

Abstract
Pten is a multifunctional tumor suppressor. Deletions and mutations in the Pten gene have been associated with multiple forms of human cancers. Pten is a central regulator of several signaling pathways that influences multiple cellular functions. One such function is in cell motility and migration, although the precise mechanism remains unknown. In this study, we deleted Pten in the embryonic lung epithelium using Gata5-cre mice. Absence of Pten blocked branching morphogenesis and ERK and AKT phosphorylation at E12.5. In an explant model, PtenΔ/Δ mesenchyme-free embryonic lung endoderm failed to branch. Inhibition of budding in PtenΔ/Δ explants was associated with major changes in cell migration, while cell proliferation was not affected. We further examined the role of ERK and AKT in branching morphogenesis by conditional, endodermal-specific mutants which blocked ERK or AKT phosphorylation. MEKDM/+; Gata5-cre (blocking of ERK phosphorylation) lung showed more severe phenotype in branching morphogenesis. The inhibition of budding was also associated with disruption of cell migration. Thus, the mechanisms by which Pten is required for early endodermal morphogenesis may involve ERK, but not AKT, mediated cell migration.
Keywords: Lung morphogenesis, Branching morphogenesis, Pten, ERK, AKT, Cell migration, Endoderm
Graphical abstract
Introduction
The mammalian lung is a highly precise branched structure consisting of epithelial airways and alveoli as well as mesenchymal stroma and vascular networks. Each of the latter components is essential to proper respiratory functions. The formation of the lungs involves the structuring of epithelial tissues into complex but highly organized tubular networks that transport gases. This process is generally referred to as “branching morphogenesis”. In mammalian lung, branching morphogenesis starts from formation of an anlagen and invagination of the epithelium to branch initiation and outgrowth. The branching pattern is controlled genetically and gives rise to successive rounds of branched structures in a predictable manner.
The formation of lung via branching morphogenesis can generally be subdivided into a series of steps: formation of anlagen; formation of primary bud; branch initiation; branch outgrowth; organization of successive branching events and differentiation of proximal-distal structures. To generate this arborized epithelial network, each individual step depends on proper execution of cell proliferation, migration, apoptosis and changes in cell shape. The pattern of arborization depends on positive regulation by growth factors and receptors to promote proliferation and migration and negative regulation to inhibit excessive budding and bifurcation. How potential positive and negative signals are integrated to control cell movement, while retaining the essential character of an invasive epithelium, remains unclear.
A sequence of induction of factors was found. The implication of these factors in lung development was based on their association with specific lung defects in knockout mouse models. The major factors found regulating the branching are Fibroblast Growth Factors (FGFs), Transforming Growth Factor-β (TGF-β) superfamily, Wnt and Sonic Hedgehog. Recently Pten has been identified to control lung morphogenesis (1). PTEN has a dual specificity protein phosphatase and lipid phosphatase activity that can dephosphorylate serine, threonine and tyrosine residues. This positions PTEN as a critical negative regulator of PI3K/AKT signaling pathway (2). Deletion of Pten in mouse models revealed that PTEN is critical for animal development. Pten null embryos die early during embryogenesis (3). Thus, much of our current knowledge regarding the roles of PTEN in development is acquired from animals with tissue-specific Pten deletion using the Cre-LoxP system. The functions of PTEN in individual tissues include stem cell self-renewal and proliferation, cell differentiation and migration, organ size control, and the hormone-regulated organogenesis (4).
The mitrogen-activated protein kinase (MAPK) /extracellular signal-regulated kinase (ERK) cascade is conserved in mammals and controls early developmental processes, including determination of morphology, organogenesis, synaptic plasticity and growth (5). Signal transmission via this cascade is initiated by the activation of cell surface receptors by extracellular ligands, which results in the phosphorylation and activation of the MAPK/ERK kinases. Activation of the MAPK/ERK pathway leads to transcriptional control of genes important for cell proliferation and differentiation (6). Evidence is provided that MAPK/ERK kinases influence the cells’ motility machinery. Inhibition of ERK activity causes decreased cell migration on extracellular matrix protein (7).
In the current study, we assessed the direct impact of epithelial Pten inactivation on lung morphogenesis. The results demonstrate that Pten controls early stage branching morphogenesis by regulating epithelial cell migration, without major or measurable changes in cell proliferation. This function is mediated via phosphorylation of ERK, which is also essential for embryonic lung epithelial cell migration.
Materials & Methods
Mouse Lines
PtenΔ/Δ mice were generated by crossing Ptenfl/fl (8) with Gata5-Cre (9) mice. MEKDN/+, R26StopFL-Pik3ca and mT/mG mice were purchased from The Jackson Laboratory. MEKDN/+; Gata5-Cre and R26StopFL-Pik3ca; Gata5-Cre mice were generated by crossing MEKDN/+ and R26StopFL-Pik3ca with Gata5-Cre mice.
Lung culture and lung endodermal explant culture
Whole embryonic lungs were dissected at gestational stage E11.5. In each Grobstein Falcon dish, two to three lungs were placed on filters (Millipore, Bedford, MA) that were placed on top of a stainless steel grid. The filters were in close contact with BGJb (GIBCO, Carlsbad, CA) growth medium supplemented with penicillin (100U/ml) and streptomycin (100μg/ml), with 20μM MEK inhibitor U0126 (Promega, Madison, WI) or same amount of DMSO as control. Heparin beads (Sigma) were incubated with FGF10 (10μl of a 50ng/μl solution; R&D Systems, MN) at 37°C for 2hr, grafted onto lung explants and cultured for 48 hr. The lungs were incubated under optimal humidity in 95% air/5% CO2. At indicated times, the lung explants were either homogenized in Trizol (GIBCO) for RNA isolation or fixed in 4% Paraformaldehyde (PFA).
For mesenchyme-free endodremal lung explant culture, distal lung tips were isolated as previously described (10). Briefly, lungs from mutant or control embryos were dissected at E13.5 and treated with Dispase (50U/ml, BD Biosciences, Franklin Lakes, NJ) at 4°C for 20 min. Epithelial buds of distal lung tips were then isolated by removing mesenchyme with tungsten needles and embedded into growth factor-reduced Matrigel (Fisher Scientific, Fremont, CA), diluted 1:1 in culture medium (50% DMEM: 50% Ham’s F12, 0.05U/ml penicillin, 0.05mg/ml streptomycin). After polymerization of the Matrigel at 37°C, explants were covered with culture medium with FGF10 and cultured at 37°C at 5% CO2 for various lengths of time as indicated. FGF10-saturated beads were prepared with acrylic beads with immobilized heparin (Sigma). Beads were rinsed with PBS three times, manually selected under dissection microscope and then soaked in 50ng/μl recombinant human FGF10 at 37°C for 1hr. The beads were then washed in DMEM medium for 1hr at 37°C before use (11). MEK inhibitor U0126 was purchased from Promega.
Migration assay
Mesenchyme-free explants were treated with 0.25% trypsin/EDTA and DNase I(1:1) at 37°C for 15min. E18.5 lung epithelial cells were isolated according to Corti et al. (12). The lungs were carefully chopped into pieces and treated with dispase at 37°C for 15min. DMEM was added to terminate the reaction. The cell suspension was filtered through 100μm, 40μm and 20μm filters. Then the epithelial cells were plated on culture dish (BD Bioscience) and culture in DMEM and 10% FBS at 37°C. Cell monolayers were wounded with a plastic tip. Cell migration was followed for 6hr at 37°C and photographed.
