‐terminus (Figure 4). Indeed, the model shows that the C‐terminal M28 residue of the orexin‐B20–28 peptide is located well inside the binding pocket, whereas the N‐terminal N20 residue is oriented outside of the pocket. The complex OX1 receptor/orexin‐B20–28 was embedded into a membrane, and the resulting system was submitted to MD simulation (see Supporting Information Fig. S3, which depicts a side view of the simulated system). The global fold of the complex is stable during the production as shown by the values of the RMSDs (Supporting Information Fig. S4). Indeed, the backbone RMSD of the TM domains (Supporting Information Fig. S4), the interface helix (amino acids 363–372) (Supporting Information Fig. S5) and the ß‐sheet (amino acids 183–204) (Supporting Information Fig. S5) of the OX1 receptor fluctuate, as a function of time, between 0.5 and 2 Å, except for TM5 in the complex. In fact, the C‐terminus of TM5 slightly unwound during the last 50 ns of the simulation. Figure 4 depicts the C‐terminus of orexin‐B inserted into the binding pocket delineated by all the TM domains of the OX1 receptor except TM1. Most of orexin‐B contacts involve van der Waals interactions and some hydrogen bonds (from Y224 OH group to backbone carbonyl of T27 and from N318 NH2 group to backbone carbonyl of G24) (Figure 5). In contrast, one ionic bond was identified between K120 and E184, one hydrogen bond between W145 and the backbone carbonyl of A52 and one hydrogen bond between T341 and the backbone carbonyl of A338 (Figure 5). It should be noted that the orexin‐B20–28 peptide inserted into the OX1 receptor adopts a helical conformation for residues H21 to T27 (Supporting Information Fig. S6).
Figure 4.
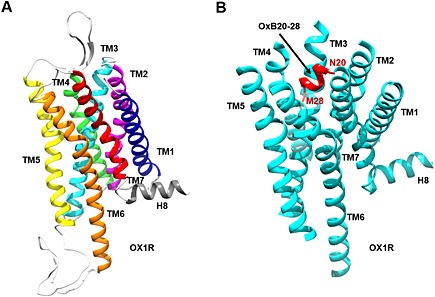
Representation of the 3D model of the OX1 receptor (OX1R) receptor (A) docked to the C‐terminus of orexin‐B (OxB; B). (A) 3D model of human OX1R based on X‐ray structure of OX2R. TM domains 1 to 7 are coloured dark blue, purple, turquoise, light green, yellow, orange and red respectively. (B) 3D model of human OX1R (turquoise) docked to sequence 20–28 of orexin‐B (red). Intracellular loops and ECLs are not represented.
Figure 5.
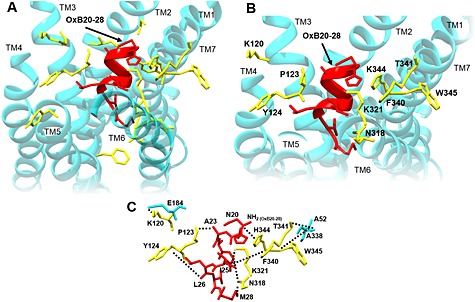
Upside‐down view of the 3D model of human OX1 receptor (OX1R) docked to the C‐terminus of orexin‐B (OxB). (A) Side chains of residues (yellow sticks) of the receptor 3D model (turquoise) in the surroundings of sequence 20–28 of orexin‐B (red). All the represented residues (yellow sticks) were substituted to alanine. (B) 3D model of human OX1R (turquoise) docked to the C‐terminus of orexin‐B (red). OX1R residues for which the alanine substitution resulted in an inhibition of the ability of orexin‐B to bind to the receptor and to induce cellular apoptosis are indicated (yellow). (C) Putative interactions (ionic and van der Walls) between residues of OX1R (yellow), which were mutated and residues of orexin‐B (red). Residues in turquoise represent native OX1R residues.
