

Abstract
Understanding the role of SCN8A in epilepsy and behavior is critical in light of recently identified human SCN8A epilepsy mutations. We have previously demonstrated that Scn8amed and Scn8amed-jo mice carrying mutations in the Scn8a gene display increased resistance to flurothyl and kainic acid-induced seizures; however, they also exhibit spontaneous absence seizures. To further investigate the relationship between altered SCN8A function and epilepsy, we introduced the SCN1A-R1648H mutation, identified in a family with generalized epilepsy with febrile seizures plus (GEFS+), into the corresponding position (R1627H) of the mouse Scn8a gene. Heterozygous R1627H mice exhibited increased resistance to some forms of pharmacologically and electrically induced seizures and the mutant Scn8a allele ameliorated the phenotype of Scn1a-R1648H mutants. Hippocampal slices from heterozygous R1627H mice displayed decreased bursting behavior compared to wild-type littermates. Paradoxically, at the homozygous level, R1627H mice did not display increased seizure resistance and were susceptible to audiogenic seizures. We furthermore observed increased hippocampal pyramidal cell excitability in heterozygous and homozygous Scn8a-R1627H mutants, and decreased interneuron excitability in heterozygous Scn8a-R1627H mutants. These results expand the phenotypes associated with disruption of the Scn8a gene and demonstrate that an Scn8a mutation can both confer seizure protection and increase seizure susceptibility.
Keywords: Nav1.6, Nav1.1, Audiogenic seizure, voltage sensor, sodium channel, GEFS+, Dravet Syndrome, interneuron
INTRODUCTION
Voltage-gated sodium channels (VGSCs) are important regulators of neuronal excitability and are responsible for the initiation and propagation of action potentials in neurons. Given their fundamental role in neuronal communication, disruptions in VGSC function can lead to a host of pathophysiological conditions. Most notably, mutations in the four VGSCs that are primarily expressed in the CNS—SCN1A (Nav1.1), SCN2A (Nav1.2), SCN3A (Nav1.3), and SCN8A (Nav1.6)—are responsible for several types of idiopathic epilepsy. Specifically, SCN1A mutations lead to genetic epilepsy with febrile seizures plus (GEFS+) and Dravet syndrome (DS) (Claes et al., 2001; Escayg et al., 2000), SCN2A mutations cause benign familial neonatal-infantile seizures (Heron et al., 2002), mutations in SCN3A have been identified in patients with partial epilepsy (Estacion et al., 2010; Holland et al., 2008; Vanoye et al., 2014), and SCN8A mutations are responsible for some cases of epileptic encephalopathies (EIEE13) (Carvill et al., 2013; Vaher et al., 2013; Veeramah et al., 2012).
Based on the VGSC mutations that have been identified to date, it is clear that distinct seizure and behavioral outcomes can result from different mutations in the same VGSC gene, with the magnitude and direction of the observed phenotypes potentially reflecting specific biophysical changes caused by the mutation. For example, within the spectrum of SCN1A epilepsies, the most severe condition, DS, often results from null mutations in the SCN1A gene, while amino acid substitutions that change the biophysical properties of the channel can give rise to less severe forms of epilepsy such as GEFS+ (Claes et al., 2009; Escayg and Goldin, 2010; Lossin, 2009).
SCN8A mutations were recently identified in several patients with epileptic encephalopathies (de Kovel et al., 2014; Estacion et al., 2014; O’Brien and Meisler, 2013; Vaher et al., 2013; Veeramah et al., 2012). Functional analyses initially suggested that increased channel activity was the likely biophysical consequence of these mutations (Estacion et al., 2014; Veeramah et al., 2012). However, the recent identification of potential loss-of-function SCN8A mutations in this disorder indicates that epilepsy might result from a range of alterations in SCN8A activity (de Kovel et al., 2014). The relationship between SCN8A dysfunction and disease outcome is also complex as illustrated by the cognitive and motor deficits, but not epilepsy, that were previously observed in a family with a SCN8A loss-of-function truncation mutation (Trudeau et al., 2006).
The introduction of clinically relevant VGSC mutations into mouse models has proven critical to our understanding of the functional consequences of distinct VGSC mutations on seizure and behavioral phenotypes. Mice expressing human SCN1A mutations recapitulate many of the clinical features of DS and GEFS+ (Martin et al., 2010; Yu et al., 2006), and have led to the “loss of inhibition” model of DS (Yu et al., 2006). Similarly, mice with loss-of-function Scn8a mutations exhibit behavioral alterations that are consistent with some psychiatric findings in patients (McKinney et al., 2008; Papale et al., 2010). Interestingly, we demonstrated that mutations that reduce the activity of Scn8a in the mouse confer resistance to induced seizures (Hawkins et al., 2011; Martin et al., 2007), but result in the generation of absence epilepsy (Papale et al., 2009). Recently, the SCN8A N1768D gain-of-function mutation, identified in a patient with epileptic encephalopathy, was introduced into the mouse Scn8a gene. Mice expressing this mutation exhibited spontaneous seizures, reduced lifespan, impaired motor coordination and deficits in social interaction (Wagnon et al., 2014).
To gain insight into the spectrum of seizure and behavioral outcomes that can arise when the same mutation is expressed in different VGSC genes, we generated a mouse mutant in which the well-characterized SCN1A mutation R1648H, first identified in a family with GEFS+ (Escayg et al., 2000), was knocked into the corresponding location in the mouse Scn8a gene. The mutant line (Scn8a-R1627H) was evaluated for seizure susceptibility, spontaneous seizure generation, behavioral deficits, and neuronal excitability.
MATERIAL AND METODS
Generation of Construct and Electrophysiology in Xenopus oocytes
For expanded methods refer to Supplemental Materials and Methods. We introduced the Scn1a-R1648H mutation plus two additional silent substitutions into exon 26 of the Scn8a cDNA plasmid for expression in Xenopus laevis oocytes. Sodium currents were recorded by two-electrode voltage-clamp.
Generation of mice expressing the Scn8a R1627H mutation
A targeting construct consisting of a 4.5-kb 5′ arm of homology, the R1627H substitution, a neomycin cassette flanked by FLP1 recombinase (Fft) sites, and a 4-kb 3′ arm of homology, was electroporated into 129X1/SvJ-derived PAT-5 embryonic stem (ES) cells at the University of Michigan Transgenic Core. PCR analysis and Southern blotting were performed to identify correctly targeted ES cells.
Animal breeding and maintenance
All experimental procedures were performed in accordance with the guidelines of Emory University and the University of California, Irvine Institutional Animal Care and Use Committees. All mice were housed in a temperature and humidity controlled vivarium and provided drinking water and food ad libitum. R1627H targeted mice were crossed to mice carrying FLP recombinase to remove the neomycin cassette included in the targeting vector. Male mutants were then crossed to female C57BL6/J wild-type (WT) mice for three generations. Heterozygous males and females from this generation (N3) were crossed to generate WT, heterozygous (Scn8aRH/+), and homozygous (Scn8aRH/RH) mice (N3F1) for experiments. A cohort of N3F1 mice were monitored, weighed, and videotaped weekly from P10 to P120 in order to assess locomotor function and to make general assessments of health and survival. Male Scn8aRH/+ mice (N3 generation) were also crossed to female C3/HeJ mice for three generations to generate progeny for EEG analysis.
To generate mice carrying both the Scn8a-R1627H and the Scn1a-R1648H mutations, Scn8aRH/+ (N3) females were first crossed to Scn1aRH/+ (N10) males to produce Scn8aRH/+/Scn1aRH/+ offspring. Scn8aRH/+/Scn1aRH/+ males were then crossed to N10 Scn1aRH/+ females to produce WT, Scn8aRH/+, Scn1aRH/+, Scn8aRH/+/Scn1aRH/+, Scn1aRH/RH, and Scn8aRH/+/Scn1aRH/RH offspring.
Genotyping of mutants
Genotyping of R1627H mutant mice was performed by PCR analysis of DNA extracted from tail biopsies. A PCR product spanning the R1627H position was amplified using the following primers: (RH F, AAG ACA GGT TAT CTG TGT AAA CTG; RH R, AAT CGG TTT TGT CTG CAA GAC TGG) to produce a 600bp (R1627H) or a 500bp (WT) product. Genotyping of Scn1a R1648H mutant mice was performed as previously reported (Martin et al., 2010).
Protein extraction and Western blot analysis
Protein extraction and Western blot analysis was performed as previously described (Makinson et al., 2014). Detailed methods can be found in Supplemental Materials and Methods.
Animal behavior
All behavioral procedures were conducted between the hours of 10:00 AM and 3:00 PM to minimize possible circadian rhythm effects.
Locomotor activity was measured by photobeam breaks of individually housed mice (San Diego Instruments, La Jolla, CA, USA) over a 24-hour period. Ambulations were counted as consecutive beam breaks.
Rotarod performance was assessed by measuring the latency to fall from an accelerating rotating rod (Columbus Instruments, Rotamex-5 1.3). Each mouse was trained to walk on the rod three times per day for three days. Training trials were 5 minute duration at 5 r.p.m., with each training trial separated by 60 minutes. Mice that fell from the rod during the training trial were placed back on the rod. Following training, rod acceleration was increased in 0.2 r.p.m. increments every second starting at 0 r.p.m. and reaching a maximum of 50 r.p.m. Falls were detected by photobeam break.
