

Abstract
This study reports a first successful demonstration of a single channel proton 3D and 2D high-throughput ultrafast magic angle spinning (MAS) solid-state NMR techniques in an ultra-high magnetic field (1020 MHz) NMR spectrometer comprised of HTS/LTS magnet. High spectral resolution is well demonstrated.
Keywords: solid-state NMR, Proton detection, Structure Elucidation, Ultra-high field, Ultrafast MAS
Graphical Abstract
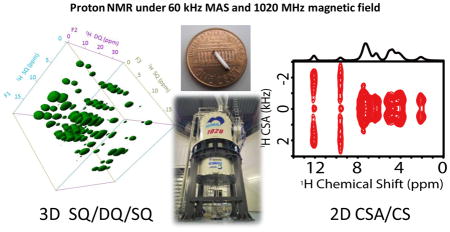
Solid-state NMR has been a valuable technique for high-resolution studies on non-soluble and even on non-crystalline chemical and biological materials.1–5 Though the challenges posed by molecular size, mobility, and physical and chemical nature of a system can be overcome, the major factors that severely limit wide-spread applications of solid-state NMR spectroscopy are poor spectral resolution and sensitivity.6 Therefore, there has been considerable interest in the development of high-resolution and high-throughput solid-state NMR techniques.7 Particularly, the development of proton-based techniques under ultrafast magic angle spinning (MAS) conditions, which effectively utilize the very high sensitivity of protons by dramatically suppressing the dipole-dipole interactions among protons, has been the focus of recent research.8–20 Furthermore, there are extensive efforts on the development of ultra-high field magnets for high-resolution NMR studies.21–24 The traditional LTS (low-Tc superconductors) magnet has an upper limit of magnetic field strength around 1 GHz, which was achieved in 2009. On the other hand, the recent achievement by combining the HTS (high-Tc superconductors) innermost coils and LTS outer coils has resulted in a magnetic field up to 24 T (with 1020 MHz 1H resonance frequency) with good magnetic field stability and spatial homogeneity.23,24 While there are numerous challenges in the construction of stable HTS/LTS ultra-high field magnets (≥1.0 GHz), there are also tremendous difficulties in performing multidimensional NMR experiments in general and solid-state NMR experiments in particular utilizing such magnets. To be more specific, the issues related to the stability/magnetic field drift and magnetic field inhomogeneity with these magnets do not typically favor proton solid-state NMR experiments. Nevertheless, extensive efforts were made to overcome these limitations in order to carry out the present study. While an external 2H lock as discussed later was used to reduce the magnetic field drift, the magnetic field inhomogeneity was controlled by a passive shimming approach. Subsequently, we successfully demonstrate herein the unique advantages of combining the high spectral resolution, rendered by fast MAS (60 kHz) and ultra-high magnetic field (24 T), to perform multidimensional single-channel proton solid-state NMR experiments. Specifically, a three-dimensional SQ/DQ/SQ experiment14 that correlates the single-quantum (SQ), double-quantum (DQ), and SQ frequencies of protons at 1020 MHz is demonstrated on a powder sample of L-Histidine·HCl·H2O at 60 kHz MAS. Since the span of chemical shift anisotropy (CSA) tensor is proportional to the magnetic field strength, it is shown that the high magnetic field enables easy measurement of proton CSA tensors. To the best of our knowledge, this is the first successful demonstration of single-channel proton high-resolution multidimensional solid-state NMR experiments at the highest magnetic field (24 T) so far.