Proliferation assay
Subsequent to culturing, endodermal explants were treated with bromodeoxyuridine (BrdU) reagent at 1μl per 400μl medium for 3hr. The explants were fixed, dehydrated and paraffin embedded. The sections were then re-hydrated and labeled by BrdU staining kit (Invitrogen, Carlsdad, CA) and Hematoxylin/Eosin staining. The sections were then photographed. The BrdU positive cells as well as the total number of cells (Hematoxylin/Eosin positive cells) in the explants per section were counted manually using photomicrograph of tissue taken at 40X on a Zeiss Microscope. The percent of labeled cells was calculated.
Cell death analysis
The cultured endodermal explants were fixed, dehydrated and embedded in paraffin and 5μm sections were prepared. The “In Situ Cell Death Detection Kit, Fluoresein” kit (Roche Allied Science, Indianapolis, IN) was used for detection and quantification of cell apoptosis at single cell level, based on labeling of DNA strand breaks (TUNEL technology). The sizes of labeled areas and whole sections were calculated for the percentage of cell death. 5 sections from each treatment were analyzed.
Immunohistochemistry
5μm tissue sections were prepared. Subsequent to deparaffinization, the sections were hydrated, heated in 10mM citrate buffer (pH6.0) and treated with 10% H2O2 in methanol for 20min and blocked with 10% of nonimmune serum. Sections were then incubated with primary antibodies at 4°C overnight. Biotinylated secondary antibody and streptavidin-peroxidase conjugate (Vector Laboratories, Inc.) were used to detect the bound antibodies. The sections were developed with diaminobenzidine. GFP and Ki67 primary antibody was purchased from Thermo Scientific.
RNA extraction and Real-time PCR
Total RNA was isolated from lungs or endodermal explants using Trizol (GIBCO). The cDNA was synthesized from 1μg total RNA by following the protocol of the SuperScript™ First-Strand Synthesis Kit (Invitrogen). Real-time PCR was performed by a LightCycler System (Roche). Primer sets for following genes were used for Real-time PCR: Cdc42: 5′-TGCTCTGCCCTCACACAG-3′ and 5′-GGCTCTTCTTCGGTTCTGG-3′; Rac1: 5′-AGATGCAGGCCATCAATGT-3′ and 5′-GAGCAGGCAGGTTTTACCAA-3′; RhoA: 5′-GAATGACGAGCACACGAGAC-3′ and 5′-TCCTGTTTGCCATATCTCTGC-3′.
Western Blot Analysis
Cells or lung tissues were harvested and frozen in liquid nitrogen. Protein extracts were prepared in RIPA buffer (Sigma, St. Louis, MO) by homogenization, and equal amounts of protein were separated on 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA). Proteins were transferred onto Immobilon-P transfer membranes (Millipore Corp. Billerica, MA) and analyzed by western blotting using antibodies recognizing the following proteins: ERK−P, ERK, AKT−P, Pten and PI3 Kinase p110 (Cell Signaling Technology, Danvers, MA), and α-tubulin (BioGenex, San Ramon, CA).
RESULTS
Epithelial-specific deletion of Pten in the murine lung
To determine the potential role of epithelial Pten signaling in lung morphogenesis, we used a Gata5-Cre mouse line (9) to generate Pten deletion in the lung epithelium (Figure 1, Panel A). The pattern, efficiency and cell type specificity of the Gata5-Cre mouse line in mediating LoxP-dependent DNA excision in the lung was determined by crossing with mT/mG reporter mice (Figure 1, Panel B). mT/mG mice possess LoxP sites on either side of a membrane-targeted tdTomato (mT) cassette and express strong red fluorescence in all tissues and cell types. When bred to Cre recombinase expressing mice, the resulting offspring have the mT cassette deleted in the Cre expressing tissue(s), allowing expression of the membrane-targeted EGFP (mG) cassette located just downstream. In E13.5 Gata5-Cre; mT/mG double transgenic embryos, EGFP activity was found to be uniform and limited to the endodermally derived lung epithelium (Figure 1, Panel B, a&b). Immuno-histochemistry (IHC) with an anti-GFP antibody showed similar epithelial cell type specificity (Figure 1, Panel B, c&d). To test the efficiency of Pten deletion in PtenΔ/Δ lung, we measured the mRNA level of Pten by real-time PCR and expression of Pten by IHC in E18.5 lung. The results showed that the ablation of Pten caused significant decrease of Pten (Supplemental Figure 2).
Figure 1. Generation of PtenΔ/Δ; Gata5-cre mice.
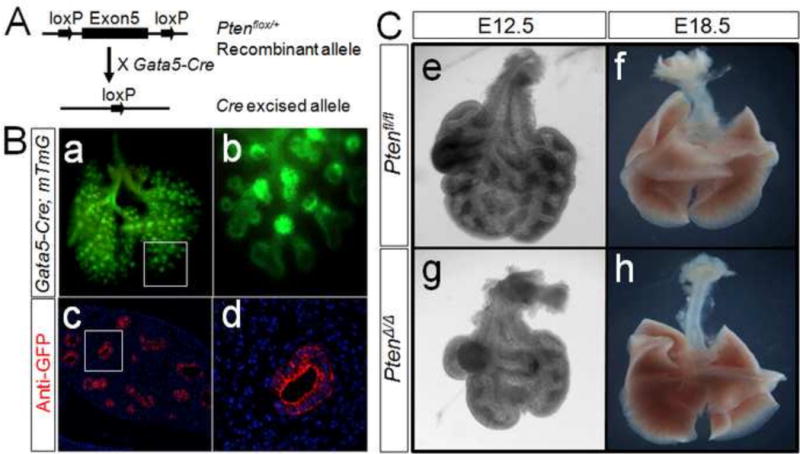
Panel A: PtenΔ/Δ; Gata5-Cre was generated by crossing Ptenfl/fl with Gata5-Cre mice. Panel B: Gata5-Cre-mediated recombination in E12.5 lung was analyzed by GFP labeling in Gata5-Cre; mT/mG lung (a&b). Anti-GFP antibody showed specific staining in epithelial cells (c&d). On E12.5, PtenΔ/Δ; Gata5-Cre lung appeared smaller in size and less branching (Panel C). On E18.5, there was no difference in branching morphogenesis between mutant and control lung (Panel C, f&h).
Inactivation of Pten in mice, referred to simply as PtenΔ/Δ, caused abnormal branching of the endoderm in the lung of E12.5 embryos (Figure 1, Panel C, e–h). Compared with the controls, the PtenΔ/Δ lungs were consistently smaller in overall size and contained fewer branches (Figure 1, Panel C, e&g). However, as development proceeds there appeared to be compensation for these early defects. By E18.5 the lungs were indistinguishable from the control (Figure 1, Panel C, f&h). And, the newborn PtenΔ/Δ mice did not appear to have difficulty breathing (data not shown).
Pten is required for cell migration in early lung
To analyze the impact of Pten on endodermal branching, we examined the response of lung mesenchyme-free endoderm to FGF10. Mesenchyme-free nascent endodermal tissue can be isolated from mouse embryonic lungs of E12.5 gestation and explanted in Matrigel for various studies (8). Culturing of endodermal explants requires the presence of FGF10, which is necessary and sufficient to direct branching morphogenesis (10). In the following studies, all control and PtenΔ/Δ explant culture contained 400ng/ml of FGF10 ligand for survival and induction of branching morphogenesis as described previously (8). Figure 2 shows the results of these studies at times 0 and 48 hours. As expected, FGF10 had a robust impact on induction of branching morphogenesis in E12.5 control lung endoderm (Figure 2, Panel A, a&c). Without exception, inactivation of Pten in PtenΔ/Δ explants strongly inhibited FGF10-induced branching (Figure 2, Panel A, b&d).
