

Abstract
Background and Purpose
Dipeptidyl peptidase 4 (DPP4) is an aminopeptidase that is widely expressed in different cell types. Recent studies suggested that DPP4 plays an important role in tumour progression in several human malignancies. Here we have examined the mechanisms by which up‐regulation of DPP4 expression causes epithelial transformation and mammary tumourigenesis.
Experimental Approach
Expression of DPP4 and the peptidylprolyl cis/trans isomerase, NIMA‐interacting 1 (PIN1), and the cytotoxic effects of combined treatment with sitagliptin and juglone were investigated by immunohistochemistry, immunoblotting, real‐time PCR, TUNEL and soft agar assays, using MCF7 cells. The effects of sitagliptin on tumour development in vivo were studied in the syngeneic 4T1 metastatic breast cancer model.
Key Results
Activity of the transcription factor E2F1 induced by EGF was enhanced by DPP4, thus increasing PIN1 expression. Furthermore, DPP4 enhanced MEK/ERK and JNK/c‐Jun signalling induced by EGF, inducing AP‐1 activity and epithelial cell transformation. In contrast, DPP4 silencing or DPP4 inhibition in MCF7 cells inhibited PIN1 expression via E2F1 activity induced by EGF, decreasing colony formation and inducing DNA fragmentation. In the syngeneic 4T1 metastatic breast cancer model, DPP4 overexpression increased tumour development, whereas treatment with sitagliptin and/or juglone suppressed it. Consistent with these observations, DPP4 levels were positively correlated with PIN1 expression in human breast cancer.
Conclusions and Implications
DPP4 promoted EGF‐induced epithelial cell transformation and mammary tumourigenesis via induction of PIN1 expression, suggesting that sitagliptin targeting of DPP4 could be a treatment strategy in patients with breast cancer.
Abbreviations
- AP‐1
activator protein‐1
- DPP‐4
dipeptidyl peptidase 4
- GLP‐1
glucagon‐like peptide‐1
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- T‐LBL
T cell lymphoblastic leukaemia
- T‐ALL
T cell acute lymphoblastic leukaemia
- T2DM
type 2 diabetes mellitus
Tables of Links
| LIGANDS |
|---|
| 1G244 |
| EGF |
| GLP‐1 |
| Sitagliptin |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Type 2 diabetes mellitus (T2DM) is associated with a modestly increased risk of postmenopausal breast cancer (Larsson et al., 2007; Onitilo et al., 2014). T2DM is characterized by insulin resistance and hyperinsulinaemia (Weyer et al., 2001). Apart from its metabolic effects, insulin has mitogenic effects that are mediated through the insulin‐like growth factor‐1 and insulin receptors (Frasca et al., 2008). Epidemiological studies have demonstrated that insulin resistance and hyperinsulinaemia are related to an increased risk of epithelial malignancy, including cancer of the breast, prostate, colon and kidney (Lipscombe et al., 2006; Frasca et al., 2008). Furthermore, diabetes is associated with markedly increased mortality in women with breast cancer (Lipscombe et al., 2008). Therefore, there is considerable interest in strategies that reduce both the risk of developing breast cancer and the mortality in diabetic subjects.
Dipeptidyl peptidase 4 (DPP4) is a cell surface aminopeptidase, which was originally characterized as a T cell differentiation antigen, CD26 (Fleischer, 1994), and it has been reported to be present on epithelial cells of various organs, including the lung, liver, kidney, intestine, prostate and placenta (Mizutani et al., 1985; Heike et al., 1988; Nemoto et al., 1999). As DPP4 cleaves the first two amino acids from peptides with penultimate L‐proline or L‐alanine residues, this enzyme is capable of degrading several bioactive peptides, cytokines and several chemokines, such as CXCL12 (SDF‐1α) and CCL5 (RANTES) (Oravecz et al., 1997; Mentlein, 1999; Nemoto et al., 1999). DPP4 inhibitors, such as sitagliptin, vildagliptin and saxagliptin, have been developed for their ability to inhibit degradation of glucagon‐like peptide‐1 (GLP‐1), one of the incretin hormones; GLP‐1 is critical for glucose homeostasis and represents a therapeutic target in T2DM (Aschner et al., 2006; Drucker and Nauck, 2006; Deacon and Holst, 2009). Furthermore, DPP4 plays key roles in the control of growth, differentiation and signal transduction in many cellular systems by modulating the activity of peptide factors (Hanski et al., 1985; Loster et al., 1995). Abnormalities in the expression pattern and/or catalytic function of peptidases result in altered peptide activation or inactivation, contributing to the disruption of normal cellular homeostasis, neoplastic transformation or tumour progression (Varona et al., 2010).
A novel phosphorylation signalling regulator, the peptidylprolyl cis/trans isomerase, PIN1, sits at the crossroads of many signalling pathways that control cell proliferation and transformation (Lu and Zhou, 2007). PIN1 is the only mammalian enzyme known to specifically catalyse the cis/trans isomerization of Ser–Pro or Thr–Pro peptide bonds (Lu and Zhou, 2007). The effects of PIN1‐induced isomerization on target proteins are diverse and include altering their stability and localization, as well as modifying their interactions with other proteins (Lu and Zhou, 2007; Liou et al., 2011). PIN1‐induced conformational changes can have profound effects on the function of many substrates, including p53, cyclin D1, c‐Jun, MAPK kinase 1 (MEK1), NF‐κB and STAT3. These actions result in PIN1 playing important roles in many cellular processes, such as cell cycle progression and differentiation (Wulf et al., 2001; Zheng et al., 2002; Ryo et al., 2003; Fukuchi et al., 2006; Khanal et al., 2010; Kim et al., 2014). Significantly, PIN1 is a target gene for the transcription factor E2F1 which is strongly overexpressed in breast cancer, and its expression is closely correlated with tumour grade and cyclin D1 expression level in tumours (Wulf et al., 2001; Ryo et al., 2002). Importantly, up‐regulation of PIN1 elevated cyclin D1 expression by activating the c‐jun/activator protein‐1 (AP‐1) and β‐catenin/T cell factor transcription factors (Ryo et al., 2001; Wulf et al., 2001). Although many details of PIN1 overexpression in breast cancer have been elucidated, the influence of DPP4 on PIN1 overexpression and its significance in oncogenesis remain largely unknown.
The aim of this study was to investigate the role of DPP4 in the transformation of epithelial cells and in breast epithelial tumourigenesis and to define the molecular mechanism by which DPP4 governs PIN1 overexpression. We demonstrated that DPP4 regulated PIN1 expression by enhancing EGF‐induced activation of the transcription factor E2F1, up‐regulating MEK/ERK and JNK/c‐Jun signalling and of AP‐1 activity. More importantly, PIN1 overexpression induced by DPP4 leads not only to moderate cell transformation in epithelial cells but also to enhance breast epithelial tumourigenicity. In contrast, the inhibition of DPP4 by pharmacological means, specifically by sitagliptin treatment, suppressed EGF‐induced epithelial cell transformation and mammary epithelial tumourigenesis via inhibition of PIN1 expression. These results indicate that DPP4 acts upstream of PIN1 signalling and plays an essential role in mammary tumourigenesis through activation of AP‐1.
Methods
Cell culture and small interfering RNAs (siRNAs)
MCF7 human breast cancer cells were cultured in DMEM supplemented with 10% FBS. Murine 4T1 metastatic breast cancer cells were maintained in RPMI supplemented with 10% FBS. Murine JB6 Cl41 normal epithelial cells were cultured in MEM supplemented with 5% FBS. All cell lines were cultured and maintained at 37°C in humidified air containing 5% CO2. The DNA transfection of cells was performed using the jetPEI cationic polymer transfection reagent. The human genes, DPP4 (GenBank accession number: NM_001935) and PIN1 (GenBank accession number: NM_006221), were silenced by transfecting cells with the ON‐TARGETplus SMART siRNA pool‐specific or nonspecific‐control pool double‐stranded RNA oligonucleotides (Dharmacon, Chicago, IL, USA), using Lipofectamine® 2000 reagent (Invitrogen).
Immunoblot analysis
Cells were lysed in RIPA buffer. The proteins were resolved by SDS‐PAGE and transferred onto PVDF membrane. For detecting chemiluminescence, an ImageQuant™ LAS 4000 imaging system (GE Healthcare Biosciences, Pittsburgh, PA, USA) was used.
