

Abstract
Tumor cells disseminate into compartments that are poorly accessible from circulation, which necessitates high doses of systemic chemotherapy. However, the effectiveness of many drugs, such as the potent topoisomerase I poison SN-38, are hampered by poor pharmacokinetics. To deliver SN-38 to lymphoma tumors in vivo, we took advantage of the fact that healthy lymphocytes can be programmed to phenocopy the biodistribution of the tumor cells. In a murine model of disseminated lymphoma, we expanded autologous polyclonal T cells ex vivo under conditions that retained homing receptors mirroring lymphoma cells, and functionalized these T cells to carry SN-38–loaded nanocapsules on their surfaces. Nanocapsule-functionalized T cells were resistant to SN-38, but mediated efficient killing of lymphoma cells in vitro. Upon adoptive transfer into tumor-bearing mice, these T cells served as active vectors to deliver the chemotherapeutic into tumor-bearing lymphoid organs. Cell-mediated delivery concentrated SN-38 in lymph nodes at levels 90-fold greater than free drug systemically administered at 10-fold higher doses. The live T cell delivery approach reduced tumor burden significantly after two weeks of treatment and enhanced survival under conditions where free SN-38 and SN-38-loaded nanocapsules alone were ineffective. These results suggest that tissue-homing lymphocytes can serve as specific targeting agents to deliver nanoparticles into sites difficult to access from the circulation, and thus improve the therapeutic index of chemotherapeutic drugs with unfavorable pharmacokinetics.
Introduction
Lymphomas are a heterogeneous family of hematological cancers that are generally sensitive to a wide variety of cytotoxic drugs. However, survival rates vary between subtypes, and their disseminated nature poses a challenge for therapy – tumor cell detection in an increased number of lymph nodes and extranodal sites is directly associated with more advanced staging and poorer prognosis (1). Being derived from immune cells that normally recirculate among lymphoid organs, lymphomas seed tumors in multiple organs and surgery cannot prevent the spread of disease. One route by which lymphomas evade chemotherapy in circulation is through entry into lymph nodes by transmigration across the high endothelial venules. Lymph nodes are a common tissue site for metastasis of many tumors, but systemic administration of some cytotoxic drugs has been shown to generate barely detectable levels of drug in the lymph nodes of cancer patients (2).
A second issue is permeation of drug throughout a tumor mass. For example, in breast cancer patients given intravenous doxorubicin, the drug was found to only permeate tumor cells in close proximity to blood vessels (3). Pharmacological studies have highlighted many obstacles to drug accumulation in tumors, including rapid metabolism or excretion and the physical barrier of the tumor stroma (4, 5). Administered by traditional intravenous routes, toxic systemic doses may be required to overcome these bottlenecks and achieve therapeutically relevant drug concentrations in the tumor. In addition, some tissue compartments, such as the lymph nodes, may serve as tumor sanctuaries, out of reach of normally effective drugs.
To overcome these issues, nanoparticles have been engineered to regulate the timescale of drug circulation in vivo, as well as to improve accumulation of drug payloads in tumors (6–8). Modification of particle material properties, surface chemistry, and microenvironment responsiveness can facilitate particle escape from scavenging by the reticuloendothelial system, as well as extend payload retention and, thus, increase the time between doses. Nanoparticle drug delivery has shown the best efficacy in preclinical models of solid tumors with leaky vasculature, where the enhanced permeation and retention (EPR) effect is most active. However, nanoparticles often become trapped in the matrix just outside tumor vessels, and thus fail to reach tumor cells distal from the vasculature. In addition, the EPR effect is heterogeneous and may be completely lacking in some tumors (9), including lymphoma subtypes that do not exhibit abnormal angiogenesis (10, 11).
We hypothesized that the efficacy of chemotherapy could be enhanced while reducing off-target toxicity if autologous lymphocytes were used as carriers to target drug-loaded nanoparticles to the lymphoid tissue sites where lymphomas home. Because the normal function of lymphocytes is to migrate throughout lymphoid tissues in search of antigen, we reasoned that polyclonal T cells, which express lymph node homing receptors but do not specifically recognize tumor cell antigens, could serve as effective chaperones for targeting of chemotherapy drugs to tumor-ridden lymphoid organs. By homing into these tumor sanctuary sites and distributing throughout the tissue, each nanoparticle-carrying cell would serve as a local micro-depot of drug to dose surrounding tumor cells. The use of polyclonal T cell carriers would be attractive for clinical implementation, because tumor antigen-specific T cells can only be isolated from a subset of cancer patients (12) and by contrast large numbers of T cells (250–500 million) can be obtained from a single leukapheresis and quickly expanded as much as 5000-fold using established clinical procedures in adoptive cell therapy (13).
To test this concept in a murine model of Burkitt’s lymphoma, we prepared controlled-release lipid nanocapsules (NCs) loaded with the potent topoisomerase I poison SN-38. These NCs were covalently conjugated to the plasma membrane of in vitro-activated primary T cells expanded under conditions promoting retention of CD62L and CCR7 receptor expression required for lymph node homing. T cells conjugated to SN-38-releasing NCs were used as live vectors to transport NCs systemically into lymphoid organs where lymphoma cells are enriched. SN-38 NC-functionalized T cells rapidly reduced tumor burden in multiple anatomical sites and significantly extend survival compared to systemic drug therapy in an aggressive transplanted Eμ-myc Arf−/− lymphoma model. These results suggest that autologous lymphocytes with engineered tissue-specific homing receptors can serve as effective chaperones for drug delivery to systemic cancer sites.
Results
Poor uptake of systemically administered SN-38 in sites of lymphoma dissemination
To model Burkitt lymphoma, we employed malignant B-cells isolated from double-transgenic Eμ-myc Arf−/− mice, in which the Eμ promoter drives c-myc oncogene overexpression in B cells and the Arf tumor suppressor is deleted, thus inactivating the tumor suppressor p53 (14). To track tumor distribution and growth kinetics in multiple tissues, Eμ-myc Arf−/− cells were transduced with dual green fluorescent protein (GFP) and firefly luciferase reporters (hereafter, Eμ-myc cells) (15). Consistent with prior studies (14), when transplanted into wild type recipients, Eμ-myc cells established systemic disseminated disease, accumulating in peripheral lymph nodes, bone marrow, and spleen (Fig. 1A). This pattern of tumor cell dissemination was accompanied by the expression of a suite of chemokine and adhesion receptors associated with normal lymphocyte trafficking, including the lymph node homing receptors CD62L and CCR7, bone marrow homing receptors CXCR4 and α4, β1 and β2 integrins, and the mucosal homing receptor α4β7 (fig. S1).
Fig. 1. SN-38 is a potent cytotoxic agent against Eμ-myc lymphoma cells, but fails to access sites of lymphoma dissemination in vivo.