Figure 2. Deletion of Pten affects endodermal morphogenesis by inhibiting cell migration.
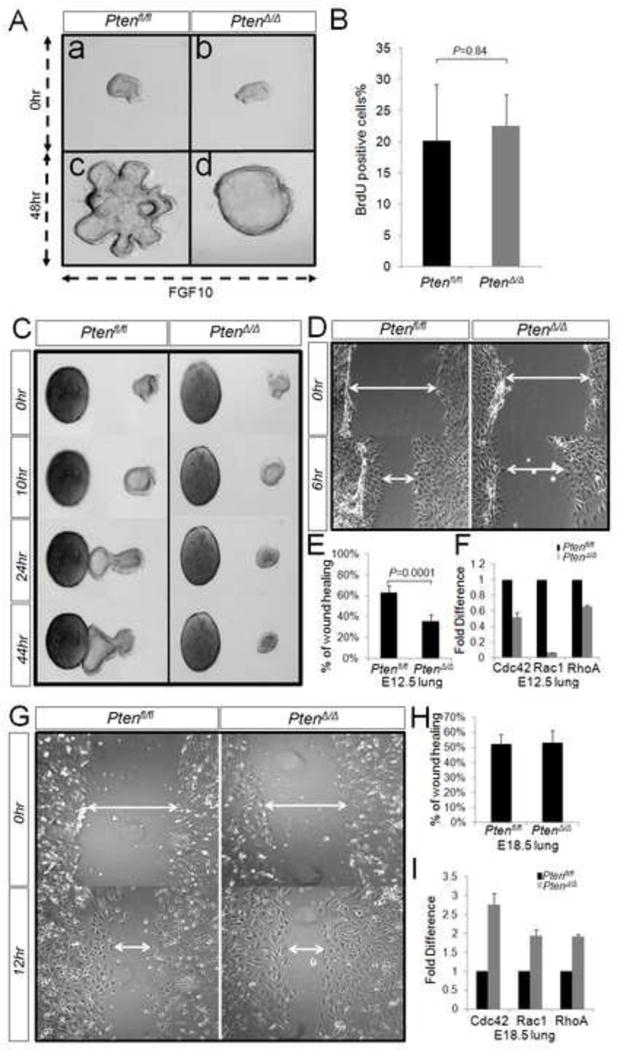
Isolated endodermal explants from E12.5 mouse embryos were cultured in presence of FGF10. In PtenΔ/Δ explant, FGF10-induced branching morphogenesis is blocked (Panel A). No significant difference was observed in the ratio of BrdU positive cells (in total cells) between the control and PtenΔ/Δ explants. P=0.84 (Panel B). Endodermal explants from both PtenΔ/Δ and control lungs are treated with FGF10-soaked beads. The control explants move towards FGF10 source after 10 hrs. In opposite, PtenΔ/Δ explants show no sign of movement (Panel C). Cells isolated from endodermal explants were cultured and scratched to create a wound. The migration of the cells was photography at 0 hr and 6 hr after wounding (Panel D). The percentage of wound healing was calculated from the mean of 20 wound widths per condition with SD measured at 6 hr (Panel E). By real-time PCR analysis, the expression of RhoA, Rac1 and Cdc42 is reduced largely in E12.5 PtenΔ/Δ lungs (Panel F). In contrast, the epithelium cells from E18.5 PtenΔ/Δ lung showed no difference in cell migration vs control (Panel G&H). The expression pattern of RhoA, Rac1 and Cdc42 in E18.5 PtenΔ/Δ lung is opposite from E12.5 (Panel I).
Lung branching morphogenesis requires cell proliferation and migration. To examine whether inhibition of FGF10-induced endodermal branching morphogenesis observed in PtenΔ/Δ explants is accompanied by changes in cellular proliferation, we used the BrdU labeling approach. The ratio of BrdU positive cells as a fraction of total cells was determined by manual counting of multiple samples. The analysis showed no significant differences in cellular proliferation between control and PtenΔ/Δ explants (Figure 2, Panel B).
To address whether inactivation of Pten affected cell migration, we cultured the endodermal explants with directional FGF10 signaling. The control and PtenΔ/Δ explants were placed 215μm to 260μm away from FGF10-saturated beads. Control explants showed clear chemotaxis in the direction of the FGF10 beads as early as approximately 10 hours of culturing. Within the same distance from FGF10 signaling source, the PtenΔ/Δ explants showed no evidence of chemotaxis (Figure 2. Panel C). We also isolated and cultured the epithelial cells from mesenchyme-free explants. The monolayer of epithelial cells from control or PtenΔ/Δ explants was injured by a pipette tip, and cell migration to close the wound was analyzed. After 6 hours, cells from control explants had undergone a 63% wound closure. In contrast, cells from PtenΔ/Δ explants showed a mere 36% wound closure (Figure 2. Panel D&E). This observation indicated that Pten is required for cell migration. Rho family GTPases, particularly RhoA, Rac1 and Cdc42, are key regulators of cell polarization and directional migration (14). Recent data proposed that RhoA functions as an initiator of membrane protrusion by promoting actin polymerization, whereas Rac1 and Cdc42 might actually facilitate protrusion by regulating cell adhesion (15). Consistent with our earlier result, quantitative PCR analysis showed significantly decreased expression of RhoA, Rac1 and Cdc42 in E12.5 PtenΔ/Δ lung, when compared to controls (Figure 2, Panel F).
To compare with the early stages of lung development, we also analyzed migration of cell extracted from E18.5 lungs. No differences were observed in isolated epithelial cell monolayers from control or PtenΔ/Δ lungs. Both exhibited 52–53% wound closer within a 12 hour period, suggesting similar abilities in cell migration between the control and the PtenΔ/Δ lung cells (Figure 2, Panel G&H). Also, in contrast to E12.5 lungs, the expression of RhoA, Rac1 and Cdc42 was increased in E18.5 PtenΔ/Δ lungs as compared to that of control lungs (Figure 2, Panel I).
Pten deletion in endoderm affects ERK and AKT phosphorylation
Both ERK and AKT pathways have been reported to be regulated by Pten (9, 16). Activation of ERK kinase 1/2 positively regulated airway epithelial cell migration in vitro (17). AKT is also well known as an important regulator of cell survival, growth and cell migration in various organs (18). To determine the mechanisms of Pten-regulated endodermal morphogenesis, we analyzed the levels of activated (phospho) ERK and AKT in developing lungs. By western-blot analysis, we found that activation (phosphorylation) of ERK and AKT was reduced in PtenΔ/Δ lung during the window (E12.5) in which defects in branching were observed (Figure 3, Panel A; Figure 1, Panel C). The reductions of phospho-ERK and phospho-AKT levels were also confirmed in PtenΔ/Δ lung explants (Figure 3, Panel B). Importantly, phospho-ERK and phospho-AKT levels were increased in E18.5 PtenΔ/Δ lungs compared to controls (Figure 3, Panel A).
Figure 3. The expression patterns of phospho-ERK and phospho-AKT in PtenΔ/Δ lungs.