Before fast MAS NMR measurements at 1020 MHz spectrometer, the magnetic field drift was reduced to minimum with an external 2H lock by using a D2O micro probe.24 However, the temporal field drift could still be around 0.17 ppm / 10 hours in comparison to 0.001 ppm / 10 hours for a solution probe with an internal lock. Actually, such a drift may also contribute to the line broadening observed for protons. However, the higher sensitivity rendered by the high natural-abundance and gyromagnetic ratio of protons greatly reduces the experimental time, and thus minimizes the effect of field drift on the spectrum. 1D ultrafast MAS proton spectra of L-Histidine·HCl·H2O obtained at 1020 and 600 MHz utilizing 1 mm rotors are compared in Figure 1. It is clearly shown that the peaks in the high-field region (< below 7 ppm) at 600 MHz with 60 kHz MAS display significant broadening due to residual 1H-1H dipolar interactions, whereas this broadening is largely suppressed at 1020 MHz under the same MAS rate leading to a higher proton resolution. More interestingly, the resolution of these peaks at 1020 MHz is comparable to that attained at 600 MHz under 120 kHz MAS using 0.75 mm rotor (Table S1). The line broadening observed for resonances 1 and 2 at 600 MHz and 120 kHz MAS should be attributed to poor shimming and increased frictional heating with larger temperature gradient as the spectrum under 110 kHz MAS (Figure S1) shows resolution comparable to that observed at 1020 MHz under 60 kHz MAS. The poor shimming of 120 kHz MAS probe results from the use of prototype stator carrying metal parts that can induce B0 inhomogeneity. It is worthwhile to mention here that 120 kHz MAS probe is still at its developmental stage and we hope to build this probe with well-optimized features anytime soon. The resolution enhancement at ultra-high magnetic field mainly results from two factors: (1) the increased chemical shift range (in Hz) results in larger separation of peaks, and (2) the better suppression of 1H-1H dipolar couplings by MAS when the chemical shift difference (in Hz) is larger. Both factors act simultaneously to improve the resolution of peaks below 7 ppm, which are associated with strong 1H-1H dipolar interactions. Furthermore, the resolution enhancement above 7 ppm is not significant, which is due to the presence of relatively weak dipolar couplings as compared to H2O and CH2. In addition, the presence of anisotropic bulk magnetic susceptibility (ABMS)25 can also induce inhomogeneous broadening on the order of 1 to 2 ppm for compounds containing π electrons.
Figure 1.

1D ultrafast MAS proton spectra of L-Histidine·HCl·H2O.
Encouraged by the feasibility of 1H detection and higher spectral resolution at 1020 MHz, we performed a 3D proton SQ/DQ/SQ experiment to show the higher resolution enabled by the high magnetic field using the pulse sequence given in Figure S2. The 3D SQ/DQ/SQ proton spectrum of L-Histidine·HCl·H2O obtained at 1020 MHz and 60 kHz MAS is shown in Figure 2; the 3D spectrum obtained at 600 MHz under the same experimental conditions is given in Figure S3. The spectral resonances are all well-resolved along all three dimensions. To better understand the resonance patterns in the 3D spectrum, 2D spectra projected from the 3D spectrum are shown in Figure 3. More 2D DQ/SQ1 (F2/F1) slices taken at different chemical shift values along the SQ2 (F3) dimension are given in Figure S4. Remarkably, rich structural information could be extracted from DQ/SQ1 (F2/F1), DQ/SQ2 (F2/F3) and SQ1/SQ2 (F1/F3) spectra. The SQ1/SQ2 correlation spectra (Figure 3A and 3D) directly provide the proton proximity information, which in principle is similar to a normal 2D 1H/1H dipolar-coupling based correlation spectrum.15 It is