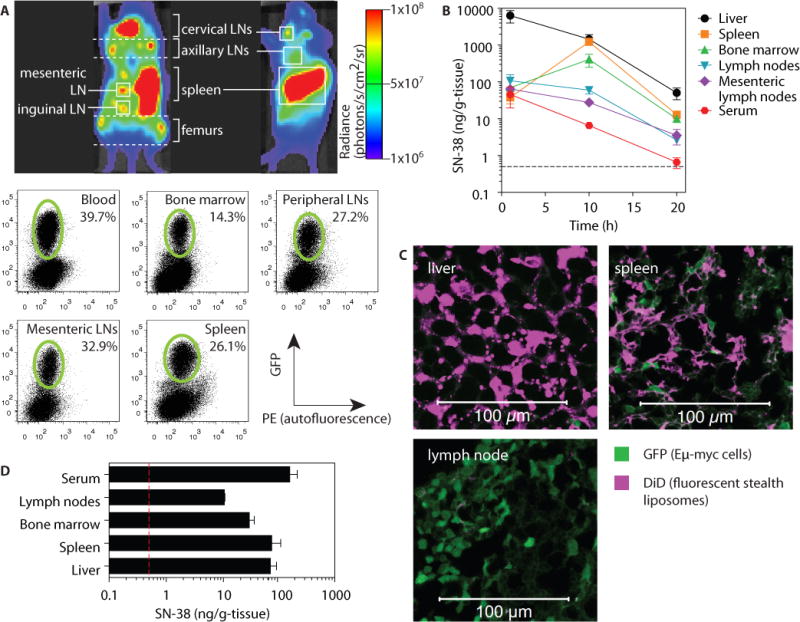
C57BL/6J recipients were injected i.v. with 1×106 luciferase- and GFP-expressing Eμ-myc cells. (A) On day 21 post inoculation, lymphoma biodistribution was imaged by IVIS in intact animals (shown are representative ventral and side views), followed by flow cytometry to detect GFP+ tumor cells (green gates). (B) On day 17 post inoculation, free SN-38 (10 mg/kg) was injected i.v. into tumor-bearing mice and tissue drug concentrations were measured by HPLC over time. Data are means ± s.e.m. (n=3/group). (C) On day 17 post-inoculation, tumor-bearing mice were i.v. injected with empty fluorescent liposomes (36.3 mg/kg lipid). Tissues were collected 24 h following injection for histology. (D) On day 17 post-inoculation, tumor-bearing mice were i.v. injected with SN-38-containing liposomes (1 mg/kg SN-38). Tissues were collected 24 h following injection for HPLC analysis. All data are representative of 1 of 2 independent experiments (n = 3 animals per group). Dashed lines in (B, D) denote limit of detection (0.5 ng/g tissue) for SN-38 by HPLC.
As an advanced chemotherapy strategy for treatment of lymphoma, we focused on SN-38, the active form of the camptothecin derivative irinotecan. Although 1000-fold more potent than irinotecan (16), SN-38 exhibits poor pharmacokinetics in vivo, with a 7-minute half-life in the bloodstream and rapid clearance through the liver (17). Consistent with these prior reports, following i.v. injection we found the majority of the drug was in the liver within 1 h (Fig. 1B). SN-38 showed some accumulation in the spleen and bone marrow at 10 h post-injection, but was rapidly cleared from these compartments. Less than 0.05% of the injected dose was detected in the lymph nodes at any time point (Fig. 1B).
A common strategy to alter the biodistribution of chemotherapy agents is encapsulation in nanoparticles (18). Following intravenous administration of fluorescently labeled stealth liposomes (120±24 nm in diameter) to Eμ-myc tumor-bearing mice, some liposomes were found in the liver and spleen at 24 h. By contrast, no particles were detected in the lymph nodes (Fig. 1C). Systemic injection of stealth liposomes carrying SN-38 (136±11 nm diameter, 4.8±1.5 μg SN-38/μmol lipid) into tumor-bearing mice resulted in high levels of SN-38 in the blood, spleen and liver, but accumulation of drug in LNs was 10-fold lower than these organs and indistinguishable from the result obtained with systemic free drug administration (Fig. 1D). Thus, free drug or liposome formulations that promote accumulation of chemotherapy agents in tumors by the EPR effect failed to access tumor-ridden lymph nodes efficiently, and thus these tissues may serve as a survival niche for lymphoma cells in the face of chemotherapy.
Generation of T cells as chaperones for SN-38 delivery
We recently described a strategy to enhance the functionality of tumor-specific T cells via conjugation of cytokine- or small molecule drug-releasing nanoparticles to the plasma membrane of adoptively transferred lymphocytes; adjuvant drugs released from cell-bound particles provided autocrine stimulation to the carrier T cells to support their antitumor activity in vivo (19, 20). The failure of free or liposome-formulated SN-38 to effectively reach lymphoid organs led us to test whether a similar “pharmacyte” strategy could be used for paracrine delivery of chemotherapy to tumor cells, employing the intrinsic tissue-homing pattern of lymphocytes rather than specific antigen recognition as a means to deliver drugs to sites of lymphoma dissemination (Fig. 2A). For this approach to succeed, several conditions needed to be met: (i) the tropism of the carrier cell needed to match as closely as possible the tissue distribution of the target tumor cells; (ii) the chaperone T cell needed to be resistant to SN-38 to avoid death of the carrier cell prior to arrival in target tissues; and (iii) the lymphocytes needed to carry a dosage of SN-38-NCs sufficient to kill lymphoma cells, which were expected to be in excess of the chaperone T cells.
Fig. 2. IL-2/rapamycin-expanded T cells express homing receptors to traffic to lymphoma sites and are resistant to SN-38 toxicity.
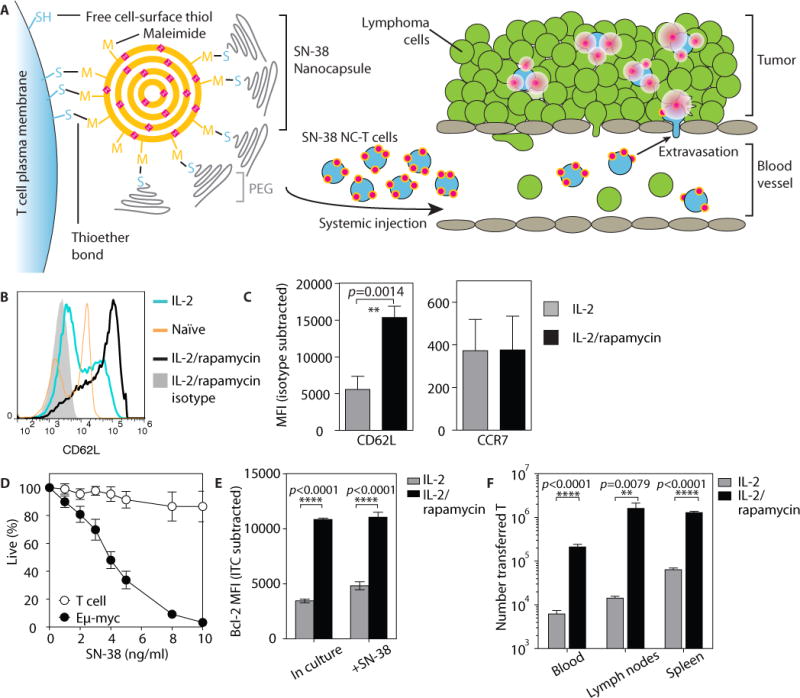
(A) Schematic of T cell functionalization and cell-mediated delivery of SN-38 NCs into tumors. (B and C) Polyclonal T cells from C57BL/6J mice were primed with concanavalin A and IL-7 for 2 days, then expanded in IL-2 with or without rapamycin for 2 days and analyzed for expression of tissue homing receptors by flow cytometry. Shown are representative staining histograms (B) and quantification markers (C) of n=7 replicate cultures from 2 independent experiments. Data are means ± s.e.m., **p<0.01, by t-test. (D) Eμ-myc cells or IL-2/rapamycin-expanded T cells were cultured in vitro with SN-38 at indicated doses, and viability was assessed by flow cytometry after 24 h. Data are means ± s.e.m of pooled n=4–8 replicate cultures from 4 independent experiments. (E) IL-2- or IL-2/rapamycin-cultured T cells were taken directly from culture or incubated with 20 ng/ml SN-38 for 12 h, then stained for intracellular Bcl-2 expression and analyzed by flow cytometry. Data are representative plots for n=3 cultures per condition, from 1 of 3 independent experiments. ****p<0.0001 by two-way ANOVA with Bonferroni post-test. (F) IL-2- or IL-2/rapamycin-cultured Thy1.1+ T cells (40×106) were adoptively transferred into C57BL/6J mice (n=6–7/group). The number of Thy1.1+ T cells in blood, spleen and lymph nodes were enumerated by flow cytometry 2 days post-transfer. Data are means ± s.e.m from 1 of 2 independent experiments. **p<0.01, ****p<0.0001 by t-test.