Site‐directed mutagenesis analysis of the receptor
Based on the distance measured between the side‐chain residues of the OX1 receptor TM domains and those of the orexin‐B20–28 peptide, we selected residues within the OX1 receptor‐TMs and orexin‐B20–28. All residues of the OX1 receptor fulfilling this condition were mutated to alanine by site‐directed mutagenesis (Table 2 and Figure 5). All mutants were functionally expressed in CHO‐S cells (not shown) and tested for their abilities to bind [125I]‐orexin‐A and to induce apoptosis response, as mentioned above (Table 2). Mutants I98A, C99A, P101A, V121A, Y211A, F220A, V319A, L320A, K321A and Y348A were able to bind [125I]‐orexin‐A and to induce apoptosis with an IC50 in the nm range, which is similar to the IC50 determined for the wt OX1 receptor, for example, IC50mut/IC50wt <5 and EC50mut/EC50wt < 5 (Table 2 and Figure 5). Of note, the substitution of Y348 by an alanine residue induced a slight inhibition of the OX1 receptor interaction with the peptide ligand and of apoptosis (Table 2). Inversely, the K321A mutant has altered binding parameters, whereas its ability to induce apoptosis was similar to the wt OX1 receptor (Table 2). The Y124A, N318A, F340A and T341A mutants displayed much lower affinity for [125I]‐orexin‐A than the wt OX1 receptor with a ratio IC50mut/IC50wt < 25 (Table 2). As shown in Table 2, these mutants inhibited apoptosis, with a ratio EC50mut/EC50wt < 25. It should be noted that the IC50 for [125I]‐orexin‐A binding for these mutants was very similar to the EC50 determined for the apoptosis response (Table 2). However, substitution of K120, P123, H344 and W345 residues by an alanine residue had a strong impact on receptor affinity and the apoptotic response (Table 2 and Figure 5), indicating that these residues play a critical role in the interaction of OX1 receptors with orexins and/or the structure of the OX1 receptor. Indeed, the Y124A, F340A and T341A mutants displayed a ratio IC50mut/IC50wt > 10 and EC50mut/EC50wt > 10 (Table 2), the W345A mutant displayed a ratio IC50mut/IC50wt > 60 and EC50mut/EC50wt > 60 (Table 2), whereas the K120A, P123A and H344A mutants displayed a ratio IC50mut/IC50wt > 400 and EC50mut/EC50wt > 400.
Discussion and conclusion
Several studies in recent years have shown that GPCRs represent new promising targets for the therapeutic treatment of various cancers (Lappano and Maggiolini, 2011). Among the large GPCR family, the OX receptors, a class A GPCR expressed in the hypothalamus, also display pro‐apoptotic properties in cancer cell lines (Laburthe and Voisin, 2012). Our group demonstrated in the last few years that the OX1 receptor is not expressed in normal colon tissue but is ectopically expressed in colon cancers where orexins bound to OX1 receptors induce: (i) robust mitochondrial apoptosis (El Firar et al., 2009); and (ii) significant inhibition of tumour growth in nude mice xenografted with cancer cell lines (Voisin et al., 2011). These effects are mediated via a novel mechanism involving (i) the presence of two ITIM sequences in the OX1 receptor that are tyrosine phosphorylated upon receptor binding of orexins (Voisin et al., 2008); and (ii) the recruitment to tyrosine phosphorylated sites and activation of the tyrosine phosphatase, SHP‐2, which is responsible for mitochondrial apoptosis involving cytochrome c release from mitochondria to cytosol and caspase‐3 and caspase‐7 activation (El Firar et al., 2009). In this context, the determination of the pharmacophore involved in the pro‐apoptotic properties of the orexin peptide represents a key step for the design of new molecules with therapeutic interest. Although some studies regarding the pharmacophore determination of orexins to mobilize intracellular Ca2+ have been reported (Darker et al., 2001; Lang et al., 2004; German et al., 2013; Heifetz et al., 2013), no systematic evaluation of the pro‐apoptotic function of every residue of orexin‐B has been performed. In the present study, we have synthesized a total of 28 single alanine or leucine mutants of orexin‐B and have analysed their biological properties by binding assay, ability to stimulate intracellular IP3, and thereby Ca2+ release, and cellular apoptosis measurements in CHO‐S cell clones expressing recombinant human OX1 receptors. Our data provide critical new information on the key orexin‐B amino acid residues that play a role in the pro‐apoptotic function of the peptide.