shown that a finite pulse radiofrequency driven dipolar recoupling (fp-RFDR)15,26 mixing time of 3.73 ms is sufficient to accomplish a total correlation of proton resonances in L-Histidine·HCl·H2O. In addition, the DQ/SQ2 (Figure 3B and 3E) spectra give proximity information at a higher resolution compared to that rendered by the 2D SQ1/SQ2 correlation spectrum due to double spectral width in the DQ dimension. Furthermore, new cross peaks are observed in the 2D DQ/SQ1 (Figure 3C and 3F) spectra in comparison to the 2D DQ/SQ2 spectra given in Figure 3B and 3E. In fact, these new cross peaks indicate the proximity between a spin (e.g. spin A) and a certain other pair of spins (e.g. spins B and C) that are close enough to induce a DQ signal through the recoupled dipolar coupling. The spin A is indicated by the isotropic chemical shift along SQ1 dimension, while spins B and C are indicated by the chemical shift along the DQ dimension, which equals to the sum of the isotropic chemical shifts of spins B and C. Actually, better resolved 2D DQ/SQ1 spectra could be sliced at a specific chemical shift as given in Figure S4, as they only provide local information related to the protons at the specific chemical shift. Such cross peaks are important for distance measurements in determining molecular structures, especially when there are protons that are overlapped and cannot be distinguished. More details about this 3D pulse sequence could be found in Ref.[14] Though the signal-to-noise (S/N) ratio is similar in the spectra obtained at 600 and 1020 MHz NMR spectrometers, the spectral resolution is definitely better at 1020 MHz. In principle, the S/N ratio at 1020 MHz should be better than 600 MHz, however, the fact that we observed similar S/N ratio can be attributed to the use of prototype probe at 1020 MHz, which is not optimized well in comparison to the commercial probes. On the other hand, the effects of chemical shift offset are more pronounced at 1020 MHz. However, in the 3D experiments, we utilized a strong radiofrequency (RF) field for the rectangular 90°/180° pulses to overcome the offset effects. Besides, the implementation of super-cycled fp-RFDR15 and BABA-XY1627 sequences could also overcome the chemical shift offset effect as well as the RF field inhomogeneity. Herein, our results successfully demonstrate the feasibility of obtaining high-resolution multidimensional solid-state NMR spectra at an ultra-high magnetic field NMR spectrometer. In fact, the difficulties faced in accomplishing the higher sensitivity at 1020 MHz are not surprising. Therefore, the development of novel composite pulses28 or offset-compensating short pulses are worthwhile for further MAS NMR applications, not to mention about the need for super phase cycling schemes for the pulse sequences. Moreover, we would like to add here that similar resolution in the 3D proton SQ/DQ/SQ experiment is also possible at 600 MHz if we spin the sample at ultrafast MAS rates (≥ 95 kHz) using 0.75 mm rotor (Figure S5). However, the main drawbacks of such measurement are the rise in the sample temperature due to spinning at such high rates, and the use of 2.75 times less sample volume in comparison to 1.0 mm rotors.
Figure 2.

3D SQ/DQ/SQ spectrum of L-His·HCl·H2O obtained at 1020 MHz under 60 kHz MAS. 16 t1 and t2 increments, 8 scans, and a 6 s recycle delay were used (total experimental time = 13.65 hrs.). A 3.73 ms fp-RFDR mixing was utilized for magnetization exchange, and the BABA-XY16 sequence with 133.3 μs was used for excitation/reconversion.
Figure 3.

2D SQ1/SQ2 (A,D), DQ/SQ2 (B,E) and DQ/SQ1 (C,F) spectral projection extracted from the 3D spectra at 600 MHz (A,B,C) and 1020 MHz (D,E,F). Both experiments were performed under 60 kHz MAS.
