

Abstract
Background and Purpose
Kv1.3 potassium channels are promising pharmaceutical targets for treating immune diseases as they modulate Ca2+ signalling in T cells by regulating the membrane potential and with it the driving force for Ca2+ influx. The antimycobacterial drug clofazimine has been demonstrated to attenuate antigen‐induced Ca2+ oscillations, suppress cytokine release and prevent skin graft rejection by inhibiting Kv1.3 channels with high potency and selectivity.
Experimental Approach
We used patch‐clamp methodology to investigate clofazimine's mechanism of action in Kv1.3 channels expressed in HEK293 cells.
Key Results
Clofazimine blocked Kv1.3 channels by involving two discrete mechanisms, both of which contribute to effective suppression of channels: (i) a use‐dependent open‐channel block during long depolarizations, resulting in accelerated K+ current inactivation and (ii) a block of closed deactivated channels after channels were opened by brief depolarizations. Both modes of block were use‐dependent and state‐dependent in that they clearly required prior channel opening. The clofazimine‐sensitive closed‐deactivated state of the channel was distinct from the resting closed state because channels at hyperpolarized voltages were not inhibited by clofazimine. Neither were channels in the C‐type inactivated state significantly affected. Kv1.3 channels carrying the H399T mutation and lacking C‐type inactivation were insensitive to clofazimine block of the closed‐deactivated state, but retained their susceptibility to open‐channel block.
Conclusions and Implications
Given the prominent role of Kv1.3 in shaping Ca2+ oscillations, the use‐dependent and state‐dependent block of Kv1.3 channels by clofazimine offers therapeutic potential for selective immunosuppression in the context of autoimmune diseases in which Kv1.3‐expressing T cells play a significant role.
Abbreviations
- CLF
clofazimine
- FDA
Food and Drug Administration
- IPI
interpulse intervals
- WT
wild type
Tables of Links
| TARGETS |
|---|
| Kv1.3 channel |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
The shaker‐related Kv1.3 potassium channel (Grissmer et al., 1990) is a voltage‐gated ion channel that has a relatively limited expression pattern and is primarily found in various cells of the immune system (Cahalan et al., 2001; Gutman et al., 2003; Wulff et al., 2003a; Cahalan and Chandy, 2009), including subsets of T and B lymphocytes, natural killer cells, macrophages and microglia. It activates rapidly upon transmembrane depolarization and then inactivates in a time‐dependent manner by a mechanism called C‐type inactivation (Kurata and Fedida, 2006). This slow process of closing the channel was originally thought to involve the C‐terminus (Hoshi et al., 1991) but has since been shown to involve structural changes in the selectivity filter within the external mouth of the pore due to constriction (López‐Barneo et al., 1993) or dilation of the permeation pathway (Hoshi and Armstrong, 2013).
Kv1.3 channels serve at least two functions in T cells: (i) they stabilize the resting membrane potential and (ii) they participate in shaping oscillatory changes in membrane potential following antigen stimulation, which fuel oscillations in intracellular Ca2+ concentration through store‐operated Ca2+ release activated (CRAC) channels due to alterations in driving force for Ca2+ influx (Hoth and Penner, 1992; Lewis and Cahalan, 1995; Parekh and Penner, 1997; Cahalan et al., 2001; Cahalan and Chandy, 2009; Feske et al., 2012). The latter role is particularly important in effector memory T cells (Sallusto et al., 1999) as these cells massively upregulate the expression of Kv1.3 channels (Chandy et al., 2004; Wulff and Pennington, 2007; Cahalan and Chandy, 2009; Wulff et al., 2009). Autoreactive memory T cells have been implicated in the pathogenesis of important autoimmune diseases, including multiple sclerosis (Wulff et al., 2003b, 2009; Rus et al., 2005), type 1 diabetes (Beeton et al., 2006) and psoriasis (Nguyen et al., 2010). Due to its prominent role in the activation and function of human T cells, the Kv1.3 potassium channel has emerged as one of the most promising targets for developing novel immunosuppressants (Chandy et al., 2004; Beeton et al., 2006; Wulff et al., 2009). As a result, extensive efforts have been made to discover and develop small molecule inhibitors of Kv1.3 channels as immunosuppressants and immunomodulators (Nguyen et al., 2010), as this may enable specific targeting of effector memory T cells without causing wide‐spread immunosuppression.
Chemical library screens have identified several small‐molecule compounds with significant potency, including the iminodihydroquinoline CP‐339818 (Nguyen et al., 1996), the benzylpiperidine UK‐78282 (Hanson et al., 1999), the cyclohexyl‐substituted benzamides (Miao et al., 2003) and the sulfamidebenzamidoindanes (Wulff et al., 2003a). Natural products and polypeptide toxins were also found to potently block Kv1.3, including the triterpenoid correolide (Felix et al., 1999), the candelalides (Singh et al., 2001), as well as the polypeptide scorpion toxins charybdotoxin (Sands et al., 1989; Miller, 1995), margatoxin (Bartok et al., 2014), heterometrus spinnifer toxin (Rashid et al., 2014), noxiustoxin (Drakopoulou et al., 1995), kaliotoxin (Meki et al., 2000), Autoimmune Drug 1 from WenXin group (ADWX‐1) (Han et al., 2008) and (Arg22)‐Stichodactyla helianthus toxin (Norton et al., 2004; Beeton et al., 2011). Some of these compounds can block Kv1.3 with high potency in the pM to nM range (Cahalan and Chandy, 2009; Rashid et al., 2014); however, most of them are not overly selective and may block other K+ channels as well. In addition, the blocking mechanism of most of the aforementioned Kv1.3 inhibitors appears to impede K+ permeation through channel pore occlusion rather than state‐dependent mechanisms. Only a few inhibitors have been demonstrated to inhibit Kv1.3 in a state‐dependent manner, namely, the benzylpiperidine UK‐78282 (Hanson et al., 1999) and the phenoxyalkoxypsoralens Psora‐4 and PAP‐1 (Schmitz et al., 2005). They act by preferentially binding to the inactivated state.
Another promising small‐molecule inhibitor with good potency and favourable selectivity profile for Kv1.3 is clofazimine, an antimycobacterial drug that has been used in the treatment of leprosy since the early 1960s (Cholo et al., 2012). This drug emerged in an IL‐2 reporter gene bioassay screen of a chemical library of mostly Food and Drug Administration (FDA)‐approved clinical therapeutics (Ren et al., 2008). Clofazimine demonstrated efficacy as an inhibitor of T cell receptor‐mediated Ca2+ signalling and suppressed transcriptional activation of the IL‐2 gene in human T cells. Patch‐clamp analysis of T cells revealed that the drug selectively blocked the Kv1.3 channel activity in these cells, thereby affecting the amplitude and frequency of intracellular Ca2+ oscillations, and leading to the inhibition of the calcineurin/nuclear factor of activated T cells (NFAT) signalling pathway (Ren et al., 2008). Clofazimine was also effective in preventing human T cell‐mediated skin graft rejection in a reconstituted mouse model of skin transplantation (Ren et al., 2008). In addition, clofazimine's inhibitory effects on Kv1.3 has made it an attractive candidate for therapeutic use in cancer, because Kv1.3 channels are also located in the mitochondrial inner membrane of certain cancer cells, where they participate in apoptotic signalling (Leanza et al., 2012, 2013, 2014).
In this study, we determined the mechanism of block associated with clofazimine on human Kv1.3 channels that were stably expressed in HEK293 cells. We demonstrated that clofazimine blocks Kv1.3 in a unique manner involving two discrete mechanisms that both contribute to effective use‐dependent and state‐dependent suppression of Kv1.3 channels.