Stride length was determined by measuring the distance between paw prints as mice walked across a 5 cm wide 60 cm long corridor. Mice were trained daily for 5 consecutive days to walk down the corridor. On training days, each mouse was placed at the beginning of the corridor and the home cage was placed on its side at the other end of the corridor. Once the mouse traversed the length of the corridor to the home cage, it was allowed to remain for at least 5 minutes before the next training trial. Three training trials were conducted on each day. Mouse forepaws were dipped in black ink so that a print would be left on white paper as the mouse traversed the corridor. Strides were measured from the tip of one paw print to the base of the next paw print on the same side. Stride length was measured in instances in which at least 5 consecutive uninterrupted strides occurred. Average stride length was based on the analysis of at least 15 strides for each mouse.
Open field performance was measured by individually placing each mouse in an opaque Plexiglas box (61 cm × 61 cm) for 5 minutes. The center of the open field was set 15 cm from the each side of the box. The latency to enter the center of the open field, the time spent in the center, and the number of entries into the center were measured.
The forced swim task was performed by placing each mouse in a beaker (diameter 30 cm) filled with water 27°C ±1°C. The time spent struggling, defined as movements in which forelimbs broke the surface of the water, versus floating, defined as minimum movement required to remain afloat, were scored for the first 6 minutes.
The tail suspension task was performed by suspending each mouse from its tail at a height of 15 cm above a surface. Struggling was defined as forelimb and hindlimb movement or twisting movements. Mice were considered to be immobile if their forelimb and hindlimbs were not moving. Swinging was not scored as movement. Behavior was scored for the first 6 minutes.
The novel object recognition task was conducted as we previously described (Papale et al., 2010). Briefly, mice were acclimated to an open-field box for 10 minutes each day for five consecutive days. On the sixth day each animal was placed in the open-field for 5 minutes and then returned to the home cage for 2 minutes. Mice were then returned to the open field where they were given 5 minutes to explore three objects placed in three of the four corners of the box, at least 10 inches from the edges. Mice were returned to their home cage for either 5 or 20 minutes. Mice were then returned to the open-field for 5 minutes where one object had changed position (novel location), one object had been exchanged for a different object (novel object), and one object was the same (familiar object). The time spent exploring each object during the second exposure was recorded.
Video recordings of open field, forced swim, tail suspension, and novel object recognition tasks were analyzed using Any-Maze video tracking software (Stoelting Co. IL).
The elevated-plus maze consisted of four 30 cm arms (two open and two enclosed), suspended 76 inches above the floor. The test began with the mouse oriented towards an open arm. Each mouse was given 5 minutes to freely explore the apparatus during which time beam breaks were recorded to track movement and location. The percentage of time spent in the open arms was recorded and used as a measure of anxiety as mice naturally prefer the enclosed arms and anxiolytics have been shown to increase the time rodents spend in the open arm (Pellow et al., 1985). The percentage of time spent exploring the open arms was calculated by dividing the time spent in the open arms by the combined time spent in the open and closed arms. Total distance traveled in the apparatus was also recorded.
Sucrose preference was assessed by providing two food cups to each individually housed mouse. One cup contained between 1–2 g of standard rodent diet (Purina, Lab Diet 5001) the second cup contained 1–2 g of high sucrose diet (AIN-76A Rodent Tablets, TestDiet). The amounts of the high sucrose and standard diets consumed over a 1 hour period was determined by subtracting the post-feeding weight of the food from the pre-feeding weight of the food. This procedure was repeated on 3 consecutive days. The reported sucrose preference values were determined on the third day.
The Y-maze task was performed by placing each mouse at the end of one arm of the Y-maze and allowing free exploration of the apparatus for 8 minutes. The Y-maze consists of three identical arms and a center zone. Spontaneous alternations were recorded.
Continuous video-electrocorticogram (EEG) monitoring
Video-EEG recordings to detect cortical seizure activity were collected and analyzed as we previously described (Dutton et al., 2012; Martin et al., 2007; Papale et al., 2009). EEG recordings from the inferior colliculus were collected as follows. Mice were anesthetized with isoflurane (2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-thane). Two sterile 0–80×3/32 sterile screw electrodes (Vintage Machine Supplies, Medina, OH) were implanted in the skull at the following coordinates from Bregma (anteroposterior (AP) 1.5 mm and mediolateral (ML) 1.2 mm; AP 1.0 mm and ML 1.2 mm). Stainless steel depth electrodes (diameter 0.25 mm, length 1.5 mm; Plastics One, Roanoke, VA) were implanted at the following coordinates (AP −3.5 mm, ML ±1.5 mm, depth 1.2 mm from the surface of the skull). Two fine-wire electrodes were implanted in the neck muscle for electromyography (EMG) acquisition. Electrodes were covered with dental acrylic following implantation. After a minimum of five days for recovery from surgery, mice were placed in the recording chamber and connected to the EEG acquisition system via a flexible tether and commutator (Dragonfly inc.). Animals were allowed to freely move and were provided food and water ad libitum. EEG recordings were sampled at 200 Hz. Signals were digitized, amplified and processed by Stellate Harmonie EEG system (Natus Medical, Inc.). EEG traces were manually scored as previously described (Papale et al., 2013).
Seizure induction
Flurothyl seizure induction was performed as we previously described (Martin et al., 2007). Latencies to the first myoclonic jerk (MJ) and generalized tonic-clonic seizure (GTCS) were recorded. The MJ presents as a jerk of the upper body sometimes accompanied by tail-limb clonus. The GTCS was defined by complete loss of postural control and clonus of all limbs. Mice were also observed for the presence of hindlimb extension immediately following the GTCS.
Partial psychomotor seizures were evoked using the 6 Hz paradigm. Briefly, mice were administered a corneal analgesic 30 minutes prior to stimulation. Corneal electrical stimulation (6-Hz, 3 sec, 14 mA to 30 mA) was applied. Resulting seizures were scored according to the following modified Racine scale: 1 = staring, 2 = forelimb clonus, 3 = rearing and falling.
Audiogenic seizures were examined following auditory stimulation using either key ringing or a high-intensity tone. For the key ringing method, each mouse was placed in a clear Plexiglas box and keys were shaken for 5 minutes approximately 25 cm above the mouse. Shaking a key bundle was found to produce a broad-spectrum acoustic stimulus (13–85 kHz) at an intensity ranging from 50–80 dB, as determined by a 1/4″ Brüel & Kjaer microphone (Brüel & Kjaer, Denmark). For the high-intensity tone method, a wide range speaker (Model RT1.3, HiVi inc., Arcadia, CA) was used to produce a 12 kHz, 80 dB tone. Each mouse was exposed to this tone for 5 min. Audiogenic seizures were defined by wild-running behavior progressing to loss of posture and forelimb and hindlimb clonus, sometimes followed by tonic hindlimb extension.
Electrophysiology
Mice P16–28 were deeply anesthetized with halothane, rapidly decapitated, and their brains were removed. Horizontal hippocampal slices were cut 350 μm thick with a vibratome (VT1000S; Leica Systems, Germany) in ice-cold sucrose-containing artificial cerebrospinal fluid (sACSF) (in mM: 85 NaCl, 75 sucrose, 2.5 KCl, 25 glucose, 1.25 NaH2PO4, 4 MgCl2, 0.5 CaCl2, and 24 NaHCO3). Slices were incubated in oxygenated normal ACSF (in mM: 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, and 10 glucose) for 30 minutes to 1 hour at 27°C. All solutions used in preparation and recording were oxygenated by bubbling 95% O2–5% CO2.
Current clamp recordings were obtained using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA), digitized with a Digidata 1322A digitizer (Molecular Devices), and data were acquired and analyzed with pClamp 10.2 software (Molecular Devices). Signals were sampled at 25 kHz and filtered at 10 kHz. The pipette solution contained the following (in mM): 126 K-gluconate, 4 KCl, 10 HEPES, 4 Mg-ATP, 0.3 Tris-GTP, and 10 Phospho-creatine, pH 7.2. The bath solution contained the following (in mM): 126 NaCl, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 2 Mg Cl2, 26 NaHCO3, and 10 glucose, pH 7.3. Whole-cell recordings were obtained with access resistance <25 MΩ, and cells were held at −70 mV for all experiments. Cells were visualized using infrared DIC illumination under 40x magnification. Hyperpolarizing current injections of −10 pA and −30 pA were used to calculate cellular impedance. Firing patterns were recorded in response to 2-second depolarizing current injections in 20-pA increments, starting at 10 pA, up to 350 pA.