To generate large populations of lymphocytes capable of targeting SN-38 to lymphoid organs, we first established an ex vivo T cell priming protocol that allowed robust expansion of primary T cells while retaining key homing receptors required for lymphoid tissue trafficking. Both mouse and human T cells can be rapidly expanded to large numbers in vitro by polyclonal TCR triggering followed by culture in interleukin-2 (IL-2). However, following TCR stimulation, CD62L is rapidly shed/downregulated, resulting in decreased T cell homing to lymph nodes, mediated in part by mTOR signaling (21). To counteract these effects, we expanded primary T cells isolated from C57BL/6J mice in the presence of IL-2 and the mTOR inhibitor rapamycin, which has been shown to preserve CD62L and CCR7 expression during IL-2-induced growth and proliferation of T cells (21). As expected, IL-2 expanded both CD4+ and CD8+ T cells with an activated CD25+CD44+CD69+ phenotype (fig. S2, A and B), regardless of whether rapamycin was present. However, only T cells co-treated with rapamycin retained high levels of CD62L (Fig. 2, B and C). IL-2/rapamycin-treated T cells also expressed the integrins α4β7, β1, and β2 and the chemokine receptor CXCR4 (fig. S2C), thus imitating the homing receptor repertoire of Eμ-myc cells.
Eμ-myc cells were sensitive to SN-38-induced apoptosis in vitro at concentrations as low as 2 ng/ml and were essentially eradicated at 10 ng/ml (Fig. 2D). In contrast, IL-2/rapamycin-expanded T cells were minimally affected over the same concentration range. This selective activity of SN-38 towards Eμ-myc cells is consistent with previous reports of tumor cells having increased sensitivity to topoisomerase I poisons (22). These results suggest a therapeutic window in which T cells could carry therapeutic doses of SN-38 without undergoing apoptosis themselves.
Both sustained T cell receptor signaling and IL-2 withdrawal promote apoptosis in T cells (23); rapamycin counteracts this by increasing levels of the anti-apoptotic protein Bcl-2 (24). Consistent with these reports, IL-2/rapamycin-treated T cells had higher Bcl-2 expression, as compared to T cells expanded only in IL-2, and this expression difference was maintained in the presence of SN-38 (Fig. 2E), suggesting that IL-2/rapamycin T cells would preferentially survive in vivo. Indeed, when we transferred IL-2- or IL-2/rapamycin-expanded T cells into naïve hosts and analyzed the biodistribution of the transferred cells 2 days later, we observed between 10- and 100-fold more viable IL-2/rapamycin T cells in the blood, spleen and lymph nodes compared to IL-2 T cells (Fig. 2F). Altogether, these data demonstrate that combined IL-2/rapamycin “programming” yields expanded T cells that are resistant to SN-38 treatment, exhibit lymphoid tissue homing, and maintain effective survival in vivo.
To enable T cell-mediated delivery of highly hydrophobic SN-38 over a period of days, we entrapped the drug in multilamellar lipid nanocapsules (NCs) (25) designed to covalently react with T cell surface thiols (Fig. 2A). NCs were formed by a variation on the synthesis of interbilayer-crosslinked multilamellar vesicles we previously described: in brief, a co-solution of phosphatidylglycerol lipid, maleimide-headgroup lipid, and SN-38 formed precursor vesicles, which were fused together and each liposome wall was covalently crosslinked to others to form multilamellar lipid capsules (Fig. 3A and fig. S3, A and B). The resulting SN-38 NCs had a mean diameter of 340±12 nm and entrapped 14.3 μg SN-38 per mg lipid, which was completely released over 3 days in vitro (Fig. 3B).
Fig. 3. T cells conjugated with SN-38 NCs kill bystander lymphoma cells but not the T cells themselves.
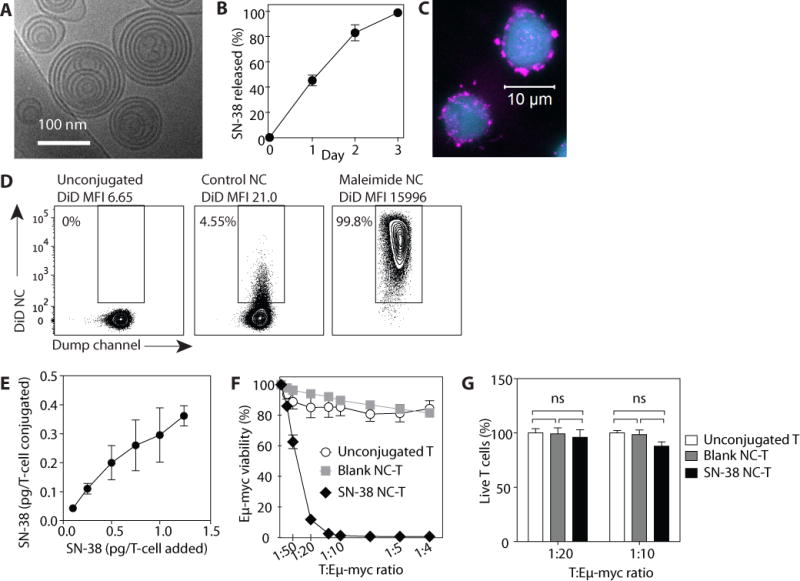
(A) Cryo-EM image of SN-38 NCs. (B) Kinetics of SN-38 release from NCs at 37°C in 10% serum. Data are means ± s.e.m (n=8). (C) T cells stained with CFSE (blue) and conjugated to fluorescently labeled NCs (pink) were imaged by confocal microscopy. (D) T cellls were conjugated to fluorescent DiD NCs either lacking maleimide-lipid (control NCs) or containing maleimide-headgroup lipids (maleimide NCs), washed, and then analyzed by flow cytometry. (E) T cells were conjugated with SN-38 NCs over a range of NC:cell ratios, then lysed to measure the final conjugated amount of SN-38. (F and G) Eμ-myc cells were co-cultured with unmodified T cells, empty (“blank”) NC-conjugated T cells, or SN-38 NC-conjugated T cells at indicated T cell:lymphoma cell ratios. Viability of Eμ-myc cells (F) and T cells (G) were measured by flow cytometry 24 h later. Data are means ± s.e.m (n=3–8 samples/group). n.s., not significant, two-way ANOVA with Bonferroni post-test. All data are representative of 2–3 independent experiments.