Methods
Cell culture and transfection
The drug/ion channel nomenclature used in this article conforms to British Journal of Pharmacology's concise guide to Pharmacology (Alexander et al., 2013). The pRc plasmids containing the entire coding sequence for hKv1.3 wild‐type (WT) gene and the sequence for the mutant channel H399T (both a generous gift from Dr Heike Wulff, UC Davis) were stably transfected into HEK293 cells. These cells showed average current amplitudes at +40 mV of 5 nA for the WT and 10 nA for the mutant. HEK293 cells stably expressing hKv1.3 WT and H399T mutant channels were cultured at 37°C with 10% CO2 in DMEM supplemented with 10% fetal bovine serum and G418 (500 µg·mL−1).
Electrophysiology
Patch‐clamp experiments were performed in the whole‐cell configuration at room temperature. Patch pipettes had typical resistances in the range of 1.8 to 2.5 MΩ. Data were acquired with an EPC‐9 amplifier and PatchMaster data acquisition software (HEKA, Lambrecht, Germany). Currents were filtered at 2.9 kHz and digitized at 100 µs intervals. Because Kv1.3‐mediated currents were typically several nA in magnitude and endogenous currents in HEK293 cells were rather small (typically <200 pA at +40 mV), no leak subtraction or P/n protocols were used. To avoid voltage errors and kinetic distortions, we limited data analysis to cells in which series resistance remained below 10 MΩ for the duration of the experiment. Cells were kept in standard external solution containing (mM): 140 NaCl, 2.8 KCl, 1 CaCl2, 2 MgCl2, 10 glucose and 10 HEPES‐NaOH. To assess voltage‐dependence of activation, external KCl was increased to 5 mM to augment tail current amplitudes. The internal pipette solution contained (mM): 140 K‐glutamate, NaCl 8 mM, MgCl2 1 mM, 10 HEPES‐KOH and 10 K‐EGTA. In all solutions, the pH was adjusted to 7.3 and the osmolarity to 290–310 mOsmol⋅L−1. In some experiments that required extended measurements of 30 min or more, we substituted intracellular K‐glutamate with KF, as KF helped stabilize recordings. Data comparison between these two solutions showed no difference in channel behaviour. External solution changes were made using the SmartSquirt delivery system (AutoMate Scientific, San Francisco CA, USA). clofazimine (Sigma) was dissolved in Dimethyl sulfoxide (DMSO) in a stock concentration of 10 mM and typically applied at a supramaximal concentration 10 μM, as determined from the IC50 obtained by fitting a dose–response equation:
where I min and I max are the maximal and minimal current amplitudes, x is the concentration, IC50 is the concentration of half‐maximal inhibition and nH is the Hill coefficient.
Pulse protocols and data analysis
Data were analysed with FitMaster (HEKA) and IgorPro (WaveMetrics, Lake Oswego, OR, USA). Ionic currents were typically recorded from a holding potential of –80 mV and step depolarized pulses to +40 mV with a typical pulse duration of 500 ms. Interpulse intervals of 30 s were used to reduce accumulation of C‐type inactivation.
Steady‐state activation was studied by measuring the peak tail current amplitudes generated when hyperpolarizing the membrane to –120 mV after a 20 ms step depolarization to various voltages between –70 and +80 mV. Data were normalized to maximum peak conductance at +80 mV (G max) and fit to a two‐state Boltzmann distribution:
| (1) |
where G max is the maximum conductance, V 50 is the half‐maximal conductance voltage and k is a slope factor.
To study steady‐state inactivation, cells were held at –80 mV and subjected to pre‐pulse potentials from −120 to –50 mV for 2 min before subjected to a +40 mV test pulse for 20 ms. Normalized peak currents were plotted versus pre‐pulse potentials to determine the fraction of current inactivated during the prepulse, and curves were fitted by the Boltzmann function:
| (2) |
where I max is the current recorded at +40 mV after the most hyperpolarizing prepulse (–120 mV) and V and k are the same as described previously.
Recovery from inactivation was studied by a dual‐pulse protocol in which the first pulse to +40 mV for 10 s was designed to inactivate all of the current and the second pulse of 20 ms to +40 mV was delivered after variable delays to assess the degree of recovery from inactivation. Dual pulses were spaced 30 s to enable recovery. Normalized peak current amplitudes were plotted and fitted to a single‐exponential function:
| (3) |
where A is the difference between the current amplitudes at time 0 (the end of the long depolarizing pulse) and the peak current elicited by the test pulse, t is the time and τ is the time constant. Similarly, C‐type inactivation of currents during long depolarizations was fitted by a monoexponential function.
Results
To characterize the mechanism of clofazimine's block, we established stable cell lines of HEK293 cells overexpressing the human WT Kv1.3 channel as well as a mutant version (H399T) that exhibits greatly reduced C‐type inactivation.
Basic properties of WT and H399T mutant Kv1.3 channels
We first performed basic characterizations of the recombinant channels and found WT Kv1.3 currents to reproduce the well‐known features of Kv1.3 channels in native or other heterologous systems (Vennekamp et al., 2004; Schmitz et al., 2005). WT Kv1.3 currents activated rapidly in response to depolarizing pulses in a voltage‐dependent manner (Figure 1A, left panel). During long depolarizations (Figure 1A, right panel), the outward K+ current decreased due to inactivation, a common property associated with Kv1.3 channels and referred to as C‐type inactivation (Hoshi et al., 1991; López‐Barneo et al., 1993; Hoshi and Armstrong, 2013).
Figure 1.
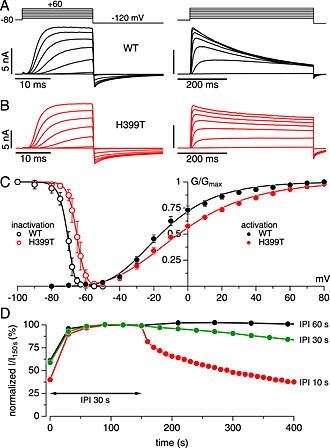
Basic properties of WT and H399T Kv1.3 channels expressed in HEK293 cells. (A) Examples of raw outward K+ currents evoked by increasing step depolarizations from –60 to +60 mV from a holding potential of –80 mV, followed by a hyperpolarizing pulse to –120 mV to assess tail currents. Left panel shows 20 ms depolarizing steps, and the right panel shows 500 ms depolarizations to illustrate C‐type inactivation. To augment tail currents, the external solution contained 5 mM KCl instead of the 2.8 mM. (B) Same as (A) but recorded from cells expressing the H399T mutant Kv1.3 channels. Note the strongly reduced C‐type inactivation during long depolarizing steps. (C) Activation and steady‐state inactivation behaviour of WT and H399T Kv1.3 currents. Activation data points (filled symbols) reflect mean peak tail currents (n = 4 for WT and n = 7 for H399T) at –120 mV after 20 ms step depolarizations to various potentials (see left panels in (A) and (B)). Data were fit using a Boltzmann function, yielding V half values of –16 mV for WT and –5 mV for the mutant. Steady‐state inactivation data (open symbols) reflect peak currents evoked by 20 ms depolarizing steps to +40 mV preceded by 2 min pre‐pulses to various potentials (n = 5 each for WT and H399T). Data were fit using a Boltzmann function, yielding V half values of –70 mV for WT and –65 mV for the mutant. (D) Time course of average normalized peak K+ currents evoked by 500 ms step depolarization to +40 mV from a holding potential of –80 mV at IPI of 60 s (n = 4), 30 s (n = 4) and 10 s (n = 3) after an initial stabilization of 120 s at IPI 30 s.