Analysis of the 28 single alanine or leucine substitutions indicated that 12 residues in native orexin‐B could not be changed without a significant decrease either in (i) the binding affinity for the OX1 receptor; (ii) the ability to stimulate IP3 production; or (iii) the ability to induce a pro‐apoptotic response in transfected cells. These important residues were distributed mainly at the C‐terminal end of the peptide chain, except for L11 and L15, which are located in the peptide middle region. These results are in good agreement with previous observations indicating that the C‐terminal end of orexin‐B is crucial for both the binding to the OX1 receptor and the mobilization of intracellular Ca2+ (Lang et al., 2004). Moreover, the deletion of the first five residues of orexin‐B had no impact on the ability of the truncated peptide to bind to OX1 receptors, induce IP3 production and trigger cellular apoptosis. In contrast, the deletion of one more residue (orexin‐B7–28) strongly reduced the peptide activity, suggesting that the sequence 6–28 is essential for peptide function. Thus, we identified seven residues, that is, L11, L15, A22, G24, I25, L26 and M28, as critical for binding to human OX1 receptors and subsequent induction of cell apoptosis. It is quite interesting to note that all these residues in orexin‐B are conserved in orexin‐A, except for M28, which is a homolog to L33 in orexin‐A. Moreover, the analysis of the side chain of these residues indicated mainly the presence of hydrophobic residues (L11, L15, A22, G24, and I25 and L26) and also the presence of one non‐polar residue (M28). Inversely, the substitution to alanine or leucine of the Q16, A17, S18, N20 and T27 residues had only a weak impact on binding affinity and Ca2+ mobilization, although it affected the induction of apoptosis. The structure determination of orexin‐B by NMR had previously revealed the presence of two α‐helices at position L7–G19 (Helix I) and A23–M28 (Helix II), connected by a flexible loop (Lee et al., 1999). Residues S18 and N20 are located in the flexible loop, while T27 is found in Helix II. Alanine substitution of S18, N20 and T27 strongly abolished peptide‐induced apoptosis in CHO‐S–OX1 receptors, but the ability of these mutants to bind OX1 receptors was not affected by alanine substitution of S18 and only slightly affected by alanine substitution of the N20 and T27 residues. These observations suggest that these three residues might play a key role in the induction of apoptosis mediated by orexins binding to their cognate receptors. The double substituted S18A/N20A and S18A/T27A peptides exhibited a slightly altered ability to interact with OX1 receptors, indicating that substituting two of the three critical residues do not substantially alter affinity compared with single substitution of N20 and T27 residues. Although the possibility that the double substitutions have a structural impact on orexin‐B molecules cannot be excluded. Moreover, these double substitutions totally abolished the ability of the two peptides to induce cellular apoptosis, suggesting that these peptides could be partial agonists/antagonists. Indeed, these two peptides partially antagonized the inhibition of cellular growth by orexin‐B through a competitive mechanism. Thus, both the flexible loop and the C‐terminal Helix II play a crucial role in the peptide main activity.
Sequence analysis of class A GPCRs revealed that there is a large diversity in the length and residue composition of the extracellular loops (ECLs). ECLs, and more particularly ECL2, which links TM4 and TM5, displayed different structures, including helices (e.g. in some aminergic or adenosine receptors) or β‐sheets (e.g. peptide‐binding receptor) (Venkatakrishnan et al., 2013). A unique feature of the extracellular region of class A GPCRs is the presence of a disulfide bridge between a cysteine residue located in TM3 and a cysteine residue located in ECL2 (Fanelli and De Benedetti, 2011). The disulfide bridge has been shown to be involved in GPCR stability and activity (De Graaf et al., 2008). Indeed, the TM3‐ECL2 disulfide bridge stabilized the extracellular side of TM3 close to the binding pocket and limited the conformational change of this region during receptor activation (Preininger et al., 2013). The recent determination of the structure of the OX2 receptor (Yin et al., 2015), which shares 64% sequence identity with the OX1 receptor, allowed us to develop a homology 3D model of the OX1 receptor that was subsequently used for ligand docking studies. The use of GPCR models in combination with site‐directed mutagenesis represents an effective tool to study both ligand binding and functional properties. The OX1 receptor model exhibited an eighth helix (H8) at the top of the C‐terminal sequence between residues G363 and S374. The existence of H8 is frequently observed in class A GPCRs, except for the CXCR4, NTS1 receptor and PAR1 in which this region is unstructured (Venkatakrishnan et al., 2013). The structural and/or functional role of H8 in GPCRs remains conjectural, although some reports indicated a role in coupling and activation of G proteins (Rasmussen et al., 2011a).