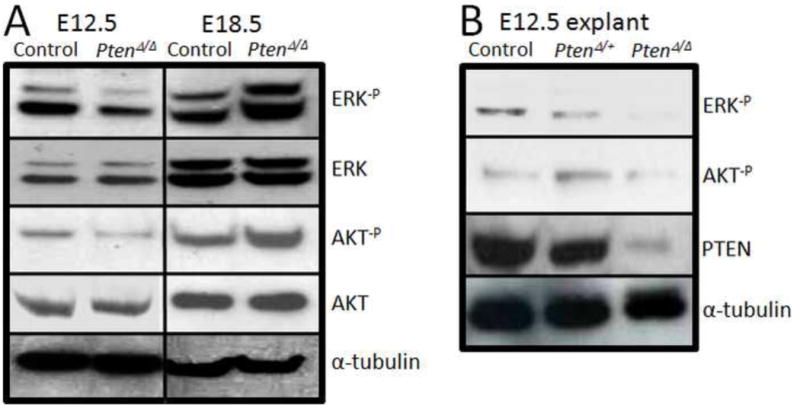
Panel A: Comparing with control lungs, the level of ERK and AKT phosphorylations is reduced in E12.5 PtenΔ/Δ lungs but it is increased in E18.5 PtenΔ/Δ lungs. Panel B: In consistent with in vivo data, the level of ERK and AKT phosphorylation is reduced in PtenΔ/Δ endodermal explants.
The role of phospho-ERK in branching morphogenesis
As decrease of phospho-ERK and phospho-AKT in the mutant lungs occurred only during the early stage when branching morphogenesis was affected, we hypothesized that this reduction in E12.5 lungs may be responsible for the defect in branching morphogenesis (Figure 1, g). To test this hypothesis we generated mice with over-expression of a dominant negative MEK1 (dnMEK1) via deletion of a stop cassette through the activity of Gata5-Cre (Figure 4, Panel A). By western-blot analysis, we confirmed that the level of phospho-ERK was significantly reduced in mutant lungs (Figure 4, Panel B). On day E12.5, the MEKDN/+; Gata5-Cre lungs were much smaller and showed less branching (Figure 4, Panel C). However, the airway epithelium in the mutant lungs appeared multilayered compared to controls. To examine whether reduced ERK activation affected cell proliferation or cell death, anti-Ki67 and TUNEL staining were employed. Interestingly, there was no significant difference found between mutant and control lungs (Supplemental Figure 1). By quantitative PCR analysis, RhoA, Rac1 and Cdc42 were significantly decreased in E12.5 MEKDN/+; Gata5-Cre lung as compared to control lungs (Figure 4, Panel D). These data suggested that the observed branching morphogenesis defects in MEKDN/+; Gata5-Cre lungs might be caused by reduced cell migration, without discernible or measurable changes in cell proliferation or apoptosis. Consistent with the in vivo data, the mesenchyme-free endodermal explants from MEKDN/+; Gata5-Cre lung exhibited significant reduction in branching morphogenesis after 48 hours of FGF10 treatment in vitro (Figure 5, Panel A). In addition, BrdU analysis, revealed no differences in the ratios of BrdU positive cells to total cell counts between mutant and control explants (Figure 5, Panel B). To further analyze the impact of phospho-ERK on lung morphogenesis, we used embryonic whole lung explants. E11.5 embryonic mouse lungs were excised from mouse embryos and cultured in a serum-free medium (8) in presence or absence of 4ng/ml MEK inhibitor. The result showed MEK inhibitor had a profound inhibitory impact on branching morphogenesis (Figure 5, Panel C, c vs d) and blocked ERK phosphorylation (Figure 5, Panel D).
Figure 4. Generation of MEKDN/+; Gata5-cre mice.
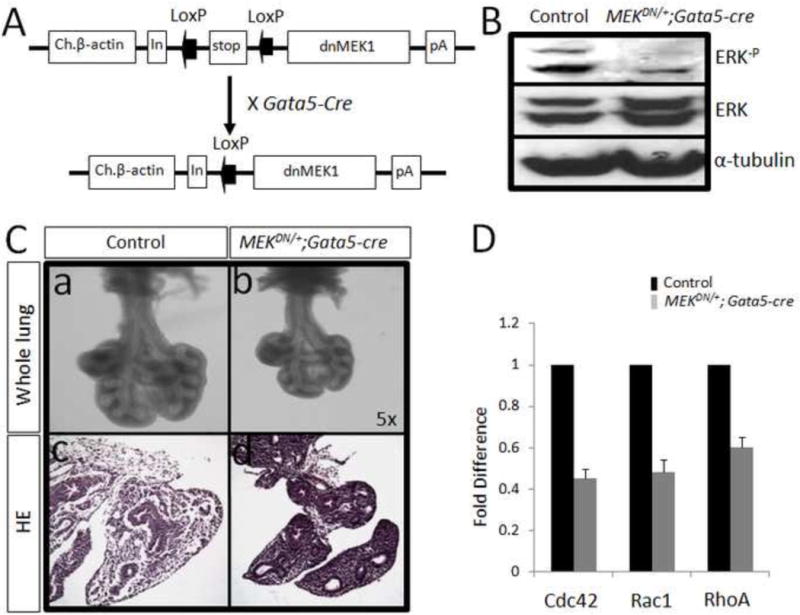
MEKDN/+; Gata5-Cre mice were generated by crossing MEKDN/+ mice with Gata5-Cre mice (Panel A). By western blot analysis, the level of ERK phosphorylation was dramatically reduced in MEKDN/+; Gata5-Cre mice (Panel B). Panel C: On day E12.5, MEKDN/+; Gata5-Cre lungs appeared smaller in overall size and less branching, although the thickness of epithelial airway appears different. Panel D: By real-time PCR analysis, the expression of RhoA, Rac1 and Cdc42 is reduced largely in E12.5 PtenΔ/Δ lungs.
Figure 5. Reduction of ERK phosphorylation blocks branching morphogenesis.
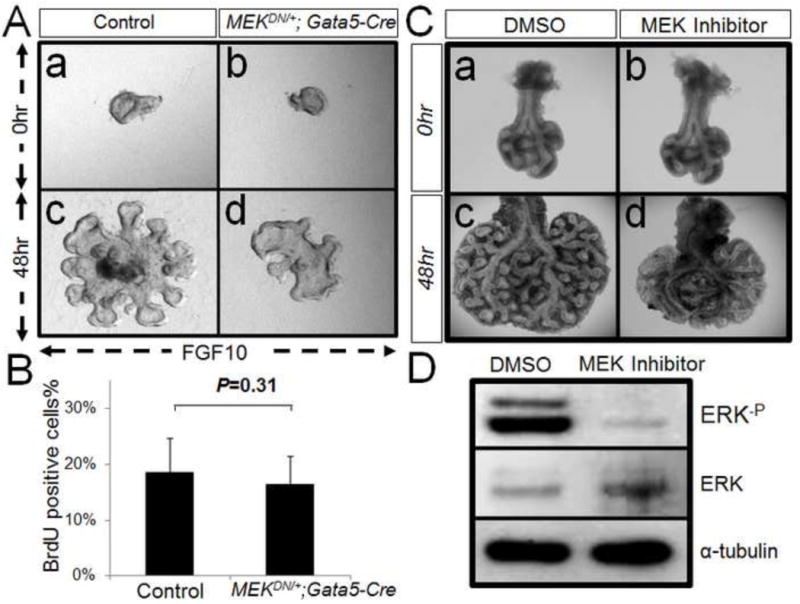
Panel A: Endodermal explants from E12.5 control and MEKDN/+; Gata5-Cre lungs are cultured in presence of FGF10. After 48 hr, the control explants undergo robust morphogenesis (a&c). Whereas MEKDN/+; Gata5-Cre explants shows reduction of branching (b&d). Panel B: By BrdU analysis, the ratio of proliferating cells in total cells shows no difference between control and MEKDN/+; Gata5-Cre explants, P=0.31. Panel C: Lungs from E12.5 mice are cultured and treated with MEK inhibitor vs DMSO as control. After 48 hr, the control lung undergoes branching morphogenesis (a&c). Whereas MEK inhibitor-treated lung shows significant reduction in branching (b&d). Panel D: Western-blot result shows that MEK inhibitor can fully block ERK phosphorylation in treated lung.
