

Abstract
The reelin gene is conserved across many vertebrate species, including humans. The protein product of this gene plays several important roles in early brain development and regulation of neural network plasticity of a matured brain structure. With an extended structure of 3461 amino acid sequences, consisting of eight reelin repeats, the human reelin sequence stands out as an exceptional model for evolutionary studies. In this study, sequence analysis of the human reelin and its homologues and reelin sequences from 104 other species is described in detail. Interesting sequence conservation patterns of individual repeats have been highlighted. Sequence phylogeny of the reelin sequences indicates a pattern similar to the evolution of the species, thereby serving as a highly conserved family for evolutionary purposes. Multiple sequence alignment of different reelin domain repeats, derived from homologues, suggests specific functions for individual repeats and high sequence conservation across reelin repeats from different organisms, albeit with few unusual domain architectures. A three-dimensional structural model of the full-length human reelin is now available that provides clues on residues at the dimer interface.
Keywords: reelin protein, glycoprotein, domain repeats, phylogeny, domain architecture, neurogenesis, 3D modeling
Introduction
Neuronal migration is an important part of brain development leading to the formation of heterogeneous functional regions in adults. There are many proteins that participate in the early development of the mammalian central nervous system and brain.1,2 In this study, we focus on reelin, a glycoprotein, which plays an important role in early brain development and also extends its functionality in regulating the structural plasticity of neural networks in adulthood and aging.2,3 It plays an important role in the proper migration and layering of the Purkinje cells into embryonic cortical plate during the early stages of the cerebellum development.2 It was first discovered as a gene product absent in reeler mouse,4 where behavioral abnormalities were observed due to the malformations of the cerebral cortex.
The human reelin gene is mapped on to chromosome 7q22 and is ~450 kbp long. The reelin protein is composed of ~3461 residues3 and is divided into three main regions with respect to its primary structure. The N-terminal signal peptide is followed by an F-spondin-like domain of ~200 amino acid residues.3 This region is further followed by an unknown region of ~400 residues with no sequence similarity to known domains.5,6 The third region is a series of reelin domain repeats of 300–350 residues each. Each reelin repeat is further divided into three regions: subrepeat A, an epidermal growth factor (EGF) domain, and subrepeat B. The subrepeat A of reelin shows weak but significant sequence similarity to subrepeat B.6
Reelin, the protein that is defective in reeler mice, is secreted by Cajal–Retzius cells in the cortical marginal zones and cerebellar granule cells. Reelin interacts with its lipoprotein receptors, namely apolipoprotein E receptor-2 (ApoER2) or very low-density lipoprotein receptor (VLDLR) on the target neurons.3 The binding of reelin to these receptors results in tyrosine phosphorylation3,7 of intracellular disabled-1 (DAB1), an adapter protein by the Src family kinases: Fyn and Src. Tyrosine-phosphorylated DAB1 associates with several adaptor proteins, playing distinct roles in neuronal positioning and appropriate morphogenesis of cortical and subcortical structures. The disruption of reelin, DAB1, or both ApoER2 and VLDLR leads to the formation of the reeler phenotype, indicating that the interaction of these proteins plays an important role in the formation and plasticity of the neural networks (see D’Arcangelo’s study for a recent review8).
The role of reelin in the modulation of synaptic plasticity relates to its role in several brain disorders, including schizophrenia, autism, and Alzheimer’s disease,7,9 in the case of humans and has become an important aspect of exploration in the recent past. An autosomal recessive form of lissencephaly is characterized with severe abnormalities of the cerebellum, hippocampus, and brainstem maps to chromosome 7q22 and is associated with two independent mutations in the human gene encoding reelin (RELN). In both the mutations, the reelin gene is truncated at the carboxyl terminus.10 The importance of reelin repeats 5 and 6 has been documented earlier – where cerebellar hypoplasia patients contain reelin mutants with the deletion of repeats 5–8.10 The biological importance of reelin has prompted us to investigate the evolutionary profile and domain architecture of reelin by the phylogeny of human reelin with its homologues in other model organisms. Three-dimensional (3D) modeling of the full-length human reelin gene has been possible using an integrated approach of homology modeling and superposition of individual domains guided by known tomography data. The relatively conserved subrepeat region of reelin repeats 5 and 6 is suggested to be at the dimeric interface of reelin.