As mentioned earlier, the C‐terminal end of orexin‐B had a crucial role not only in the binding activity of the peptide but also in its ability to induce a pro‐apoptotic response. To understand how the C‐terminal helix of orexin‐B interacts with OX1 receptor, we docked the C‐terminal 20–28 fragment of orexin‐B into the homology model of the OX1 receptor. We substituted independently to alanine 18 residues of the OX1 receptor (Table 2), and showed that the substitution of nine of these residues (K120, P123, Y124, N318, K321, F340, T341, H344 and W345), located in the TM2, TM3, TM6 and TM7 of the OX1 receptor, reduces the binding affinity of orexin‐B to the OX1 receptor and inhibits the ability of orexin‐B to induce apoptosis. OX1 receptor/orexin‐B20–28 complex model analysis has shown that most of these residues were located close to orexin‐B20–28. Indeed, P123 was close to A22, Y124 close to L26, N318 close to G24, K321 close to I25, F340 close to I25 and H344 close to N20 (Figure 5) suggesting that these residues could be involved in the interaction between the OX1 receptor and its ligand. In contrast, K120, T341 and W345 were clearly far from orexin‐B20–28 residues suggesting that these residues were involved in the structural maintaining of the receptor (Figure 5). Alanine substitution of P123 and H344 resulted in a strong reduction of the ability of the receptor to bind orexin‐B and to trigger an apoptotic response. These residues are located in TM3 and TM7 of OX1R and delineated the binding pocket. Based on homology modelling of the OX1 receptor using the human dopamine D3 receptor as a template, Heifetz et al. (2013) have demonstrated the important role of OX1 receptor residues in the ability of orexin‐B to mobilize intracellular calcium including D203, W206, Y215, F219, Y224, Y311 and H344. As shown in this study, the prediction of 2D interaction map suggested that H344 was in interaction of L11 and L15 located in Helix I of orexin‐B (Heifetz et al., 2013). This prediction totally contrasted with the present data indicating that H344 interacted with N20 of orexin‐B. These observed differences could be caused by the differences between the OX1 receptor 3D models used. The determination of X‐ray structure of the OX2 receptor in the presence of the dual orexin receptor antagonist, suvorexant, revealed that it lodged deep in the orthosteric site and contacted all TM domains except TM1 (Yin et al., 2015). Analysis of the OX2 receptor binding pocket reveals that N324 and H350 were in direct contact with suvorexant (Yin et al., 2015). These data suggested that the corresponding residues in the OX1 receptor, that is, N318 and H344 could play a direct role in the interaction of orexin‐B with the OX1 receptor. Assuming that the alanine substitution of H344 strongly abolished the ability of the OX1 receptor to recognize orexin‐B and to induce the pro‐apoptotic response, this residue was one of the key residues involved in the activity of the OX1 receptor. A previous report had revealed that substitution of residue Y348 did not modify the ability of the OX1 receptor to bind orexin‐A and to mobilize intracellular calcium (Heifetz et al., 2013). In this study, the substitution of Y348 induced only a weak alteration of OX1 receptor affinity and its ability to induce cellular apoptosis. However, the 2D interaction map of contact between the antagonist suvorexant and the OX2 receptor revealed that Y354 (corresponding to Y348 in OX1 receptor) is in contact with the ligand (Yin et al., 2015). The same observation was obtained for the OX2 receptor P131 residue (corresponding to P123 in OX1 receptor), indicating that OX1 receptor residues P123 and to a lesser extent Y348 could directly interact with orexin‐B. Finally, a hypothetic 3D model of the complex OX1 receptor/orexin‐B has been constructed by structural alignment of the docked orexin‐B20–28 with full sequence of orexin‐B (Supporting Information Fig. S7). The resulting model showed that the Helix I of orexin‐B was mainly located along the partially β‐sheet structured ECL2, which could act as an anchorage site for L11 and L15 located in the middle part of orexin‐B.
As mentioned earlier, the orexins/OX1 receptor complexes have pro‐apoptotic properties mediated by SHP‐2 recruitment via phosphorylation of tyrosine residues (Y83 and Y358) located in TM2 and TM7 (Laburthe and Voisin, 2012). This mechanism accounted for the dual participation of the Gq β/γ dimer and OX1 receptor ITIM sites (Laburthe and Voisin, 2012). Agonist‐mediated activation of the GPCRs involved inward movements of TM5 and TM6 relative to the rest of TM bundle as observed for the β2 adrenoceptor (Rasmussen et al., 2011b) and M2 (muscarinic) acetylcholine receptor (Kruse et al., 2013). Moreover, it was demonstrated that the binding of agonist peptide to the GPCR promoted the conformational change inducing a destabilization of the inactive state and a stabilization of the active state, leading in the case of the OX1 receptor to the production of IP3 and recruitment of SHP‐2. Recent data indicated that the orexin‐B and also orexin‐A peptides binding altered noncovalent interactions between TM3 and TM6 leading to a decrease in the stability of the inactive state of the receptor (Heifetz et al., 2013). In the present work, orexin‐B and OX1 receptor mutants, which modified their activities in terms of binding, production of IP3 and stimulation of the apoptosis, probably altered the ability to destabilize the inactive state of the receptor. In contrast, orexin‐B mutants S18A, N20A and T27A, which were only altered in their abilities to induce apoptosis, partially promoted the active state but did not interact with the transmission switch residues located in the TM domain and involved in the unmasking of tyrosine residues responsible for SHP‐2 recruitment.