The H399T mutant channels activated in much the same way (Figure 1B, left panel), but showed very little C‐type inactivation over 500 ms (Figure 1B, right panel). Analysis of tail currents revealed similar activation properties of WT and H399T channels with half‐maximal activation voltages (V50) of –15 and –5 mV, respectively (Figure 1C, filled symbols). Steady‐state inactivation was assessed by short test pulses to +40 mV that were preceded by 2 min pre‐pulses to varying potentials and yielded V50 values for WT and H399T channels of –70 and –65 mV, respectively (Figure 1C, open symbols). Based on the previously mentioned characteristics, we selected –80 mV as our standard holding potential to minimize resting activity of channels and +40 mV test pulses to achieve nearly full activation of channels.
The time constant for Kv1.3 to recover from C‐type inactivation is typically 8–10 s (Figure 6), which requires extended interpulse intervals (IPI) in order to avoid accumulation of C‐type inactivation. Given the presence of C‐type inactivation, we assessed the frequency dependence of Kv1.3 currents during repeated depolarizations to +40 mV for 500 ms by varying the IPI (Figure 1D). We first let the currents stabilize using an IPI of 30 s, which is the standard interval used in the majority of studies on Kv1.3. Then we either kept the IPI at 30 s or adjusted it to 60 or 10 s. Increasing the IPI to 60 s produced very stable currents at the expense of fewer data points, whereas shorter IPI increased resolution but resulted in significant decay of Kv1.3 current amplitudes due to significant accumulation of C‐type inactivation. Maintaining the standard IPI at 30 s caused only a small decrease of Kv1.3 currents over time and represents a good compromise between current stability, kinetic resolution and total duration of individual experiments.
Figure 6.
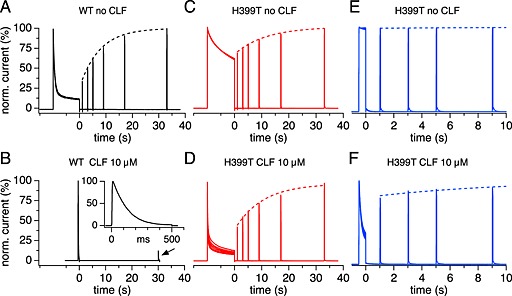
Recovery from inactivation in WT and H399T mutant channels in the absence and presence of clofazimine (CLF). Superimposed current records of cells subjected to double‐pulse protocols of a long conditioning step depolarization followed by a 100 ms test pulse timed with progressively increasing delays (IPIs of double pulses were 30 s). The dotted lines represent exponential fits to the peak currents obtained by the test pulses. In clofazimine experiments, cells were pre‐incubated for 10–20 min to achieve a steady‐state block before delivering the pulse protocols. (A) Recovery of WT channels in the absence of clofazimine had a time constant of 8 s (n = 4). (B) WT channels exposed to 10 μM clofazimine required only 500 ms initial depolarizations to completely block currents (expanded inset) and current amplitudes did not appreciably recover even after 30 s delay (n = 4). (C) In the absence of clofazimine, H399T mutant channels showed some slow C‐type inactivation over the initial 10 s depolarization. Recovery from this C‐type inactivation had a time constant of 11 s (n = 5). (D) H399T mutant channels exposed to clofazimine showed strong reduction of the current during the initial depolarization due to open‐channel block, but 40% of the channels recovered very quickly within 1 s, and the rest of the channels recovered with a time constant of 10 s (n = 5). (E) In H399T cells without clofazimine, a conditioning step pulse duration of 500 ms exhibited no C‐type inactivation, and subsequent test pulses remained at this level (n = 4). (F) H399T mutant channels exposed to clofazimine showed strong reduction of current even during 500 ms initial depolarization due to open‐channel block, but most of the channels recovered very quickly within 1 s, and the rest of the channels recovered with a time constant of 10 s (n = 4).
Kinetics of clofazimine block in Kv1.3 channels
We next investigated the pharmacological effects of clofazimine using the protocol illustrated in Figure 1, where cells were held at –80 mV and repeatedly stimulated by 500 ms step depolarizations to +40 mV spaced 30 s apart (Figure 2A). Before the application of clofazimine, we allowed current amplitudes to stabilize for several minutes. Clofazimine was then applied, and peak current amplitudes were normalized to the start of the clofazimine application (Figure 2A). Application of 10 μM clofazimine resulted in a slow progressive block of peak current amplitudes reaching average steady‐state levels of 83% block after 600 s. The level of steady‐state block varied somewhat due to variabilities in Kv1.3 current amplitudes and variable degrees of contaminating outward currents at +40 mV (e.g. leak, Cl−, non‐Kv1.3 K+ and transient receptor potential melastatin, member 7 (Yu and Kerchner, 1998; Nadler et al., 2001)).
Figure 2.

Kinetics of Kv1.3 block by acute application of clofazimine (CLF). (A) Time course of average normalized peak K+ currents evoked by 500 ms step depolarization to +40 mV from a holding potential of –80 mV (IPI 30 s) in the absence (control, n = 4) and the presence of 10 μM clofazimine (n = 5). (B) Examples of raw outward K+ currents obtained at the three time points labelled in (A) by step depolarizations to +40 mV from a holding potential of –80 mV in the absence (control) and the presence of 10 μM clofazimine. (C) Time constants of K+ current inactivation during the depolarization derived from mono‐exponential fits to the current decay at the three time points labelled in (A) and (B) in control and clofazimine‐treated cells.
The high‐resolution K+ currents evoked by voltage steps and used in the analysis in Figure 2A are illustrated in Figure 2B. They illustrate that control Kv1.3 currents decreased over time (labelled as traces 1, 2 and 3 corresponding to the points shown in Figure 2A), whereas application of 10 μM clofazimine caused a significant inhibition of peak current amplitudes (Figure 2B). Concurrent with the decrease in peak current amplitudes, clofazimine also accelerated the inactivation rates of K+ currents during the voltage pulse. We determined the inactivation rates in control and clofazimine‐treated cells at three different experimental times by fitting the current decays with a single exponential function and confirmed that the time constants of inactivation became shorter as clofazimine inhibited the current (Figure 2C).
Clofazimine block requires channel opening
We next determined whether clofazimine blocks Kv1.3 channels in the closed or open state. First, the Kv1.3 channels were activated during a series of control step pulses to +40 mV without clofazimine. Then, step pulses were suspended to return the channels into the closed state at a holding potential of –80 mV for 5 or 10 min before resuming test pulses (Figure 3A). In control cells (no clofazimine exposure during the 5 or 10 min pause), resumed stimulation initially produced slightly increased Kv1.3 current amplitudes (point 1) that gradually decreased in subsequent pulses (Figure 3A and C, left panel). This behaviour probably reflects the recovery of channels from residual C‐type inactivation during the stimulus‐free waiting period, followed by resumption of regularly spaced depolarizations. In cells that were exposed to 1 or 10 μM clofazimine during the 5 min pause, the first pulse showed no block of peak current when compared with the last control pulse in the absence of clofazimine (point C), although there was a slight decrease compared with the amplitude of the first pulse in control cells. This indicates that very little clofazimine block occurred while channels were closed. However, the subsequent traces (points 2 and 3) showed significant and dose‐dependent inhibition (Figure 2A–C).
Figure 3.