Field recordings were performed as previously described (Makinson et al., 2014). Slices were held at 33°C and perfused (at 2 mL/min) with oxygenated ACSF containing reduced Mg++, 0.5 mM. Recording pipettes (2–3 MΩ) were pulled from borosilicate glass with a P-87 Flaming-Brown puller (Sutter Instruments, Novato, CA). Electrophysiological recordings were performed using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA) and a Digidata 1322A digitizer (Molecular Devices). Data were acquired and analyzed with pClamp 10.2 software (Molecular Devices). Pipettes were filled with 150 mM NaCl and positioned in the CA3 stratum pyramidal layer. ACSF with elevated potassium was prepared by supplementing standard ACSF with 3 M KCl to raise the potassium concentration to 8.5 mM. The experimental paradigm consisted of a control recording for 2 min in standard ACSF, followed by 15–25 min with 8.5 mM K+ ACSF perfusion, during which burst activity was recorded. This was followed by a 7-minute washout with standard ACSF. Population spikes were elicited in the CA3 pyramidal layer by stimulation of the mossy fiber tract with a tungsten wire. Population spikes were evoked in physiological 2.5 mM [K+]O before and after exposing the slice to 8.5 mM [K+]O. Slices in which population spike amplitudes did not change more than 20% after exposing the slice to 8.5 mM [K+]O were used for analysis.
Auditory Brainstem Nuclei Response
Mice were anesthetized with ketamine and xylazine (i.p., 100 and 10 mg/kg respectively) and placed in a heated (25°C) sound attenuating booth (Industrial Acoustics Company, Bronx, NY). Auditory brainstem responses (ABRs) were recorded using Tucker Davis Technologies BioSigRP© software running on a System 3 platform equipped with an RX5 Pentusa Base Station connected to subdermal electrodes via an RA4LI low impedance headstage (TDT, Alachua, FL, USA). Sets of tone pips at different frequencies were presented in a random order to each animal to avoid possible ordering effects.
A subdermal needle electrode was placed over the skull vertex as the active lead. The ground was placed ventral lateral to the left external pinna and the reference was placed ventral lateral to the right external pinna. The bioelectric signals were sampled at 25 kHz, bandpass filtered between 100 Hz – 3000 Hz, amplified 200,000 times and averaged over 500 consecutive responses, following a previously established ABR screening protocol in mice (Zheng et al., 1999). To determine the ABR threshold, we reduced the stimulus intensity in 10 dB steps and then 5 dB steps until the lowest intensity at which the dominant ABR wave was visible.
Auditory stimuli
Calibrated stimuli were generated using TDT SigGenRP© software and presented through BioSigRP© software via a TDT RX6 digital signal processor at a sample rate of 195 kS/s. Sounds were attenuated by a TDT PA5 programmable attenuator and played from an Infinity EMIT tweeter placed 90° to the right side of the animal. Absolute sound pressure levels (SPL) of sound stimuli were measured prior to ABR recording experiments using a calibrated ¼” Bruel and Kjaer (B&K, Denmark) Type 4139 microphone with a Type 2669 preamplifier. Pure tone pips of 3 ms duration with 1.5 ms cos2 rise/fall times were presented for 5, 8, 10, 12, 15, and 24 kHz at a rate of 21 per second.
Immunohistochemistry
Following auditory stimulation, mice were transcardially perfused with ice-cold 4% paraformaldehide (PFA). Brains were extracted and stored in 30% sucrose for 5 days before slicing. Coronal slices (45 μm) were cut using a cryostat (Leica, Germany). Every fifth slice was washed in phosphate buffered saline (PBS) and incubated overnight in primary rabbit anti-c-Fos antibody (Abcam, 1:5,000). Slices were washed in PBS and incubated in horseradish peroxidase conjugated goat anti-rabbit secondary antibody (GE, 1:5,000) for 30 minutes. Staining was performed using the Vectastain Elite ABC System (Vector). Images were collected and stereological analysis performed using the Microbrightfield stereology system (MBF Bioscience, Williston, VT).
Statistics
All behavioral tests and latencies to flurothyl-induced seizures were analyzed using a one-way analysis of variance (ANOVA) followed by Dunnett’s or Tukey’s post hoc tests. Binary seizure outcomes were analyzed using the χ2 test. Weight data was analyzed using a two-way analysis of variance, genotype X time. ABRs were analyzed by two-way ANOVA or by the Student’s t-test. Electrophysiology results were analyzed by Student’s t-test or ANOVA (one or two-way) followed by Dunn’s or Holm-Sidak correction.
RESULTS
The R1627H mutation alters recovery from inactivation, use-dependent inactivation, and persistent current
The effects of a mutation may not be the same in two different VGSCs either because the mutation has different effects on the biophysical properties of the two channels, or because the channels have different physiological functions. To determine how the R1627H mutation altered the properties of Nav1.6 channels, WT and mutant channels were first expressed in Xenopus oocytes and examined by two-electrode voltage clamping. The channels were expressed with the β1 subunit because β1 might differentially modulate WT and mutant channels, as we and others have previously observed for mutations that cause GEFS+ (Spampanato et al., 2004). Sample sodium current traces through WT Scn8a and R1627H channels are shown in Fig. 1A and B, respectively. The R1627H mutation altered a number of properties in the presence of β1.
FIGURE 1.

Characterization of the biophysical properties of R1627H channels in Xenopus oocytes. A and B. Sample two-electrode voltage clamp recordings of currents through wild-type Nav1.6 (A) and R1627H channels (B) with co-expression of β1 during depolarizations between −50 and +50 mV in 10 mV increments. C. The R1627H mutation leads to a depolarizing shift in the V1/2 of voltage-dependent activation (Student’s t test, P < 0.05). D. R1627H channels have accelerated recovery from inactivation (two-way ANOVA, Holm-Sidak correction, P < 0.05). E. At holding potentials from −30 to +40 mV, R1627H channels exhibit increased persistent current (two-way ANOVA, Holm-Sidak correction, P < 0.05). F. The R1627H mutation decreased use-dependent inactivation at 30 Hz (two-way ANOVA, Holm-Sidak correction, P < 0.05).
Mutant channels demonstrated a significantly more depolarized V1/2 of activation (−15±1mV) compared to WT channels (−20±2mV) (Student’s t-test, P < 0.05, n = 5 per group, Fig. 1C). The mutation also led to faster recovery from inactivation (Fig. 1D) and decreased use-dependent inactivation (Fig. 1F), which correlates with faster recovery and increased persistent current (two-way ANOVA, P<0.05, Fig. 1E). The biophysical changes caused by R1627H in Nav1.6 are predicted to have variable effects on neuronal excitability, with the positive shift in voltage-dependence leading to a decrease in excitability and the faster recovery from inactivation and increased persistent current leading to an increase in excitability.
Targeted knock-in of the R1627H mutation into the mouse Scn8a gene
To determine the effects of the R1627H mutation in neurons and on the animal, we generated a mouse model by knocking in the human SCN1A R1648H GEFS+ mutation into the corresponding position (R1627) of the mouse Scn8a gene. Western blotting was performed on whole brain membrane enriched samples from homozygous Scn8aRH/RH, heterozygous Scn8aRH/+, and WT littermates (n = 3 per genotype). Protein levels of Nav1.1, Nav1.2, Nav1.3, and Nav1.6 were found to be comparable between WT, Scn8aRH/+ and Scn8aRH/RH littermates, indicating that knock-in of the R1627H mutation did not lead to altered VGSC protein levels (Supplemental Fig. 1).
Survival, weight, and video monitoring of the R1627H line
Mice were monitored and weighed weekly from P10 to P120 to examine locomotor function and to make general assessments of health and survival. R1627H mice were born in approximately the expected Mendelian ratio (1:2:1) (WT = 24, Scn8aRH/+ = 36, Scn8aRH/RH = 28) and had a normal lifespan. However, homozygous Scn8aRH/RH mutants gained weight more slowly so that by 14 weeks of age the average weights of male and female Scn8aRH/RH mice were 5.6% (males) and 9.5% (females) less than WT and Scn8aRH/+ sex-matched littermates, respectively (two-way ANOVA, P < 0.05, n = 11–18). While WT and Scn8aRH/+ mice appeared visibly normal, tremors and uncoordinated gait were observed in Scn8aRH/RH mutants by the third postnatal week and persisted throughout the life of the animal. The hind legs of Scn8aRH/RH mice were rotated outward, possibly to provide a wider base for postural support, and repetitive “high stepping” movements often preceded locomotion. These findings are consistent with recessive motor abnormalities that have been reported in other Scn8a mutant mouse lines (Kohrman et al., 1996; Sprunger et al., 1999).
Scn8aRH/RH mutant mice have reduced motor function
Recessive motor dysfunction has been described in other mouse models of Scn8a dysfunction. Therefore, we tested whether the Scn8a-R1627H mutation also results in altered motor function, even though we did not observe any motor abnormalities in mice with the Scn1a R1648H mutation (Martin et al., 2010). Motor function was assessed by measuring 24 hr locomotor activity, rotarod performance, and stride length. Significantly reduced locomotor activity was observed in Scn8aRH/RH mice compared to Scn8aRH/+ and WT littermates in the first 5 hours of the recording period and at the beginning of the dark cycle, when mice are most active (two-way ANOVA, genotype X time, P < 0.05, n = 9–12, Fig. 2A). Average latency to fall from the rotarod was not significantly different between WT and Scn8aRH/+; however, significantly reduced latencies were observed in Scn8aRH/RH mutants (one-way ANOVA, P < 0.05, n = 7–18, Fig. 2B). Shorter stride lengths were also observed in Scn8aRH/RH mice when compared to Scn8aRH/+ and WT littermates (one-way ANOVA, P < 0.001, n = 6–10, Fig. 2C–D).