Conclusions
The present ‘Ala‐scan’ study of orexin‐B, associated with the development of a global 3D model of the OX1 receptor and site‐directed mutagenesis, demonstrated that the C‐terminal end of orexin‐B and seven residues located in the TM domains of the OX1 receptor are important for the induction of cellular apoptosis mediated by the orexin‐B/OX1 receptor interaction. As the OX1 receptor is aberrantly expressed in colon cancer and its activation by exogenous orexins results in robust apoptosis and strong inhibition of tumour development in preclinical animal models (Voisin et al., 2011), the design of full agonist peptide or non‐peptide molecules represents a major challenge in new therapeutic approaches for the treatment of digestive cancers. The knowledge of the structure–function relationship of orexin‐B and its receptor towards their pro‐apoptotic responses advanced by this study represents a key step in the development of such molecules.
Author contributions
P. N. carried out the alanine scanning of orexin‐B experiments including binding tests, apoptosis and IP3 measurement and participated to the construction of OX1 receptor mutants and their characterization. P. C. performed OX1 receptor mutants construction and their characterization; T. V. contributed to the study design and co‐analysed results with A. C. N. J. built the OX1 receptor and OX1 receptor–orexin‐B models and performed MD simulations. A. C. supervised all experiments, developed the OX1 receptor 3D model, and wrote the manuscript.
Conflict of interest
The authors disclose no conflict of interest.
Supporting information
Figure S1A and S1B Specific 125I‐OxA binding to CHO‐SOX1R cells in the presence of increasing concentrations of unlabeled OxB and OxB analogs as indicated in figure (left) and determination of the stimulation of IP3 production (middle) or the inhibition of cellular growth induced (right) by increasing concentrations of OxB and OxB analogs in CHO‐S‐OX1R cells. CHO‐S‐OX1R cells were incubated with the indicated concentrations of OxB ( ) and OxB analogs (
) and OxB analogs ( ). Left, results are expressed as the percentage of radioactivity specifically bound in the absence of added unlabeled peptide; Middle, results are expressed as the percentage of maximal IP3 production; Right, results are expressed as the percentage of total viable cells. Each point is the mean of three separate experiments. For the sake of clarity, standard errors are not indicated. They were always below 15% of mean values.
). Left, results are expressed as the percentage of radioactivity specifically bound in the absence of added unlabeled peptide; Middle, results are expressed as the percentage of maximal IP3 production; Right, results are expressed as the percentage of total viable cells. Each point is the mean of three separate experiments. For the sake of clarity, standard errors are not indicated. They were always below 15% of mean values.
Figure S2 Antagonism S18A/N20A and S18A/T27A was analysed by a Schild plot in which antagonism was expressed by the dose ratios of OxB needed to produce half‐maximal responses in the absence and the presence of different concentrations of S18A/N20A (left panel) or S18A/T27A (right panel). Dose ratios were estimated graphically in three separate experiments as shown in figure 3C and 3D. The regression line was calculated by the method of the least square.
Figure S3 Snapshot representation of the system composed of the OXR1‐OxB complex embedded in a POPC bilayer and water and ions. OXR1 and OxB are represented as a yellow and a red ribbon, respectively. The POPC lipids are in blue sticks without the hydrogen atoms for sake of clarity. Potassium and chloride ions are represented as Van derWaals spheres colored in blue and orange, respectively. Water molecules are in red.
Figure S4 Molecular dynamic simulation of the complex OX1R/OxB20‐28. Conformational drift from initial structure during the simulation (100 ns) measured as backbone rmsd for the complex OX1R/OxB20‐28. Values for TM1 to TM7 as indicated in the figure, were represented.