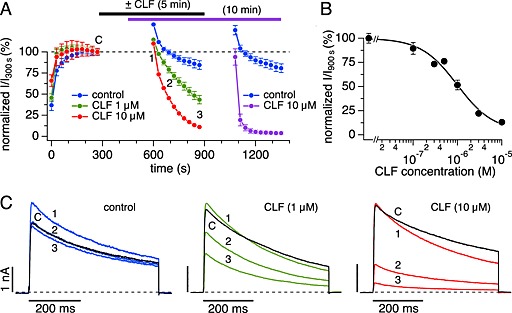
Kinetics of Kv1.3 block accelerate after stimulus‐free pre‐incubation with clofazimine (CLF). (A) Time course of average normalized peak K+ currents evoked by step depolarization to +40 mV (IPI 30 s) from a holding potential of –80 mV (current amplitude at 300 s was set to 100%). Data represent two experimental sets, showing the initial stabilization phase, followed by stimulus‐free pre‐incubation phase of 5 min (blue, green and red symbols) and a second data set in which pre‐incubation period was 10 min (blue and purple; initial stabilization phase of this set from 0 to 450 s not shown). Control data were in the absence of clofazimine (n = 3 for both sets). Data with 10 μM clofazimine pre‐incubation times of 5 min (red, n = 3) or 10 min (purple, n = 5) show the acceleration in blocking kinetics with pre‐incubation time. Data set with 1 μM clofazimine and 5 min pre‐incubation (green, n = 3) show dose‐dependence. (B) Dose–response curve for clofazimine‐mediated inhibition of Kv1.3 currents based on 5 min clofazimine pre‐incubations as shown in (A). Data points represent average normalized current amplitudes (n = 3–5) measured at point 3 in (A). Data were fit with a dose–response equation with IC50 = 1 μM and Hill coefficient = 1.2. (C) Examples of raw outward K+ currents obtained before pre‐incubation (point C, black traces) and three time points labelled in (A) after resuming stimulation by step depolarizations to +40 mV from a holding potential of –80 mV. Panels reflect K+ currents in the absence of clofazimine (blue) and after pre‐incubation for 5 min with 1 μM clofazimine (green) or 10 μM clofazimine (red).
Using this experimental protocol, we established the dose–response behaviour on Kv1.3 channels in this cell line at 900 s (point 3 in Figure 3A) and arrived at half‐maximal inhibition concentration (IC50) value of 1 ± 0.3 μM and a Hill coefficient of 1.2 (Figure 3B). This IC50 value is slightly higher than those previously reported for clofazimine inhibition of native Kv1.3 currents in Jurkat cells (0.3 μM) and heterologous mouse Kv1.3 in L292 cells (0.5 μM; (Ren et al., 2008)). The reason for this is probably the fact that the clofazimine block had not quite reached steady‐state after 5 min incubation. Indeed, with a longer incubation time of 10 min, the current was blocked very rapidly within a few pulses, much faster than the speed at which clofazimine typically blocked currents during constant pulsing (Figure 2) or 5 min incubation (Figure 3A). Taken together, these results suggest that clofazimine was able to reach the channel proteins in the membrane during the pre‐incubation time and inhibition then occurred rapidly as the channels opened and the drug interacted with its binding site.
Clofazimine block does not require C‐type inactivation
A depolarization of 500 ms, as employed in the previously mentioned experiments, will sequentially transfer Kv1.3 channels from the resting closed state to the open state and then into the inactivated state. To distinguish whether the clofazimine block developed from the open state and/or inactivated state, we performed similar experiments as those illustrated in Figure 3 but used shorter pulse durations of 20 ms. This still fully activated Kv1.3 currents but avoided C‐type inactivation (Figure 4B, see also Figure 1). In control cells stimulated by depolarizing pulses to +40 mV for 20 ms, C‐type inactivation was largely absent and resulted in only minor reduction in peak current amplitude (Figure 4A). In cells exposed to clofazimine during the waiting period, the same protocol resulted in the rapid block of Kv1.3 currents (Figure 4A and B). This would indicate that the clofazimine‐mediated block of Kv1.3 needs channel opening but does not require C‐type inactivation. Interestingly, the short pulses did not reveal significant inactivation within the 20 ms the channels were open, yet the peak currents evoked by subsequent pulses were reduced. Hence, the channel block, while requiring initial channel opening, must have proceeded during the IPI phase while the membrane was repolarized and channels were deactivated and closed.
Figure 4.
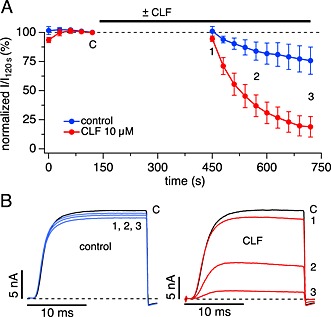
Clofazimine (CLF) block of Kv1.3 does not require C‐type inactivation. (A) Time course of average normalized peak K+ currents evoked by short (20 ms) step depolarization to +40 mV (IPI 30 s) from a holding potential of –80 mV in the absence of clofazimine (control, n = 6) and after 5 min pre‐incubation with 10 μM clofazimine (n = 5). (B) Examples of raw outward K+ currents obtained before pre‐incubation (point C, black traces) and three time points labelled in (A) after resuming stimulation by step depolarizations to +40 mV from a holding potential of –80 mV. Panels reflect K+ currents in the absence of clofazimine (control) and after pre‐incubation with 10 μM clofazimine.
Clofazimine blocks open channels in Kv1.3 mutant H399T that lacks C‐type inactivation
As shown in Figures 2 and 3, clofazimine not only reduced the overall K+ current amplitudes over time but also accelerated the current inactivation during 500 ms long depolarizations above and beyond the reduction caused by intrinsic C‐type inactivation. This could be due to a use‐dependent open‐channel block or to enhanced C‐type inactivation caused by the drug. We investigated this question in mutant Kv1.3 channels in which C‐type inactivation is diminished through a single amino acid substitution of the positively charged histidine 399 in the outer vestibule of Kv1.3 by threonine (H399T) (Dreker and Grissmer, 2005). The analysis of raw membrane currents evoked by depolarizing pulses to +40 mV in cells expressing WT Kv1.3 (Figure 5B, left panel) revealed that clofazimine inhibited the current both at the peak and at the end of the depolarizing voltage step (Figure 5A, left and right panels, respectively). In contrast, clofazimine failed to inhibit Kv1.3 peak currents in the mutant H399T channel (Figure 5A, left panel). However, clofazimine was still able to affect the currents of H399T mutant channels in that the normally non‐inactivating currents of the mutant exhibited a marked clofazimine‐mediated inactivation (Figure 5B, right panel), causing a gradual decline of the currents measured at the end of the depolarizing voltage pulse (Figure 5A, right panel). This indicates that clofazimine causes a use‐dependent open‐channel block rather than enhanced C‐type inactivation. However, this block is not equivalent to the cumulative inhibition of peak currents observed in WT channels, since peak currents of the mutant channels were not affected, and the block mostly reversed during the repolarization between depolarizing pulses.
Figure 5.
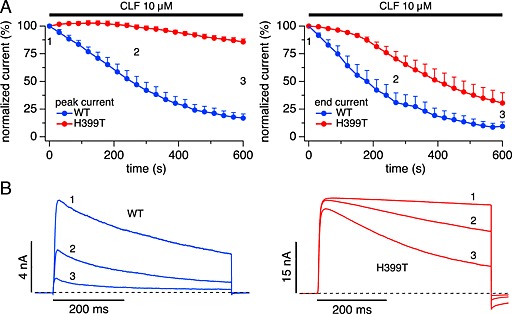
Mutant Kv1.3 H399T loses peak current inhibition but retains open‐channel block by clofazimine (CLF). (A) Time course of average normalized K+ currents evoked by step depolarization to +40 mV (IPI 30 s) from a holding potential of –80 mV in cells expressing wild‐type (WT, n = 5) and mutant H399T channels (red, n = 4). Application of 10 μM clofazimine had little effect on H399T peak currents (left panel) but inhibited current amplitude at the end of the pulse (right panel). (B) Examples of raw outward K+ currents obtained at the 3 time points labelled in (A) evoked by step depolarizations to +40 mV from a holding potential of –80 mV. Panels reflect K+ currents in cells expressing WT and H399T mutant channels.