MTT assay
The MTT assay was used to check cell viability. In brief, cells (1 × 104) were seeded in 96‐well plates with 100 μL of cell suspension in each well and incubated at 37°C in humidified air containing 5% CO2. After 24 h in culture, cells were incubated with different concentrations of sitagliptin for various periods. The cells were then treated with 5 mg∙mL−1 MTT solution (10 μL per well) and incubated for 4 h, the purple formazan formed by the live cells was dissolved in 0.04 N HCl in isopropanol (100 μL per well), and the absorbance was measured at 450 nm.
Anchorage‐independent cell transformation (soft agar) assay
Briefly, cells (8 × l03) were treated with different concentrations of sitagliptin, juglone and/or EGF in 1 mL of 0.3% basal medium Eagle agar containing 10% FBS. Cultures were incubated at 37°C in humidified air containing 5% CO2 for 14 days, and the cell colonies were scored using an Axiovert 200 M fluorescence microscope and axiovision software (Carl Zeiss, Thornwood, NY, USA).
Reporter gene promoter assay
In order to analyse promoter transcriptional activity, the firefly luciferase reporter gene assay was performed using lysates from MCF7 cells transfected with FOS‐luc, JUN‐luc, AP‐1‐responsive‐luc, PIN1‐luc or E2F1‐luc promoter‐reporter gene constructs. The Renilla luciferase control reporter vector, pRL‐TK (Promega), was cotransfected into each cell line, and the Renilla luciferase activity generated by this vector was used to normalize the results with respect to transfection efficiency. Cell lysates were mixed with luciferase assay II reagent, and firefly luciferase light emission was measured using the GloMax®‐Multi Detection System (Promega); Renilla luciferase substrate was then added to enable normalization of the firefly luciferase data.
Detection of apoptosis
The induction of apoptosis was assessed by TUNEL staining and detected with an In Situ Cell Death Detection Kit (Roche Life Science, Indianapolis, IN, USA), according to the manufacturer's instructions. Briefly, 2 × 105 cells were cultured for 24 h in six‐well plates. The cells were then starved for 24 h and treated with sitagliptin for 6 h. Treated cells were washed with PBS and fixed with Cytofix/Cytoperm™ (BD Biosciences, San Diego, CA, USA) at 4°C for 20 min. Cells were stained with 50 μL TUNEL solution at 37°C for 1 h, then washed twice with PBS and fixed. DNA fragmentation was detected using an Axiovert 200 M fluorescence microscope and quantified using the axiovision software (Carl Zeiss).
Animals and experimental design
All animal care and experimental procedures complied with local guidelines and were approved by the Animal Experiments Committee of Chosun University. All studies are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 50 mice were used in the experiments described here. Six‐week‐old female BALB/c mice (18–20 g body weight) were obtained from Samtako Co (Osan, Gyeonggi‐do, South Korea), acclimatized for 1 week and kept in a clean room with a cycle of 12 h light/12 h darkness; the temperature was 22°C, and the humidity was 40–60%. A standard diet of rodent pellets and tap water (membrane filter‐purified and autoclaved) was provided ad libitum. Mice were anaesthetized via intramuscular injection of Zoletil (30 mg∙kg−1; Virbac, Carros, France). Mice were randomly divided into two or four groups of 10 animals each; murine 4T1 metastatic breast cancer cells were injected into the mammary gland of the mice with or without 10 mM sitagliptin and/or 100 μM juglone and allowed to grow until tumours formed. The tumour volume was calculated using the formula: V = (ab 2)/2, in which ‘a’ is the longest dimension and ‘b’ the shortest dimension of the tumour.
Tumour samples
Informed consent was obtained from all the patients, and research protocols were approved by the ethics committee of Chosun University Hospital. The breast tissues that were selected for immunohistochemical staining were collected from a group of 60 patients with breast cancer (age range: 42–72 years). The normal breast group included patients with mammary infiltrating duct carcinoma who had undergone mastectomy with adjuvant hormone therapy and had no subsequent local recurrence or metastasis within 5 years, and the breast cancer group included patients with mammary infiltrating duct carcinoma who had undergone mastectomy with adjuvant hormone therapy and subsequently developed bone metastases.
Immunohistochemical analysis
All tumours investigated in the study were tested using anti‐DPP4 and anti‐PIN1 antibodies. Immunolocalization for each antibody was performed using a Polink‐2 Plus HRP mouse 3,3′‐diaminobenzidine detection system (GBI Labs, Bothell, WA, USA), according to the supplier's protocol. Slides were incubated for 1 h with anti‐DPP4 or anti‐PIN1 antibodies in a moist chamber at 37°C. In place of the primary antibody, normal goat serum was used as the negative control. Distinct cytoplasmic staining for DPP4 or PIN1 was considered to indicate positive immunoreactivity.
Chromatin immunoprecipitation
The crosslinking of proteins to DNA was accomplished by the addition of 1% formaldehyde for 10 min to cultured cells (1 × 107 cells) at 37°C. After sonication, the chromatin was immunoprecipitated with 1 µg of anti‐E2F1 antibody or control mouse IgG for 16 h at 4°C. After elution and reversal of cross links, DNA was isolated and analysed by PCR. PCR products were visualized on a 1.5% agarose gel with SYBR® Gold nucleic acid gel stain. The following primers were used for PCR: human PIN1, sense: 5′‐GGTTAGCTTTGGACATCT GTGG‐3′ and antisense: 5′‐GCCTTCTATTGGGTAGAAGAAAGG‐3′.
Data analysis
Results are expressed as the mean ± SEM of triplicate measurements from three independent experiments
Fisher's exact test with two‐sided P values (P < 0.001) was used to analyse correlations between expression of DPP4 and PIN1 in breast cancer patients. Data from the MTT assay, the reporter gene promoter assay and the soft agar assay were analysed using unpaired Student's t‐tests; values of P < 0.05 were considered significant. Statistical calculations were carried out using the prism software for Macintosh, version 5.0 (GraphPad Software, La Jolla, CA, USA).
Materials
DMEM, Eagle's minimal essential medium (MEM), RPMI and FBS were purchased from Invitrogen (Carlsbad, CA, USA). Sitagliptin phosphate monohydrate, juglone and 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) were procured from Sigma‐Aldrich (St Louis, MO, USA). 1G244 was purchased from AK Scientific (Union City, CA, USA). The Dual‐Luciferase® Reporter Assay kit was purchased from Promega (Madison, WI, USA). Ez‐Chip kit, PVDF membrane, anti‐E2F1 antibody and anti‐His antibody were from EMD Millipore (Bedford, MA, USA). EGF was from Calbiochem‐Novabiochem (San Diego, CA, USA). Antibodies against MEK1/2, ERK1/2, JNK1/2, cyclin D1 and cleaved PARP and against phosphorylated MEK1/2, ERK1/2, JNK1/2 and c‐Jun (Ser 63) were purchased from Cell Signaling Technology (Beverly, MA, USA); antibodies against DPP4, PIN1 and c‐Jun and goat anti‐mouse IgG and HRP‐conjugated goat anti‐rabbit IgG were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The jetPEI® cationic polymer transfection reagent was from Polyplus‐transfection (New York, NY, USA).
Results
Sitagliptin suppresses EGF‐induced epithelial transformation and mammary tumourigenesis
In order to understand the effect of DPP4 overexpression on cell transformation, we established normal murine epithelial cells (JB6 Cl41 cells) stably overexpressing DPP4 (DPP4‐JB6) and mock‐transfected cells (mock‐JB6) (Figure 1A). Using these cell lines, we examined differences in EGF‐promoted cell transformation in a soft agar matrix. The mock‐JB6 and DPP4‐JB6 cells were treated separately with 10 ng∙mL−1 EGF in soft agar matrix and incubated at 37°C in a 5% CO2 incubator for 14 days. As a result, DPP4‐JB6 cells showed significantly enhanced formation of EGF‐promoted colonies compared with mock‐JB6 cells (Figure 1B). An increase was evident not only in colony number but also in colony size (Figure 1C), which suggested that DPP4 could be involved in neoplastic cell transformation as a positive regulator. Given that DPP9 and its homologue DPP8, the proteases of the DPP4 gene family, associate with H‐Ras, a key signal molecule of the EGF receptor signalling pathway (Yao et al., 2011), we further examined whether DPP8 and DPP9 might regulate neoplastic transformation of JB6 Cl41 cells. We found that overexpression of DPP8 or DPP9 in JB6 Cl41 cells had no significant effects on the cell transformation compared with mock‐JB6 cells in soft agar matrix (Supporting Information Fig. S1). Given the ability of DPP4 to enhance cell transformation promoted by EGF, we examined whether inhibition of DPP4 contributed to suppression of breast tumourigenesis. We first assessed the inhibition of DPP4 activity using sitagliptin, a DPP4 inhibitor. The intracellular DPP4 activity was significantly and concentration‐dependently inhibited by treatment of sitagliptin for 24 h in MCF7 cells (IC 50 = 1.33 mM, Supporting Information Fig. S2). Furthermore, treatment with sitagliptin significantly and concentration‐dependently inhibited proliferation of MCF7 cells (Figure 1D). In addition, sitagliptin treatment concentration‐dependently inhibited colony numbers as well as colony size of MCF7 cells in soft agar (Figure 1E and 1F). We further examined the role of DPP8 and DPP9 in breast tumourigenesis and found that overexpression of DPP8 or DPP9 did not enhance cell proliferation and colony formation in MCF7 cells (Supporting Information Fig. S3). Consistent with these results, treatment with a specific inhibitor of DPP8 and DPP9, 1G244 (IC 50 = 0.16 μM, Supporting Information Fig. S4A), had no significant inhibitory effect on the cell proliferation and colony formation of MCF7 cells (Supporting Information Fig. S4B–4D). Next, the effects of sitagliptin on tumour development in vivo were studied in a syngeneic murine 4T1 metastatic breast cancer model. We found that sitagliptin treatment significantly and concentration‐dependently inhibited proliferation of 4T1 cells (Figure 1G). In addition, representative tumour images demonstrated that sitagliptin significantly suppressed mammary gland tumour development (Figure 1H and 1I).