Following crosslinking, sufficient maleimide groups remained on the particle surfaces to allow conjugation of nanocapsules to T cell surface proteins; residual maleimide groups were quenched with PEG-thiol (Fig. 2A). SN-38 NCs were then stably conjugated to the surfaces of T cells and retained following washing (Fig. 3C), whereas maleimide-free (control) NCs showed minimal non-specific binding to T cells (Fig. 3D). Titration of the NC:cell ratio showed that T cells could be readily coupled with NCs carrying up to ~0.4 pg SN-38 per cell (Fig. 3E).
T cells functionalized with SN-38-releasing nanocapsules kill lymphoma cells in vitro
To test the capacity of SN-38-carrying T cells to deliver drug in trans to lymphoma cells, we cultured unmodified cells, T cells conjugated with empty NCs, or T cells conjugated with SN-38-loaded NCs (SN-38 NC-T cells, 0.2 pg SN-38/cell) for 24 h with Eμ-myc cells, and viability was assessed by flow cytometry. Co-culture of Eμ-myc cells with unmodified T cells or T cells decorated with empty NCs (blank NC-T) did not affect tumor cell viability, but SN-38 NC-T cells elicited tumor cell killing at ratios as low as 1 SN-38 NC-T cell per 20 lymphoma cells (Fig. 3F). In this same co-culture assay, SN-38 NC-T cells showed viability comparable to T cells that were unmodified or conjugated with empty NCs, demonstrating that T cells remained resistant to SN-38 even when drug-loaded capsules were directly conjugated to their plasma membranes (Fig. 3G). Thus, particle-decorated T cells are capable of carrying doses of SN-38 that are therapeutically relevant for clearing surrounding lymphoma cells, without causing acute toxicity to the carrier cell.
Delivery of SN-38 to lymphoid organs in vivo by nanoparticle-decorated T cells
We next tested whether T cells could transport drug-loaded nanoparticles into lymphoma-infiltrated organs in vivo. Following injection into mice bearing established Eμ-myc tumors, luciferase+Thy1.1+ T cells conjugated with SN-38 NCs trafficked to the spleen, lymph nodes and bone marrow (Fig. 4A). Flow cytometry analysis showed substantial NC-T celll accumulation in each of these lymphoid organs by 20 h, reaching peak levels by ~40 h with kinetics similar to unmodified T cells, suggesting that NC conjugation did not impair the survival or trafficking of the transferred T cells (Fig. 4B). Twenty-four h after transfer, NC-T cells dispersed throughout the lymph node, in proximity to Eμ-myc cells (Fig. 4C). When conjugated with fluorescent particles, transferred T cells recovered from lymph nodes were uniformly positive for NC fluorescence, demonstrating retention of their particle cargo during homing and a lack of particle transfer to other cells in the lymph node (Fig. 4D).
Fig. 4. SN-38 NC-conjugated T cells traffic into lymphoma-bearing lymph nodes in vivo and sustain elevated intranodal SN-38 levels over time.
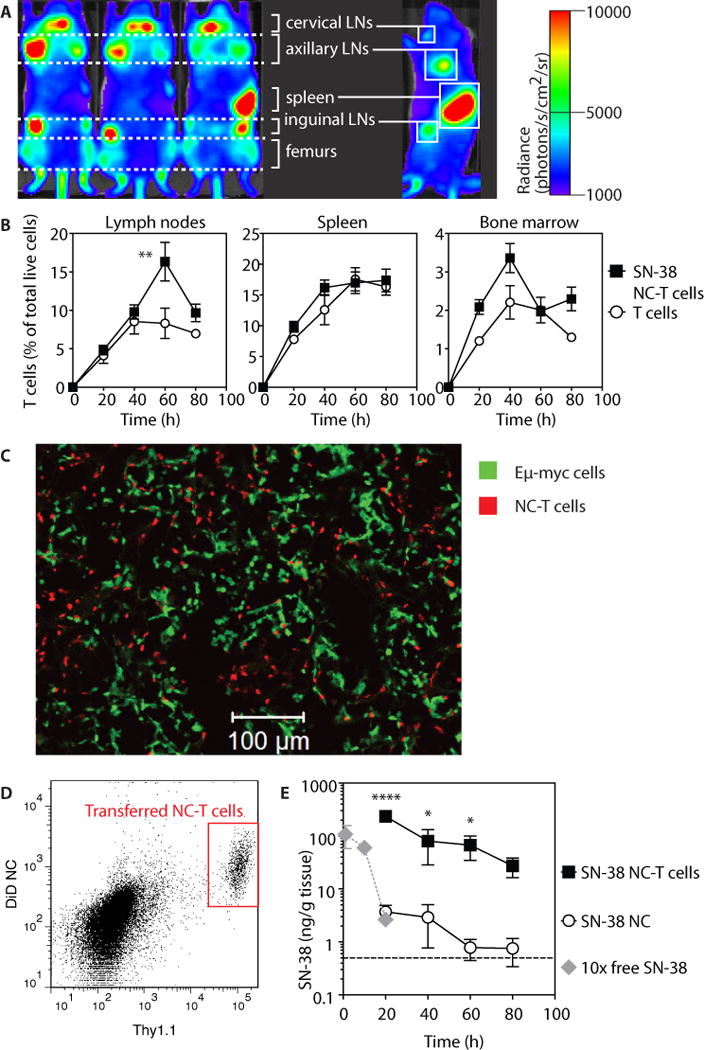
C57BL/6J mice (n=3–5/group) were injected i.v. with 1×106 Eμ-myc cells, and 17 days later received i.v. injection of 2×108 luciferase+ SN-38-carrying NC-T cells (1 mg/kg equivalent SN-38), control unmodified T cells, or an equivalent dose of free nanocapsules. (A) T cell biodistribution was assessed by whole-animal bioluminescence 38 h post-transfer. (B) Kinetics of T cell and SN-38 NC-T cell accumulation in tissues assessed by flow cytometry. Data are means ± s.e.m (n=4–12 animals/group per timepoint), pooled from 5 independent experiments. **p<0.01 versus T cells alone, by two-way ANOVA with Bonferroni post-test. (C) Mice (n=4/group) were injected with SN-38 NC-conjugated fluorescently labeled T cells, and tumor (Eμ-myc)-bearing lymph nodes were harvested at 24 h post-injection for histological analysis. Representative image from 1 of 2 experiments is shown. (D) Retention of fluorescently labeled SN-38 NCs by T cells (Thy1.1+) in tumor-bearing lymph nodes was measured by flow cytometry 15 h post-transfer. Plot is from 1 of 2 representative experiments. (E) Animals were treated with free SN-38 NCs, SN-38 NC-T cells, or free SN-38 at a dose 10-fold greater than the dose in the NCs. Tumor-bearing lymph nodes were harvested at various times after treatment for HPLC analysis. Data are means ± s.e.m (n=3–5 animals/group per timepoint), pooled from 4 independent experiments. Dashed line denotes limit of SN-38 detection (0.5 ng/g tissue). *p<0.05, ****p<0.0001 versus SN-38 NCs, by two-way ANOVA with Bonferroni post-test.