The unique advantage of an ultrahigh magnetic field could also be well demonstrated with the measurement of proton CSAs as shown in Figure 4. As the span of CSA linearly increases with the magnetic field, the amplified CSAs are easily and accurately measured at high magnetic field; this is especially more beneficial when CSA is small like that for aliphatic protons. Figure 4A shows the 2D proton anisotropic/isotropic chemical shift (CSA/CS) correlation spectrum obtained at 1020 MHz on L-Tyrosine·HCl sample using the pulse sequence given in Figure S6, whereas the 2D CSA/CS correlation spectrum at 600 MHz is given in Figure S7. Symmetry-based sequence R1887 with a phase-alternating composite π pulse (270°0–90°180) was adapted to recouple CSA interactions under ultrafast MAS, and used to obtain undistorted CSA lineshapes and to determine relative orientation of different CSA tensors.16, 29 Also, such phase-alternating composite π pulse could well overcome the RF field inhomogeneity and achieve remarkable CSA recoupling efficiency.16 Figure 4B compares the proton CSA lineshapes obtained at 600 and 1020 MHz, which were obtained from the spectral slices taken at the isotropic chemical shift (9.6 ppm) corresponding to the benzene-bonded OH group. Obviously, with the magnetic field increasing from 14.1 to 24.0 T, the splitting between the two singularities in the CSA lineshape is almost doubled. In fact, such splitting is proportional to the anisotropic chemical shift parameter δaniso = |δzz-δiso|,30 where the δiso is the isotropic chemical shift, and the principal components of the chemical shift tensor are defined as |δzz-δiso| ≥ |δxx-δiso| ≥ |δyy-δiso|. By comparing the experimental spectra with SIMPSON numerical simulations,31,32 all the principal components of the CSA tensor could easily be determined. The CSA parameters for the benzene-bonded OH from the simulation are determined to be δaniso = 20.0 ppm and η = (δxx-δyy) / δaniso = 0.5, (Figure S8). Here it should be pointed out that a DC balance, wherein the average of the final 1/8th FID points in t1 is subtracted from the total data points to remove DC offset effects, was applied prior to the Fourier transformation in the t1 dimension in order to suppress the zero-frequency peak. However, when the CSA is very small, the splitting may be absent in the CSA lineshape.30 Under such condition the zero-frequency peak may play a significant role, rendering it difficult to extract accurate CSA values. Fortunately, the high magnetic field will greatly amplify the CSA and thus the splitting in the CSA lineshape will alleviate the effect of zero-frequency peak on the extraction of the principal components of CSA tensors. On the one hand high magnetic field amplifies the CSA, but at the same time it also magnifies the higher-order cross-terms (CSAxCSA, CSAxCS) that can no longer be neglected in the recoupled CSA Hamiltonian with R-symmetry based sequences. Simulated lineshapes representative of a single spin CSA Hamiltonian at 1020 and 600 MHz (Figure S9) clearly highlight this aspect. As seen from Figure S9, the CSA lineshape at 1020 MHz is more distorted in comparison to 600 MHz due to the recoupling of the higher-order CSA cross-terms. Finally, it should be pointed out that the single channel symmetry-based CSA recoupling sequence will also recouple the heteronuclear dipolar couplings, because both CSA and heteronuclear dipolar coupling have a space rank of 2 and a spin rank of 1.33 Therefore, it may suffer some limitation on the measurement of amide proton CSAs due to the presence of 14N-1H dipolar couplings unless 14N decoupling is achieved.
Figure 4.

Proton CSA measurement under ultrafast MAS on L-Tyrosine·HCl sample. (A) 2D CSA/CS correlation spectrum under 60 kHz MAS at 1020 MHz. (B) 1H CSA lineshapes from 2D spectra extracted at the chemical shift of 9.6 ppm and obtained from a 600 MHz spectrometer at 70 kHz MAS (black) and a 1020 MHz spectrometer at 60 kHz MAS (red).