Methodology
Sequence searches and similarity analysis
Reelin protein sequences were identified using sequence searches against the National Center for Biotechnology Information nonredundant (nr) protein dataset11 using the Position-Specific Iterative Basic Local Alignment Search Tool12 with human reelin as the query sequence with an E-value cutoff of 0.0001 and default parameter settings. The hits were filtered based on the presence of the reelin domain architecture. In order to detect the domain boundaries within the reelin sequence, CD-search13 was performed against the Conserved Domain Database14 for each of the query sequence.
Multiple sequence alignment
Multiple sequence alignments were constructed using Clustal W15 in order to measure the sequence-level similarity of each repeat over evolutionary timescale across homologues. The alignments were refined manually by using Jalview16 where required. The signature of central EGF domain of reelin repeat can easily be identified by the presence of conserved cysteine (Cys) columns in the multiple sequence alignment. The alignments of individual repeats are represented by the color codes on the basis of conservation patterns (Supplementary Figs. 1–9).
Phylogenetic analysis
The phylogenetic trees were generated using the neighbor-joining method17 in MEGA 4.0.18 The percentage of replicate trees in which the associated sequences cluster together in the bootstrap test (1000 replicates) were calculated, and branches with <50% bootstrap cutoff were collapsed. The evolutionary distances were computed using the Poisson correction method and are in the units of number of amino acid substitutions per site. All positions containing alignment gaps and missing data were eliminated only in pairwise sequence comparisons (pairwise deletion option).
Modeling full-length human reelin gene
A unique and integrated approach was used to model the full-length reelin gene where the individual repeat pairs were modeled, followed by the superposition of these pairs. Further segments of the protein were separately modeled, and the entire structure was put together in various steps as described in the following sections.
Modeling of the reelin repeats
Individual two-domain reelin repeats, such as R1–R2, R2–R3, R3–R4, R4–R5, R5–R6, R6–R7, and R7–R8, were modeled using R5–R6 domain structures in 2E26 as the template using MODELLER,19 generating 20 models for each repeat pair. Sequence identity of R5–R6 Protein Data Bank (PDB) entry with two reelin repeats (2E26) with R1–R2, R2–R3, R3–R4, R4–R5, R5–R6, R6–R7, and R7–R8 reelin repeats is 35%, 35%, 34%, 36%, 97%, 38%, and 35%, respectively. The best model of each of the repeat pairs, based on MODELLER energy, was then superposed based on their common domain. For example, for R1–R2 and R2–R3, the common domain is R2. The superposed coordinates of each of the repeat pairs of domains were saved in a similar manner, and the merged coordinates that have R1–R8 were renumbered and then used as template for modeling all the eight domains together. This helped in connecting the terminal residues of the repeats. The generated model was further energy-minimised in a cubic water box using OPLS force field using GROMACS.20
Modeling of F-spondin and unique region domains
The F-spondin and unique region were modeled separately using MODELLER with 2ZOT and 3 A7Q as the templates, respectively, and docked using HADDOCK.22 This docked structure from HADDOCK with a Z-score of −1.1 was further used as a template to model the F-spondin–unique region together.
Combining the F-spondin and unique region with the reelin repeats R1–R8 using HADDOCK
The best 3D model corresponding to F-spondin and unique region was then docked with the model of R1–R2 domain using HADDOCK with constraints on the terminal residue. A total of 198 HADDOCK-suggested docked poses could be clustered into two major clusters, which represent 99% of the water-refined models. The lowest scoring structure from cluster 1 with the highest number of structures and a Z-score of −1.4 was further considered for the generation of the model.