Figure 1.
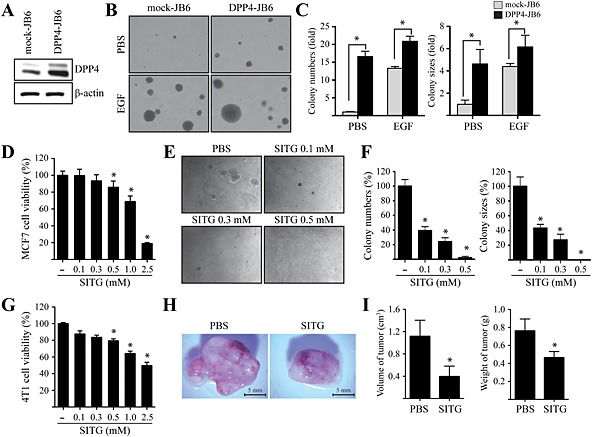
Effects of sitagliptin on EGF‐induced neoplastic cell transformation and epithelial breast tumourigenesis. (A) Mock‐transfected (mock‐JB6) or DPP4‐overexpressing (DPP4‐JB6) JB6 cells were harvested, and proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted in order to detect DPP4. (B and C) Mock‐JB6 or DPP4‐JB6 cells were treated with or without 10 ng∙mL−1 EGF in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 days. The colonies from three separate experiments are photographed (B). The average number of colonies was calculated, and colony size was measured under a microscope (C). Columns represent the means ± SD of triplicate samples. * P < 0.05, significantly different from control cells. (D) MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of sitagliptin (SITG), as indicated. Cell viability was measured by the MTT assay. Data shown are the means ± SD, from triplicate experiments. * P <0.05, significantly different from control cells. (E and F) MCF7 cells were exposed to various concentrations of sitagliptin in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 days. The colonies from three separate experiments are photographed (E). The average number of colonies was calculated (F). Columns represent the means ± SD of triplicate samples. * P < 0.05, significantly different from control cells. (G) 4T1 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of sitagliptin, as indicated. Cell viability was measured by the MTT assay, as described in Methods. Data shown are the means ± SD, from triplicate experiments. * P<0.05, significantly different from control cells. (H and I) 4T1 cells were treated or not with 10 mM sitagliptin. Treated cells were injected into the mammary glands of BALB/c mice (n = 20) and allowed to grow until tumours formed (14 days). Representative pictures of tumours (H) and tumour volume and weight (I) are shown. Columns represent the means ± SD of triplicate samples. * P < 0.05, significantly different from controls (injected with cells mock treated with PBS only).
DPP4 induces AP‐1 activity via up‐regulation of MEK/ERK and JNK/c‐Jun signalling stimulated by EGF
Given the role of DPP4 as a positive regulator of EGF‐induced cell transformation, we examined whether DPP4 might regulate the MEK/ERK and JNK/c‐Jun signalling pathways. The results showed that DPP4 overexpression in MCF7 cells increased the phosphorylation of MEK1/2 and ERK1/2 (Figure 2A), as well as JNK1/2 and c‐Jun (Figure 2B), compared with control cells. In contrast, the phosphorylation of MEK1/2 and ERK1/2 (Figure 2C), as well as JNK1/2 and c‐Jun (Figure 2D), was decreased by DPP4 silencing in MCF7 cells compared with control cells. Consistent with these results, we observed that the EGF‐induced phosphorylation of MEK1/2, ERK1/2, JNK1/2 and c‐Jun was enhanced by DPP4 overexpression in MCF7 cells (Figure 2E), whereas it was inhibited by DPP4 silencing (Figure 2F), suggesting that DPP4 might play an important role in EGF‐induced breast cell proliferation and mammary tumourigenesis via the activation of MEK/ERK and JNK/c‐Jun signalling pathways. The AP‐1 transcription factor is a dimeric complex of homodimers or heterodimers of Jun, Fos, activating transcription factor and musculoaponeurotic fibrosarcoma protein family members that are activated by MAPK signalling pathways (Karin, 1995; Eferl and Wagner, 2003). In order to determine the effects of DPP4 on AP‐1 activity, we first examined the effects of DPP4 on the c‐fos and c‐jun promoters. The EGF‐induced transcriptional activity of the c‐fos and c‐jun promoters was significantly enhanced by DPP4 overexpression in MCF7 cells in a concentration‐dependent manner (Figure 2G and 2H). To further confirm that DPP4 regulated the AP‐1‐responsive transcriptional activity induced by EGF, we assessed EGF‐induced AP‐1 activity in the presence or absence of sitagliptin in MCF7 cells. As expected, sitagliptin treatment inhibited EGF‐induced AP‐1 activity in MCF7 cells (Figure 2I), which suggests that EGF‐induced AP‐1‐responsive transcriptional activity was regulated by DPP4 signalling.
Figure 2.
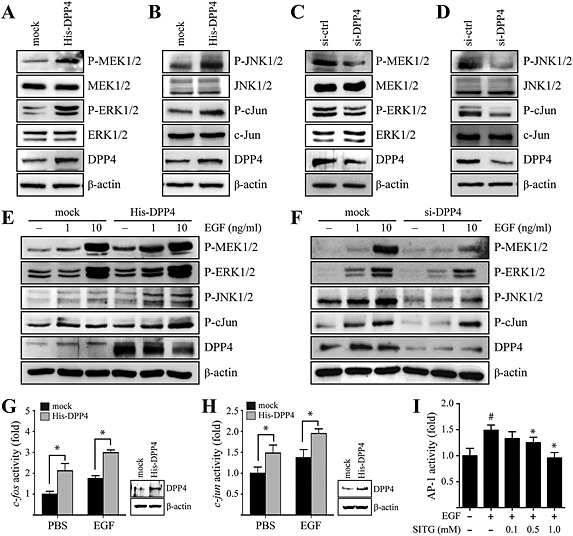
DPP4 enhances MEK/ERK and JNK/c‐Jun signalling stimulated by EGF in MCF7 cells. (A and B) Cells were transfected with a construct expressing histidine‐tagged DPP4 (His‐DPP4) or mock transfected with empty vector (mock plasmid). At 48 h after transfection, the cells were harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (C and D) Constructs expressing control (siRNA‐control) or DPP4‐specific (siRNA‐DPP4) siRNAs were transfected into MCF7 cells. At 48 h after transfection, the cells were harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (E) Cells were transfected with His‐DPP4 or mock plasmid. At 24 h after transfection, the cells were serum starved, exposed to the indicated concentration of EGF for 30 min, harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (F) siRNA‐control or siRNA‐DPP4 constructs were transfected into MCF7 cells. At 24 h after transfection, the cells were serum starved, exposed to the indicated concentration of EGF for 30 min, harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (G and H) His‐DPP4 or mock plasmid were co‐transfected with the luciferase promoter‐reporter constructs, FOS‐luc (G) or JUN‐luc (F), and the pRL‐TK (Renilla luciferase control reporter) vector into host cells. At 24 h after transfection, the cells were serum starved and then exposed or not exposed to 1 ng∙mL−1 EGF for 24 h. The firefly luciferase activity was determined in cell lysates and normalized to the Renilla luciferase activity. Columns represent the means ± SD of triplicate samples. * P < 0.05, significantly different from control (mock) cells. (I) Cells were co‐transfected with an AP‐1‐responsive luciferase promoter‐reporter plasmid and the pRL‐TK vector. At 24 h after transfection, the cells were serum starved, treated with the indicated concentration of sitagliptin (SITG) for 24 h and then exposed or not exposed to 1 ng∙mL−1 EGF for 24 h. The firefly luciferase activity was determined in cell lysates, normalized to the Renilla luciferase activity and is expressed relative to control cells. Columns represent the means ± SD of triplicate measurements from two experiments. *P < 0.05, significantly different from cells treated with EGF only.