In conclusion, we successfully demonstrated the first use of 1020 MHz NMR spectrometer with a hybrid HTS/LTS magnet configuration in carrying out a single-channel multidimensional proton solid-state NMR experiments at fast MAS. The fast MAS could greatly average out the very large 1H-1H dipolar couplings, while the high field could expand the chemical shift range. As a result, the proton spectral resolution is largely enhanced. Due to the high natural abundance and gyromagnetic ratio, high-resolution and high-sensitivity proton solid-state NMR spectroscopy could provide abundant information about molecular structures and dynamics. In particular, the 3D SQ/DQ/SQ spectrum and the 2D spectral slices show high spectral resolution rendered by the combination of high magnetic field, fast MAS, and proton-detection, and reveal dipolar-coupling based chemical shift correlation of protons. Such 3D spectrum not only provides the proximity information between a pair of proton spins, but also gives the information of proximity between a spin and a certain pairs of spins. In addition, the 2D anisotropic/isotropic correlation experiment under high field and fast MAS enables accurate measurement of CSA tensors, which can provide deep insights into hydrogen bonding interactions in numerous chemical and biological systems. Overall, the elegant single-channel high-resolution proton multidimensional solid-state NMR experiments under fast MAS and high field enable the assignment of proton chemical shift resonances, and thus allow high-resolution and high-throughput structure and dynamics studies for a variety of chemical and biological materials in solid-state that may not be amenable for traditional solution NMR or X-ray crystallographic techniques.
Supplementary Material
Highlights.
Proton 2D and 3D solid-state NMR experiments are demonstrated at 1020 MHz.
HTS/LTS hybrid magnet provides enough stability for long term measurements.
Resolution enhancement and amplified 1H CSA are attained at 1020 MHz.
Acknowledgments
This work was supported by the funds from National Institutes of Health (GM084018 and GM095640 to A. R.). This work is also supported by SENTAN, JST in Japan. We thank JEOL RESONANCE Inc. for the technical support, and we would like to dedicate this paper to the late Dr. Tsukasa Kiyoshi who started this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schmidt-Rohr K, Spiess HW. Multidimensional solid-state NMR and polymers. Academic Press; Berlin: 1994. [Google Scholar]
- 2.McDermott AE, Polenova T. Solid State NMR Studies of Biopolymers. John Wiley & Sons; 2012. [Google Scholar]
- 3.Mehring M. High resolution NMR spectroscopy in solids. Springer-Verlag; Berlin: 1976. [Google Scholar]
- 4.a) Tycko R. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hong M, Zhang Y, Hu F. Annu Rev Phys Chem. 2012;63:1–24. doi: 10.1146/annurev-physchem-032511-143731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramamoorthy A, editor. NMR spectroscopy of biological solids. CRC Press; 2010. [Google Scholar]