Building the entire structure
The best docked pose of reelin N-terminal fragment (containing F-spondin–unique region, R1–R2 domains, derived from HADDOCK) and the energy minimized structure of R1–R8 were then superposed based on their common domain, which is reelin repeat R1–R2. The superposed coordinates were used as template for a further MODELLER run to link the terminal residues of the unique region and the R1–R8 model. The final structure, including all the domains, F-spondin-unique region, and reelin repeats 1–8, was further energy-minimized using GROMACS20 in a water box with OPLS force field.22
Results and Discussion
Reelin homologues show conserved domain architectures
A total of 170 homologues of the human reelin protein were identified in 104 organisms, using sensitive sequence search approaches (Supplementary Table 1). Most of the reelin sequences that were identified belong to the phylum Chordata, although few homologues could be identified in Mollusca and Arthropoda. These sequences share a lengthy distribution ranging from 2985 to 3945 amino acids, with some of the members highlighting unusual domain architectures when compared to the human reelin that is 3461 amino acids long. The human reelin, which is an extracellular glycoprotein, comprises an N-terminal region followed by eight unique reelin repeats.4 Each reelin repeat is composed of two related subrepeats, A and B, separated by an EGF-like motif. The C-terminal region of reelin contains a stretch of positively charged amino acids. This domain architecture was found to be mostly conserved across the homologues of the human reelin (see http://caps.ncbs.res.in/download/reelin/reelin_DA.xls). However, exceptionally, the reelin sequence from Myotis brandtii (long-living Brandt’s bat) shares a sequence identity of 96% with the human reelin but retains two additional other domains after the eight reelin repeats. These two domains belong to the sulfate transporter family (pfam00916) and sulfate transporter and anti-sigma factor antagonist domain of Sulfate-permease-like sulfate transporters (cd07042). Similarly, reelin homologue from Crassostrea gigas (Pacific cupped oyster) is 3738 amino acids long and retains an additional reelin domain between the reelin repeat 7 and reelin repeat 8 of the human reelin. The isoforms of human reelin, as a result of alternate splicing (Cra_b and Cra_c lacking the reelin repeat 8), are present in Cra_a (Fig. 1). These isoforms have been found to carry important functions, though they are truncated.23
Table 1.
Motifs conserved across all the reelin domains (R1–R8) in subrepeat A and subreapeat B.
| SUBREPEAT A | SUBREPEAT B |
|---|---|
| WXXD | WXXD |
| RXXQ | RXXQP |
| WXR | PXXA |
| SXXXGXXW | |
| LXF |
Figure 1.

The graphical representation of the unusual domain architecture present in the reelin homologues: reelin repeats (blue), stas_transporter repeats (orange), reeler domain (gray), and unique region (green).
Evolutionary analysis of reelin across various genomes suggests high conservation concurrent with taxonomic hierarchy
Phylogenetic trees were constructed using the neighbor-joining method using full-length sequences as well as the individual reelin repeats across the species. It was observed that reelin proteins were classified according to the class level of the taxonomy in the trees constructed using the full-length reelin proteins in Figure 2. This is also reflected in the phylogenies constructed using single reelin domain repeats. The phylogenies of individual domains are shown in Supplementary Figures 10–17. The cladogram suggests that the reelin proteins and the reelin repeats might have originated after the evolution of Arthropoda, as only one homologue of reelin is present in this family. The closest to this was observed to be Actinopterygii followed by Amphibia, Aves, Reptilia, and then Mammalia, following the universally accepted evolution of species. Because this protein plays a major role in neurogenesis, the evolution of reelin protein can be correlated with the brain development with the evolution of higher organisms. The observed evolutionary distance is also reflected at the amino acid sequence of these proteins, as the identity of the proteins decrease with the increase in distance along with the evolution of these organisms.
Figure 2.
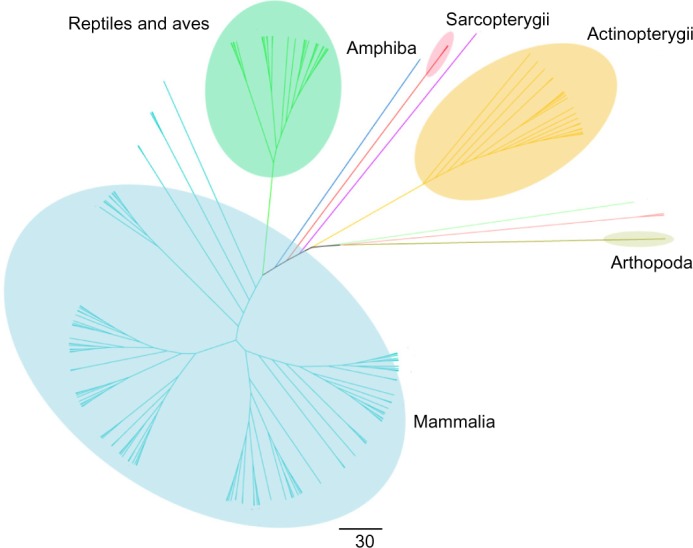
Cladogram of full-length reelin protein sequences using the neighbor-joining method. The cladogram suggests that the reelin proteins and the reelin domains might have originated in the Arthropoda that has the longest branch length as observed in the phylogenetic trees constructed using the full-length reelin sequences.