DPP4 enhances PIN1 and cyclin D1 expression stimulated by EGF
Given that PIN1, which specifically recognizes phospho‐Ser/Thr–Pro motifs on its target proteins, interacts with MEK1 (Namgoong et al., 2010), JNK1 (Park et al., 2012) and c‐Jun (Wulf et al., 2001), we next examined whether DPP4 might regulate MEK/ERK and JNK/c‐Jun signalling via PIN1. Reporter gene assays using the human PIN1 promoter demonstrated an important role for DPP4 in PIN1 expression. PIN1 promoter activity was increased by DPP4 overexpression (Figure 3A), whereas DPP4 silencing inhibited PIN1 promoter activity (Figure 3B) in MCF7 cells, indicating that endogenous DPP4 may affect PIN1 expression. We then analysed PIN1 mRNA levels following overexpression or knockdown of DPP4 in MCF7 cells. Real‐time PCR analysis showed that the PIN1 mRNA level was significantly increased by DPP4 overexpression (Figure 3C) but was decreased by DPP4 silencing in MCF7 cells (Figure 3D). Consistent with these results, overexpression of DPP4 in MCF7 cells resulted in up‐regulation of PIN1 expression, together with an increase in cyclin D1 levels (Figure 3E). However, knockdown of DPP4 in MCF7 cells decreased the expression of PIN1 and cyclin D1 (Figure 3F). Therefore, we further examined the effect of DPP4 on EGF‐induced PIN1 and cyclin D1 levels. The results showed that overexpression of DPP4 enhanced PIN1 as well as cyclin D1 expression induced by EGF in MCF7 cells (Figure 3G). In contrast, knockdown of DPP4 and sitagliptin treatment inhibited PIN1 and cyclin D1 expression induced by EGF respectively (Figure 3H and 3I).
Figure 3.
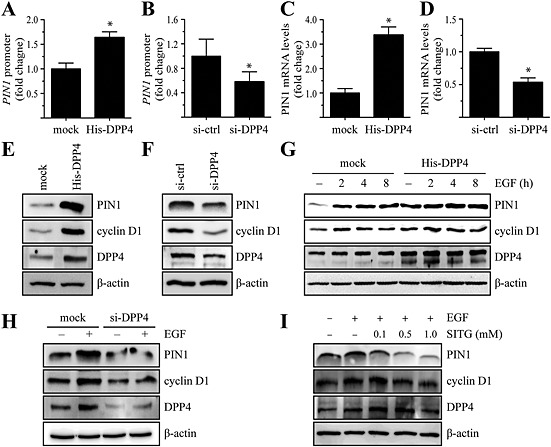
DPP4 regulates PIN1 expression via activation of E2F1 in MCF7 cells. (A and B) The PIN1‐luc luciferase promoter‐reporter construct and the pRL‐TK (Renilla luciferase control reporter) vector were cotransfected with constructs expressing histidine‐tagged DPP4 (His‐DPP4) (A) or DPP4‐specific siRNA (siRNA‐DPP4) (B). After 48 h, the firefly luciferase activity was determined in cell lysates and normalized to Renilla luciferase activity. Columns represent the means ± SD of triplicate samples.*P < 0.05, significantly different from control cells. (C and D) Cells were transfected with His‐DPP4 (C) or siRNA‐DPP4 (D). Levels of PIN1 mRNA were assessed by real‐time PCR analysis. The signal intensity corresponding to each mRNA was densitometrically determined and normalized to GAPDH mRNA. Columns represent the means ± SD of triplicate measurements from two experiments. *P < 0.05, significantly different from mock‐transfected (C) or siRNA‐control‐transfected (D) cells. (E and F) Cells were transfected with His‐DPP4 (E) or siRNA‐DPP4 (F). At 48 h after transfection, the cells were harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (G) Cells were transfected with His‐DPP4 or mock transfected, serum starved, treated with 1 ng∙mL−1 EGF for the indicated times, harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (H) siRNA‐control or siRNA‐DPP4 constructs were transfected into MCF7 cells. At 48 h after transfection, the cells were serum starved, treated with 1 ng∙mL−1 EGF for the indicated time, harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted. (I) Cells were serum starved, treated with the indicated concentration of sitagliptin (SITG) for 24 h, then exposed or not exposed to 1 ng∙mL−1 EGF for 8 h, harvested and lysed. Proteins in whole cell lysates were separated by SDS‐PAGE and immunoblotted.
DPP4 induces PIN1 expression via increased association of E2F1 in the PIN1 promoter
Given that PIN1 expression is mediated by the transcription factor E2F1 and enhanced by the Neu and Ras oncogenic proteins (Ryo et al., 2002), we next examined the effects of DPP4 on the E2F1 promoter. The E2F1 promoter activity was increased by DPP4 overexpression (Figure 4A), whereas DPP4 silencing inhibited E2F1 promoter activity (Figure 4B) in MCF7 cells. To further confirm that DPP4 regulated E2F1 transcriptional activity stimulated by EGF, we assessed EGF‐stimulated E2F1 promoter activity in the presence or absence of sitagliptin in MCF7 cells. The results showed that sitagliptin treatment inhibited EGF‐stimulated E2F1 promoter activity in MCF7 cells (Figure 4C). To examine whether DPP4 might regulate binding of E2F1 to the PIN1 promoter, we next performed chromatin immunoprecipitation assays in MCF7 cells. DPP4‐overexpressing or DPP4‐silenced MCF7 cells were immunoprecipitated with an anti‐E2F1 antibody. DNA from the immunoprecipitates was PCR amplified using primers flanking the three E2F1‐binding sites in the PIN1 promoter region shown in Figure 4D. Our data showed that DPP4 overexpression increased binding of E2F1 in the PIN1 promoter (Figure 4E). In contrast, knockdown of DPP4 in MCF7 cells decreased the E2F1 binding in the PIN1 promoter (Figure 4F). These results indicate that DPP4 regulates PIN1 gene expression through association of the E2F1 in the PIN1 promoter.
Figure 4.
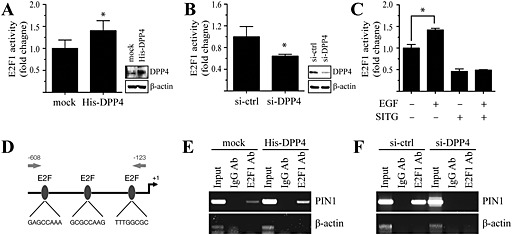
DPP4 regulates E2F1 activity to enhance binding of E2F1 in the PIN1 promoter in MCF7 cells. (A and B) The E2F1‐luc luciferase promoter‐reporter plasmid and the pRL‐TK vector were cotransfected with His‐DPP4 (A) or siRNA‐DPP4 (B). After 48 h, the firefly luciferase activity was determined in cell lysates and normalized to Renilla luciferase activity. Columns represent the means ± SD of triplicate samples. *P < 0.05, significantly different from control cells. (C) Cells were cotransfected with an E2F1‐luc luciferase promoter‐reporter plasmid and the pRL‐TK vector. At 24 h after transfection, cells were serum starved, treated with 1 mM sitagliptin (SITG) for 24 h and then exposed or not exposed to 1 ng∙mL−1 EGF for 24 h. The firefly luciferase activity was determined in cell lysates, normalized to Renilla luciferase activity and is expressed relative to control cells. Columns represent the means ± SD of triplicate measurements from two experiments. *P < 0.05, significantly different as indicated. (D) Human PIN1 promoter sequence with putative E2F1‐binding site. (E and F) Cell lysates from His‐DPP4 (E) or si‐DPP4 (F) transfected MCF7 cells were subjected to chromatin IP using anti‐E2F1 or IgG antibodies, and the recovered DNA was PCR amplified with primers specific for E2F1‐binding sites in the PIN1 promoter as shown in Figure 2D.