To determine whether phospho-ERK mediates embryonic lung branching morphogenesis via regulating cell migration, we conducted the following two experiments. First, in mesenchyme-free endoderm culture experiments, both mutant and control endoderm explants were treated with directional FGF10 signaling as described earlier. As expected, the control explants showed chemotaxis in the direction of FGF10 within 18 hrs of treatment. In contrast, chemotaxis was blocked in mutant explants (Figure 6, Panel A). Secondly, monolayers of isolated epithelial cells from mesenchyme-free explants were wounded to test cell migration. After 12 hours, cells from control explants had undergone a 66% wound closure. In contrast, cells from MEK inhibitor treated cells showed only a mere 20% wound closure (Figure 6. Panel B&C).
Figure 6. Deletion of ERK phosphorylation reduces cell migration.
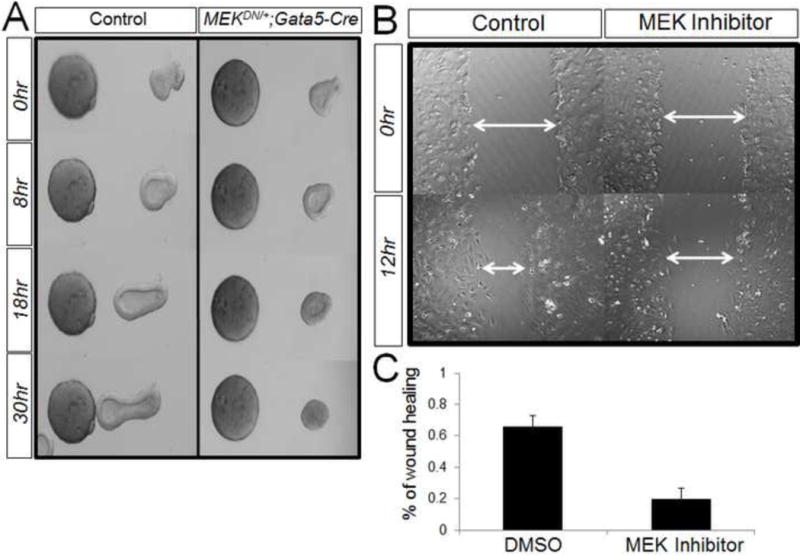
Panel A: Endodermal explants from both control and MEKDN/+; Gata5-Cre lungs were treated with FGF10-soaked beads. The control explants move towards FGF10 source after 8 hr culture. In opposite, MEKDN/+; Gata5-Cre explants show no sign of movement. Panel B: Epithelial cells from E12.5 lungs were cultured and wounded. After 12 hour, the control cells show 66% wound healing, where as MEK inhibitor treated cells show 20% (Panel C).
The role of phospho-AKT in branching morphogenesis
We also generated R26StopFL-Pik3ca; Gata5-Cre (referred to here as P110fl/fl; Gata5-Cre) mice to over-express P110. P110 is a constitutively active form of the mouse catalytic P110α subunit of PI3K (19). Interestingly, phosphorylation of AKT was completely blocked in P110fl/fl; Gata5-Cre lungs (Figure 7. Panel A&B). This made the P110fl/fl; Gata5-Cre a perfect model to examine the impact of phospho-AKT on branching morphogenesis. Compared with the controls, Phospho-AKT-deficient P110fl/fl; Gata5-cre mice appeared normal in overall size and airway branching at E12.5 (Figure 7. Panel C). In mesenchyme-free explants culture, the mutant explants displayed normal branching morphogenesis (Figure 7. Panel D). Both mutant and control explants showed clear chemotaxis in the direction of FGF10 beads (Figure 7. Panel E). By real-time PCR analysis, there was no significant change detected in the expression of RhoA, Rac1 and Cdc42 between P110fl/fl; Gata5-Cre lungs and controls (Figure 7, Panel F).
Figure 7. Reduction of AKT phosphorylation does not affect lung morphogenesis and cell migration.
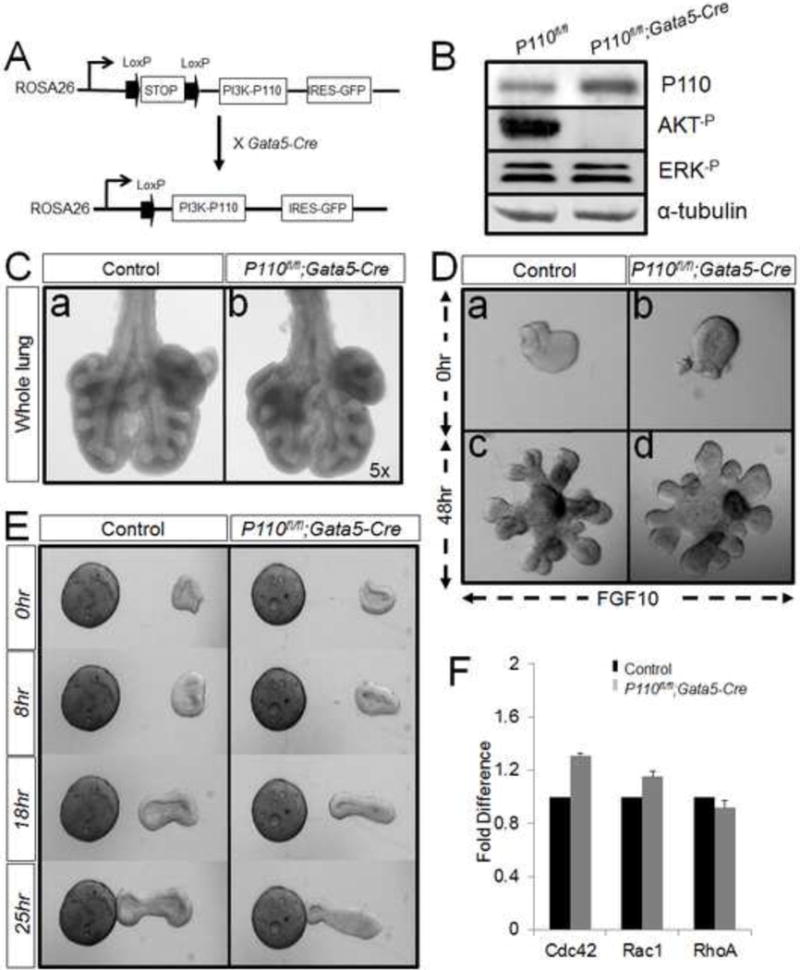
Panel A: Generation of P110fl/fl; Gata5-cre mice. Panel B: Western-blot result shows that phospho-AKT is significantly reduced in P110fl/fl; Gata5-Cre lung. Panel C: On day E12.5, P110fl/fl; Gata5-Cre lung shows no defects in overall size and branching morphogenesis. Panel D: In mesenchyme-free explants culture, P110fl/fl; Gata5-Cre explants undergo budding as control explants after treated with FGF10. Both mutant and control explants show clear chemotaxis in the direction of the FGF10 beads (Panel E). Panel F: Comparing with control lungs, the lungs from E12.5 P110fl/fl; Gata5-Cre show no difference in RhoA, Rac1 and Cdc42 expressions.