Evolutionary analysis of the reelin domains suggests that individual reelin domains are not due to simple domain duplication events
The similarity of the individual reelin repeats across these homologues is higher than the similarity of the domains within a species (Fig. 3). A combined multiple sequence alignment of the all eight reelin repeats, with their homologues from all the 167 species, was constructed to delineate if there are particular pairs of reelin repeats that are more closely related than the others. The phylogenetic tree derived from such an alignment shows distinct clusters for each of the reelin domains reaching the root of the tree. This polyploidy suggests that the individual reelin domains are not derived from simple domain duplications (Fig. 4), and each of these domains has evolved independently. This is interesting in terms of the functional importance of each of these individual repeats given the wide roles of these proteins in an organism.
Figure 3.

Percentage identity matrix. Matrix representing the average percentage identity across and between the reelin repeats from 167 species. It is seen that the sequence identity across the diagonal that represents the identity between the same repeat is higher than the identity among the different repeats.
Figure 4.
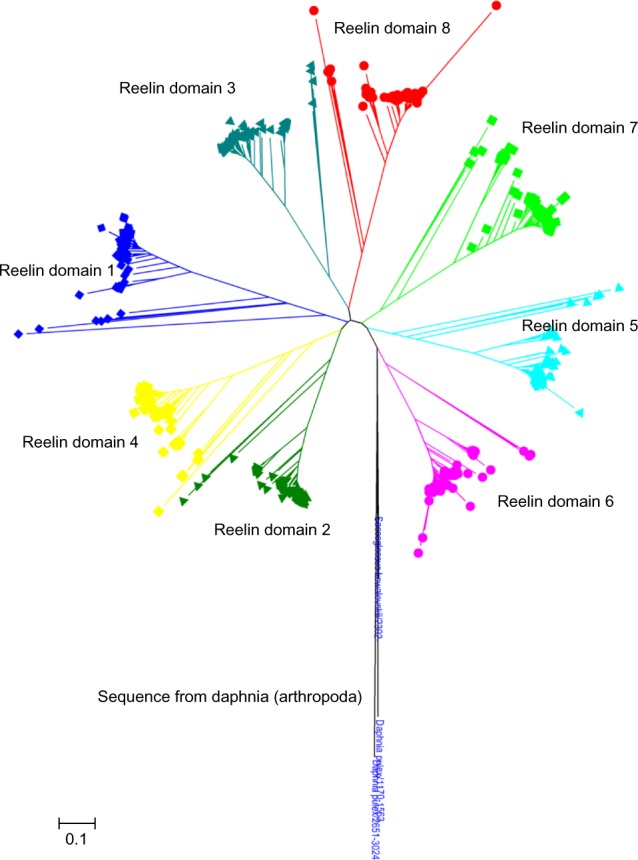
Cladogram of reelin domains (R1–R8) using the neighbor-joining method. The individual domains are clustered together and are colored differently. The cladogram suggests that the reelin domains have evolved independently.
Sequence features of reelin repeats reveal higher sequence conservation of subrepeat B
The individual reelin domains share high sequence similarity across their homologues compared to the sequence identity between the repeats. Although this prevails, it was observed that there are certain residues that are conserved across all the domains R1–R8, and this conservation suggests that these residues are ideally important for the structural organization of the repeats (Supplementary Fig. 1). Such highly conserved residues were mapped to the secondary structure regions of the reelin repeats (Fig. 5). Also, interestingly, it was observed that residues in the subrepeat B of the reelin repeats are more highly conserved compared to the residues in the subrepeat A. The presence of certain conservation of motifs was observed in each of these repeats (Table 1), and interestingly, WXXD motif and the RXXQ motifs located at the terminal end of the repeats were found conserved in both the repeats.
Figure 5.