PIN1 affects sitagliptin‐induced apoptotic signalling in MCF7 cells
As DPP4 induced PIN1 expression, we next assessed the effect of sitagliptin on the viability of PIN1‐overexpressing MCF7 (PIN1‐MCF7) cells. The results showed that PIN1‐MCF7 cells were more resistant to sitagliptin than GFP vector mock‐transfected MCF7 (GFP‐MCF7) cells (Figure 5A). We further examined whether knockdown of PIN1 could potentiate sitagliptin sensitivity in MCF7 cells. Treatment with sitagliptin reduced the viability of MCF7 cells by 72%, whereas PIN1 silencing increased the sensitivity of MCF7 cells to sitagliptin by 87% (Figure 5B). We examined the effects of PIN1 overexpression on sitagliptin‐induced cell death via PARP cleavage. Treatment of GFP‐MCF7 cells with sitagliptin concentration‐dependently induced cleavage of PARP, whereas sitagliptin‐induced cleavage of PARP was attenuated in PIN1‐MCF7 cells (Figure 5C). In contrast, sitagliptin induced increased cleavage of PARP following PIN1 silencing in MCF7 cells, compared with untreated MCF7 cells (Figure 5D). Consistent with these results, sitagliptin‐induced DNA fragmentation was also increased in PIN1‐specific siRNA‐transfected MCF7 cells compared with non‐specific control siRNA‐transfected cells, as measured by the TUNEL assay (Figure 5E).
Figure 5.
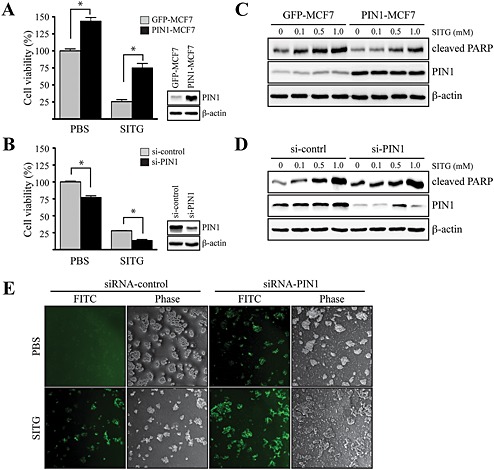
Inhibitory effects of sitagliptin on PIN1‐induced cell viability in MCF7 cells. (A) MCF7 cells, mock transfected (GFP‐MCF7) or overexpressing PIN1 (PIN1‐MCF7), were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with 1 mM sitagliptin (SITG) for 24 h. Cell viability was measured by the MTT assay, as described in Methods. Data shown are the means ± SD, from triplicate experiments. * P<0.05, significantly different as indicated. (B) MCF7 cells were transfected with constructs expressing control (siRNA‐control) or PIN1‐specific (siRNA‐PIN1) siRNAs. At 24 h after transfection, the cells were treated with 1 mM sitagliptin for 24 h. Cell viability was measured by the MTT assay, as described in Methods. Data are represented as the mean ± SD, as determined from triplicate experiments. (C) GFP‐MCF7 or PIN1‐MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were serum starved for 24 h, exposed to the indicated concentration of sitagliptin for 48 h, harvested and lysed, and the lysate was immunoblotted. (D) siRNA‐control or siRNA‐PIN1 constructs were transfected into MCF7 cells. At 24 h after transfection, the cells were serum starved for 12 h, exposed to the indicated concentration of sitagliptin for 48 h, harvested and lysed, and the lysate was immunoblotted. (E) siRNA‐control or siRNA‐PIN1 constructs were transfected into MCF7 cells. After 30 h, the cells were serum starved for 12 h and then exposed or not exposed to 1 mM sitagliptin for 24 h, and DNA fragmentation induced by sitagliptin was measured.
DPP4 abundance is positively correlated with PIN1 expression in human breast cancer tissues and promotes epithelial cell transformation
To understand the pathological relevance of DPP4 in breast tumourigenesis, immunohistochemistry was performed on human normal and breast cancer tissues using an anti‐DPP4 antibody. Of the 20 normal breast samples, 16 had a low amount of DPP4, whereas 11 of the 20 breast cancer samples contained a high amount of DPP4 (Figure 6A), indicating that DPP4 expression was significantly increased in breast cancer. To further investigate the relationship between DPP4 and PIN1 in human breast cancer, we performed immunohistochemistry using specific antibodies against DPP4 and PIN1 on human breast cancer tissues. Of the six breast cancer samples that had a low amount of DPP4, four also showed lower expression of PIN1, whereas all the 11 breast cancer samples that contained a high amount of DPP4 correspondingly had higher expression of PIN1 (Figure 6B). Given the ability of DPP4 to induce epithelial cell transformation via the induction of PIN1 expression, we examined whether inhibition of DPP4 contributes to the suppression of breast tumourigenesis induced by PIN1. The results showed that PIN1 overexpression significantly increased the number of MCF7 cell colonies formed in a soft agar matrix (Figure 6C). However, treatment with sitagliptin significantly attenuated the number of colonies in PIN1‐overexpressing as well as mock‐transfected MCF7 cells (Figure 6C). The increased PIN1 and DPP4 expression (Figure 6B) in breast cancer tissues supports our hypothesis that breast tumourigenesis promoted by DPP4 may occur as a result of activation of MAPK signalling through PIN1. Therefore, we examined whether combined treatment with sitagliptin and the PIN1 inhibitor, juglone, increased the anti‐umourigenic effect of sitagliptin. Co‐treatment of MCF7 cells, with sitagliptin and juglone, significantly and concentration‐dependently inhibited the number of colonies formed in soft agar compared with treatment with sitagliptin alone (Figure 6D). Subsequently, the effects of co‐treatment with sitagliptin and juglone on tumour development in vivo were studied in the syngeneic mouse 4T1 metastatic breast cancer model. Representative tumour images demonstrated that a profound reduction in the weight and volume of tumours was mediated by co‐treatment with sitagliptin and juglone, compared with sitagliptin alone or the PBS control (Figure 6E and 6F).
Figure 6.
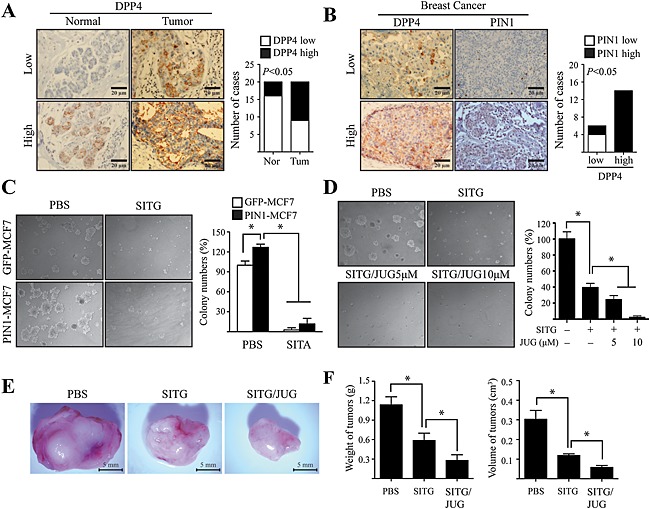
PIN1 overexpression induced by DPP4 is associated with human breast tumourigenesis. (A) Representative samples showing the results of immunohistochemical analysis of human normal breast tissue (Nor) and infiltrating ductal carcinoma of the breast (Tum), performed using an anti‐DPP4 antibody. Correlation was analysed using Fisher's exact test. (B) Representative samples showing the results of immunohistochemical analysis of infiltrating ductal carcinoma of the breast, performed using anti‐DPP4 and anti‐PIN1 antibodies on adjacent sections of samples. Correlations were analysed using Fisher's exact test. (C) MCF7 cells, mock transfected (GFP‐MCF7) or overexpressing PIN1 (PIN1‐MCF7), were treated with 1 mM sitagliptin (SITG) in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 days. The colonies from three separate experiments are photographed, and the average number of colonies was calculated. Columns represent the means ± SD of triplicate samples. P < 0.05, significantly different from control cells. (D) MCF7 cells were treated with 1 mM sitagliptin with/without additional treatment with juglone (JUG), dose dependently in soft agar matrix, and incubated at 37°C in a 5% CO2 atmosphere for 14 days. The colonies from three separate experiments are photographed, and the average number of colonies was calculated. Columns represent the means ± SD of triplicate samples. P < 0.05, significantly different from control cells. . (E and F) 4T1 cells were treated or not treated with 10 mM sitagliptin, alone (SITG) or in combination with 100 μM juglone (SITG/JUG). Treated cells were injected into the mammary glands of BALB/c mice (n = 30) and allowed to grow until tumours formed (14 days). Representative pictures of tumours (E) and tumour volumes and weights (F) are shown. Columns represent the means ± SD of triplicate samples. *P < 0.05, significantly different from control group (injected with cells mock‐treated with PBS only).