FIGURE 2.

Recessive motor impairment in R1627H mice. A. Locomotor activity of Scn8a-R1627H mice in a new cage was measured over a 24-hour period. A significant deficit in locomotor activity was detected between Scn8aRH/RH mice and WT littermates (two-way ANOVA, genotype X time, P < 0.05). No significant differences were detected between Scn8aRH/+ and WT littermates (P > 0.05). Inset graph shows reduced locomotor activity of Scn8aRH/RH mice during the initial period in a novel environment (P < 0.05). White and black bars under the X-axis represent the light-dark cycle. n = 9–12. B. Rotarod performance was reduced in Scn8aRH/RH (red) but not WT (black) or Scn8aRH/+ (blue) mice (one-way ANOVA, P < 0.001, Dunnett’s post hoc, P < 0.05, n = 7–18). C. Representative example of paw prints showing reduced stride length in Scn8aRH/RH mice. Black lines show relative stride length. D. Stride lengths were found to be significantly reduced in Scn8aRH/RH but not Scn8aRH/+ mice when compared to WT littermates (one-way ANOVA, P < 0.001; Dunnett’s post hoc, P < 0.05). Error bars represent SEM, n = 6–10.
The R1627H mutation is not associated with alterations in anxiety, depressive-like behavior, or learning and memory
Scn8a mutations have been associated with alterations in anxiety in mice (McKinney et al., 2008; Sawyer et al., 2014) and psychiatric and cognitive deficits in humans (Trudeau et al., 2006; Wang et al., 2008; Wasserman et al., 2005). However, anxiety levels, as measured by the percentage of time spent in the open arm or the number of entries into the open arm of an elevated plus maze, were comparable between Scn8aRH/+, Scn8aRH/RH and WT littermates (Table 1). Likewise, no significant differences were observed between the three genotypes in the time spent in the center zone or the number of entries into the center of the open field (Table 1). In the tail suspension task, Scn8aRH/RH mice spent significantly more time struggling and less time immobile, when compared to Scn8aRH/+ and WT littermates (one-way ANOVA, P < 0.05, n = 8–10); however, no differences in time spent struggling or floating were observed in the forced swim task (one-way ANOVA, P > 0.05, n = 10 per group, Table 1). Likewise, preference for sucrose was comparable between Scn8aRH/RH, Scn8aRH/+ and WT littermates (one-way ANOVA, P > 0.05, n = 8–12, Table 1), providing no evidence for depressive-like behavior in the mutant mice.
TABLE 1.
Summary of survival and behavioral assessment of the R1627H line.
| Category | Test | # of Animals | Measurement | Genotype | Average | s.e.m. | p | Post hoc Test | Comparison | Significance |
|---|---|---|---|---|---|---|---|---|---|---|
| Survival | Survival | WT = 24 RH/+ = 36 RH/RH = 28 |
4 mo % mortality | WT | 0.0 | - | - | - | - | - |
| RH/+ | 0.0 | - | - | - | ||||||
| RH/RH | 0.0 | - | - | - | ||||||
| Anxiety | Elevated Plus Maze | WT = 10 RH/+ = 12 RH/RH = 10 |
% time in open arm | WT | 25.1 | 3.8 | 0.84 | - | - | - |
| RH/+ | 26.9 | 2.6 | - | - | ||||||
| RH/RH | 28.6 | 5.5 | - | - | ||||||
| Open arm entries | WT | 13.3 | 2.5 | 0.73 | - | - | - | |||
| RH/+ | 12.1 | 1.6 | - | - | ||||||
| RH/RH | 10.8 | 2.4 | - | - | ||||||
| Distance traveled (cm) | WT | 106.2 | 8.1 | 0.64 | - | - | - | |||
| RH/+ | 115.9 | 7.3 | - | - | ||||||
| RH/RH | 104.6 | 12.6 | - | - | ||||||
| Open Field | WT = 8 RH/+ = 12 RH/RH = 10 |
Time in center (sec) | WT | 5.3 | 5.5 | 0.05 | - | - | - | |
| RH/+ | 7.9 | 8.5 | - | - | ||||||
| RH/RH | 1.1 | 2.0 | - | - | ||||||
| Number of entries into the center | WT | 2.8 | 2.1 | p < 0.01 | Dunnett’s Test | - | - | |||
| RH/+ | 3.6 | 2.5 | WT vs. RH/+ | p > 0.05 | ||||||
| RH/RH | 0.6 | 0.7 | WT vs. RH/RH | p > 0.05 | ||||||
| Latency to first center entry (sec) | WT | 67.3 | 86.4 | 0.23 | - | - | - | |||
| RH/+ | 132.0 | 93.9 | - | - | ||||||
| RH/RH | 65.6 | 108.6 | - | - | ||||||
| Depression-like | Forced Swim | WT = 10 RH/+ = 10 RH/RH = 10 |
Time swimming (sec) | WT | 90.1 | 10.3 | 0.57 | - | - | - |
| RH/+ | 83.6 | 11.2 | - | - | ||||||
| RH/RH | 99.5 | 8.6 | - | - | ||||||
| Time floating (sec) | WT | 122.2 | 22.3 | 0.2 | - | - | - | |||
| RH/+ | 121.5 | 24.5 | - | - | ||||||
| RH/RH | 71.2 | 15.8 | - | - | ||||||
| Tail Suspension | WT = 10 RH/+ = 15 RH/RH = 10 |
Time struggling (sec) | WT | 170.7 | 8.4 | p < 0.01 | Dunnett’s Test | - | - | |
| RH/+ | 171.8 | 10.6 | WT vs. RH/+ | p > 0.05 | ||||||
| RH/RH | 247.7 | 7.2 | WT vs. RH/RH | p < 0.05 | ||||||
| Time immobile (sec) | WT | 229.5 | 5.0 | p < 0.05 | Dunnett’s Test | - | - | |||
| RH/+ | 220.7 | 12.9 | WT vs. RH/+ | p > 0.05 | ||||||
| RH/RH | 164.1 | 8.5 | WT vs. RH/RH | p < 0.05 | ||||||
| Sucrose Preference | WT = 11 RH/+ = 10 RH/RH = 11 |
Sucrose preference (sucrose g/total g) *100 | WT | 96.9 | 2.9 | 0.64 | - | - | - | |
| RH/+ | 97.0 | 1.9 | - | - | ||||||
| RH/RH | 89.5 | 10.7 | - | - | ||||||
| Learning/Memory | Novel Object Recognition (20 min exposure) | WT = 8 RH/+ = 10 RH/RH = 12 |
Novel object preference | WT | 59.7 | 4.9 | 0.77 | - | - | - |
| RH/+ | 62.1 | 6.8 | - | - | ||||||
| RH/RH | 55.1 | 10.9 | - | - | ||||||
| Novel position preference | WT | 56.2 | 4.9 | 0.41 | - | - | - | |||
| RH/+ | 62.6 | 6.8 | - | - | ||||||
| RH/RH | 64.8 | 10.9 | - | - | ||||||
| Novel Object Recognition (5 min exposure) | WT = 8 RH/+ = 10 RH/RH = 12 |
Novel object preference | WT | 69.6 | 10.3 | 0.923 | - | - | - | |
| RH/+ | 58.5 | 8.8 | - | - | ||||||
| RH/RH | 66.4 | 7.8 | - | - | ||||||
| Novel position preference | WT | 61.9 | 10.8 | 0.136 | - | - | - | |||
| RH/+ | 60.6 | 5.9 | - | - | ||||||
| RH/RH | 70.9 | 13.0 | - | - | ||||||
| Y-Maze | WT = 10 RH/+ = 12 RH/RH = 10 |
% correct rotations | WT | 59.3 | 2.2 | 0.031 | Dunnett’s Test | - | - | |
| RH/+ | 56.8 | 1.4 | WT vs. RH/+ | p > 0.05 | ||||||
| RH/RH | 64.2 | 6.4 | WT vs. RH/RH | p > 0.05 | ||||||
| Distance traveled (cm) | WT | 331.4 | 19.3 | 0.021 | Dunnett’s Test | - | - | |||
| RH/+ | 352.3 | 18.7 | WT vs. RH/+ | p > 0.05 | ||||||
| RH/RH | 264.3 | 27.11 | WT vs. RH/RH | p > 0.05 |
Mice carrying the R1627H mutation and WT littermates were evaluated for survival and were subjected to a battery of behavioral tests in order to evaluate anxiety, depressive-like, and learning and memory related behaviors.
We previously reported modest improvement in spatial memory in mice with the Scn8amedjo mutation (Papale et al., 2010). However, mice carrying the R1627H mutation were found to perform comparably to WT littermates in the novel object recognition task (one-way ANOVA, P > 0.05, n = 8–12, Table 1). Spatial learning was also comparable between the three genotypes according to performance in the Y-maze task (one-way ANOVA, P > 0.05, n = 10–12, Table 1).