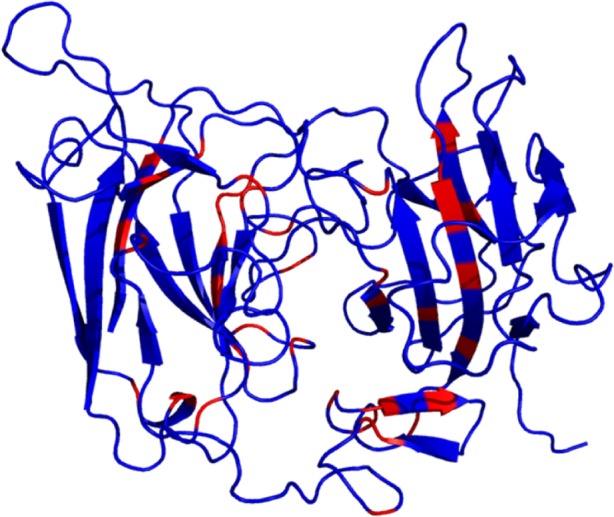
Conserved residues across all reelin domains: the conserved residues across all the reelin domains (R1–R8) are represented in red.
Apart from these universally conserved residues, the residues that are conserved within each repeat could impart a specific function to that reelin domain. In order to identify these residues, an alignment was generated for each of these repeats across the homologues. The sequence logo representation of these alignments is provided in Supplementary Figures 2–9. It was observed that a number of charged and aromatic residues are conserved in a class-specific manner on the surface of these proteins, suggesting their importance in complex formation and ligand interaction (Supplementary Fig. 18).
Model of the full-length reelin domains could lead to prediction of dimer interface
Structures of three reelin repeats have been determined using X-ray crystallography till date. The crystal structure of reelin repeat 3, which is the first ever structure of a reelin domain, was resolved at a resolution of 2.05 Å.7 The structure indicated that the three subdomains (ie, subrepeat A, EGF, and subrepeat B) are arranged in a horseshoe-like manner, making intimate contact with one another. Subsequently, the structure of R5 and R6 domains together has also been determined by the similar group revealing identical subdomain arrangement. Considering that the R3–R6 fragment of the reelin protein represents the functionally active unit within the reelin protein, the authors further examined the electron tomographic images of the R3–R6 fragment, and it was observed that the fragment exhibits an elongated rod-like structure. Based on this available information, a possible model for the full-length protein of the human reelin has been attempted. Individual repeat pairs (ie, R1–R2, R2–R3, R3–R4, R4–R5, R5–R6, R6–R7, R7–R8) were modeled using the crystal structure of R5–R6 domains of the mouse reelin (PDB ID: 2E26) as the template. These models of individual repeat pairs were validated using Ramachandran map, Verify3D,24 and ProSA25 (Supplementary Table 2). These repeats were further superimposed on the common repeat present in each pair to connect all the reelin repeats (see Methodology for details). The N-terminal region of the protein that holds the F-spondin and unique region was modeled using the ab initio method, and these domains were concatenated with the reelin repeats using the successive steps of rigid-body superposition (Supplementary Fig. 19). The resultant structure was further minimized in a water box. A total of 85% of the residues of any of the two-domain models and indeed that of the template structure were within fully allowed regions of the Ramachandran map. The final model (as shown in Fig. 6) was verified using Ramachandran map, which showed that 75% of the residues were present in fully allowed regions and (and 95.6% after including the partially allowed regions), suggesting that the arrived structure is a reasonably good model (Supplementary Fig. 20).
Figure 6.

Model of the full-length reelin protein: reelin repeats are represented in orange, F-spondin domain in green, and unique region in purple.
R5 and R6 domains are involved in dimer formation
It is known that functional reelin exists as a multimer formed by interchain disulfide bond(s) as well as through noncovalent interactions. The role of Cys 2101 in the R5–R6 domains in the dimerization of the reelin protein has been described earlier.26 The available full-length reelin model has helped in the identification of a putative dimer interface based on this information (Supplementary Table 3). A putative dimer interface, as predicted by CPORT,27 on the modeled protein of these domains is shown in Figure 7A. The conservation of these residues was analyzed using the alignments generated in this study, and it was interestingly noted that many of the predicted dimer interface residues are indeed conserved among the vertebrate sequences (Fig. 7B). This could suggest that the dimerization of reelin sequences is more specific to the vertebrate species. Additionally, it was also observed that several of the putative conserved interface residues are mapped to sub-repeat B and, in particular, the beta-sheet that is almost perpendicular to the jellyroll fold. An electrostatic map of the two domains is further represented in Figure 7C. The availability of full-length reelin model has enabled us to realize residues that will line all along the putative dimer interface for the first time. However, dimer modeling by introducing an interchain disulfide, reported at Cys 1201, was not feasible in a parallel orientation. This suggests that there could be slight conformational change to present these Cys residues at the time of dimer formation. This study will also enable docking its known interactors, such as ApoER2 and VLDLR, perhaps involving domains R3 and R4 or R7 and R8, as R5–R6 is implicated in dimer formation. More questions remain to be answered, such as how reelin binding promotes receptor clustering and if there could be additional receptors for reelin.