To determine the impact of T cell-mediated NC transport on tissue levels of the chemotherapy cargo, we measured SN-38 concentrations in tumor-bearing lymph nodes. Consistent with the T cell/NC biodistribution, SN-38 accumulation was greatly increased in tumor-bearing lymph nodes when delivered via T cell-bound NCs, reaching concentrations 63-fold greater than free NCs at 20 h and remaining at high levels for at least 4 days (Fig. 4E).
Enhanced efficacy of SN-38 chemotherapy by T cell-mediated drug delivery
We next tested the therapeutic efficacy of pharmacyte-mediated drug delivery against these aggressive disseminated lymphoma tumors. Five days after tumor inoculation, mice were treated with SN-38 as free drug i.v., SN-38 NCs, or SN-38 NC-T cellls (4 doses total given every 3 days, 1 mg/kg SN-38 per dose for all groups). Mice treated with saline or free SN-38 had high Eμ-myc tumor burdens in the blood, bone marrow, spleen and lymph nodes (Fig. 5A). Imaging revealed that free SN-38 at this dose did not suppress tumor growth at any point during the therapy (Fig 5B). We also confirmed that transfer of IL-2/rapamycin-treated T cells alone without nanocapsule conjugation had no effect on tumor progression (fig. S4). By contrast, mice treated with SN-38-loaded NCs had a significant reduction of tumor burden in all of these compartments, with the total tumor burden reduced by 5.1-fold (Fig. 5A, B). However, animals treated with SN-38 NC-T cells showed the most dramatic tumor eradication, exhibiting a 60-fold reduction in tumor burden on day 16 relative to the free drug or untreated animals (Fig. 5A, B). These results confirm that the lymph nodes are an important growth niche for Eμ-myc cells, and that increasing the local SN-38 concentration by T cell delivery suppressed tumor growth.
Fig. 5. T cell-mediated delivery of nanocapsules improves the therapeutic efficacy of SN-38 without toxicity.
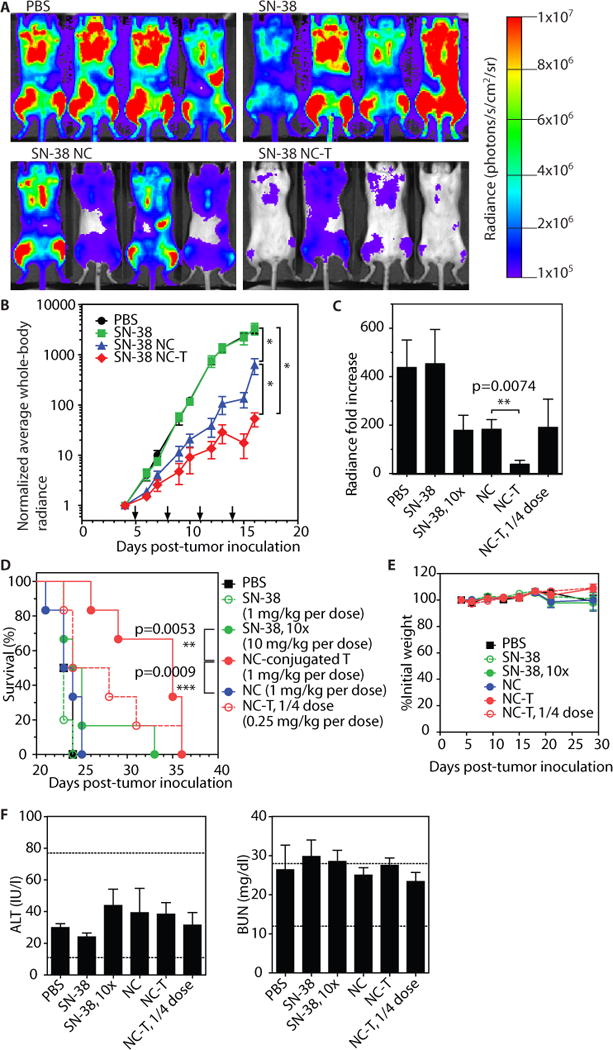
(A–B) Albino C57BL/6J mice (n=5 animals/group) were inoculated with 1×106 Eμ-myc cells on day 0, then received 4 treatments of free SN-38, free SN-38 NCs, or 2×108 NC-T cells (1 mg/kg SN-38 in each group) as indicated (arrows on panel B). Shown are representative results from 1 of 2 independent experiments. (A) Bioluminescence images of tumor burden on day 16. (B) Tumor burden as assessed by Eμ-myc bioluminescence (normalized to signal at the start of therapy) over time. *, p<0.01 by two-way ANOVA with Bonferroni post-test on day 16. (C–F) Albino C57BL/6J mice (n=5–6 animals/group) were inoculated as in (A), then received free SN-38 at 1 mg/kg or 10 mg/kg, free SN-38 NCs at 1 mg/kg, or 2×108 or 5×107 NC-T cells (1 mg/kg or 0.25 mg/kg SN-38, respectively) every 3 days, starting on day 5, for a total of 7 treatments. Shown are representative results from 1 of 2 independent experiments. (C) Normalized total body bioluminescence measured on day 15. **p<0.01, by t-test. (D) Overall survival. ***p<0.001 by log-rank test. (E, F) Animals were inoculated and treated as in (C), shown are representative results from 1 of 3 independent experiments. (E) Weights of animals normalized individually to the pre-therapy weight. (F) On day 28, serum was collected for ALT and BUN measurement; dashed lines indicate reference healthy ranges. Data shown are means ± s.e.m., n=4–6 animals/group.
To evaluate the efficacy of the therapy within the context of a wider dose range, we carried out survival studies comparing the same four treatments as well as two new treatments: a 10-fold higher dose of free SN-38 (10 mg/kg) and a one-quarter dose of 50×106 NC-T cells per dose (corresponding to 0.25 mg SN-38/kg). Each group was treated 7 times, once every 3 days, and then followed for overall survival. On day 15, tumor burdens were measured by whole body imaging (Fig. 5C). As before, free SN-38 at 1 mg/kg did not impact tumor burden, but increasing the dose of free SN-38 10-fold still did not achieve efficacy comparable to drug-carrying T cells. In fact, the ¼ cell dose NC-T cell treatment (¼ the drug dose as the free SN-38 group) had approximately the same tumor burden at this time point as the 10X free SN-38 group – implying that T cell-mediated drug delivery enhanced the potency of SN-38 by at least 40-fold (Fig. 5C).