Discussion and conclusions
In this study, we have demonstrated that DPP4 regulates PIN1expression via induction of the transcription factor, E2F1. Furthermore, overexpression of DPP4 alone is sufficient to induce normal epithelial cells to display transformed properties and to enhance the transformed phenotype of epithelial cells that is induced by EGF. In contrast, inhibition of DPP4 by sitagliptin suppresses the EGF‐induced transformed phenotype, and combined treatment with sitagliptin and juglone, the latter a PIN1 inhibitor, significantly enhances sensitivity to sitagliptin with respect to inhibition of the colony formation of breast cancer cells and mammary gland tumour development. This is the first study to demonstrate that DPP4 up‐regulates PIN1 expression via E2F1 and thereby plays an essential role in EGF‐induced mammary tumourigenesis via PIN1.
The enzyme DPP4 is a well‐characterized glycoprotein, which regulates the activities of mitogenic growth factors and neuropeptides (Boonacker and Van Noorden, 2003). Studies have examined DPP4 expression across a wide spectrum of malignancies in an attempt to elucidate its potential role in tumour development (Iwata and Morimoto, 1999). In addition to the fact that DPP4 status may be altered in certain malignancies, DPP4 can affect growth, development and aggressive behaviour in a range of human malignancies (Havre et al., 2008). Several studies have observed loss or alteration of DPP4 in hepatocellular carcinoma (Stecca et al., 1997), melanoma (Roesch et al., 2006), epithelial ovarian carcinoma (Kajiyama et al., 2002) and non small cell lung cancer (Dimitrova et al., 2012). Furthermore, overexpression of DPP4 reduced invasive and metastatic potential through the up‐regulation of other important enzymes, such as E‐cadherin and tissue inhibitors of matrix metalloproteinases (Kajiyama et al., 2002; Kajiyama et al., 2003). In contrast, DPP4 also appears to have an important role in the development of selected haematological malignancies (Farag et al., 2013). Higher levels of DPP4 are expressed in chronic lymphocytic leukaemia B cells compared with their normal resting B cell counterparts (de Andrade et al., 2009). In addition, DPP4 expression is mainly found in aggressive subtypes of non‐Hodgkin lymphoma, such as T cell lymphoblastic lymphoma (T‐LBL)/T cell acute lymphoblastic leukaemia (T‐ALL) and T cell CD30+ anaplastic large cell lymphoma (Ribatti et al., 2013). Furthermore, the expression of DPP4 in T‐LBL/T‐ALL was found to be associated with significantly worse survival (Carbone et al., 1995). Similarly, in T cell large granular lymphocytic leukaemia, the presence of DPP4 is associated with a more aggressive clinical course (Havre et al., 2009). Apart from its expression on the tumour cell surface, DPP4 can also be found in the serum, and its levels are correlated with disease status and tumour behaviour in colorectal cancer (Cordero et al., 2000). While DPP4 expression seems to be associated with decreased progression in certain cancers, up‐regulation of DPP4 in others is associated with a more aggressive clinical course. This seemingly contrasting effect of DPP4 in different tumour types is probably explained in part by the multifunctional nature of this enzyme.
We report here, for the first time, that overexpression of DPP4 can play an important role in the transformation of mammary epithelial cells and in breast tumourigenesis. We showed that DPP4 overexpression in MCF7 cells strongly induced PIN1 expression via E2F1 activation and that this resulted in the activation of MEK/ERK and JNK/c‐Jun signalling pathways. In addition, knockdown of DPP4 in MCF7 cells lowered the PIN1 expression induced by EGF, indicating that PIN1 expression depends on DPP4 signalling via the E2F1 transcription factor. Moreover, treatment with sitagliptin, a DPP4 inhibitor, strongly suppressed MEK/ERK and JNK/c‐Jun signalling induced by EGF, via inhibition of PIN1 expression. Finally, in vivo results using the syngeneic murine 4T1 metastatic breast cancer model showed that DPP4 overexpression induced mammary tumourigenesis, whereas inhibition of DPP4 activity significantly reduced it. Although the role of DPP4 in tumourigenesis has been demonstrated, the physiological significance of DPP4 in breast cancer is not yet elucidated. To further understand the role of DPP4 in breast tumourigenesis, immunohistochemical analysis was performed on human breast cancer tissues, using anti‐DPP4 and anti‐PIN1 antibodies. We found that DPP4 levels were positively correlated with PIN1 expression in breast cancer tissues, which indicates that DPP4 functions as an important mediator of breast tumourigenesis via induction of PIN1 expression.
Phosphorylation of proteins on Ser/Thr–Pro is a key regulatory mechanism in controlling cell proliferation and transformation (Hunter, 1998; Zhou et al., 1999; Alt et al., 2000). The conformation and function of many phosphorylated proteins are regulated by the phosphorylation‐specific prolyl isomerase, PIN1 (Lu and Zhou, 2007). Interestingly, PIN1 is highly overexpressed in cancers of many human tissues and cells, including breast cancer cells (Ryo et al., 2001). It was known that PIN1 expression is regulated by the transcription factor E2F through its binding in the PIN1 promoter, which is enhanced by Neu or Ras (Ryo et al., 2002). Besides, E2F1 has been found to be a good prognostic or predictive marker of breast cancer because E2F1 significantly correlates with histological grade, stage, and metastatic status of breast tumour. (Zhang et al., 2000). We have shown that DPP4 is able to enhance binding of E2F1 in the PIN1 promoter for up‐regulation of PIN1 protein levels, suggesting that DPP4 signalling could have an important role in breast tumourigenesis, together with E2F1‐activated PIN1 overexpression. In addition, PIN1 regulates MAPK and STAT3 signalling pathways in breast tumour development through its interaction with MEK1 (Khanal et al., 2010), JNK1 (Park et al., 2012), c‐Jun (Wulf et al., 2001) and STAT3 (Lufei et al., 2007). These reports support our hypothesis that high levels of DPP4 regulate MAPK signalling pathways via PIN1, leading to tumour progression in breast cancer. Therefore, we examined the molecular events that are mediated by PIN1 in DPP4‐overexpressing or DPP4‐silenced MCF7 cells. Our present data provided the first evidence that DPP4 regulated PIN1‐induced signalling in such a way as to increase the phosphorylation of MEK1/2 and JNK1/2, thus resulting in increased AP‐1‐responsive promoter activity and neoplastic cellular transformation.
In conclusion, our studies have demonstrated that DPP4 signalling can activate expression of PIN1 and that overexpression of PIN1 in epithelial cells enhances their transformed phenotype. In contrast, inhibition of DPP4, by treatment of cells with sitagliptin, dramatically reduced both the cell proliferation and the transformed cellular phenotype. Importantly, the direct inhibition of PIN1 by juglone enhanced the inhibitory effect of sitagliptin on PIN1 via DPP4. Sitagliptin treatment is currently being used clinically in patients with T2DM (Aschner et al., 2006; Deacon and Holst, 2009) and can improve glucose homeostasis due to its ability to inhibit GLP‐1 degradation and enhance insulin secretion. These results suggest a model in which DPP4 signalling can activate expression of PIN1, which in turn enhances the phosphorylation of MEKs and JNKs, leading to cell proliferation and transformation. These findings raise the possibility that sitagliptin could offer a strategy for the treatment of mammary epithelial tumours in patients with T2DM.
Author contributions
H. J. C, J. Y. K, G. K and H. J. Y performed the research. S. C. L performed the research and analysed the data. H. S. C designed the research study, analysed the data and wrote the paper.
Conflict of interest
None.
Supporting information
Figure S1 Comparison of neoplastic transformation of JB6 Cl41 cells with overexpression of DPP4, DPP8 and DPP9. (A) Mock‐transfected (mock), DPP8‐overexpressing (His‐DPP8) and DPP9‐overexpressing (His‐DPP9) JB6 Cl41 cells were harvested, and proteins in whole‐cell lysates were separated by SDS‐PAGE and immunoblotted with anti‐His antibody against His‐DPP8 and His‐DPP9, respectively. (B) Mock, DPP4‐overexpressing (His‐DPP4), His‐DPP8, or His‐DPP9 JB6 Cl41 cells were seeded in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (left), and then the average number of colonies was calculated (right). Columns represent the mean of triplicate samples; bars show S. D. P < 0.05, compared to mock cells.