Figure 7.
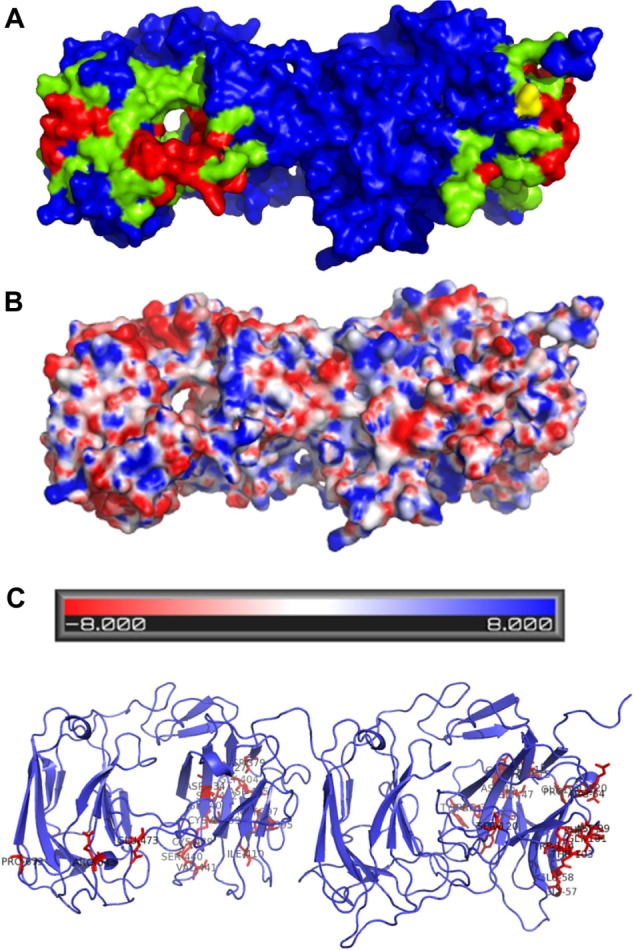
Putative dimer interface and its electrostatic map. (A) The predicted dimer interface of the R5 and R6 domains using CPORT: the active residues involved in the interaction are represented in red color and the passive residues in green color. Cysteine involved in disulfide formation is represented in yellow. (B) Representation of the electrostatic map of the domains predicted using Adaptive Poisson-Boltzmann Solver (APBS). (C) Representation of the conservation of the predicted interface residues.
Conclusion
The reelin protein, with its unique domain architecture consisting of eight reelin repeats, stands out as an exceptional model for evolutionary studies, as it is well conserved across the species, including humans. Sequence similarity shows that each repeat has an average 88% identity to its corresponding repeat in other species and an average 33% identity with other repeats within the protein sequence. Such sequence similarity values and phylogenetic trends suggest that individual reelin domains are not tandem repeats but retain specific functional roles. This is further confirmed by the biomedical importance of deletion mutants. Phylogenetic analysis shows that the reelin proteins and the reelin domains might have originated in Arthropoda and retain its consistent presence in mammals. We present 3D model of the full-length human reelin and suggest key residues at the putative dimer interface region. Although full-length reelin is known to exist as a disulfide-linker dimer, proteolytically cleaved reelin is known to exist as a multimer. The availability of 3D model of the full-length reelin provides avenues for further studies in these directions.
Acknowledgments
The authors would like to thank the National Center for Biological Sciences Tata Institute of Fundamental Research (TIFR) for the infrastructural support.
Footnotes
ACADEMIC EDITOR: J. T. Efird, Associate Editor
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 794 words, excluding any confidential comments to the academic editor.
FUNDING: Authors disclose no external funding sources.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: RS. Analyzed the data: SAM, MM. Wrote the first draft of the manuscript: SAM, MM. Contributed to the writing of the manuscript: SAM, MM. Agree with manuscript results and conclusions: RS, MM, SAM. Jointly developed the structure and arguments for the paper: RS. Made critical revisions and approved the final version: RS. All authors reviewed and approved the final manuscript.