Scn8aRH/RH and Scn8aRH/+ mice do not exhibit spontaneous seizures
We previously observed spontaneous absence seizures in three Scn8a mouse lines (Papale et al., 2009). To determine if the R1627H mutation also leads to seizure generation, ten days of continuous video-EEG recordings were collected from WT, Scn8aRH/+, and Scn8aRH/RH mice. No seizures were detected in any mice (n = 4–6). Based on previous work, the C3H/HeJ genetic background is known to be more permissive to the generation of absence seizures (Beyer et al., 2008). Therefore, we crossed Scn8aRH/+ mice to the C3H/HeJ background for five generations and then repeated the EEG analysis. No absence seizures were observed over five days of continuous video-EEG recordings collected from Scn8aRH/+ mutants and WT littermates at this generation (n = 5–6).
Scn8aRH/+ but not Scn8aRH/RH mice are resistant to 6 Hz- and flurothyl-induced seizures
Average latency to the flurothyl-induced myoclonic jerk (MJ) and the generalized tonic-clonic seizure (GTCS) were significantly increased in Scn8aRH/+ mice when compared to Scn8aRH/RH and WT littermates (one-way ANOVA, P < 0.05, Fig. 3A). However, the severity of the seizures, as assessed by the presence of hindlimb extension and mortality following the GTCS, was reduced in both Scn8aRH/+ and Scn8aRH/RH mice when compared to WT littermates (χ2, P < 0.05, Fig. 3B). Similarly, Scn8aRH/+ but not Scn8aRH/RH mice were found to be resistant to 6 Hz-induced psychomotor seizures when compared to WT littermates (Fig. 3C), and the severity of the observed 6 Hz-induced seizures was lower in both Scn8aRH/+ and Scn8aRH/RH mice when compared to WT littermates at current intensities of 20, 22, and 24 mA (one-way ANOVA, P < 0.0001, Dunnett’s post hoc, P < 0.001, Fig. 3D).
FIGURE 3.

Effects of the R1627H mutation on flurothyl- and 6 Hz-induced seizure susceptibility. A. Scn8aRH/+ but not Scn8aRH/RH mice have longer latencies to flurothyl-induced seizures when compared to WT littermates (one-way ANOVA, MJ and GTCS, Tukey post hoc, * P < 0.05, n = 11–12). B. Seizure severity, as measured by hindlimb extension (HE) and death, were reduced in Scn8aRH/+ and Scn8aRH/RH mice (χ2, *P < 0.05). C. Scn8aRH/+ mice are more resistant to 6 Hz seizures compared to Scn8aRH/RH and WT littermates. D. Both Scn8aRH/+ and Scn8aRH/RH mice experience less severe seizures in response to stimulus intensities of 20, 22, and 24 mA compared to WT littermates (one-way ANOVA, P < 0.0001, Dunnett’s post hoc, P < 0.001, n = 8–10).
Scn8a mutant mice have reduced hippocampal bursting activity in the presence of high extracellular potassium
To examine the susceptibility of the CNS network to seizure-like activity, extracellular recordings were performed in the CA3 region of hippocampal slices from WT, Scn8aRH/+ and Scn8aRH/RH mice in the presence of high extracellular potassium (Fig. 4A). We previously observed reduced hippocampal bursting in Scn8amed/+ mice that carry a loss of function Scn8a mutation using the high [K+] seizure model (Makinson et al., 2014) and reduced hippocampal network excitability is thought to be a key component of Scn8a-related seizure resistance (Blumenfeld et al., 2009; Makinson et al., 2014). We found that the percentage of slices exhibiting bursting was significantly lower in Scn8aRH/+ mice (1/8, 12.5%) compared to WT (5/5, 100%) and Scn8aRH/RH mice (4/7, 57.1%) (Fig. 4B). The average latency to the onset of bursting was comparable between slices from WT mice (6.1 min) and Scn8aRH/RH mutants (6.3 min). However slices from Scn8aRH/+ did not show bursting even upon prolonged (> 20 min) exposure to elevated potassium, except for 1 slice (1/8) that did show bursting with a latency of 20 min (Fig. 4C). The intra-burst spiking frequency was reduced in that one Scn8aRH/+ slice (32.0 Hz) and also in slices from Scn8aRH/RH mice (49.3 Hz) when compared to WT slices (61.5 Hz) (Fig. 4D). These results indicate that slices from heterozygous Scn8a R1627H mice are resistant to seizure-like activity compared to WT, and slices from homozygous mice are intermediate between heterozygous and WT in their susceptibility to seizure-like activity.
FIGURE 4.

Effects of the R1627H mutation on hippocampal bursting. A. Example of bursting observed during field recording from WT CA3 when bath solution is switched from regular ACSF to high K+ ACSF. Bursting stops soon after high K+ ACSF is replaced with regular ACSF. B,The percentage of slices that exhibited bursting in the presence of high K+ was WT (5/5, 100%), Scn8aRH/+ (1/8, 12.5%), and Scn8aRH/RH (4/7, 57.1%, *P < 0.05, Fisher Exact Test). C. The latency to the occurrence of bursting activity in the presence of high potassium (K+) was increased in slices from Scn8aRH/+ but not WT or Scn8aRH/RH littermates. D. The frequency of intra-burst high amplitude spiking activity was reduced in Scn8aRH/+ compared to WT and Scn8aRH/RH littermates. (B and C, data are average ± SEM, *P < 0.05, one-way ANOVA, Holm-Sidak post-hoc test).
Scn8a mutant mice have altered hippocampal cell firing
Based on oocyte data (Fig. 1), the effects of the R1627H mutation on sodium channel recovery and persistent current would be predicted to increase neuronal activity, whereas the effects on the voltage-dependence of activation would be predicted to decrease activity. Therefore, we examined the effect of the mutation on network excitability by current clamp in hippocampal slices. Consistent with the predictions based on the biophysical properties of the channel in oocytes, the R1627H mutation altered action potential firing in CA3 pyramidal neurons in a complex manner. Scn8aRH/+ neurons fired more action potentials than WT and Scn8aRH/RH neurons, and Scn8aRH/RH neurons fired more action potentials than WT. These differences were statistically significant (two-way ANOVA, P < 0.05, n = 21–28, Fig. 5A–B). The action potential height measured from threshold was significantly decreased in Scn8aRH/RH neurons compared to WT and Scn8aRH/+ (two-way ANOVA, P < 0.05, n = 21–28, Table 2), and the action potential amplitude half-width was increased (broadened) for both Scn8aRH/RH and Scn8aRH+ compared to WT (two-way ANOVA, P < 0.05, n = 21–28, Table 2). The threshold for firing an action potential was slightly higher for the mutants compared to WT, but the differences were not significantly different (two-way ANOVA, P > 0.05, n = 21–28, Table 2). Overall, pyramidal neurons from Scn8aRH/+ and Scn8aRH/RH mutants demonstrated greater excitability compared to WT. Hippocampal interneurons recorded from sub-pyramidal layers of CA3 in Scn8aRH/+ mice fired fewer action potentials compared to WT (two-way ANOVA, P < 0.05, n = 6–8, Fig. 5D), while no significant difference was observed between interneuron firing of Scn8aRH/RH and WT neurons. Furthermore, no differences were observed in interneuron action potential properties (two-way ANOVA, P > 0.05, n = 6–8, Table 2). The resting membrane potentials of pyramidal neurons and interneurons across all genotypes were approximately −70 mV and −65 mV, respectively. There were no significant differences in the cell impedance of pyramidal and inhibitory neurons.
FIGURE 5.

Firing properties of CA3 neurons from R1627H mice. A. Sample traces from CA3 pyramidal neurons of WT, Scn8aRH/+ and Scn8aRH/RH mice at depolarizing current injections of 30 and 190 pA from a resting potential of −70mV. B. Average number of action potentials (AP#) plotted against current injection (pA) (Error bars represent SEM, n = number of cells ; *P < 0.05, two-way ANOVA, Holm-Sidak correction). C. Sample traces from CA3 interneurons of WT, Scn8aRH/+ and Scn8aRH/RH mice at depolarizing current injections of 30 and 190 pA from a resting potential of −70 mV. B. Average number of action potentials (AP#) plotted against current injection (pA). Error bars represent SEM, n = number of cells, *P < 0.05 two-way ANOVA, Holm-Sidak correction.
TABLE 2.
Intrinsic properties of hippocampal neurons from R1627H mice.
| AP properties of pyramidal cells | AP properties of interneurons | |||||||
|---|---|---|---|---|---|---|---|---|
| Amplitude (mV) | Threshold (mV) | Half-width (ms) | N | Amplitude (mV) | Threshold (mV) | Half-width (ms) | N | |
| WT | 87 ± 2 | −45 ± 1 | 2 ± 0.1 | 21 | 80 ± 5 | −49 ± 2 | 2 ± 0.2 | 8 |
| RH/+ | 88 ± 2 | −46 ± 1 | 2.3 ± 0.1* | 28 | 81 ± 4 | −43 ± 2 | 2.5 ± 0.1 | 8 |
| RH/RH | 78 ± 3* | −46 ± 2 | 2.7 ± 0.1* | 23 | 80 ± 4 | −45 ± 3 | 2.6 ± 0.4 | 6 |
Summary of intrinsic membrane properties of hippocampal pyramidal cells and interneurons obtained by current clamp recording (one-way ANOVA, Holm-Sidak test).