Clofazimine locks Kv1.3 channels in a closed state
Following C‐type inactivation, Kv1.3 channels slowly return to a closed state from which they can be re‐activated. Figure 6 illustrates the recovery rates from C‐type inactivation in WT channels with a high degree of C‐type inactivation and in mutant H399T channels with a low degree of C‐type inactivation. The pulse protocol consisted of two depolarizing steps to +40 mV spaced 1–38 s apart. The first pulse had a fixed duration of 10 s to achieve nearly complete C‐type inactivation, while the second pulse lasted 100 ms to assess the level of recovery from C‐type inactivation relative to the preceding pulse. To prevent accumulation of C‐type inactivation between the paired pulses, a 30‐s interval was imposed after each pair of pulses. For clofazimine experiments, the same cells were pre‐incubated for 10–20 min to achieve a steady‐state block and the same double‐pulse protocol described previously was repeated.
In the absence of clofazimine, WT Kv1.3 currents exhibited strong inactivation during the 10 s depolarizing pulse and recovered with a time constant of 8 s (Figure 6A). Clofazimine completely inhibited Kv1.3 currents within just 500 ms probably due to the combination of C‐type inactivation and open‐channel block, obviating the need for long depolarizations (Figure 6B). Once inactivated, the currents did not recover significantly even after 30 s (12% recovery), indicating that clofazimine essentially locked the channels in the inactivated and/or blocked state (Figure 6B). While 500 ms depolarization of the mutant H399T currents showed no significant inactivation (Figure 6E), long depolarizations of 10 s caused partial inactivation (Figure 6C), which recovered with a time constant of 11 s, similar to WT currents.
We next analysed the effect of clofazimine on recovery from C‐type inactivation using identical protocols (Figure 6D and F). In cells expressing mutant H399T Kv1.3 channels, clofazimine greatly enhanced the current decay during the 500 ms depolarization, but this effect was rapidly reversed (Figure 6F), reflecting the recovery from open‐channel block. When applying very long depolarizations of 10 s clofazimine caused nearly complete inactivation of the H399T current due to the combination of C‐type inactivation (as in Figure 6C) and open‐channel block (as in Figure 6F). However, 40% of the channels recovered very quickly within 1 s, and the rest of the channels recovered with a time constant of 10 s, similar to the recovery rates seen in Figure 6C without clofazimine. The fast recovery likely reflects the recovery from the open‐channel block and the slower kinetics likely reflects recovery from residual C‐type inactivation of the mutant channels. Interestingly, the portion of C‐type inactivated mutant channels fully recovered over 30–40 s (Figure 6D), unlike the persistently inhibited WT channels (Figure 6B). Together, these data indicate that clofazimine exerts two modes of inhibition on Kv1.3: (i) a readily reversible block of the open channel affecting both WT and H399T mutant channels during long step depolarizations and (ii) a persistent block of WT channels that have either undergone C‐type inactivation or are otherwise in some closed state and prevent Kv1.3 from re‐opening. The mutant H399T channels are not persistently inhibited, even when mutant channels are driven into the C‐type inactivated state with very long depolarizations.
If clofazimine locked Kv1.3 channels in the inactivated or closed state, it might affect the reversibility of the clofazimine block when attempting to wash out clofazimine. We tested this in WT and H399T‐expressing cells by using the same protocol as shown in Figure 3, where cells were exposed to clofazimine for 5 min in the absence of depolarizing pulses (to enable binding of the compound) and subsequently stimulated by depolarizing voltage pulses that completely suppressed WT Kv1.3 currents within a few pulses (Figure 7A). After complete block of the current, cells were perfused with clofazimine‐free external solution, but this wash out resulted in very little recovery of current. In fact, significant amounts of clofazimine were probably still present as the initially recovered current was rapidly re‐blocked. In contrast, the same experimental protocol applied to H399T mutant channels, which exhibit very little C‐type inactivation, caused a progressive inhibition of currents during the depolarizing pulses due to the clofazimine‐mediated open‐channel block, but this block was readily reversible upon wash out of clofazimine (Figure 7B). This behaviour is consistent with the hypothesis that clofazimine indeed locks Kv1.3 channels in the inactivated or otherwise closed state, possibly due to stronger binding with reduced dissociation constant or due to conformational changes of the channel that occlude the binding site of clofazimine and hinder the wash out of the compound.
Figure 7.
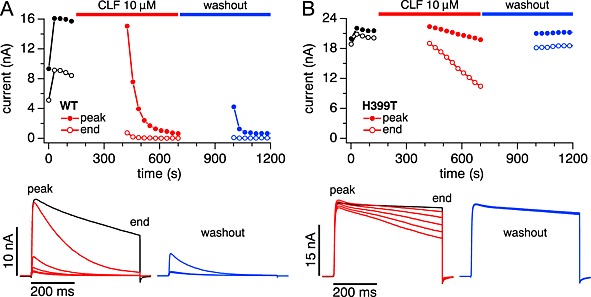
Recovery from block by washout is absent in WT and fast in H399T mutant channels. Cells were pre‐incubated with 10 μM clofazimine (CLF) for 5 min before resuming stimulation and allowing the currents to be blocked for another 5 min. Then stimulation was suspended and clofazimine was washed out by perfusing cells with standard external solution. After 5 min of washout, stimulation was resumed to assess the degree of recovery. (A) Representative example (n = 7) of the time course of block and recovery in WT Kv1.3 currents (top panel) and raw K+ currents (bottom panels) during the block phase (red) and washout phase (blue). Note that clofazimine does not appear to actually wash out, because the small fraction of channels that recovered during the washout phase were immediately re‐blocked. (B) Same as (A) but for H399T mutant Kv1.3 in a representative cell (n = 4). Here, washout appears to remove clofazimine from the channels since no re‐block occured (blue traces).
Clofazimine does not affect inactivated channels
The previous experiments suggested that clofazimine can access resting closed channels without blocking them. In order to inhibit Kv1.3, clofazimine requires the channels to open. This then leads to a block of open channels during the depolarization as well as the persistent inhibition of those channels that enter the inactivated or possibly some other closed state. Finally, we tested whether clofazimine could act on channels that were already in the inactivated state, which is the mechanism of action that has previously been proposed for the Kv1.3 inhibitor PAP‐1 (Schmitz et al., 2005).
Adopting the same stimulation protocol (Schmitz et al., 2005), Kv1.3 channels were driven into the inactivated state by a train of five depolarizing pulses to +40 mV for 2 s (Figure 8), followed by 5 min pause at the holding potential of –80 mV. During this pause, Kv1.3 channels completely recovered from C‐type inactivation, and the second train of pulses produced an increased response compared with the first. In control cells, this protocol could be repeated several times without any significant decrease in the response to the first pulse of the subsequent train (Figure 8A and C). To test clofazimine effects on C‐type inactivated channels, cells were immediately exposed to 10 μM clofazimine right after completion of five pulses of the second train, and a further set of five pulses was appended to ensure that most channels remained in the inactivated state. Thereafter, cells remained exposed to clofazimine during the 5 min waiting period before the third train of pulses was delivered to test whether clofazimine was able to bind and block inactivated channels. Interestingly, most of the channels could be reactivated after the waiting period during the first pulse of the third train. The rest of the pulses in the third train failed to evoke any current whatsoever, and waiting another 5 min for recovery failed to produce reactivate significant current. The slightly reduced current of the first pulse of the third train compared with controls could have been due to clofazimine‐mediated block of some channels that opened during six to ten pulses of the second train. This suggests that clofazimine does not affect C‐type inactivated channels very strongly and requires channels to first open before it can lock them in the closed state.