Supplementary Material
Supplementary Table 1. List of the reelin homologues.
Supplementary Table 2. Structure validation and quality of the models. Ramachandran plot, Verify 3D, and ProSA scores of individual modeled reelin domain pairs.
Supplementary Table 3. List of residues in the putative dimer interface predicted by CPORT web sever. The conserved residues are highlighted in bold. The residue numbering is according to the PDB of R5 and R6 domains, and UniProt residue numbering is according to the full-length human reelin gene.
Supplementary Figure 1. Sequence logo of the alignment with the reelin domains (R1–R8).
Supplementary Figure 2. Sequence logo of reelin protein repeat 1. The subrepeat A and subrepeat B of first repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 3. Sequence logo of reelin protein repeat 2. The subrepeat A and subrepeat B of second repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 4. Sequence logo of reelin protein repeat 3. The subrepeat A and subrepeat B of third repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 5. Sequence logo of reelin protein repeat 4. The subrepeat A and subrepeat B of fourth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 6. Sequence logo of reelin protein repeat 5. The subrepeat A and subrepeat B of fifth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 7. Sequence logo of reelin protein repeat 6. The subrepeat A and subrepeat B of sixth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 8. Sequence logo of reelin protein repeat 7. The subrepeat A and subrepeat B of seventh repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 9. Sequence logo of reelin protein repeat 8. The subrepeat A and subrepeat B of eighth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 10. Phylogeny of reelin first repeat using the neighbor-joining algorithm.
Supplementary Figure 11. Phylogeny of reelin second repeat using the neighbor-joining algorithm.
Supplementary Figure 12. Phylogeny of reelin third repeat using the neighbor-joining algorithm.
Supplementary Figure 13. Phylogeny of reelin fourth repeat using the neighbor-joining algorithm.
Supplementary Figure 14. Phylogeny of reelin fifth repeat using the neighbor-joining algorithm.
Supplementary Figure 15. Phylogeny of reelin sixth repeat using the neighbor-joining algorithm.
Supplementary Figure 16. Phylogeny of reelin seventh repeat using the neighbor-joining algorithm.
Supplementary Figure 17. Phylogeny of reelin eighth repeat using the neighbor-joining algorithm.
Supplementary Figure 18. Conserved residues of individual domains mapped to the structure. The conserved residues are represented in gray.
Supplementary Figure 19. Root-meet-square deviation values of superposition of domain pairs for the construction of the full-length reelin proteins.
Supplementary Figure 20. Ramachandran plot for the modeled full-length reelin protein. Blue squares represent nonglycine (non-Gly) residues in allowed regions, red squares represent non-Gly residues in disallowed regions, and blue triangles represent Gly residues.
REFERENCES
- 1.Curran T, D’Arcangelo G. Role of reelin in the control of brain development. Brain Res Brain Res Rev. 1998;26:285–94. doi: 10.1016/s0165-0173(97)00035-0. [DOI] [PubMed] [Google Scholar]
- 2.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–9. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- 3.Yasui N, Nogi T, Takagi J. Structural basis for specific recognition of reelin by its receptors. Structure. 2010;18:320–31. doi: 10.1016/j.str.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 4.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–23. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 5.Yasui N, Nogi T, Kitao T, Nakano Y, Hattori M, Takagi J. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc Natl Acad Sci U S A. 2007;104:9988–93. doi: 10.1073/pnas.0700438104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ichihara H, Jingami H, Toh H. Three novel repetitive units of reelin. Brain Res Mol Brain Res. 2001;97:190–3. [Google Scholar]
- 7.Nogi T, Yasui N, Hattori M, Iwasaki K, Takagi J. Structure of a signaling-competent reelin fragment revealed by X-ray crystallography and electron tomography. EMBO J. 2006;25:3675–83. doi: 10.1038/sj.emboj.7601240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Arcangelo G. Reelin in the years: controlling neuronal migration and maturation in the mammalian brain. Adv Neurosci. 2014;2014:19. [Google Scholar]
- 9.Fatemi SH. Reelin glycoprotein: structure, biology and roles in health and disease. Mol Psychiatry. 2004;10:251–7. doi: 10.1038/sj.mp.4001613. [DOI] [PubMed] [Google Scholar]