- 6.a) Fujiwara T, Ramamoorthy A. Annu Rep NMR Spectrosc. 2006;58:155–175. [Google Scholar]; b) Tycko R. Acc Chem Res. 2013;46:1923–1932. doi: 10.1021/ar300358z. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Maly T, Debelouchina GT, Bajaj VS, Hu K-N, Joo C-G, Mak Jurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, Griffin RG. J Chem Phys. 2008;128:052211. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Castellani F, Rossum BV, Diehl A, Schubert M, Rehbein K, Oschkinat H. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]; b) Lewandowski JR, Halse ME, Blackledge M, Emsley L. Science. 2015;348:578–581. doi: 10.1126/science.aaa6111. [DOI] [PubMed] [Google Scholar]; c) Wickramasinghe NP, Parthasarathy S, jones CR, Bhardwaj C, Long F, Kotecha M, Mehboob S, Fung LWM, Past J, Samoson A, Ishii Y. Nat Meth. 2009;6:215–218. doi: 10.1038/nmeth.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Cho G, Wu Y, Ackerman JL. Science. 2003;300:1123–1127. doi: 10.1126/science.1078470. [DOI] [PubMed] [Google Scholar]
- 8.Ishii Y, Tycko R. J Magn Reson. 2000;142:199–204. doi: 10.1006/jmre.1999.1976. [DOI] [PubMed] [Google Scholar]
- 9.Reif B, Griffin RG. J MagnReson. 2003;160:78–83. doi: 10.1016/s1090-7807(02)00035-6. [DOI] [PubMed] [Google Scholar]
- 10.Paulson EK, Morcombe CR, Gaponenko V, Dancheck B, Byrd RA, Zlim KW. J Am Chem Soc. 2003;125:15831–15836. doi: 10.1021/ja037315+. [DOI] [PubMed] [Google Scholar]
- 11.Wiench JW, Bronnimann CE, Lin VS-Y, Pruski M. J Am Chem Soc. 2007;129:12076–12077. doi: 10.1021/ja074746+. [DOI] [PubMed] [Google Scholar]
- 12.Khitrin AK, Fung BM. J Magn Reson. 2001;152:185–188. doi: 10.1006/jmre.2001.2388. [DOI] [PubMed] [Google Scholar]
- 13.a) Zhang R, Damron J, Vosegaard T, Ramamoorthy A. J Magn Reson. 2015;250:37–44. doi: 10.1016/j.jmr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang R, Ramamoorthy A. J Chem Phys. 2015;142:204201. doi: 10.1063/1.4921381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Pandey MK, Nishiyama Y, Ramamoorthy A. Sci Rep. 2015;5:11810. doi: 10.1038/srep11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Nishiyama Y, Zhang R, Ramamoorthy A. J Magn Reson. 2014;243:25–32. doi: 10.1016/j.jmr.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang R, Nishiyama Y, Sun P, Ramamoorthy A. J Magn Reson. 2015;252:55–66. doi: 10.1016/j.jmr.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pandey MK, Malon M, Ramamoorthy A, Nishiyama Y. J Magn Reson. 2015;250:45–54. doi: 10.1016/j.jmr.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Nishiyama Y, Malon M, Gan Z, Endo Y, Nemoto T. J Magn Reson. 2013;230:160–164. doi: 10.1016/j.jmr.2013.02.015. [DOI] [PubMed] [Google Scholar]; b) Nishiyama Y, Endo Y, Nemoto T, Utsumi H, Yamauchi K, Hioka K, Asakura T. J Magn Reson. 2011;208:44–48. doi: 10.1016/j.jmr.2010.10.001. [DOI] [PubMed] [Google Scholar]; c) Nishiyama Y, Malon M, Ishii Y, Ramamoorthy A. J Magn Reson. 2014;244:1–5. doi: 10.1016/j.jmr.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a) Reif B. J Magn Reson. 2012;216:1–12. doi: 10.1016/j.jmr.2011.12.017. [DOI] [PubMed] [Google Scholar]; b) Linser R, Dasari M, Hiller M, Higman V, Fink U, Lopez del Amo J-M, Markovic S, Handel L, Kessler B, Schmieder P, Oesterhelt D, Oschkinat H, Reif B. Angew Chem Int Edit. 2011;50:4508–4512. doi: 10.1002/anie.201008244. [DOI] [PubMed] [Google Scholar]; c) Linser R, Fink U, Reif B. J Magn Reson. 2008;193:89–93. doi: 10.1016/j.jmr.2008.04.021. [DOI] [PubMed] [Google Scholar]
- 19.a) Zhou DH, Shah G, Mullen C, Sandoz D, Rienstra CM. Angew Chem. 2009;121:1279–1282. doi: 10.1002/anie.200801029. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM. J Am Chem Soc. 2007;129:11791–11801. doi: 10.1021/ja073462m. [DOI] [PubMed] [Google Scholar]