Figure S2 Inhibition of intracellular DPP4 activity after treatment of sitagliptin inMCF7 cells.MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of sitagliptin, as indicated, for 24 h. DPP4 activities measured by a fluorometric assay using Gly‐Pro‐AMC, as described in Methods. Columns represent the mean of triplicate samples; bars show standard deviation of the mean (S.D.). P < 0.05, compared to control cells.
Figure S3 Effects of overexpressing of DPP8 and DPP9 on the proliferation and colony formation of MCF7 cells. (A) Mocktransfected (mock), DPP8‐overexpressing (His‐DPP8) or DPP9‐overexpressing (His‐DPP9) MCF7 cells were harvested, and proteins in whole‐cell lysates were separated by SDSPAGE and immunoblotted with anti‐His antibody against His‐DPP8 and His‐DPP9, respectively. (B) Mock, His‐DPP8 or His‐DPP9 MCF7 cells were seeded and maintained at 37°C in a 5% CO2 atmosphere for 72 d. Cell proliferation was estimated using a 5‐bromo‐2‐deoxyuridine (BrdU) assay. (C and D) Mock, His‐DPP8, or His‐DPP9 MCF7 cells were seeded in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (C), and then the average number of colonies was calculated (D). Columns represent the mean of triplicate samples; bars show S.D.
Figure S4 Effects of 1G244 on the proliferation and colony formation of MCF7 cells. (A) Inhibition of intracellular DPP8 activity after treatment of 1G244 in MCF7 cells. MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of 1G244, as indicated, for 24 h. DPP8 activities measured by a Ala‐Pro‐AMC, as described in Methods. Columns represent the mean of triplicate samples; bars show S. D. P < 0.05, compared to control cells. (B) MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of 1G244, as indicated. Cell viability was estimated using a MTTassay. Columns represent the mean of triplicate samples; bars show S.D. P < 0.05, compared to control. (C and D)MCF7 cells were exposed to various concentrations of 1G244 in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (C), and then average number of colonies was calculated (D). Columns represent the mean of triplicate samples; bars show S.D.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute, funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C2004). This study also received support from a research grant funded by the Sunchon Research Center for Natural Medicine.
Choi, H. J. , Kim, J. Y. , Lim, S.‐C. . , Kim, G. , Yun, H. J. , and Choi, H. S. (2015) Dipeptidyl peptidase 4 promotes epithelial cell transformation and breast tumourigenesis via induction of PIN1 gene expression. British Journal of Pharmacology, 172: 5096–5109. doi: 10.1111/bph.13274.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt JR, Cleveland JL, Hannink M, Diehl JA (2000). Phosphorylation‐dependent regulation of cyclin D1 nuclear export and cyclin D1‐dependent cellular transformation. Genes Dev 14: 3102–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams‐Herman DE et al. (2006). Effect of the dipeptidyl peptidase‐4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 29: 2632–2637. [DOI] [PubMed] [Google Scholar]
- Boonacker E, Van Noorden CJ (2003). The multifunctional or moonlighting protein CD26/DPPIV. Eur J Cell Biol 82: 53–73. [DOI] [PubMed] [Google Scholar]
- Carbone A, Gloghini A, Zagonel V, Aldinucci D, Gattei V, Degan M et al. (1995). The expression of CD26 and CD40 ligand is mutually exclusive in human T‐cell non‐Hodgkin's lymphomas/leukemias. Blood 86: 4617–4626. [PubMed] [Google Scholar]
- Cordero OJ, Ayude D, Nogueira M, Rodriguez‐Berrocal FJ, de la Cadena MP (2000). Preoperative serum CD26 levels: diagnostic efficiency and predictive value for colorectal cancer. Br J Cancer 83: 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade CF, Bigni R, Pombo‐de‐Oliveira MS, Alves G, Pereira DA (2009). CD26/DPPIV cell membrane expression and DPPIV activity in plasma of patients with acute leukemia. J Enzyme Inhib Med Chem 24: 708–714. [DOI] [PubMed] [Google Scholar]
- Deacon CF, Holst JJ (2009). Saxagliptin: a new dipeptidyl peptidase‐4 inhibitor for the treatment of type 2 diabetes. Adv Ther 26: 488–499. [DOI] [PubMed] [Google Scholar]
- Dimitrova M, Ivanov I, Todorova R, Stefanova N, Moskova‐Doumanova V, Topouzova‐Hristova T et al. (2012). Comparison of the activity levels and localization of dipeptidyl peptidase IV in normal and tumor human lung cells. Tissue Cell 44: 74–79. [DOI] [PubMed] [Google Scholar]
- Drucker DJ, Nauck MA (2006). The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet 368: 1696–1705. [DOI] [PubMed] [Google Scholar]
- Eferl R, Wagner EF (2003). AP‐1: a double‐edged sword in tumorigenesis. Nature reviews . Cancer 3: 859–868. [DOI] [PubMed] [Google Scholar]
- Farag SS, Srivastava S, Messina‐Graham S, Schwartz J, Robertson MJ, Abonour R et al. (2013). In vivo DPP‐4 inhibition to enhance engraftment of single‐unit cord blood transplants in adults with hematological malignancies. Stem Cells Dev 22: 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischer B (1994). CD26: a surface protease involved in T‐cell activation. Immunol Today 15: 180–184. [DOI] [PubMed] [Google Scholar]
- Frasca F, Pandini G, Sciacca L, Pezzino V, Squatrito S, Belfiore A et al. (2008). The role of insulin receptors and IGF‐I receptors in cancer and other diseases. Arch Physiol Biochem 114: 23–37. [DOI] [PubMed] [Google Scholar]
- Fukuchi M, Fukai Y, Kimura H, Sohda M, Miyazaki T, Nakajima M et al. (2006). Prolyl isomerase Pin1 expression predicts prognosis in patients with esophageal squamous cell carcinoma and correlates with cyclinD1 expression. Int J Oncol 29: 329–334. [PubMed] [Google Scholar]
- Hanski C, Huhle T, Reutter W (1985). Involvement of plasma membrane dipeptidyl peptidase IV in fibronectin‐mediated adhesion of cells on collagen. Biol Chem Hoppe Seyler 366: 1169–1176. [DOI] [PubMed] [Google Scholar]
- Havre PA, Abe M, Urasaki Y, Ohnuma K, Morimoto C, Dang NH (2009). CD26 expression on T cell lines increases SDF‐1‐alpha‐mediated invasion. Br J Cancer 101: 983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havre PA, Abe M, Urasaki Y, Ohnuma K, Morimoto C, Dang NH (2008). The role of CD26/dipeptidyl peptidase IV in cancer. Front Biosci 13: 1634–1645. [DOI] [PubMed] [Google Scholar]
- Heike M, Mobius U, Knuth A, Meuer S, Meyer zum Buschenfelde KH (1988). Tissue distribution of the T cell activation antigen Ta1. Serological, immunohistochemical and biochemical investigations. Clin Exp Immunol 74: 431–434. [PMC free article] [PubMed] [Google Scholar]
- Hunter T (1998). Prolyl isomerases and nuclear function. Cell 92: 141–143. [DOI] [PubMed] [Google Scholar]
- Iwata S, Morimoto C (1999). CD26/dipeptidyl peptidase IV in context. The different roles of a multifunctional ectoenzyme in malignant transformation. J Exp Med 190: 301–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiyama H, Kikkawa F, Khin E, Shibata K, Ino K, Mizutani S (2003). Dipeptidyl peptidase IV overexpression induces up‐regulation of E‐cadherin and tissue inhibitors of matrix metalloproteinases, resulting in decreased invasive potential in ovarian carcinoma cells. Cancer Res 63: 2278–2283. [PubMed] [Google Scholar]
- Kajiyama H, Kikkawa F, Suzuki T, Shibata K, Ino K, Mizutani S (2002). Prolonged survival and decreased invasive activity attributable to dipeptidyl peptidase IV overexpression in ovarian carcinoma. Cancer Res 62: 2753–2757. [PubMed] [Google Scholar]
- Karin M (1995). The regulation of AP‐1 activity by mitogen‐activated protein kinases. J Biol Chem 270: 16483–16486. [DOI] [PubMed] [Google Scholar]
- Khanal P, Namgoong GM, Kang BS, Woo ER, Choi HS (2010). The prolyl isomerase Pin1 enhances HER‐2 expression and cellular transformation via its interaction with mitogen‐activated protein kinase/extracellular signal‐regulated kinase kinase 1. Mol Cancer Ther 9: 606–616. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160 (7): 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Kim G, Kim JY, Yun HJ, Lim SC, Choi HS (2014). Interleukin‐22 promotes epithelial cell transformation and breast tumorigenesis via MAP3K8 activation. Carcinogenesis 35: 1352–1361. [DOI] [PubMed] [Google Scholar]
- Larsson SC, Mantzoros CS, Wolk A (2007). Diabetes mellitus and risk of breast cancer: a meta‐analysis. Int J Cancer 121: 856–862. [DOI] [PubMed] [Google Scholar]