Figure S5 Molecular dynamic simulation of the complex OX1R/OxB20‐28. Conformational drift from initial structure during the simulation (100 ns) measured as backbone rmsd for the complex OX1R/OxB20‐28. Values for Interface helix, ß1‐ß2 and OxB20‐28 as indicated in the figure, were represented.
Figure S6 Stability of the secondary structure of OxB20‐28 as a function of time during the MD trajectory. The X axis represents the MD trajectory time (in ns), while the residues are shown on the Y‐axis. Color indicates secondary structure elements: alpha helical structure is in pink, 3‐10 helical structure in blue, turn in green and random coil in white. The graph was generated using VMD timeline tool. (Humphrey, W., Dalke, A. and Schulten, K., “VMD ‐ Visual Molecular Dynamics”, J. Molec. Graphics, 1996, vol. 14, pp. 33–38.).
Figure S7 Putative 3D‐model of human OX1R (turquoise) docked to sequence of OxB (red). The full sequence (1–28) of OxB was structurally aligned to 3D model of the complex OX1R/OxB6‐28 using Chimera software (http://www.cgl.ucsf.edu/chimera/)
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This study was supported by INSERM U1149/The Inflammation Research Center (CRI), The Institut National du Cancer (INCA) (translational project N°2013‐213) and the Ligue contre le Cancer (RS 12/75‐64). The authors wish to thank Dr Martine Torres for the critical reading of the manuscript and editorial help.
Nicole, P. , Couvineau, P. , Jamin, N. , Voisin, T. , and Couvineau, A. (2015) Crucial role of the orexin‐B C‐terminus in the induction of OX1 receptor‐mediated apoptosis: analysis by alanine scanning, molecular modelling and site‐directed mutagenesis. British Journal of Pharmacology, 172: 5211–5223. doi: 10.1111/bph.13287.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol. 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The Concise Guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol. 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandre D, Hautot C, Mehio M, Jeandel L, Courel M, Voisin T et al. (2014). The orexin type 1 receptor is overexpressed in advanced prostate cancer with a neuroendocrine differentiation, and mediates apoptosis. Eur J Cancer. 50: 2126–33. [DOI] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics. 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO (1959). Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 14: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi S, Egashira S, Matsuda M, Iwaasa H, Kanatani A, Ohkubo M et al. (2003). Development of an orexin‐2 receptor selective agonist, [Ala(11), D‐Leu(15)]orexin‐B. Bioorg Med Chem Lett. 13: 111–113. [DOI] [PubMed] [Google Scholar]
- Benkert P, Biasini M, Schwede T (2011). Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 27: 343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T et al. (2014). SWISS‐MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42 (Web Server issue): W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceraudo E, Hierso R, Tan YV, Murail S, Rouyer‐Fessard C, Nicole P et al. (2008). Spatial proximity between the VPAC1 receptor and the amino terminus of agonist and antagonist peptides reveals distinct sites of interaction. FASEB J. 26: 2060–71. [DOI] [PubMed] [Google Scholar]
- Ceraudo E, Murail S, Tan YV, Lacapère JJ, Neumann JM, Couvineau A et al. (2012). The vasoactive intestinal peptide (VIP) alpha‐helix up to C terminus interacts with the N‐terminal ectodomain of the human VIP/pituitary adenylate cyclase‐activating peptide 1 receptor: photoaffinity, molecular modeling, and dynamics. Mol Endocrinol. 22: 147–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darker JG, Porter RA, Eggleston DS, Smart D, Brough SJ, Sabido‐David C et al. (2001). Structure‐activity analysis of truncated orexin‐A analogues at the orexin‐1 receptor. Bioorg Med Chem Lett. 11: 737–40. [DOI] [PubMed] [Google Scholar]
- De Graaf C, Foata N, Engkvist O, Rognan D (2008). Molecular modeling of the second extracellular loop of G‐protein coupled receptors and its implication on structure‐based virtual screening. Proteins 71: 599–620. [DOI] [PubMed] [Google Scholar]
- El Firar A, Voisin T, Rouyer‐Fessard C, Ostuni MA, Couvineau A, Laburthe M (2009). Discovery of a functional immunoreceptor tyrosine‐based switch motif in a 7‐transmembrane‐spanning receptor: role in the orexin receptor OX1R‐driven apoptosis. FASEB J. 23: 4069–80. [DOI] [PubMed] [Google Scholar]