- 20.Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G. J Magn Reson. 2015;253:36–49. doi: 10.1016/j.jmr.2015.01.003. [DOI] [PubMed] [Google Scholar]; Laage S, Sachleben JR, Steuernagel S, Pierattelli R, Pintacuda G, Emsley L. J Magn Reson. 2009;196:133–141. doi: 10.1016/j.jmr.2008.10.019. [DOI] [PubMed] [Google Scholar]; Bertini I, Emsley L, Lelli M, Luchinat C, Mao J, Pintacuda G. J Am Chem Soc. 2010;132:5558–5559. doi: 10.1021/ja100398q. [DOI] [PubMed] [Google Scholar]; Knight MJ, Felli IC, Pierattelli R, Bertini I, Emsley L, Herrmann T, Pintacuda G. J Am Chem Soc. 2012;134:14730–14733. doi: 10.1021/ja306813j. [DOI] [PubMed] [Google Scholar]; Knight MJ, Pell AJ, Bertini I, Felli IC, Gonnelli L, Pierattelli R, Herrmann T, Emsley L, Pintacuda G. Proc Natl Acad Sci USA. 2012;109:11095–11100. doi: 10.1073/pnas.1204515109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu R, Brey WW, Shetty K, Gor’kov P, Saha S, Long JR, Grant SC, Chekmenev EY, Hu J, Gan Z, Sharma M, Zhang F, Logan TM, Brüschweller R, Edison A, Blue A, Dixon IR, Markiewicz WD, Cross TA. J Magn Reson. 2005;177:1–8. doi: 10.1016/j.jmr.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Hashi K, Shimizu T, Goto A, Kiyoshi T, Matsumoto S, Wada H, Fujito T, Hasegawa K-i, Yoshikawa M, Miki T, Ito S, Hamada M, Hayashi S. J Magn Reson. 2002;156:318–321. doi: 10.1006/jmre.2002.2559. [DOI] [PubMed] [Google Scholar]
- 23.Yanagisawa Y, Nakagome H, Tennmei K, Hamada M, Yoshikawa M, Otsuka A, Hosono M, Kiyoshi T, Takahashi M, Yamazaki T, Maeda H. J Magn Reson. 2010;203:274–282. doi: 10.1016/j.jmr.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Hashi K, Ohki S, Matsumoto S, Nishijima G, Goto A, Deguchi K, Yamada K, Noguchi T, Sakai S, Takahashi M, Yanagisawa Y, Iguchi S, Yamazaki T, Maeda H, Tanaka R, Nemoto T, Suematsu H, Miki T, Saito K, Shimizu T. J Magn Reson. 2015;256:30–33. doi: 10.1016/j.jmr.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 25.a) Zorin VE, Brown SP, Hodgkinson P. J Chem Phys. 2006;125:144508. doi: 10.1063/1.2357602. [DOI] [PubMed] [Google Scholar]; b) Kubo A, Spaniol TP, Terao T. J Magn Reson. 1998;133:330–340. doi: 10.1006/jmre.1998.1473. [DOI] [PubMed] [Google Scholar]
- 26.a) Bennett AE, Griffin RG, Ok JH, Vega S. J Chem Phys. 1992;96:8624–8627. [Google Scholar]; b) Ishii Y. J Chem Phys. 2001;114:8473–8483. [Google Scholar]
- 27.Saalwächter K, Lange F, Matyjaszewski K, Huang C-F, Graf R. J Magn Reson. 2011;212:204–215. doi: 10.1016/j.jmr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 28.Levitt MH. Prog Nucl Magn Reson Spectrosc. 1986;18:61–122. [Google Scholar]
- 29.Pandey MK, Nishiyama Y. Solid State Nucl Magn Reson. 2015;70:15–20. doi: 10.1016/j.ssnmr.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Miah HK, Bennett DA, Iuga D, Titman JJ. J Magn Reson. 2013;235:1–5. doi: 10.1016/j.jmr.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Bak M, Rasmussen JT, Nielsen NC. J Magn Reson. 2000;147:296–330. doi: 10.1006/jmre.2000.2179. [DOI] [PubMed] [Google Scholar]
- 32.Tošner Z, Andersen R, Stevensson B, Edén M, Nielsen NC, Vosegaard T. J Magn Reson. 2014;246:79–93. doi: 10.1016/j.jmr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Levitt MH. Symmetry-based pulse sequences in magic-angle spinning solid-state NMR. In: Grant DM, Harris RK, editors. Encycl Nucl Magn Reson. Vol. 9. Wiley; Chichester: 2002. pp. 165–196. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.