Despite the early slowing of tumor growth achieved by treatment with 10X free SN-38 or free NCs, tumor growth control decayed over the course of therapy (as seen for free NCs in Fig. 5B), and the median survival times of these groups were not significantly different from untreated controls (~24 days, Fig. 5D). Increasing the free drug dose 10X only increased the survival of 1 out of 6 animals. By contrast, animals with lymphoma receiving SN-38 NC-T cells showed significant increases in lifespan, with median survival extended to 35 days. Infusion of a 4-fold lower dose of SN-38 NC-T cells also modestly extended survival of 50% of the cohort, although the overall median survival time was not statistically different than the controls (Fig. 5D).
To assess possible toxicities from SN-38 NC-T cell therapy, we tracked animal weights and liver enzymes, but saw no weight loss at any time point in any group and found that alanine aminotransferase (ALT), blood urea nitrogen (BUN), and other serum measurements fell in the range of healthy animals (Fig 5E; fig. S5). Thus, NC-T therapy significantly improved the efficacy of SN-38 without increasing the risk of adverse side effects.
Discussion
SN-38 is representative of a large class of potent chemotherapy drugs with limited in vivo efficacy owing to very poor pharmacokinetics and toxicity. Nanoparticle formulation has been pursued as a strategy to overcome these issues, and several SN-38 formulations have been developed – including liposomes, PLGA nanoparticles, and micelles – some of which are in clinical trials for colorectal and other solid cancers (26). However, nanoparticle delivery faces its own challenges, as tumor accumulation is dependent on the presence of a leaky tumor vasculature (the EPR effect), and particles often become trapped perivascularly in tumors. Autochthonous Eμ-myc Arf+/+ tumors developing in lymph nodes show higher densities of blood vessels and lymphatic vessels compared to normal nodes, but these vessels are not permissively leaky to systemically injected dyes (27). Similar observations of increased angiogenesis have been made in patient samples, but are inconsistent across lymphoma subtypes (10, 11). Within the transplanted Eμ-myc Arf−/− model used in our studies, we did not observe an EPR effect in tumor-ridden lymph nodes with “stealth” liposomes (Fig. 1) or with SN-38 NCs (Fig. 4). Thus, nanoparticle-based delivery of SN-38, or similar drugs, would be insufficient to reach lymphoid tissue-homing lymphomas or leukemias.
To overcome these hurdles, here we demonstrated a strategy for active targeting of chemotherapy to disseminated tumors, using lymphocytes as living chaperones to deliver drug-loaded nanocapsules to tumor sites. By expanding T cells under conditions that maintained chemokine and adhesion receptors necessary for lymphoid tissue homing, we were able to generate large numbers of lymphocyte chaperones with highly specific tumor-targeting tropism. These T cell chaperones were resistant to SN-38, even with high doses of drug-loaded particles bound directly to the cell membrane. Although T cell homing occurs over days, even low numbers of NC-T cells entering tumors at early times after transfer could initiate therapeutic responses, as seen from the high concentration (20-fold above the EC90) of SN-38 in tumor-bearing lymph nodes at 20 h post-transfer (Fig. 4). The substantially lower efficacy of free SN-38 NCs, which effectively accumulated in one lymphoma residence site (the spleen) but not lymph nodes, suggests increased lymph node delivery is key to the efficacy of this approach. This enhanced delivery of SN-38 to tumors yielded therapeutic benefits at modest doses of SN-38—doses that had no effect in free drug form and limited efficacy in NCs only. We demonstrated a 12-day extension of survival using T cell-mediated SN-38-NC delivery, at a cumulative dose of only 7 mg/kg SN-38. This efficacy was far greater than 70 mg/kg free SN-38, due to the poor pharmacokinetics of the free drug. However, even with irinotecan, the water-soluble FDA-approved analog of SN-38, a cumulative 100 mg/kg dose was required to extend survival by a maximum of 9 days in this model (28). Doxorubicin, another cytotoxic chemotherapeutic, achieved a 14–15 day survival extension at 10 mg/kg (28), but this is the maximal lifetime tolerated dose and causes potentially lethal myocardial damage. By contrast we saw no evidence for toxicity with SN-38 NC-T cells, suggesting that the therapeutic window for higher levels of drug dosing with this approach is much wider than for traditional chemotherapy.
The intrinsic trafficking ability of host cells to infiltrate disease sites and act as self-directed vectors for therapeutics is being explored in a number of contexts, most commonly employing phagocytic cells as drug carriers (29, 30). Monocytes, macrophages, and mesenchymal stem cells readily phagocytose nanoparticles and microparticles, and particle-loaded cells have been used as transporters for gold nano-shells for photothermal tumor ablation (31), chemotherapy- or imaging agent-loaded particles for tumor treatment (32, 33), and antiretroviral drugs for treatment of HIV infection (34). A limitation of such approaches is that they can only be applied to cell-permeable drug cargos or agents that provide a function from within the carrier cell. By contrast, the plasma membrane-conjugation approach described here allows particles to deliver cargos, such as biologics, that are not membrane-permeable and must access cell surface receptors of nearby target cells (19).
We used polyclonal T cells as nanoparticle carriers, employing lymphocytes that express homing receptors to traffic to the lymphoid organs where tumor cells reside, but which do not target tumor antigens explicitly. The pronounced therapeutic effects shown here using non-antigen-specific cells demonstrate the utility of organ-specific targeting in this disease model (as opposed to tumor cell-specific targeting). The effectiveness of polyclonal T cells facilitates clinical implementation, because isolation of endogenous tumor antigen-specific T cells is only possible for a fraction of cancer patients (12). However, in diseases such as melanoma where tumor-specific T cells are more readily obtained or cancers where genetically engineered artificial antigen receptors can be safely introduced [e.g. leukemias (35)], our approach could be combined with tumor antigen-specific T cells.
Although significant tumor cell killing was seen at a T cell:tumor cell ratio of 1:20 in vitro, complete eradication required ratios of at least ~1:10. Owing to in vivo confounding factors, such as lymph flow, secreted factors, and stromal cells that augment Eμ-myc tumor growth and survival (36, 37), we aimed to transfer enough T cells to reach at least 1:5 T cell:tumor ratios in the nodes. Although the cell numbers needed to reach this density of pharmacytes in lymph nodes is high, it is within the range of cell doses that have been used in clinical trials of adoptive T cell therapy for cancer. Using the FDA guidelines for determining dose conversions between species (38), the equivalent of our 2×108 cell dosing for a human patient would be ~4.6×1010 lymphocytes. Early adoptive T cell therapy clinical trials treated melanoma patients with >2×1011 total tumor-infiltrating lymphocytes in the first course alone, with some patients receiving up to 5 total courses (39). More recent trials have used up to 1.5–1.6×1011 total cells per patient (40, 41). These numbers reflect the large quantity of T cells required to successfully eliminate large tumor burdens even when relying on antigen-specific recognition for tumor elimination, and suggest that the number of SN-38 NC-conjugated T cells required for human patients would be both technologically and clinically reasonable to achieve. This cell dosing could be substantially reduced with the development of NCs loaded more efficiently with drug cargo, but our objective here was to demonstrate proof of concept in a cell dose that has been used in patients. We also expect this approach would have substantial synergy with traditional chemotherapy dosing, using the “pharmacyte” approach to eradicate residual disease in lymphoid tissue sanctuaries rather than as a sole treatment regimen– this is an area for future study. Further efficacy with this strategy in patients might also be expected because human lymphomas exhibit a broad range of phenotypes, while the Eμ-myc tumors used here with constitutive expression of Myc and deletion of Arf model the most aggressive subtypes of these cancers (42).