Figure 8.

Clofazimine (CLF) does not block channels in the C‐type inactivated state. Trains of high‐frequency depolarizations (2 s duration, 1 s IPI, holding potential: –80 mV and depolarization voltage: +40 mV) spaced by 5 min inter‐train intervals. (A) Representative control experiment superimposing the currents evoked by each of the four trains of stimuli (illustrated by the stimulus protocol at the top). Note that repeated trains behave very similarly in that the first response of each train employed fully recovered Kv1.3 channels. (B) Same as (A) but exposing cells to clofazimine during six to ten pulses of the second stimulus train. The first train caused C‐type inactivation of Kv1.3 channels. After a 5 min pause, channels recovered from C‐type inactivation and the second train behaved like the first. The additional five pulses of the second train maintained C‐type inactivation while 10 μM clofazimine was applied. After this 10‐pulse train, clofazimine was allowed to interact with inactivated channels. The third train revealed that most of the channels recovered from C‐type inactivation and were not affected by clofazimine. Once these channels opened, they were immediately blocked, and all subsequent pulses were essentially flat and did not significantly recover for the fourth train. (C) Average peak currents for each pulse normalized to the peak amplitude of the first pulse in the first train (n = 8 for control and n = 7 for clofazimine‐treated cells). Data points are colour coded to match the raw current traces in (A) and (B).
Discussion and conclusions
In the present study, we have characterized the inhibitory mechanism of action of clofazimine on the human potassium channel Kv1.3. This channel is of major importance in regulating the resting membrane potential as well as the membrane potential oscillations that occur following activation of a variety of immune cells (Lewis and Cahalan, 1995; Cahalan et al., 2001; Cahalan and Chandy, 2009; Feske et al., 2012), including memory T and B cells and activated macrophage/microglia cells. Given this context and the potential of clofazimine as a therapeutic option for treating autoimmune diseases, it was important to understand how clofazimine inhibits Kv1.3. The most remarkable finding of this investigation was that clofazimine interacts with and ultimately blocks Kv1.3 channels in a precise and state‐dependent manner. In order for clofazimine to inhibit Kv1.3, it requires the channel to open while resting closed or C‐type inactivated states remain largely unaffected. Once opened, clofazimine will block the channels regardless of whether they are kept activated by depolarization or deactivated by repolarization. This use‐dependent mechanism of block endows clofazimine with a unique pharmacodynamic profile that would effectively suppress hyperpolarizing K+ currents in immune cells in which Kv1.3 is upregulated.
To elucidate clofazimine's mechanism of Kv1.3 channel block, we used the HEK cell line HEK293 in which we stably expressed human Kv1.3. Heterologous expression of Kv1.3 has been shown to faithfully reproduce the behaviour of native channels (Grissmer et al., 1990; Cai et al., 1992; Deutsch and Chen, 1993; Hou et al., 2014) and our basic characterization of Kv1.3 currents, both WT and H399T mutant channels, confirm this (Figure 1). This offers a convenient and reliable way to study drug effects with minimal contaminations by other ion channel species. Indeed, the present study confirms that clofazimine blocks heterologously expressed Kv1.3 channels as previously demonstrated for native human Kv1.3 in Jurkat T cells as well as heterologous mouse Kv1.3 expressed in L929 cells (Ren et al., 2008). The IC50 value of 1 μM we established for this cell line (Figure 3) is in reasonable agreement with those determined previously (0.3 to 0.5 μM; (Ren et al., 2008)). The only notable difference we observed was the kinetics of inhibition, which was somewhat slower in the present study (~220 s to half‐maximal inhibition; Figure 2) compared with ~30 s in Jurkat cells and L292 cells (Ren et al., 2008). Possible factors affecting the drug's kinetics include different diffusional barriers such as cell membrane compositions, different protein expression levels and/or different stimulus protocols employed in the studies. We surmise that the primary determinant of the kinetics observed in the present study is caused by the traversal or membrane incorporation of the drug and binding to the channel since the actual block of the current occurred very rapidly when cells were pre‐incubated with clofazimine and subsequently stimulated (Figure 3).
The kinetic behaviour of Kv1.3 is well understood and generally believed to comprise several closed states, a strongly voltage‐dependent open state and the so‐called C‐type inactivated state (Marom and Levitan, 1994; Panyi et al., 1995; Levy and Deutsch, 1996; Hou et al., 2014):
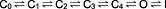
In H399T mutant Kv1.3 channels, the inactivated state is largely absent, and these channels open normally (Figure 1) without significant inactivation over 500 ms (Figures 1, 5 and 6). The data presented here indicate that clofazimine can bind to and interact with one or more of the closed states (C0–C4) of either WT or mutant Kv1.3 without impeding their transition into the open state when the membrane is depolarized. This is evident from the full availability of channels evoked by the first depolarizing pulse after pre‐incubating cells with clofazimine (Figure 3). Also clofazimine does not seem to affect the inactivated state very strongly, since exposure of inactivated channels to clofazimine did not preclude significant re‐activation (Figures 6 and 8). According to the previously mentioned state diagrams model, this would leave the open state of Kv1.3 as the state in which clofazimine causes the block. Indeed, as soon as Kv1.3 channels enter the open state (O), they cause an open‐channel block in a time‐dependent manner. This block superimposes on the intrinsic C‐type inactivation of WT Kv1.3 channels and causes accelerated inactivation of macroscopic currents (Figure 2). The open‐channel block is preserved and can be observed in isolation in mutant H399T Kv1.3 channels that have minimal C‐type inactivation (Dreker and Grissmer, 2005) (Figure 5). This open‐channel block is easily and rapidly reversible in the mutant Kv1.3 channels (Figure 6) and therefore is unlikely to reflect an alternative mechanism such as clofazimine‐mediated acceleration of C‐type inactivation in the mutant channels.
Interestingly, drug interactions with verapamil have suggested that Kv1.3 might have a second open state (Kuras and Grissmer, 2009). This study found that recovery from verapamil block was mainly due to the voltage‐dependent closing of channels (state‐dependent block), implying a second open state of the channel and concluding that the wild‐type Kv1.3 channel undergoes at least two different conformational changes before finally closing, with a low verapamil affinity in one open state and a high verapamil affinity in the other open state:

Because the open‐channel block by clofazimine is rapidly reversible in the mutant Kv1.3 (Figures 6 and 7), it appears to be distinct from the nearly irreversible block of the WT channels, requiring us to consider an additional state of block in WT channels. We must also consider that the inhibition of WT channels can proceed while channels are closed due to repolarization from short depolarizations of 20 ms in which no open‐channel block is evident (Figure 4). This would require clofazimine to inhibit Kv1.3 in a closed state that affects the WT channel but not the mutant channel, suggesting that this closed state is not one of the C0 to C4 states in the classical state diagram. We propose to add an additional closed state adjacent to the open state, which we could denote ‘deactivated closed’ (CD) with clofazimine affecting both the open and the deactivated closed state, denoted by the asterisks in the following diagram:
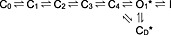
Alternatively, if Kv1.3 indeed has a second open state (Kuras and Grissmer, 2009), then the model could adopt the following form:

This model can explain the phenomenology of the clofazimine block of WT Kv1.3 channels in that clofazimine binds to the channel in any of its closed states C0–C4 without blocking it. Upon a long depolarization, the channel first enters the open state(s) and then experiences both C‐type inactivation and open‐channel block by clofazimine. In mutant channels, the C‐type inactivation is absent, so we only observe the clofazimine‐mediated open‐channel block. The proposed closed deactivated state would help explain the observation that clofazimine can block channels in their deactivated state after repolarization from short depolarizing pulses in which channels open, but neither C‐type inactivation nor open‐channel block is evident. It appears that clofazimine locks the channels in that deactivated closed state, and they do not become available for reactivation, resulting in the persistent block we observed in WT channels. For the mutant channels, we must assume that similar to the inactivated state, they cannot access the closed deactivated state and therefore are not persistently blocked by clofazimine.