- 10.Hong SE, Shugart YY, Huang DT, et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–6. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 11.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–4. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marchler-Bauer A, Bryant SH. CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 2004;32:W327–31. doi: 10.1093/nar/gkh454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchler-Bauer A, Panchenko AR, Shoemaker BA, Thiessen PA, Geer LY, Bryant SH. CDD: a database of conserved domain alignments with links to domain three-dimensional structure. Nucleic Acids Res. 2002;30:281–3. doi: 10.1093/nar/30.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–7. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 17.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–9. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 19.Eswar N, Webb B, Marti-Renom MA, et al. Comparative protein structure modeling using MODELLER. In: Board John E Coligan Al, editor. Curr Protoc Protein Sci. 2007. Chapter 2:Unit 2.9. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–18. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 21.Dominguez C, Boelens R, Bonvin AMJJ. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J Am Chem Soc. 2003;125:1731–7. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 22.Jorgensen W, Maxwell D, Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J Am Chem Soc. 1996;118:11225–36. [Google Scholar]
- 23.Lambert de Rouvroit C, Bernier B, Royaux I, et al. Evolutionarily conserved, alternative splicing of reelin during brain development. Exp Neurol. 1999;156:229–38. doi: 10.1006/exnr.1999.7019. [DOI] [PubMed] [Google Scholar]
- 24.Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 25.Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407–10. doi: 10.1093/nar/gkm290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yasui N, Kitago Y, Beppu A, et al. Functional importance of covalent homodimer of reelin protein linked via its central region. J Biol Chem. 2011;286:35247–56. doi: 10.1074/jbc.M111.242719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Vries SJ Bonvin AM. CPORT: a consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS One. 2011;6:e17695. doi: 10.1371/journal.pone.0017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. List of the reelin homologues.
Supplementary Table 2. Structure validation and quality of the models. Ramachandran plot, Verify 3D, and ProSA scores of individual modeled reelin domain pairs.
Supplementary Table 3. List of residues in the putative dimer interface predicted by CPORT web sever. The conserved residues are highlighted in bold. The residue numbering is according to the PDB of R5 and R6 domains, and UniProt residue numbering is according to the full-length human reelin gene.
Supplementary Figure 1. Sequence logo of the alignment with the reelin domains (R1–R8).
Supplementary Figure 2. Sequence logo of reelin protein repeat 1. The subrepeat A and subrepeat B of first repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 3. Sequence logo of reelin protein repeat 2. The subrepeat A and subrepeat B of second repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 4. Sequence logo of reelin protein repeat 3. The subrepeat A and subrepeat B of third repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 5. Sequence logo of reelin protein repeat 4. The subrepeat A and subrepeat B of fourth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 6. Sequence logo of reelin protein repeat 5. The subrepeat A and subrepeat B of fifth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 7. Sequence logo of reelin protein repeat 6. The subrepeat A and subrepeat B of sixth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 8. Sequence logo of reelin protein repeat 7. The subrepeat A and subrepeat B of seventh repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 9. Sequence logo of reelin protein repeat 8. The subrepeat A and subrepeat B of eighth repeat are shown here from 170 sequences. Five Cys columns in between the two subrepeats of reelin can be used to recognise the EGF motif domain.
Supplementary Figure 10. Phylogeny of reelin first repeat using the neighbor-joining algorithm.
Supplementary Figure 11. Phylogeny of reelin second repeat using the neighbor-joining algorithm.
Supplementary Figure 12. Phylogeny of reelin third repeat using the neighbor-joining algorithm.
Supplementary Figure 13. Phylogeny of reelin fourth repeat using the neighbor-joining algorithm.
Supplementary Figure 14. Phylogeny of reelin fifth repeat using the neighbor-joining algorithm.
Supplementary Figure 15. Phylogeny of reelin sixth repeat using the neighbor-joining algorithm.
Supplementary Figure 16. Phylogeny of reelin seventh repeat using the neighbor-joining algorithm.
Supplementary Figure 17. Phylogeny of reelin eighth repeat using the neighbor-joining algorithm.
Supplementary Figure 18. Conserved residues of individual domains mapped to the structure. The conserved residues are represented in gray.
Supplementary Figure 19. Root-meet-square deviation values of superposition of domain pairs for the construction of the full-length reelin proteins.
Supplementary Figure 20. Ramachandran plot for the modeled full-length reelin protein. Blue squares represent nonglycine (non-Gly) residues in allowed regions, red squares represent non-Gly residues in disallowed regions, and blue triangles represent Gly residues.