The fundamental approach for cell-mediated drug delivery demonstrated here should be generalizable to many types of cellular carriers. Lymphocytes as drug carriers offer a number of advantages. First, clinical protocols for T cell expansion are well established in the adoptive therapy field, and autologous lymphocytes are easily obtained from blood. Second, lymphocytes exhibit substantial plasticity in their expression of tissue homing markers. T cells can be induced under conditions of inflammation or disease to enter nearly every tissue, and the receptors required for T cell trafficking to the lungs, skin, gut, and brain, as well as the molecular stimuli required to induce expression of these homing markers have been characterized (43), and could potentially be used to generate lymphocyte carriers to deliver drugs to each of these organs.
In summary, we have developed here a strategy for active targeting of drugs to disease sites, using autologous lymphocytes as “Trojan horses” to deliver drug-loaded nanocapsules. A limitation of this approach is the total payload of drug that can be loaded per cell and the number of cells that can be transferred, but as shown here, the greatly enhanced efficacy of drug delivery to tumor sites allows even modest total doses of drug to be highly efficacious. Further, ongoing advances in nanoparticle design can facilitate loading of very high payloads of drug (44–46). We envision that this approach could be particularly relevant as a means to eliminate residual disease in tissue sanctuaries that are difficult to reach at safe doses and by traditional nanomedicine.
Materials and Methods
Study Design
The hypothesis was that SN-38-carrying nanoparticles attached to T cells would show greater uptake in tumor tissues and enhanced anti-tumor efficacy compared to free drug or free nanoparticle treatments. All experiments were performed independently at least twice. In vivo therapy studies were designed to evaluate the impact of active versus passive delivery of a chemotherapeutic agent to disseminated tumor sites in a syngeneic mouse model of lymphoma, and were executed with at least 5 animals per group. Prior to treatment, cumulative tumor burden was measured by whole body radiance, and animals were randomized to minimize variances between groups. The radiance value of each animal was normalized by its pre-treatment radiance. Representative data were shown for tumor therapy experiments due to variation in tumor growth kinetics between inoculations. Pooled data were shown for NC-T cell biodistribution experiments, to obtain sufficient replicates for each timepoint and condition. Data analyses were not blinded. Outliers were not excluded.
Cell-NC conjugation
Live T cells were purified by Ficoll gradient, washed in PBS, and resuspended in serum-free unsupplemented RPMI-1640 at 50×106/ml. NCs were added and incubated with gentle mixing at 4°C for 30 min. Cells were washed by pelleting and resuspending in 50 ml PBS twice. Cells were then resuspended in serum-free RPMI with 1 mg/ml PEG2000-SH, and incubated with gentle mixing at 4°C for 30 min. Cells were washed in PBS and used immediately. The amount of SN-38 NC conjugated to T cells was quantified by lysing T cell pellets in 0.1 M NaOH + 0.5% Triton X-100, pelleting at 21000g and reading the SN-38 fluorescence in the supernatant.
In vivo tumor experiments
Animals were cared for following federal, state and local guidelines. To inoculate tumors, 1×106 Eμ-myc cells were injected via the tail vein. For luciferase+GFP+ Eμ-myc Arf−/− cells, tumor burden was assessed by whole-animal bioluminescent imaging (Xenogen Spectrum 200). Animals were injected subcutaneously with 150 mg/kg D-luciferin 10 minutes prior to imaging. At the terminal timepoint, blood, spleen, lymph nodes (cervical, axillary, brachial, inguinal, mesenteric, iliac), and bone marrow (from femurs and tibia) and liver were collected. For flow cytometry analysis, tissues were mechanically dissociated into single cell suspensions, and Eμ-myc cells were gated on GFP.
Statistical analysis
Statistical analyses were performed using Graphpad Prism 5.0. All plots show mean ± s.e.m. Statistical significance threshold was set at p≤0.05. All tests assumed normal distribution and were two-sided.
Supplementary Material
Acknowledgments
Histology samples were sectioned by the Koch Institute Swanson Biotechnology Center. Eμ-myc Arf−/− cells were a gift of M. T. Hemann. Funding: This work was supported in part by the Koch Institute Support (core) Grant P30-CA14051 from the NCI, the NIH (CA140476 and CA172164), and the Dept. of Defense (W81XWH-10-1-0290). DJI is an investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: The authors have no conflicts of interest to declare.
Author contributions: BH performed all experiments and statistical analyses. WA, YZ and SBL contributed to animal experiments. SSL contributed to nanoparticle development and characterization. BH and DJI designed the experiments and wrote the paper.
Data and materials availability: No material used in this study was obtained from external parties under material transfer agreement.