Values represent average ± SEM, n = number of cells,
P < 0.05.
The Scn8a-R1627H mutation increases the lifespan and seizure thresholds of Scn1a-R1648H GEFS+ mice
The R1627H mutation in Scn8a is orthologous to the Scn1a R1648H mutation that causes GEFS+ in humans. To determine if there is a functional interaction between the two channels with orthologous mutations, we crossed the two lines to generate offspring with the following genotypes: Scn1aRH/+, Scn1aRH/RH, Scn8aRH/+, Scn1aRH/+/Scn8aRH/+, Scn1aRH/RH/Scn8aRH/+, and WT. Scn1aRH/+ and Scn1aRH/RH mice experienced 18% (2/11) and 100% (11/11) mortality by 3 months of age, respectively. In contrast, 40% of Scn1aRH/RH/Scn8aRH/+ mutants survived to at least three months of age. No mortality was observed in WT, Scn8aRH/+, and Scn1aRH/+/Scn8aRH/+ mice (Fig. 6A). As previously observed, the average latency to the flurothyl-induced GTCS was lower in Scn1aRH/+ mutants; however, latencies to the GTCS were comparable between Scn1aRH/+/Scn8aRH/+ mutants and WT littermates (Fig. 5B). In contrast, significantly lower latencies to the MJ and GTCS were still observed in Scn1aRH/RH/Scn8aRH/+ mutants (one-way ANOVA, Dunnett’s post hoc, *P < 0.05, ***P <0.001, n = 5–17, Fig. 6B). The increase in lifespan and seizure thresholds is consistent with our previous results examining the interactions between the Scn8amedjo and GEFS+ or DS Scn1a mutants (Hawkins et al., 2011; Martin et al., 2007).
FIGURE 6.

The Scn8a-R1627H mutation increases lifespan and seizure resistance in a mouse model of GEFS+ (Scn1a-R1648H). A. No mortality was observed over the first three months of postnatal development in WT, Scn8aRH/+, and Scn1aRH/+/Scn8aRH/+ mice. In contrast, Scn1aRH/+ and Scn1aRH/RH mice experienced 18% (2/11) and 100% (11/11) mortality, respectively. Scn1aRH/RH/Scn8aRH/+ mice exhibited 40% (4/10) survival. B. Scn1aRH/+ mutants (green) have lower latencies to flurothyl-induced GTCS, while Scn8aRH/+ mutants (orange) exhibit elevated latencies when compared to WT littermates. Normal latencies were restored in Scn1aRH/+/Scn8aRH/+ mice (blue). Scn1aRH/RH/Scn8aRH/+ mice (grey) have reduced latencies to the MJ and GTCS. One-way ANOVA, Dunnett’s post hoc, *P < 0.05, ***P < 0.001. Error bars represent SEM, n = 5–17.
Scn8aRH/RH mice exhibit wild running behavior in response to high intensity sound stimuli
Scn8aRH/RH mice were sometimes observed exhibiting wild-running in response to elevated sound stimuli. To further explore this behavior, Scn8aRH/+, Scn8aRH/RH, and WT littermates were exposed to tones (12 kHz, 80 dB) or broad-spectrum (13–85 kHz, 50–80 dB) acoustic stimuli. Approximately 36% (11/31) of Scn8aRH/RH mice exhibited wild-running behavior followed by loss of postural control and forelimb and hindlimb clonus in response to sound stimuli (χ2, *P < 0.05, Fig. 7A). In contrast, only one WT littermate (1/15, 7%) exhibited wild running behavior in response to the sound stimulus. Furthermore, this WT mouse progressed to tonic hindlimb extension, which was not observed in the Scn8aRH/RH mutants. No Scn8aRH/+ mice (0/10) exhibited abnormal audiogenic responses.
FIGURE 7.

Brainstem activity in Scn8aRH/RH mice following sound-induced wild-running behavior. A. Scn8aRH/RH, Scn8aRH/+, and WT mice were exposed to 5-minute duration tones (12 kHz, 80 dB) or broad-spectrum (13–85 kHz, 50–80 dB) acoustic stimuli. Aproximately 7% (1/15) of WT, 0% (0/10) of Scn8aRH/+, and 36% (11/31) of Scn8aRH/RH mice exhibited wild running behavior during sound stimulus presentation (χ2, *P < 0.05). B. Representative example of inferior colliculus (IC) and cortical EEG activity in an Scn8aRH/RH mouse before (baseline) and during abnormal behavoir. C. Representative image of c-Fos expression in the IC of a responding and non-responding Scn8aRH/RH mouse 2 hours after exposure to sound stimulus. D. Increased c-Fos immunoreactive cells were observed in the IC of responding compared to non-responding Scn8aRH/RH mice 2 hours following exposure to acoustic stimuli (Mann-Whitney U test, *P < 0.05, Error bars represent SEM, n = 3–4). E. Regional expression of c-Fos 2 hours after exposure to sound stimulus. Relative c-Fos expression: (+) low; (++) light; (+++) moderate; (++++) high. High levels of c-fos expression were observed in the superior and inferior colliculus of Scn8aRH/RH responding animals but not in non-responding Scn8aRH/RH, Scn8aRH/+ or WT littermates. Low to light levels of c-Fos was observed in the hippocampus and cortex of all animals.
To investigate whether the observed abnormal behaviors may have been the result of audiogenic seizures, we recorded EEG activity in the inferior colliculus, which has been shown to be an important site for the initiation and maintenance of audiogenic seizures (Browning, 1986; Kesner, 1966; Willott and Lu, 1980). EEG activity in Scn8aRH/RH mice was unaltered by exposure to the tone prior to the onset of the behavioral response (Fig. 7B). However at the onset of the behavioral response, we observed high amplitude - greater than twice baseline - EEG signals in the inferior colliculus of Scn8aRH/RH mice (Fig. 7B). High amplitude EEG activity was not observed in cortical EEG recordings during the sound-induced behavioral response (Fig. 7B). To provide an independent measure of neuronal activity following sound exposure, we performed immunohistochemistry to compare c-Fos expression in the cortex, hippocampus, superior colliculus and inferior colliculus of responding and non-responding Scn8aRH/RH mice as well as Scn8aRH/+ and WT controls (Fig. 7C–E). In agreement with the EEG results, low levels of c-Fos expression was observed in the cortex and hippocampus of all animals (Fig. 7D). High levels of c-Fos expresion were observed in the superior and inferior colliculus of responding Scn8aRH/RH mice but not non-responding Scn8aRH/RH, WT, or Scn8aRH/+ mice (Fig. 7D).
Scn8aRH/RH mice have reduced auditory brainstem responses (ABRs)
To test the possiblity that the sound-induced wild running behavior in Scn8aRH/RH mice may have been the result of increased hearing ability, ABRs to sound stimuli were recorded (Fig. 8). WT, Scn8aRH/+ and Scn8aRH/RH mice were exposed to 5–24 kHz tones from 110–20 dB SPL while measuring ABR responses. Scn8aRH/RH mice were found to have higher thresholds between 12–15 kHz (two-way ANOVA, genotype X frequency, Dunnett’s post hoc, *P < 0.05, Fig. 8A,C). However, no differences in ABR thresholds were observed between Scn8aRH/RH mice that were not susceptible to sound-induced wild-running behavior (non-responders) and Scn8aRH/RH mice that were found to be susceptible (responders) (Student’s t-test, P > 0.05, Fig. 8B).
FIGURE 8.
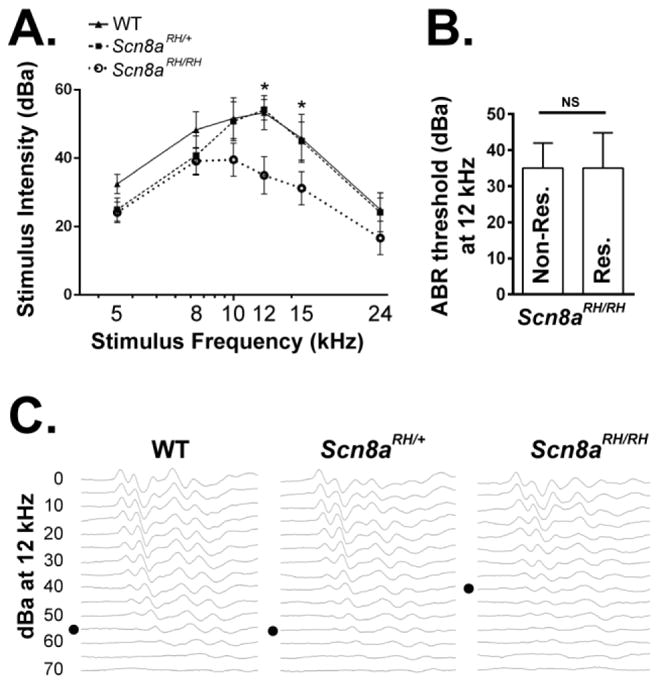
Scn8aRH/RH mice have reduced auditory brainstem nuclei responses (ABRs). A. Auditory brainstem nuclei responses (ABR) were measured in response to 5–24 kHz tones in WT, Scn8aRH/+ and Scn8aRH/RH mice. Scn8aRH/RH mice were found to have reduced auditory responses between 12–15 kHz (two-way ANOVA, Dunnett’s post hoc, *P < 0.05, n = 8–10). B. No differences in ABR thresholds at 12 kHz were observed between Scn8aRH/RH mice that did not display wild-running behavior (non-responders) and Scn8aRH/RH mice that did exhibit abnormal behavior (responders) (Student’s t test, P > 0.05, n = 4–8). C. Representative examples of ABR responses in WT and R1627H mutant mice. Black dot indicates the response threshold. Error bars represent SEM.