Discussion
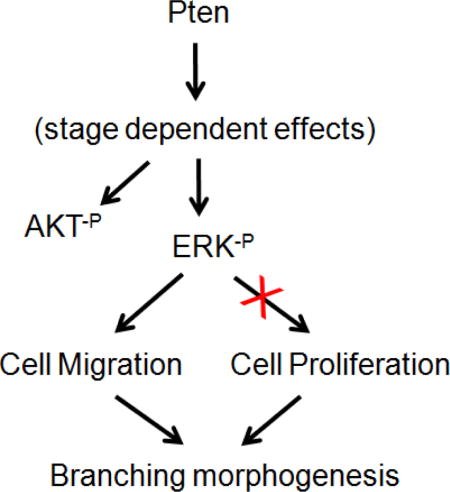
The purpose of the current study was to determine the precise role of endodermal-specific Pten in early lung branching morphogenesis. Endodermal deletion of Pten disrupted branching morphogenesis during early stage of lung development (between E11.5 and E13.5). Both in vivo, ex vivo, and cell culture experiments demonstrated that the inhibition of branching by Pten deletion is due to inhibition of cell migration, without measurable changes in cell proliferation. Since inhibition of branching morphogenesis was concomitant with changes in expression of phosphor-ERK and AKT, we therefore examined the roles of the latter molecules in embryonic lung branching and in airway epithelial cell migration. Examination of conditional deletion of phospho-ERK and AKT found that only phospho-ERK is required for cell migration and branching morphogenesis, but not phospho-AKT (Figure 8).
Figure 8. A simplified model illustrating Pten signaling in early lung branching morphogenesis.
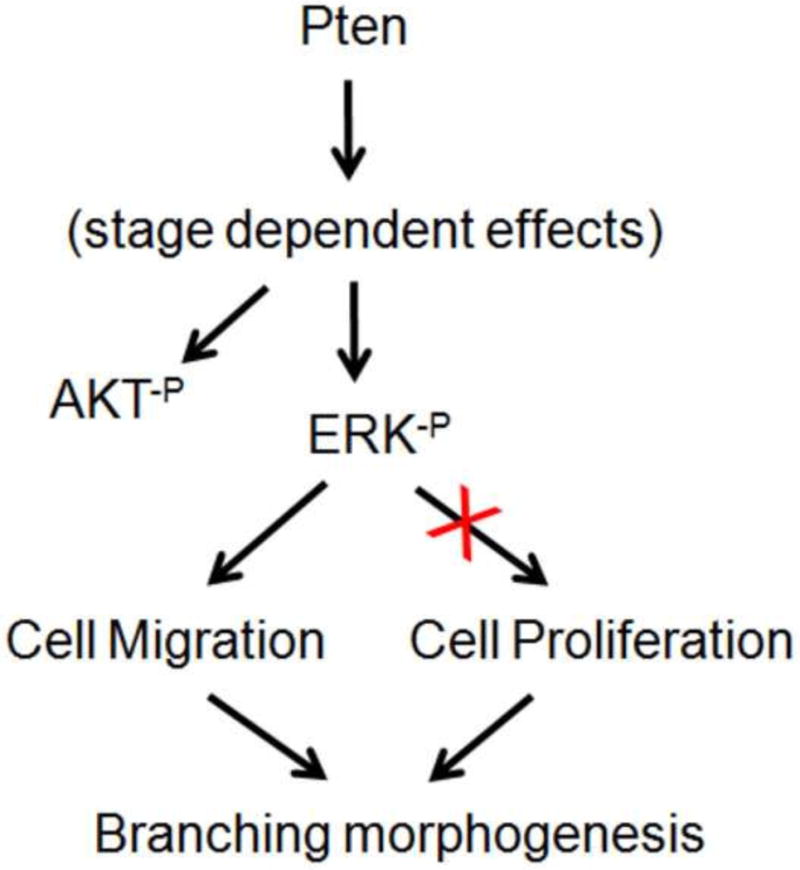
In E12.5 lung, Pten positively regulates ERK and AKT phosphorylation. The Pten-ERK pathway controls airway branching via regulation of epithelial cell migration, without measurable alterations in cell proliferation. The Pten-AKT pathway does not appear to affect epithelial cell migration.
In this study, our first aim was to determine whether Pten loss-of-function in epithelial cell impacts lung morphogenesis during development. Loss-of-function of Pten has been found in a wide range of human tumors (20). Various mouse models were generated to study the crucial role of Pten as tumor suppressor. Pten knock-out mice die early in development, but the cellular and developmental bases of lethality have not been defined. In recent years, several mouse models of epithelial-specific inactivation of Pten were made to study the roles of Pten during lung development. Surprisingly, the results from different research groups drove to different conclusions. By using SPC-rtTA/(tetO)7-Cre/Ptenfl/fl mice model, Yanagi showed that Pten was essential for normal lung morphgenesis (1), but the phenotype observed in E18 lung by Yanagi did not seem to occur in Dave’s mice (21). Comparing with their model, in our case, Gata5-Cre targets lung epithelial progenitors much early than SPC-rtTA/(tetO)7-Cre. This allows us to see changes in early stage of lung development. In this paper, by generating Ptenfl/fl; Gata5-Cre mice, we found that Pten is required for early lung branching morphogenesis by inhibiting cell migration. Interestingly, this affection was time-dependent and rescued in later embryonic stage. Prior to the current study, the roles of Pten in regulating cell migration were very different depending on the experimental materials used. Several reports showed that loss of Pten in developing tissues, such as neutrophils and B cells (22, 23), disrupted cell migration and the ability to respond to chemoattractants. However, the studies from human cancer cell lines showed opposite results (24, 25), the cells lacking Pten migrated faster. Together with our data, it suggests that the function of Pten in cell migration is dependent on the cellular context. During lung development, E12.5 and E18.5 represent two distinct developmental stages, the pseudoglandular and saccular stages, respectively. At E12.5, lung epithelial cells remain as progenitors, while at E18.5 lung epithelial cells have differentiated into multiple cell types which have distinct gene expression profiles. These intrinsic differences may underlie the stage dependent role of Pten in cell migration. Previous studies showed that mechanisms by the C2 domain of Pten and the autodephosphorylation of Pten mediate cell migration. These two mechanisms are both involved in phosphatase activity which can alter cell migration (24). In our case, whether the two mechanisms caused the differences between E12.5 and E18.5 Pten null epithelial cells is unknown.
Our second aim was to investigate which downstream target(s) of Pten in pulmonary epithelium was responsible for the Pten-regulated cell migration. By western blot analyses, we found deletion of PTEN results in decreased levels of phospho-ERK (Figure 3) in airway epithelial cells during early stage of lung development. Previous studies have demonstrated that the MEK/ERK cascade mediates FGF signaling (26), which functions as the major chemoatractants on the branching epithelium (27). These data collectively suggest a central role of MEK/ERK pathway in epithelial cell migration. We therefore generated the MEKDM/+; Gata5-Cre mice to conditionally inactivate the MEK/ERK pathway in embryonic lung epithelial cells. Examination of lung morphogenesis and epithelial cell migration in phospho-ERK mutant lungs revealed a marked reduction in number of branching and speed of cell movement. This study, for the first time, directly demonstrated the critical roles of MEK/ERK pathway in lung branching morphogenesis and in lung epithelial cell migration. In the PTEN mutant lung, deletion of PTEN decreases levels of phospho-ERK which is essential for mediating the chemotactic effectors of FGF10 on the branching epithelium.