The dual‐mode, use‐dependent block of Kv1.3 by clofazimine represents a novel mechanism of action that is distinct from the known Kv1.3 inhibitors described so far. Clofazimine exhibits genuine use‐dependence in that Kv1.3 currents exhibit accelerated inhibition of currents during a depolarization in both wild‐type Kv1.3 and in mutant channels that lack C‐type inactivation. This may be analogous to the state‐dependent block observed with verapamil (Kuras and Grissmer, 2009). In addition, clofazimine exhibits a second state‐dependent block that occurs in a presumably closed deactivated state of WT Kv1.3 channels, which channels adopt when repolarizing the membrane within a few tens of milliseconds after full activation. Although the inhibition of the closed deactivated state by clofazimine may be considered equivalent to the PAP‐1‐mediated inhibition of the C‐type inactivated state (Dreker and Grissmer, 2005), these two closed states are functionally distinct, and the two blockers may therefore have different pharmacodynamic effects. While both compounds compare favourably to simple pore blockers in that they spare Kv1.3 channel inhibition at rest, clofazimine's mode of action could offer therapeutic benefits over blockers that preferentially bind to the C‐type inactivated state since clofazimine merely requires opening of channels to suppress Kv1.3 channels, whereas the efficacy of block by, for example, PAP‐1 will probably depend on the degree of channels transitioning from the open state into C‐type inactivation in vivo. In addition, while some Kv1.3 inhibitors in preclinical development appear to have good selectivity against other K+ channels, their potential off‐target effects in therapeutic settings remain to be investigated, whereas clofazimine has gained FDA approval and is generally considered safe for clinical use (Fajardo et al., 1999; Gurfinkel et al., 2009; Cholo et al., 2012).
Taken together, the results of the present study establish the mechanism of action of clofazimine on Kv1.3 channels. The inhibition of Kv1.3 is consistent with previously observed suppression of Ca2+ oscillations in T cells by clofazimine (Ren et al., 2008) and is probably caused by the reduction in driving force Ca2+ entry in the absence of Kv1.3‐mediated hyperpolarizations. Given the prominent role of Kv1.3 in shaping Ca2+ oscillations in effector memory T and B cells as well as macrophages, dendritic cells and microglia, clofazimine may offer potential therapeutic benefits in treating immune diseases in which these cells participate.
Acknowledgements
We wish to thank Dr Heike Wulff for generously providing the human Kv1.3 and H399T cDNA and Mahealani Monteilh‐Zoller and Rebecca Kim for their expert technical assistance. We also thank Drs Stefan Heinemann, Zoltan Varga and Gyorgi Panyi for their helpful discussions. Partial support was provided through a private donation by Ms Amy Chong (RP).
Conflict of interest
All authors declare no conflict of interest.
Author contribution
M. F. and J. S. are responsible for the design, execution and interpretation of data and manuscript preparation. R. P. is responsible for the design, interpretation of data and manuscript preparation.
Faouzi, M. , Starkus, J. , and Penner, R. (2015) State‐dependent blocking mechanism of Kv1.3 channels by the antimycobacterial drug clofazimine. British Journal of Pharmacology, 172: 5161–5173. doi: 10.1111/bph.13283.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al. (2013). The concise guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartok A, Toth A, Somodi S, Szanto TG, Hajdu P, Panyi G et al. (2014). Margatoxin is a non‐selective inhibitor of human Kv1.3 K+ channels. Toxicon 87: 6–16. [DOI] [PubMed] [Google Scholar]
- Beeton C, Pennington MW, Norton RS (2011). Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm Allergy Drug Targets 10: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW et al. (2006). Kv1.3 channels are a therapeutic target for T cell‐mediated autoimmune diseases. Proc Natl Acad Sci U S A 103: 17414–17419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD, Chandy KG (2009). The functional network of ion channels in T lymphocytes. Immunol Rev 231: 59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD, Wulff H, Chandy KG (2001). Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol 21: 235–252. [DOI] [PubMed] [Google Scholar]
- Cai YC, Osborne PB, North RA, Dooley DC, Douglass J (1992). Characterization and functional expression of genomic DNA encoding the human lymphocyte type n potassium channel. DNA Cell Biol 11: 163–172. [DOI] [PubMed] [Google Scholar]
- Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD (2004). K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci 25: 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R (2012). Clofazimine: current status and future prospects. J Antimicrob Chemother 67: 290–298. [DOI] [PubMed] [Google Scholar]
- Deutsch C, Chen LQ (1993). Heterologous expression of specific K+ channels in T lymphocytes: functional consequences for volume regulation. Proc Natl Acad Sci U S A 90: 10036–10040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakopoulou E, Cotton J, Virelizier H, Bernardi E, Schoofs AR, Partiseti M et al. (1995). Chemical synthesis, structural and functional characterisation of noxiustoxin, a powerful blocker of lymphocyte voltage‐dependent K+ channels. Biochem Biophys Res Commun 213: 901–907. [DOI] [PubMed] [Google Scholar]
- Dreker T, Grissmer S (2005). Investigation of the phenylalkylamine binding site in hKv1.3 (H399T), a mutant with a reduced C‐type inactivated state. Mol Pharmacol 68: 966–973. [DOI] [PubMed] [Google Scholar]
- Fajardo TT, Abalos RM, dela Cruz EC, Villahermosa LG, Walsh DS, Cellona RV et al. (1999). Clofazimine therapy for lepromatous leprosy: a historical perspective. Int J Dermatol 38: 70–74. [DOI] [PubMed] [Google Scholar]
- Felix JP, Bugianesi RM, Schmalhofer WA, Borris R, Goetz MA, Hensens OD et al. (1999). Identification and biochemical characterization of a novel nortriterpene inhibitor of the human lymphocyte voltage‐gated potassium channel, Kv1.3. Biochemistry 38: 4922–4930. [DOI] [PubMed] [Google Scholar]
- Feske S, Skolnik EY, Prakriya M (2012). Ion channels and transporters in lymphocyte function and immunity. Nat Rev Immunol 12: 532–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissmer S, Dethlefs B, Wasmuth JJ, Goldin AL, Gutman GA, Cahalan MD et al. (1990). Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci U S A 87: 9411–9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurfinkel P, Pina JC, Ramos‐e‐Silva M (2009). Use of clofazimine in dermatology. J Drugs Dermatol 8: 846–851. [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE et al. (2003). International Union of Pharmacology. XLI. Compendium of voltage‐gated ion channels: potassium channels. Pharmacol Rev 55: 583–586. [DOI] [PubMed] [Google Scholar]