DISCUSSION
We constructed a mouse line in which the SCN1A GEFS+ mutation R1648H was introduced into the corresponding position in the mouse Scn8a gene (R1627H). The biophysical properties of the two sodium channels were altered by the mutation in similar but not identical ways. In oocytes, the R1627H mutation altered some properties of Nav1.6 function that would be predicted to increase neuronal excitability, including faster recovery from inactivation, reduced use dependence and increased persistent current. These effects are consistent with those previously observed for R1648H (Lossin et al., 2002; Spampanato et al., 2001).
Action potential firing of pyramidal neurons was increased in heterozygous and homozygous R1627H mice compared to WT. These results contrast with our previous investigation of the Scn1a-R1648H mutation in which we observed no significant changes in excitability of pyramidal neurons from mutant compared to WT mice (Martin et al., 2010). Because the Scn1a-R1648H mutation also markedly decreased action potential firing in inhibitory bipolar neurons (Martin et al., 2010) and Scn8a expression has been detected at the axon initial segments of parvalbumin positive interneurons (Lorincz and Nusser, 2008), we also recorded from CA3 interneurons of Scn8a-R1627H animals. The only difference we observed in interneuron firing was a decrease in the number of action potentials from heterozygous mutants compared to WT mice. To assess the net effect of the Scn8a-R1627H mutation on network excitability, we examined seizure-like activity (bursting) in response to elevated extracellular potassium. Slices from the Scn8aRH/+ mice exhibited less epileptiform bursting activity (Fig. 4), consistent with increased seizure resistance. Interestingly, no change in the latency to burst onset was observed between Scn8aRH/RH compared to WT slices, and more slices from Scn8aRH/RH mice displayed bursting behavior compared to Scn8aRH/+ littermates, indicating a partial loss of seizure resistance at the homozygous level (Fig. 4). Similarly Scn8aRH/+ but not Scn8aRH/RH animals were found to be resistant to flurothyl and electrically induced seizures (Fig. 3). It is not clear if the observed increase in CA3 pyramidal neuron firing and decrease in interneuron firing contributes directly to seizure resistance or if it results from compensatory responses to reduced network excitability in the heterozygous mice.
Consistent with findings from some other Scn8a mouse mutants, at the heterozygous level, the R1627H mutation increased resistance to induced seizures and compensated for the increase in seizure susceptibility resulting from the orthologous GEFS+ R1648H mutation in the Scn1a gene (Hawkins et al., 2011; Makinson et al., 2014; Martin et al., 2007).
A novel feature of the R1627H mice was their abnormal behavioral response to sound stimuli, which has not been previously reported in Scn8a mutants. Three observations suggest that the sound-induced wild running and forelimb-hindlimb clonic events represented audiogenic seizures. First, the behavioral presentation of these events, consisting of loss of posture and forelimb and hindlimb clonus, was similar to flurothyl-induced brainstem seizure behaviors (Samoriski et al., 1998). Second, following exposure to auditory stimuli, Scn8aRH/RH mice that exhibited the abnormal behavior expressed high levels of c-Fos expression in the brainstem, including the inferior colliculus, a region responsible for the initiation and maintenance of audiogenic seizures (Browning, 1986; Kesner, 1966; Willott and Lu, 1980). In contrast, c-Fos expression was not increased in the midbrain and brainstem of non-responding Scn8aRH/RH, Scn8aRH/+, or WT littermates and comparatively little c-Fos expression was observed outside of brainstem regions in Scn8aRH/RH responders following the auditory stimulus. c-Fos expression in brainstem nuclei following exposure to sound stimuli is a hallmark of audiogenic seizures (Klein et al., 2004; Kwon and Pierson, 1997; Le Gal La Salle and Naquet, 1990; Snyder-Keller and Pierson, 1992). Third, we observed increased amplitude of EEG signals in the inferior colliculus during the abnormal behavior in Scn8aRH/RH mice, indicating hypersynchronous neuronal activity in a major audiogenic seizure region. Scn8aRH/RH mice were also found to have higher ABR thresholds. This was similarly observed in the Scn8a mutant ‘Cloth-ears’, lending further support to the prediction that Scn8a dysfunction may contribute to neural hearing loss (Mackenzie et al., 2009).
Homozygous Scn8aRH/RH mutants exhibited motor impairments and reduced locomotor activity, consistent with previously described Scn8a rodent models (Dickie, 1965; Hamann et al., 2003; Kearney et al., 2002; Kohrman et al., 1996). However, in contrast to previously described Scn8a mutants, R1627H mutants did not display alterations in measures of anxiety, or learning and memory (McKinney et al., 2008; Papale et al., 2010).
Unexpectedly, R1627H mutants did not exhibit absence seizures, which were observed in mice expressing Scn8a8J, Scn8amed, and Scn8amedjo alleles (Papale et al., 2009). Across measurements of seizure susceptibility, the R1627H mutants contrast strikingly to the severe convulsive seizures and premature lethality observed in mice expressing the epileptic encephalopathy SCN8A N1768D mutation (Wagnon et al., 2014). Since electrophysiological analysis of neurons from Scn8a N1768D mutants has not yet been reported, a more direct comparison of the effect of each mutation on neuronal excitability cannot be performed. However, the striking phenotypic differences between these two mutants highlight the importance of studying mouse lines expressing different SCN8A mutations.
CONCLUSIONS
The increased seizure resistance conferred by the Scn8a R1627H mutation at the heterozygous level raises the possibility that seizure protection may be achieved by targeting specific biophysical properties of Scn8a function without elevating the risk of spontaneous seizure generation or negative behavioral outcomes. However, we report that the seizure resistance of the R1627H mutation was lost at the homozygous level and that these mice additionally were susceptible to audiogenic seizures, which coincided with increased pyramidal cell excitability but normal interneuron excitability. Together these observations indicate that Scn8a is an important regulator of excitatory as well as inhibitory cell excitability. This study expands upon the phenotypes associated with disruption of the Scn8a gene and demonstrates that an Scn8a mutation can both confer seizure protection and increase seizure susceptibility.
Supplementary Material
Highlights.
The R1627H mutation provides seizure resistance without causing absence epilepsy.
Scn8a-dependent seizure phenotypes are gene-dose dependent.
The R1627H mutation alters excitatory and inhibitory neuronal excitability in the hippocampus.
The Scn8a-R1627H mutation increases susceptibility to audiogenic seizures
Acknowledgments
We would like to thank Dr. David Weinshenker and Dr. Jason Schroeder of the Emory University Rodent Behavioral Core for assistance with assessing locomotor activity.
Research in this publication was supported by the National Institute of Neurological Disorders and Stroke (NINDS) and National Institute on Deafness and Communication Disorders (NIDCD) of the National Institutes of Health (NIH) under award numbers R01NS072221 (A.E.), R01NS048336 (A.L.G.), R01NS065187 (A.E. and A.L.G.), F31NS074717 (C.D.M), and R01DC008343 (R.L).