To date, it is still poorly understood how the ERK signaling pathway is involved in cell migration. In current study, we found that the decreased phospho-ERK level lead to reduction of Cdc42, Rac1 and RhoA in early lung embryonic epithelial cells. But the regulations or collaborations between MEK/ERK pathway and Rho family GTPases are still not clear. Previous published data in human skin fibroblasts showed that Cdc42 downregulates MMP-1 expression by inhibiting the ERK pathway (28). In transformed cells, ERK kinase selectively uncouples RhoA from one of its effecter pathways (29). Other recent study also indicated that the architecture of the Rho kinase–ERK network allows bi-directional flow of information in its default state to regulate neuronal differentiation (30). In our case, it needs further investigation to discover whether ERK regulated Rho family GTPases expression is direct interaction.
The ERK activity is stimulated by a wide variety of growth factors and mitogens (31). In PtenΔ/Δ lungs, levels of phospho-ERK is decreased in early stage but increased in late stages of lung development. The stage dependent ERK activity in Pten deficient lungs might be caused by interactions of Pten with other pathways that also regulate ERK cascade. For example, our previous results showed that TGFß/ALK5 signaling controls ERK phosphorylation, and this is negatively regulated by PTEN in E18.5 lung epithelial (9). Therefore, the balance between Pten and TGFß/ALK5 signaling (and other ERK regulators such as FGF10) may modify the overall ERK activity at different developmental stages. An activity profile of all the ERK regulators at each embryonic stage may provide a more detail explanation on the stage dependent ERK activity in PtenΔ/Δ lungs. Nevertheless, current study demonstrated that ERK activity is critical for epithelial cell migration during embryonic lung development.
To investigate whether the level of phospho-AKT affects lung morphogenesis and epithelial cell migration, we generated P110fl/fl; Gata5-Cre mouse, which constitutively active p110α. Western-blot result showed that the level of phospho-AKT in mutant lung is significantly reduced (Figure 7, Panel B). By in vivo and in vitro examination, the reduction of phospho-AKT level does not affect early airway branching and the speed of epithelial cell movement. This result suggests that, contrary to the prevailing concept, Pten-regulated cell migration is mediated by PI3K/AKT-independent pathway in embryonic lung epithelial cells. Previous studies found that many of Pten’s cellular processes are through PI3K signaling. PI3Ks are classified into three groups (class I, II and III) based on structure and substrate specificity (32). Class IA PI3Ks transduce signals encoding its catalytic subunit p110α, which is the only PI3K gene identified with common mutations in human cancer (32). Activated PI3Ks catalyze the formation of PIP3 from PIP2, and PTEN directly opposes the activity of PI3Ks by dephosphorylating PIP3 into PIP2. AKT is a centrally important downstream effector of PIP3. We have showed that the expression of p110 was largely increased in mutant lung. Surprisingly, the level of phospho-AKT in mutant lung was significantly reduced. As suggested by a review from Chalhoub (33), the PI3K signaling pathway is more complex in mammals due to the evolution of multiple family members for some effectors in the pathway and also the diversity of growth factors that transmit their signals through PI3K in context-dependent fashions. It is possible that the AKT phosphorylation is directly blocked by other factors from cross-talk pathways in mutant lung, although increased p110 may cause higher level of PIP3. It still needs further investigation to discover whether this potential signaling transition is selectively expressed in lung epithelial cells. Nevertheless, epithelial specific reduction of phospho-AKT in P110fl/fl; Gata5-Cre lungs does not appear to disrupt early lung morphogenesis. A recent study by Carter et al (34) showed that airway branching is increased in lung explants, in which AKT activity is globally blocked by pharmacological inhibitor. These indicate that the AKT activity in the mesenchymal compartment may also contribute to regulating airway branching. The mesenchymal compartment is the major source of chemotatic factors for airway branching.
A wealth of knowledge regarding Pten regulated cell migration has come from both development and cancer. Pten deletion in the developing nervous system shows the failure of neurons to complete migration (35). Whereas overexpression of Pten can inhibit glioma cell migration (24). Thus, both Pten loss and gain of function can inhibit migration in different setting. It was generally thought that Pten-regulated cell migration was through PI3K pathway. Interestingly, strong evidence now indicates that the principal mechanism by which Pten expression inhibits cell migration is PI3K-independent, and mediated through C2 domain of Pten (24). In present study, we have found Pten regulated lung epithelial cell migration is PI3K-pathway independent. Since, this effect is corrected in later stage; it is unlikely that the blocking of cell migration in early branching morphogenesis is mediated through C2 domain of Pten.
Supplementary Material
Cell proliferation and apoptosis were analyzed by anti-Ki67 (a&b) and TUNEL (c&d, arrows show the TUNEL positive cell) staining.
By real-time PCR and IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show significant decrease in Pten expressions.
By IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show no significant difference in Nkx2.1 and α-SMA expression (Panel A). The lungs from E18.5 PtenΔ/Δ show increased CCSP/CC10 and Nkx2.1 levels vs control lungs.
Cell proliferation and apoptosis are analyzed by anti-Ki67 (a&b) and TUNEL (c&d, arrows show the TUNEL positive cell) staining in E12.5 and E18.5 lungs.
By IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show significant reduction in phospho-ERK expression (a). The lungs from E18.5 PtenΔ/Δ show increased phospho-ERK level vs control lungs (b).
Supplemental Figure 6. HE staining and BrdU analysis of PtenΔ/Δ lung explants after 48hr culture.
Anti-E-Cadherin was used to analyze branching morphogenesis in E12.5 P110fl/fl; Gata5-Cre and control lungs.
Highlights.
-
➢
Pten controls early stage branching morphogenesis by regulating epithelial cell migration, but not proliferation.
-
➢
This function is mediated via phosphorylation of ERK, which is also essential for embryonic lung epithelial cell migration.
-
➢
Reduction of phosphorylation of AKT in epithelial cell does not affect branching morphogenesis and cell migration.