- Liou YC, Zhou XZ, Lu KP (2011). Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem Sci 36: 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE (2008). The impact of diabetes on survival following breast cancer. Breast Cancer Res Treat 109: 389–395. [DOI] [PubMed] [Google Scholar]
- Lipscombe LL, Goodwin PJ, Zinman B, McLaughlin JR, Hux JE (2006). Increased prevalence of prior breast cancer in women with newly diagnosed diabetes. Breast Cancer Res Treat 98: 303–309. [DOI] [PubMed] [Google Scholar]
- Loster K, Zeilinger K, Schuppan D, Reutter W (1995). The cysteine‐rich region of dipeptidyl peptidase IV (CD 26) is the collagen‐binding site. Biochem Biophys Res Commun 217: 341–348. [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ (2007). The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8: 904–916. [DOI] [PubMed] [Google Scholar]
- Lufei C, Koh TH, Uchida T, Cao X (2007). Pin1 is required for the Ser727 phosphorylation‐dependent Stat3 activity. Oncogene 26: 7656–7664. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentlein R (1999). Dipeptidyl‐peptidase IV (CD26) – role in the inactivation of regulatory peptides. Regul Pept 85: 9–24. [DOI] [PubMed] [Google Scholar]
- Mizutani S, Sumi S, Narita O, Tomoda Y (1985). Purification and properties of human placental dipeptidyl peptidase IV. Nihon Sanka Fujinka Gakkai zasshi 37: 769–775. [PubMed] [Google Scholar]
- Namgoong GM, Khanal P, Cho HG, Lim SC, Oh YK, Kang BS et al. (2010). The prolyl isomerase Pin1 induces LC‐3 expression and mediates tamoxifen resistance in breast cancer. J Biol Chem 285: 23829–23841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto E, Sugawara S, Takada H, Shoji S, Horiuch H (1999). Increase of CD26/dipeptidyl peptidase IV expression on human gingival fibroblasts upon stimulation with cytokines and bacterial components. Infect Immun 67: 6225–6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onitilo AA, Stankowski RV, Berg RL, Engel JM, Glurich I, Williams GM et al. (2014). Breast cancer incidence before and after diagnosis of type 2 diabetes mellitus in women: increased risk in the prediabetes phase. Eur J Cancer Prev 23: 76–83. [DOI] [PubMed] [Google Scholar]
- Oravecz T, Pall M, Roderiquez G, Gorrell MD, Ditto M, Nguyen NY et al. (1997). Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation, normal T cell expressed and secreted) by dipeptidyl peptidase IV (CD26)‐mediated cleavage. J Exp Med 186: 1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Lee JA, Park SG, Lee DH, Kim SJ, Kim HJ et al. (2012). A critical step for JNK activation: isomerization by the prolyl isomerase Pin1. Cell Death Differ 19: 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A (2013). The role of angiogenesis in human non‐Hodgkin lymphomas. Neoplasia 15: 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch A, Wittschier S, Becker B, Landthaler M, Vogt T (2006). Loss of dipeptidyl peptidase IV immunostaining discriminates malignant melanomas from deep penetrating nevi. Mod Pathol 19: 1378–1385. [DOI] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP (2002). PIN1 is an E2F target gene essential for Neu/Ras‐induced transformation of mammary epithelial cells. Mol Cell Biol 22: 5281–5295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP (2001). Pin1 regulates turnover and subcellular localization of beta‐catenin by inhibiting its interaction with APC. Nat Cell Biol 3: 793–801. [DOI] [PubMed] [Google Scholar]
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G et al. (2003). Regulation of NF‐kappaB signaling by Pin1‐dependent prolyl isomerization and ubiquitin‐mediated proteolysis of p65/RelA. Mol Cell 12: 1413–1426. [DOI] [PubMed] [Google Scholar]
- Stecca BA, Nardo B, Chieco P, Mazziotti A, Bolondi L, Cavallari A (1997). Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J Hepatol 27: 337–345. [DOI] [PubMed] [Google Scholar]
- Varona A, Blanco L, Perez I, Gil J, Irazusta J, Lopez JI et al. (2010). Expression and activity profiles of DPP IV/CD26 and NEP/CD10 glycoproteins in the human renal cancer are tumor‐type dependent. BMC Cancer 10: 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE et al. (2001). Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86: 1930–1935. [DOI] [PubMed] [Google Scholar]
- Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V et al. (2001). Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c‐Jun towards cyclin D1. EMBO J 20: 3459–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao TW, Kim WS, Yu DM, Sharbeen G, McCaughan GW, Choi KY et al. (2011). A novel role of dipeptidyl peptidase 9 in epidermal growth factor signaling. Mol Cancer Res 9: 948–959. [DOI] [PubMed] [Google Scholar]
- Zhang SY, Liu SC, Al‐Saleem LF, Holloran D, Babb J, Guo X et al. (2000). E2F‐1: a proliferative marker of breast neoplasia. Cancer Epidemiol Biomarkers Prev 9: 395–401. [PubMed] [Google Scholar]
- Zheng H, You H, Zhou XZ, Murray SA, Uchida T, Wulf G et al. (2002). The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 419: 849–853. [DOI] [PubMed] [Google Scholar]
- Zhou XZ, Lu PJ, Wulf G, Lu KP (1999). Phosphorylation‐dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell Mol Life Sci 56: 788806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of neoplastic transformation of JB6 Cl41 cells with overexpression of DPP4, DPP8 and DPP9. (A) Mock‐transfected (mock), DPP8‐overexpressing (His‐DPP8) and DPP9‐overexpressing (His‐DPP9) JB6 Cl41 cells were harvested, and proteins in whole‐cell lysates were separated by SDS‐PAGE and immunoblotted with anti‐His antibody against His‐DPP8 and His‐DPP9, respectively. (B) Mock, DPP4‐overexpressing (His‐DPP4), His‐DPP8, or His‐DPP9 JB6 Cl41 cells were seeded in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (left), and then the average number of colonies was calculated (right). Columns represent the mean of triplicate samples; bars show S. D. P < 0.05, compared to mock cells.
Figure S2 Inhibition of intracellular DPP4 activity after treatment of sitagliptin inMCF7 cells.MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of sitagliptin, as indicated, for 24 h. DPP4 activities measured by a fluorometric assay using Gly‐Pro‐AMC, as described in Methods. Columns represent the mean of triplicate samples; bars show standard deviation of the mean (S.D.). P < 0.05, compared to control cells.
Figure S3 Effects of overexpressing of DPP8 and DPP9 on the proliferation and colony formation of MCF7 cells. (A) Mocktransfected (mock), DPP8‐overexpressing (His‐DPP8) or DPP9‐overexpressing (His‐DPP9) MCF7 cells were harvested, and proteins in whole‐cell lysates were separated by SDSPAGE and immunoblotted with anti‐His antibody against His‐DPP8 and His‐DPP9, respectively. (B) Mock, His‐DPP8 or His‐DPP9 MCF7 cells were seeded and maintained at 37°C in a 5% CO2 atmosphere for 72 d. Cell proliferation was estimated using a 5‐bromo‐2‐deoxyuridine (BrdU) assay. (C and D) Mock, His‐DPP8, or His‐DPP9 MCF7 cells were seeded in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (C), and then the average number of colonies was calculated (D). Columns represent the mean of triplicate samples; bars show S.D.
Figure S4 Effects of 1G244 on the proliferation and colony formation of MCF7 cells. (A) Inhibition of intracellular DPP8 activity after treatment of 1G244 in MCF7 cells. MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of 1G244, as indicated, for 24 h. DPP8 activities measured by a Ala‐Pro‐AMC, as described in Methods. Columns represent the mean of triplicate samples; bars show S. D. P < 0.05, compared to control cells. (B) MCF7 cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of 1G244, as indicated. Cell viability was estimated using a MTTassay. Columns represent the mean of triplicate samples; bars show S.D. P < 0.05, compared to control. (C and D)MCF7 cells were exposed to various concentrations of 1G244 in a soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 d. The colonies from three separate experiments are photographed (C), and then average number of colonies was calculated (D). Columns represent the mean of triplicate samples; bars show S.D.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item