References AND NOTES
- 1.National Cancer Institute. PDQ® Adult Non-Hodgkin Lymphoma Treatment. Bethesda, MD: National Cancer Institute; Date last modified 02/13/2015. Available at: http://www.cancer.gov/cancertopics/pdq/treatment/adult-non-hodgkins/HealthProfessional. [Google Scholar]
- 2.Chen J, Wang L, Yao Q, Ling R, Li K, Wang H. Drug concentrations in axillary lymph nodes after lymphatic chemotherapy on patients with breast cancer. Breast Cancer Res. 2004;6:R474–477. doi: 10.1186/bcr819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lankelma J, Dekker H, Luque FR, Luykx S, Hoekman K, van der Valk P, van Diest PJ, Pinedo HM. Doxorubicin gradients in human breast cancer. Clin Cancer Res. 1999;5:1703–1707. [PubMed] [Google Scholar]
- 4.Undevia SD, Gomez-Abuin G, Ratain MJ. Pharmacokinetic variability of anticancer agents. Nat Rev Cancer. 2005;5:447–458. doi: 10.1038/nrc1629. [DOI] [PubMed] [Google Scholar]
- 5.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hubbell JA, Chilkoti A. Nanomaterials for Drug Delivery. Science. 2012;337:303–305. doi: 10.1126/science.1219657. [DOI] [PubMed] [Google Scholar]
- 7.Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615–627. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 8.Balmert SC, Little SR. Biomimetic Delivery with Micro- and Nanoparticles. Advanced Materials. 2012;24:3757–3778. doi: 10.1002/adma.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhakar U, Maeda H, Jain RK, Sevick-Muraca EM, Zamboni W, Farokhzad OC, Barry ST, Gabizon A, Grodzinski P, Blakey DC. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73:2412–2417. doi: 10.1158/0008-5472.CAN-12-4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koster A, Raemaekers JM. Angiogenesis in malignant lymphoma. Curr Opin Oncol. 2005;17:611–616. doi: 10.1097/01.cco.0000181404.83084.b5. [DOI] [PubMed] [Google Scholar]
- 11.Ribatti D, Nico B, Ranieri G, Specchia G, Vacca A. The role of angiogenesis in human non-Hodgkin lymphomas. Neoplasia. 2013;15:231–238. doi: 10.1593/neo.121962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.June C, Rosenberg SA, Sadelain M, Weber JS. T-cell therapy at the threshold. Nat Biotechnol. 2012;30:611–614. doi: 10.1038/nbt.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yee C. Adoptive T-cell therapy for cancer: boutique therapy or treatment modality? Clin Cancer Res. 2013;19:4550–4552. doi: 10.1158/1078-0432.CCR-13-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitt CA, Wallace-Brodeur RR, Rosenthal CT, McCurrach ME, Lowe SW. DNA Damage Responses and Chemosensitivity in the Emu-myc Mouse Lymphoma Model. Cold Spring Harbor Symposia on Quantitative Biology. 2000;65:499–510. doi: 10.1101/sqb.2000.65.499. [DOI] [PubMed] [Google Scholar]
- 15.Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101:640–648. doi: 10.1182/blood-2002-06-1751. [DOI] [PubMed] [Google Scholar]
- 16.Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991;51:4187–4191. [PubMed] [Google Scholar]
- 17.Atsumi R, Okazaki O, Hakusui H. Pharmacokinetics of SN-38 [(+)-(4S)-4,11-diethyl-4,9-dihydroxy-1H- pyrano[3′,4′:6,7]-indolizino[1,2-b]quinoline-3,14(4H,12H)-dione], an active metabolite of irinotecan, after a single intravenous dosing of 14C-SN-38 to rats. Biol Pharm Bull. 1995;18:1114–1119. doi: 10.1248/bpb.18.1114. [DOI] [PubMed] [Google Scholar]
- 18.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 19.Stephan MT, Moon JJ, Um SH, Bershteyn A, Irvine DJ. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat Med. 2010;16:1035–1041. doi: 10.1038/nm.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephan MT, Stephan SB, Bak P, Chen J, Irvine DJ. Synapse-directed delivery of immunomodulators using T-cell-conjugated nanoparticles. Biomaterials. 2012;33:5776–5787. doi: 10.1016/j.biomaterials.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinclair LV, Finlay D, Feijoo C, Cornish GH, Gray A, Ager A, Okkenhaug K, Hagenbeek TJ, Spits H, Cantrell DA. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 23.Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis–immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–253. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- 24.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat Mater. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172:48–61. doi: 10.1016/j.jconrel.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Ruddell A, Mezquita P, Brandvold KA, Farr A, Iritani BM. B lymphocyte-specific c-Myc expression stimulates early and functional expansion of the vasculature and lymphatics during lymphomagenesis. Am J Pathol. 2003;163:2233–2245. doi: 10.1016/S0002-9440(10)63581-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT. Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Natl Acad Sci U S A. 2008;105:9053–9058. doi: 10.1073/pnas.0803513105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth JC, Curiel DT, Pereboeva L. Cell vehicle targeting strategies. Gene Ther. 2008;15:716–729. doi: 10.1038/gt.2008.38. [DOI] [PubMed] [Google Scholar]
- 30.Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drug delivery. Expert Opin Drug Deliv. 2011;8:415–433. doi: 10.1517/17425247.2011.559457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7:3759–3765. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 32.Choi J, Kim HY, Ju EJ, Jung J, Park J, Chung HK, Lee JS, Lee JS, Park HJ, Song SY, Jeong SY, Choi EK. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33:4195–4203. doi: 10.1016/j.biomaterials.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Zhang F, Wang H, Niu G, Choi KY, Swierczewska M, Zhang G, Gao H, Wang Z, Zhu L, Choi HS, Lee S, Chen X. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials. 2013;34:1772–1780. doi: 10.1016/j.biomaterials.2012.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dou H, Destache CJ, Morehead JR, Mosley RL, Boska MD, Kingsley J, Gorantla S, Poluektova L, Nelson JA, Chaubal M, Werling J, Kipp J, Rabinow BE, Gendelman HE. Development of a macrophage-based nanoparticle platform for antiretroviral drug delivery. Blood. 2006;108:2827–2835. doi: 10.1182/blood-2006-03-012534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, June CH. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. New England Journal of Medicine. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dierks C, Grbic J, Zirlik K, Beigi R, Englund NP, Guo GR, Veelken H, Engelhardt M, Mertelsmann R, Kelleher JF, Schultz P, Warmuth M. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat Med. 2007;13:944–951. doi: 10.1038/nm1614. [DOI] [PubMed] [Google Scholar]
- 37.Rehm A, Mensen A, Schradi K, Gerlach K, Wittstock S, Winter S, Buchner G, Dorken B, Lipp M, Hopken UE. Cooperative function of CCR7 and lymphotoxin in the formation of a lymphoma-permissive niche within murine secondary lymphoid organs. Blood. 2011;118:1020–1033. doi: 10.1182/blood-2010-11-321265. [DOI] [PubMed] [Google Scholar]
- 38.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. The FASEB Journal. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR, Seipp CA, Einhorn JH, White DE. Treatment of Patients With Metastatic Melanoma With Autologous Tumor-Infiltrating Lymphocytes and Interleukin 2. J Natl Cancer Inst. 1994;86:1159–1166. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 40.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. Journal of Clinical Oncology. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G, Glass M, Johnson VE, McMannis JD, Shpall E, Prieto V, Papadopoulos N, Kim K, Homsi J, Bedikian A, Hwu WJ, Patel S, Ross MI, Lee JE, Gershenwald JE, Lucci A, Royal R, Cormier JN, Davies MA, Mansaray R, Fulbright OJ, Toth C, Ramachandran R, Wardell S, Gonzalez A, Hwu P. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18:6758–6770. doi: 10.1158/1078-0432.CCR-12-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mori S, Rempel RE, Chang JT, Yao G, Lagoo AS, Potti A, Bild A, Nevins JR. Utilization of pathway signatures to reveal distinct types of B lymphoma in the Eμ-myc model and human diffuse large B-cell lymphoma. Cancer Res. 2008;68:8525–8534. doi: 10.1158/0008-5472.CAN-08-1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13:309–320. doi: 10.1038/nri3442. [DOI] [PubMed] [Google Scholar]
- 44.Enlow EM, Luft JC, Napier ME, DeSimone JM. Potent engineered PLGA nanoparticles by virtue of exceptionally high chemotherapeutic loadings. Nano Lett. 2011;11:808–813. doi: 10.1021/nl104117p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang L, Fan TM, Borst LB, Cheng J. Synthesis and biological response of size-specific, monodisperse drug-silica nanoconjugates. ACS Nano. 2012;6:3954–3966. doi: 10.1021/nn300149c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo S, Wang Y, Miao L, Xu Z, Lin CH, Huang L. Turning a water and oil insoluble cisplatin derivative into a nanoparticle formulation for cancer therapy. Biomaterials. 2014;35:7647–7653. doi: 10.1016/j.biomaterials.2014.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.