Acknowledgments
This work was supported by NIH/NHLBI, Hastings Foundation, the Project for Extramural Scientists of State Key Laboratory of Agrobiotechnology and generous funds from China Agricultural University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, Yajima N, Hamada K, Horie Y, Kubo H, Whitsett JA, Mak TW, Nakano T, Nakazato M, Suzuki A. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest. 2007;117(10):2929–40. doi: 10.1172/JCI31854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. Review. [DOI] [PubMed] [Google Scholar]
- 3.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19(4):348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 4.Stiles B, Groszer M, Wang S, Jiao J, Wu H. PTENless means more. Dev Biol. 2004;273(2):175–84. doi: 10.1016/j.ydbio.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 5.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26(22):3113–21. doi: 10.1038/sj.onc.1210394. Review. [DOI] [PubMed] [Google Scholar]
- 6.Sanges D, Cosma MP. Reprogramming cell fate to pluripotency: the decision-making signalling pathways. Int J Dev Biol. 2010;54:11–12. 1575–87. doi: 10.1387/ijdb.103190ds. Review. [DOI] [PubMed] [Google Scholar]
- 7.Klemke RL1, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137(2):481–92. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing Y, Li C, Hu L, Tiozzo C, Li M, Chai Y, Bellusci S, Anderson S, Minoo P. Mechanisms of TGFbeta inhibition of LUNG endodermal morphogenesis: the role of TbetaRII, Smads, Nkx2.1 and Pten. Dev Biol. 2008;320(2):340–50. doi: 10.1016/j.ydbio.2008.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xing Y, Li C, Li A, Sridurongrit S, Tiozzo C, Bellusci S, Borok Z, Kaartinen V, Minoo P. Signaling via Alk5 controls the ontogeny of lung Clara cells. Development. 2010;137(5):825–33. doi: 10.1242/dev.040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124(23):4867–78. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- 11.Li TF, O’Keefe RJ, Chen D. TGF-beta signaling in chondrocytes. Front Biosci. 2005;10:681–8. doi: 10.2741/1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corti M, Brody AR, Harrison J. Isolation and primary culture of murine alveolar type II cells. Am J Respir Cell Mol Biol. 1996;14:309–315. doi: 10.1165/ajrcmb.14.4.8600933. [DOI] [PubMed] [Google Scholar]
- 13.Okamura H, Yoshida K, Morimoto H, Haneji T. PTEN expression elicited by EGR-1 transcription factor in calyculin A-induced apoptotic cells. J Cell Biochem. 2005;94(1):117–25. doi: 10.1002/jcb.20283. [DOI] [PubMed] [Google Scholar]
- 14.Iden S1, Collard JG. Crosstalk between small GTPases and polarity proteins in cell polarization. Nat Rev Mol Cell Biol. 2008;9(11):846–59. doi: 10.1038/nrm2521. [DOI] [PubMed] [Google Scholar]
- 15.Machacek M, Hodgson L, Welch C, Elliott H, Pertz O, Nalbant P, Abell A, Johnson GL, Hahn KM, Danuser G. Coordination of Rho GTPase activities during cell protrusion. Nature. 2009;461(7260):99–103. doi: 10.1038/nature08242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. Review. [DOI] [PubMed] [Google Scholar]
- 17.Bove PF1, Hristova M, Wesley UV, Olson N, Lounsbury KM, van der Vliet A. Inflammatory levels of nitric oxide inhibit airway epithelial cell migration by inhibition of the kinase ERK1/2 and activation of hypoxia-inducible factor-1 alpha. J Biol Chem. 2008;283(26):17919–28. doi: 10.1074/jbc.M709914200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enomoto A, Murakami H, Asai N, Morone N, Watanabe T, Kawai K, Murakumo Y, Usukura J, Kaibuchi K, Takahashi M. Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev Cell. 2005;9(3):389–402. doi: 10.1016/j.devcel.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, Kutok JL, Kearney JF, Otipoby KL, Rajewsky K. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139(3):573–86. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali IU, Schriml LM, Dean M. Mutational spectra of PTEN/MMAC1 gene: a tumor suppressor with lipid phosphatase activity. J Natl Cancer Inst. 1999;91(22):1922–32. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 21.Davé V, Wert SE, Tanner T, Thitoff AR, Loudy DE, Whitsett JA. Conditional deletion of Pten causes bronchiolar hyperplasia. Am J Respir Cell Mol Biol. 2008;38(3):337–45. doi: 10.1165/rcmb.2007-0182OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anzelon AN, Wu H, Rickert RC. Pten inactivation alters peripheral B lymphocyte fate and reconstitutes CD19 function. Nat Immunol. 2003;4(3):287–94. doi: 10.1038/ni892. [DOI] [PubMed] [Google Scholar]
- 23.Heit B, Robbins SM, Downey CM, Guan Z, Colarusso P, Miller BJ, Jirik FR, Kubes P. PTEN functions to ‘prioritize’ chemotactic cues and prevent ‘distraction’ in migrating neutrophils. Nat Immunol. 2008;9(7):743–52. doi: 10.1038/ni.1623. [DOI] [PubMed] [Google Scholar]
- 24.Raftopoulou M, Etienne-Manneville S, Self A, Nicholls S, Hall A. Regulation of cell migration by the C2 domain of the tumor suppressor PTEN. Science. 2004;303(5661):1179–81. doi: 10.1126/science.1092089. [DOI] [PubMed] [Google Scholar]
- 25.Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by PTEN. Biochem Soc Trans. 2005;33(Pt 6):1507–8. doi: 10.1042/BST0331507. Review. [DOI] [PubMed] [Google Scholar]
- 26.Kuslak SL, Marker PC. Fibroblast growth factor receptor signaling through MEK-ERK is required for prostate bud induction. Differentiation. 2007;75(7):638–51. doi: 10.1111/j.1432-0436.2006.00161.x. [DOI] [PubMed] [Google Scholar]
- 27.Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127(12):2695–704. doi: 10.1242/dev.127.12.2695. [DOI] [PubMed] [Google Scholar]
- 28.Deroanne CF1, Hamelryckx D, Ho TT, Lambert CA, Catroux P, Lapière CM, Nusgens BV. Cdc42 downregulates MMP-1 expression by inhibiting the ERK1/2 pathway. J Cell Sci. 2005;118(Pt 6):1173–83. doi: 10.1242/jcs.01707. [DOI] [PubMed] [Google Scholar]
- 29.Sahai E, Olson MF, Marshall CJ. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J. 2001;20(4):755–66. doi: 10.1093/emboj/20.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hensel N, Stockbrügger I, Rademacher S, Broughton N, Brinkmann H, Grothe C, Claus P. Bilateral crosstalk of rho- and extracellular-signal-regulated-kinase (ERK) pathways is confined to an unidirectional mode in spinal muscular atrophy (SMA) Cell Signal. 26(3):540–8. doi: 10.1016/j.cellsig.2013.11.027. Epub 2013 Dec 3. [DOI] [PubMed] [Google Scholar]
- 31.Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117(Pt 20):4619–28. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 32.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7(8):606–19. doi: 10.1038/nrg1879. Review. [DOI] [PubMed] [Google Scholar]
- 33.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50. doi: 10.1146/annurev.pathol.4.110807.092311. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carter E, Miron-Buchacra G, Goldoni S, Danahay H, Westwick J, Watson ML, Tosh D, Ward SG. Phosphoinositide 3-kinase alpha-dependent regulation of branching morphogenesis in murine embryonic lung: evidence for a role in determining morphogenic properties of FGF7. PLoS One. 2014 Dec 2;9(12) doi: 10.1371/journal.pone.0113555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29(4):404–11. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell proliferation and apoptosis were analyzed by anti-Ki67 (a&b) and TUNEL (c&d, arrows show the TUNEL positive cell) staining.
By real-time PCR and IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show significant decrease in Pten expressions.
By IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show no significant difference in Nkx2.1 and α-SMA expression (Panel A). The lungs from E18.5 PtenΔ/Δ show increased CCSP/CC10 and Nkx2.1 levels vs control lungs.
Cell proliferation and apoptosis are analyzed by anti-Ki67 (a&b) and TUNEL (c&d, arrows show the TUNEL positive cell) staining in E12.5 and E18.5 lungs.
By IHC analysis, comparing with control lungs, the lungs from E12.5 PtenΔ/Δ show significant reduction in phospho-ERK expression (a). The lungs from E18.5 PtenΔ/Δ show increased phospho-ERK level vs control lungs (b).
Supplemental Figure 6. HE staining and BrdU analysis of PtenΔ/Δ lung explants after 48hr culture.
Anti-E-Cadherin was used to analyze branching morphogenesis in E12.5 P110fl/fl; Gata5-Cre and control lungs.