- Fanelli F, De Benedetti PG (2011). Update 1 of: computational modeling approaches to structure–function analysis of G protein‐coupled receptors. Chem Rev. 111: PR438–535. [DOI] [PubMed] [Google Scholar]
- German NA, Decker AM, Gilmour BP, Thomas BF, Zhang Y (2013). Truncated orexin peptides: structure–activity relationship studies. ACS Med Chem Lett. 4: 1224–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotter AL, Webber AL, Coleman PJ, Renger JJ, Winrow CJ (2012). International Union of Basic and Clinical Pharmacology. LXXXVI. Orexin receptor function, nomenclature and pharmacology. Pharmacol Rev 64: 389–420. [DOI] [PubMed] [Google Scholar]
- Heifetz A, Barker O, Morris GB, Law RJ, Slack M, Biggin PC (2013). Toward an understanding of agonist binding to human orexin‐1 and orexin‐2 receptors with G‐protein‐coupled receptor modeling and site‐directed mutagenesis. Biochemistry. 52: 8246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, Im W (2008). CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem. 29: 1859–65. [DOI] [PubMed] [Google Scholar]
- Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon‐Ramirez C et al. (2010). Update of the CHARMM all‐atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 114: 7830–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K et al. (2013). Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 504: 101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laburthe M, Voisin T (2012). The orexin receptor OX(1)R in colon cancer: a promising therapeutic target and a new paradigm in G protein‐coupled receptor signaling through ITIMs. Br J Pharmacol. 165: 1678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laburthe M, Voisin T, El Firar A (2010). Orexins/hypocretins and orexin receptors in apoptosis: a mini‐review. Acta Physiol (Oxf). 198: 393–402. [DOI] [PubMed] [Google Scholar]
- Lang M, Soll RM, Durrenberger F, Dautzenberg FM, Beck‐Sickinger AG (2004). Structure‐activity studies of orexin a and orexin B at the human orexin 1 and orexin 2 receptors led to orexin 2 receptor selective and orexin 1 receptor preferring ligands. J Med Chem. 47: 1153–1160. [DOI] [PubMed] [Google Scholar]
- Lappano R, Maggiolini M (2011). G protein‐coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 10: 47–60. [DOI] [PubMed] [Google Scholar]
- Lee JH, Bang E, Chae KJ, Kim JY, Lee DW, Lee W (1999). Solution structure of a new hypothalamic neuropeptide, human hypocretin‐2/orexin‐B. Eur J Biochem. 266: 831–9. [DOI] [PubMed] [Google Scholar]
- Le Guilloux V, Schmidtke P, Tuffery P (2009). Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 10: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ et al. (1998). All‐atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 102: 3586–616. [DOI] [PubMed] [Google Scholar]
- Mackerell AD Jr, Feig M, Brooks CL 3rd. (2004). Extending the treatment of backbone energetics in protein force fields: limitations of gas‐phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 25: 1400–15. [DOI] [PubMed] [Google Scholar]
- Nicole P, Lins L, Rouyer‐Fessard C, Drouot C, Fulcrand P, Thomas A et al. (2000). Identification of key residues for interaction of vasoactive intestinal peptide with human VPAC1 and VPAC2 receptors and development of a highly selective VPAC1 receptor agonist. Alanine scanning and molecular modeling of the peptide. J Biol Chem. 275: 24003–12. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preininger AM, Meiler J, Hamm HE (2013). Conformational flexibility and structural dynamics in GPCR‐mediated G protein activation: a perspective. J Mol Biol. 425: 2288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS et al. (2011a). Crystal structure of the β2 adrenergic receptor‐Gs protein complex. Nature. 477: 549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS et al. (2011b). Structure of a nanobody‐stabilized active state of the β(2) adrenoceptor. Nature. 469: 175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet‐Benzineb P, Rouyer‐Fessard C, Jarry A, Avondo V, Pouzet C, Yanagisawa M et al. (2004). Orexins acting at native OX(1) receptor in colon cancer and neuroblastoma cells or at recombinant OX(1) receptor suppress cell growth by inducing apoptosis. J Biol Chem. 279: 45875–86. [DOI] [PubMed] [Google Scholar]
- Thompson MD, Xhaard H, Sakurai T, Rainero I, Kukkonen JP (2014). OX1 and OX2 orexin/hypocretin receptor pharmacogenetics. Front Neurosci. 57: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM (2013). Molecular signatures of G‐protein‐coupled receptors. Nature. 494: 185–94. [DOI] [PubMed] [Google Scholar]