- Hanson DC, Nguyen A, Mather RJ, Rauer H, Koch K, Burgess LE et al. (1999). UK‐78,282, a novel piperidine compound that potently blocks the Kv1.3 voltage‐gated potassium channel and inhibits human T cell activation. Br J Pharmacol 126: 1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Yi H, Yin S‐J, Chen Z‐Y Liu H, Cao Z‐J et al. (2008). Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J Biol Chem 283: 19058–19065. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Armstrong CM (2013). C‐type inactivation of voltage‐gated K+ channels: pore constriction or dilation? J Gen Physiol 141: 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1991). Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy‐terminal region. Neuron 7: 547–556. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R (1992). Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature 355: 353–356. [DOI] [PubMed] [Google Scholar]
- Hou P, Zhang R, Liu Y, Feng J, Wang W, Wu Y et al. (2014). Physiological role of Kv1.3 channel in T lymphocyte cell investigated quantitatively by kinetic modeling. PLoS One 9: e89975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras Z, Grissmer S (2009). Effect of K+ and Rb+ on the action of verapamil on a voltage‐gated K+ channel, hKv1.3: implications for a second open state? Br J Pharmacol 157: 757–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata HT, Fedida D (2006). A structural interpretation of voltage‐gated potassium channel inactivation. Prog Biophys Mol Biol 92: 185–208. [DOI] [PubMed] [Google Scholar]
- Leanza L, Henry B, Sassi N, Zoratti M, Chandy KG, Gulbins E et al. (2012). Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak‐independent death of cancer cells. EMBO Mol Med 4: 577–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leanza L, O'Reilly P, Doyle A, Venturini E, Zoratti M, Szegezdi E et al. (2014). Correlation between potassium channel expression and sensitivity to drug‐induced cell death in tumor cell lines. Curr Pharm Des 20: 189–200. [DOI] [PubMed] [Google Scholar]
- Leanza L, Trentin L, Becker KA, Frezzato F, Zoratti M, Semenzato G et al. (2013). Clofazimine, Psora‐4 and PAP‐1, inhibitors of the potassium channel Kv1.3, as a new and selective therapeutic strategy in chronic lymphocytic leukemia. Leukemia 27: 1782–1785. [DOI] [PubMed] [Google Scholar]
- Levy DI, Deutsch C (1996). Recovery from C‐type inactivation is modulated by extracellular potassium. Biophys J 70: 798–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD (1995). Potassium and calcium channels in lymphocytes. Annu Rev Immunol 13: 623–653. [DOI] [PubMed] [Google Scholar]
- López‐Barneo J, Hoshi T, Heinemann SH, Aldrich RW (1993). Effects of external cations and mutations in the pore region on C‐type inactivation of Shaker potassium channels. Recept Channels 1: 61–71. [PubMed] [Google Scholar]
- Marom S, Levitan IB (1994). State‐dependent inactivation of the Kv3 potassium channel. Biophys J 67: 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meki A, Mansuelle P, Laraba‐Djebari F, Oughideni R, Rochat H, Martin‐Eauclaire MF (2000). KTX3, the kaliotoxin from Buthus occitanus tunetanus scorpion venom: one of an extensive family of peptidyl ligands of potassium channels. Toxicon 38: 105–111. [DOI] [PubMed] [Google Scholar]
- Miao S, Bao J, Garcia ML, Goulet JL, Hong XJ, Kaczorowski GJ et al. (2003). Benzamide derivatives as blockers of Kv1.3 ion channel. Bioorg Med Chem Lett 13: 1161–1164. [DOI] [PubMed] [Google Scholar]
- Miller C (1995). The charybdotoxin family of K+ channel‐blocking peptides. Neuron 15: 5–10. [DOI] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ et al. (2001). LTRPC7 is a Mg · ATP‐regulated divalent cation channel required for cell viability. Nature 411: 590–595. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Kath JC, Hanson DC, Biggers MS, Canniff PC, Donovan CB et al. (1996). Novel nonpeptide agents potently block the C‐type inactivated conformation of Kv1.3 and suppress T cell activation. Mol Pharmacol 50: 1672–1679. [PubMed] [Google Scholar]
- Nguyen W, Howard BL, Neale DS, Thompson PE, White PJ, Wulff H et al. (2010). Use of Kv1.3 blockers for inflammatory skin conditions. Curr Med Chem 17: 2882–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton RS, Pennington MW, Wulff H (2004). Potassium channel blockade by the sea anemone toxin ShK for the treatment of multiple sclerosis and other autoimmune diseases. Curr Med Chem 11: 3041–3052. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 Database Issue: D1098–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyi G, Sheng Z, Deutsch C (1995). C‐type inactivation of a voltage‐gated K+ channel occurs by a cooperative mechanism. Biophys J 69: 896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R (1997). Store depletion and calcium influx. Physiol Rev 77: 901–930. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Huq R, Tanner MR, Chhabra S, Khoo KK, Estrada R et al. (2014). A potent and Kv1.3‐selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci Rep 4: 4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren YR, Pan F, Parvez S, Fleig A, Chong CR, Xu J et al. (2008). Clofazimine inhibits human Kv1.3 potassium channel by perturbing calcium oscillation in T lymphocytes. PLoS One 3 e4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rus H, Pardo CA, Hu L, Darrah E, Cudrici C, Niculescu T et al. (2005). The voltage‐gated potassium channel Kv1.3 is highly expressed on inflammatory infiltrates in multiple sclerosis brain. Proc Natl Acad Sci U S A 102: 11094–11099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lenig D, Förster R, Lipp M, Lanzavecchia A (1999). Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401: 708–712. [DOI] [PubMed] [Google Scholar]
- Sands SB, Lewis RS, Cahalan MD (1989). Charybdotoxin blocks voltage‐gated K+ channels in human and murine T lymphocytes. J Gen Physiol 93: 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Sankaranarayanan A, Azam P, Schmidt‐Lassen K, Homerick D, Hänsel W et al. (2005). Design of PAP‐1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol 68: 1254–1270. [DOI] [PubMed] [Google Scholar]
- Singh SB, Zink DL, Dombrowski AW, Dezeny G, Bills GF, Felix JP et al. (2001). Candelalides A–C: novel diterpenoid pyrones from fermentations of Sesquicillium candelabrum as blockers of the voltage‐gated potassium channel Kv1.3. Org Lett 3: 247–250. [DOI] [PubMed] [Google Scholar]
- Vennekamp J, Wulff H, Beeton C, Calabresi PA, Grissmer S, Hänsel W et al. (2004). Kv1.3‐blocking 5‐phenylalkoxypsoralens: a new class of immunomodulators. Mol Pharmacol 65: 1364–1374. [DOI] [PubMed] [Google Scholar]
- Wulff H, Beeton C, Chandy KG (2003a). Potassium channels as therapeutic targets for autoimmune disorders. Curr Opin Drug Discov Devel 6: 640–647. [PubMed] [Google Scholar]
- Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C et al. (2003b). The voltage‐gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J Clin Invest 111: 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Castle NA, Pardo LA (2009). Voltage‐gated potassium channels as therapeutic targets. Nat Rev Drug Discov 8: 982–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Pennington M (2007). Targeting effector memory T‐cells with Kv1.3 blockers. Curr Opin Drug Discov Devel 10: 438–445. [PubMed] [Google Scholar]
- Yu SP, Kerchner GA (1998). Endogenous voltage‐gated potassium channels in human embryonic kidney (HEK293) cells. J Neurosci Res 52: 612–617. [DOI] [PubMed] [Google Scholar]