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beyer B, Deleuze C, Letts VA, Mahaffey CL, Boumil RM, Lew TA, Huguenard JR, Frankel WN. Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Human molecular genetics. 2008;17:1738–1749. doi: 10.1093/hmg/ddn064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H, Lampert A, Klein JP, Mission J, Chen MC, Rivera M, Dib-Hajj S, Brennan AR, Hains BC, Waxman SG. Role of hippocampal sodium channel Nav1.6 in kindling epileptogenesis. Epilepsia. 2009;50:44–55. doi: 10.1111/j.1528-1167.2008.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning RA. Neuroanatomical localization of structures responsible for seizures in the GEPR: lesion studies. Life sciences. 1986;39:857–867. doi: 10.1016/0024-3205(86)90367-x. [DOI] [PubMed] [Google Scholar]
- Carvill GL, Heavin SB, Yendle SC, McMahon JM, O’Roak BJ, Cook J, Khan A, Dorschner MO, Weaver M, Calvert S, Malone S, Wallace G, Stanley T, Bye AM, Bleasel A, Howell KB, Kivity S, Mackay MT, Rodriguez-Casero V, Webster R, Korczyn A, Afawi Z, Zelnick N, Lerman-Sagie T, Lev D, Moller RS, Gill D, Andrade DM, Freeman JL, Sadleir LG, Shendure J, Berkovic SF, Scheffer IE, Mefford HC. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature genetics. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. American journal of human genetics. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes LR, Deprez L, Suls A, Baets J, Smets K, Van Dyck T, Deconinck T, Jordanova A, De Jonghe P. The SCN1A variant database: a novel research and diagnostic tool. Human mutation. 2009;30:E904–920. doi: 10.1002/humu.21083. [DOI] [PubMed] [Google Scholar]
- de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM, Slot RV, van Lieshout S, Nijman IJ, O’Brien JE, Hammer MF, Estacion M, Waxman SG, Dib-Hajj SD, Koeleman BP. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy research. 2014;108:1511–1518. doi: 10.1016/j.eplepsyres.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickie MM. Jolting. Mouse News Lett. 1965;32:44. [Google Scholar]
- Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, Escayg A. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiology of disease. 2012;49C:211–220. doi: 10.1016/j.nbd.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia. 2010;51:1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature genetics. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Estacion M, Gasser A, Dib-Hajj SD, Waxman SG. A sodium channel mutation linked to epilepsy increases ramp and persistent current of Nav1.3 and induces hyperexcitability in hippocampal neurons. Experimental neurology. 2010;224:362–368. doi: 10.1016/j.expneurol.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Estacion M, O’Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, Meisler MH. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiology of disease. 2014;69:117–123. doi: 10.1016/j.nbd.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann M, Meisler MH, Richter A. Motor disturbances in mice with deficiency of the sodium channel gene Scn8a show features of human dystonia. Experimental neurology. 2003;184:830–838. doi: 10.1016/S0014-4886(03)00290-5. [DOI] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiology of disease. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, Shevell M, Mercho S, Seni MH, Guiot MC, Mulley JC, Berkovic SF, Scheffer IE. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–852. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- Holland KD, Kearney JA, Glauser TA, Buck G, Keddache M, Blankston JR, Glaaser IW, Kass RS, Meisler MH. Mutation of sodium channel SCN3A in a patient with cryptogenic pediatric partial epilepsy. Neuroscience letters. 2008;433:65–70. doi: 10.1016/j.neulet.2007.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Buchner DA, De Haan G, Adamska M, Levin SI, Furay AR, Albin RL, Jones JM, Montal M, Stevens MJ, Sprunger LK, Meisler MH. Molecular and pathological effects of a modifier gene on deficiency of the sodium channel Scn8a (Na(v)1.6) Human molecular genetics. 2002;11:2765–2775. doi: 10.1093/hmg/11.22.2765. [DOI] [PubMed] [Google Scholar]
- Kesner RP. Subcortical mechanisms of audiogenic seizures. Experimental neurology. 1966;15:192–205. doi: 10.1016/0014-4886(66)90045-8. [DOI] [PubMed] [Google Scholar]
- Klein BD, Fu YH, Ptacek LJ, White HS. c-Fos immunohistochemical mapping of the audiogenic seizure network and tonotopic neuronal hyperexcitability in the inferior colliculus of the Frings mouse. Epilepsy research. 2004;62:13–25. doi: 10.1016/j.eplepsyres.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Kohrman DC, Smith MR, Goldin AL, Harris J, Meisler MH. A missense mutation in the sodium channel Scn8a is responsible for cerebellar ataxia in the mouse mutant jolting. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16:5993–5999. doi: 10.1523/JNEUROSCI.16-19-05993.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Pierson M. Fos-immunoreactive responses in inferior colliculi of rats with experimental audiogenic seizure susceptibility. Epilepsy research. 1997;27:89–99. doi: 10.1016/s0920-1211(97)01024-3. [DOI] [PubMed] [Google Scholar]
- Le Gal La Salle G, Naquet R. Audiogenic seizures evoked in DBA/2 mice induce c-fos oncogene expression into subcortical auditory nuclei. Brain research. 1990;518:308–312. doi: 10.1016/0006-8993(90)90988-n. [DOI] [PubMed] [Google Scholar]
- Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossin C. A catalog of SCN1A variants. Brain & development. 2009;31:114–130. doi: 10.1016/j.braindev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Mackenzie FE, Parker A, Parkinson NJ, Oliver PL, Brooker D, Underhill P, Lukashkina VA, Lukashkin AN, Holmes C, Brown SD. Analysis of the mouse mutant Cloth-ears shows a role for the voltage-gated sodium channel Scn8a in peripheral neural hearing loss. Genes, brain, and behavior. 2009;8:699–713. doi: 10.1111/j.1601-183X.2009.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Lamar T, Goldin AL, Escayg A. Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiology of disease. 2014;68:16–25. doi: 10.1016/j.nbd.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Dutt K, Papale LA, Dube CM, Dutton SB, de Haan G, Shankar A, Tufik S, Meisler MH, Baram TZ, Goldin AL, Escayg A. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. The Journal of biological chemistry. 2010;285:9823–9834. doi: 10.1074/jbc.M109.078568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Human molecular genetics. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- McKinney BC, Chow CY, Meisler MH, Murphy GG. Exaggerated emotional behavior in mice heterozygous null for the sodium channel Scn8a (Nav1.6) Genes, brain, and behavior. 2008;7:629–638. doi: 10.1111/j.1601-183X.2008.00399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in genetics. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, Letts VA, Meisler MH, Frankel WN, Escayg A. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Human molecular genetics. 2009;18:1633–1641. doi: 10.1093/hmg/ddp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Makinson CD, Christopher Ehlen J, Tufik S, Decker MJ, Paul KN, Escayg A. Altered sleep regulation in a mouse model of SCN1A-derived genetic epilepsy with febrile seizures plus (GEFS+) Epilepsia. 2013;54:625–634. doi: 10.1111/epi.12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Paul KN, Sawyer NT, Manns JR, Tufik S, Escayg A. Dysfunction of the Scn8a voltage-gated sodium channel alters sleep architecture, reduces diurnal corticosterone levels, and enhances spatial memory. The Journal of biological chemistry. 2010;285:16553–16561. doi: 10.1074/jbc.M109.090084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin P, File SE, Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. Journal of neuroscience methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- Samoriski GM, Piekut DT, Applegate CD. Regional analysis of the spatial patterns of Fos induction in brain following flurothyl kindling. Neuroscience. 1998;84:1209–1222. doi: 10.1016/s0306-4522(97)00571-x. [DOI] [PubMed] [Google Scholar]
- Sawyer NT, Papale LA, Eliason J, Neigh GN, Escayg A. Scn8a voltage-gated sodium channel mutation alters seizure and anxiety responses to acute stress. Psychoneuroendocrinology. 2014;39:225–236. doi: 10.1016/j.psyneuen.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder-Keller AM, Pierson MG. Audiogenic seizures induce c-fos in a model of developmental epilepsy. Neuroscience letters. 1992;135:108–112. doi: 10.1016/0304-3940(92)90147-y. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Escayg A, Meisler MH, Goldin AL. Functional effects of two voltage-gated sodium channel mutations that cause generalized epilepsy with febrile seizures plus type 2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21:7481–7490. doi: 10.1523/JNEUROSCI.21-19-07481.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, MacDonald BT, Levin SI, Soltesz I, Benna P, Montalenti E, Isom LL, Goldin AL, Meisler MH. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprunger LK, Escayg A, Tallaksen-Greene S, Albin RL, Meisler MH. Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Human molecular genetics. 1999;8:471–479. doi: 10.1093/hmg/8.3.471. [DOI] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. Journal of medical genetics. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaher U, Noukas M, Nikopensius T, Kals M, Annilo T, Nelis M, Ounap K, Reimand T, Talvik I, Ilves P, Piirsoo A, Seppet E, Metspalu A, Talvik T. De Novo SCN8A Mutation Identified by Whole-Exome Sequencing in a Boy With Neonatal Epileptic Encephalopathy, Multiple Congenital Anomalies, and Movement Disorders. Journal of child neurology. 2013;54:1270–1281. doi: 10.1177/0883073813511300. [DOI] [PubMed] [Google Scholar]
- Vanoye CG, Gurnett CA, Holland KD, George AL, Jr, Kearney JA. Novel SCN3A variants associated with focal epilepsy in children. Neurobiology of disease. 2014;62:313–322. doi: 10.1016/j.nbd.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. American journal of human genetics. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Human molecular genetics. 2014 doi: 10.1093/hmg/ddu470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang J, Li X, Ji J, Yang F, Wan C, Feng G, Wan P, He L, He G. SCN8A as a novel candidate gene associated with bipolar disorder in the Han Chinese population. Progress in neuro-psychopharmacology & biological psychiatry. 2008;32:1902–1904. doi: 10.1016/j.pnpbp.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Wasserman D, Geijer T, Rozanov V, Wasserman J. Suicide attempt and basic mechanisms in neural conduction: relationships to the SCN8A and VAMP4 genes. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics. 2005;133B:116–119. doi: 10.1002/ajmg.b.30128. [DOI] [PubMed] [Google Scholar]
- Willott JF, Lu SM. Midbrain pathways of audiogenic seizures in DBA/2 mice. Experimental neurology. 1980;70:288–299. doi: 10.1016/0014-4886(80)90028-x. [DOI] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nature neuroscience. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hearing research. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.