- Voisin T, El Firar A, Fasseu M, Rouyer‐Fessard C, Descatoire V, Walker F et al. (2011). Aberrant expression of OX1 receptors for orexins in colon cancers and liver metastases: an openable gate to apoptosis. Cancer Res. 71: 3341–51. [DOI] [PubMed] [Google Scholar]
- Voisin T, El Firar A, Rouyer‐Fessard C, Gratio V, Laburthe M (2008). A hallmark of immunoreceptor, the tyrosine‐based inhibitory motif ITIM, is present in the G protein‐coupled receptor OX1R for orexins and drives apoptosis: a novel mechanism. FASEB J. 22: 1993–2002. [DOI] [PubMed] [Google Scholar]
- Voisin T, Firar AE, Avondo V, Laburthe M (2006). Orexin‐induced apoptosis: the key role of the seven‐transmembrane domain orexin type 2 receptor. Endocrinology. 147: 4977–84. [DOI] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila‐Contreras EM et al. (2014). CHARMM‐GUI membrane builder toward realistic biological membrane simulations. J Comput Chem. 35: 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TR, Yang Y, Ward R, Gao L, Liu Y (2013). Orexin receptors: multi‐functional therapeutic targets for sleeping disorders, eating disorders, drug addiction, cancers and other physiological disorders. Cell Signal. 25: 2413–23. [DOI] [PubMed] [Google Scholar]
- Yin J, Mobarec JC, Kolb P, Rosenbaum DM (2015). Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature. 519: 247–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1A and S1B Specific 125I‐OxA binding to CHO‐SOX1R cells in the presence of increasing concentrations of unlabeled OxB and OxB analogs as indicated in figure (left) and determination of the stimulation of IP3 production (middle) or the inhibition of cellular growth induced (right) by increasing concentrations of OxB and OxB analogs in CHO‐S‐OX1R cells. CHO‐S‐OX1R cells were incubated with the indicated concentrations of OxB () and OxB analogs (). Left, results are expressed as the percentage of radioactivity specifically bound in the absence of added unlabeled peptide; Middle, results are expressed as the percentage of maximal IP3 production; Right, results are expressed as the percentage of total viable cells. Each point is the mean of three separate experiments. For the sake of clarity, standard errors are not indicated. They were always below 15% of mean values.
Figure S2 Antagonism S18A/N20A and S18A/T27A was analysed by a Schild plot in which antagonism was expressed by the dose ratios of OxB needed to produce half‐maximal responses in the absence and the presence of different concentrations of S18A/N20A (left panel) or S18A/T27A (right panel). Dose ratios were estimated graphically in three separate experiments as shown in figure 3C and 3D. The regression line was calculated by the method of the least square.
Figure S3 Snapshot representation of the system composed of the OXR1‐OxB complex embedded in a POPC bilayer and water and ions. OXR1 and OxB are represented as a yellow and a red ribbon, respectively. The POPC lipids are in blue sticks without the hydrogen atoms for sake of clarity. Potassium and chloride ions are represented as Van derWaals spheres colored in blue and orange, respectively. Water molecules are in red.
Figure S4 Molecular dynamic simulation of the complex OX1R/OxB20‐28. Conformational drift from initial structure during the simulation (100 ns) measured as backbone rmsd for the complex OX1R/OxB20‐28. Values for TM1 to TM7 as indicated in the figure, were represented.
Figure S5 Molecular dynamic simulation of the complex OX1R/OxB20‐28. Conformational drift from initial structure during the simulation (100 ns) measured as backbone rmsd for the complex OX1R/OxB20‐28. Values for Interface helix, ß1‐ß2 and OxB20‐28 as indicated in the figure, were represented.
Figure S6 Stability of the secondary structure of OxB20‐28 as a function of time during the MD trajectory. The X axis represents the MD trajectory time (in ns), while the residues are shown on the Y‐axis. Color indicates secondary structure elements: alpha helical structure is in pink, 3‐10 helical structure in blue, turn in green and random coil in white. The graph was generated using VMD timeline tool. (Humphrey, W., Dalke, A. and Schulten, K., “VMD ‐ Visual Molecular Dynamics”, J. Molec. Graphics, 1996, vol. 14, pp. 33–38.).
Figure S7 Putative 3D‐model of human OX1R (turquoise) docked to sequence of OxB (red). The full sequence (1–28) of OxB was structurally aligned to 3D model of the complex OX1R/OxB6‐28 using Chimera software (http://www.cgl.ucsf.edu/chimera/)
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item