

Abstract
There is an urgent need to develop nanocarriers for the treatment of glioblastoma multiforme (GBM). Using co-registered positron emission tomography (PET) and magnetic resonance (MR) images, here we performed systematic studies to investigate how a nanocarrier’s size affects the pharmacokinetics and biodistribution in rodents with a GBM xenograft. In particular, highly stable, long-circulating three-helix micelles (3HM), based on a coiled-coil protein tertiary structure, were evaluated as an alternative to larger nanocarriers. While the circulation half-life of the 3HM was similar to 110-nm PEGylated liposomes (t1/2 = 15.5 and 16.5 h, respectively), the 20-nm micelles greatly enhanced accumulation within a U87MG xenograft in nu/nu rats after intravenous injection. After accounting for tumor blood volume, the extravasated nanoparticles were quantified from the PET images, yielding ~0.77 %ID/cc for the micelles and 0.45 %ID/cc for the liposomes. For GBM lesions with a volume greater than 100 mm3, 3HM accumulation was enhanced both within the detectable tumor and in the surrounding brain parenchyma. Further, the nanoparticle accumulation was shown to extend to the margins of the GBM xenograft. In summary, 3HM provides an attractive nanovehicle for carrying treatment to GBM.
Graphical Abstract
1. Introduction
Glioblastoma multiforme (GBM) is the most common and aggressive malignant primary brain tumor, with a median patient survival of 12–15 months (1–3). Combining radiotherapy and post-surgical chemotherapy using cisplatin (4, 5), irinotecan (6–8), thalidomide (9, 10), or bevacizumab (11, 12) has only led to a limited improvement in survival rate (13, 14). The blood-brain barrier (BBB) typically limits the accumulation of therapeutics within the brain and such drugs can be deactivated by intra- and extra-cellular enzymes in the BBB. The BBB includes a range of passive and active transport mechanisms: 1) a paracellular pathway, regulated by tight junctions; 2) a lipophilic pathway, through the lipid membranes; 3) specific receptor-mediated transcytosis actuated by specific interactions with receptors on cerebral endothelial cells; and 4) non-specific adsorptive-mediated transcytosis, triggered by interactions between positively-charged species and negatively-charged lipid membranes on endothelial cells (15). The BBB is altered in the presence of diseases such as GBM and transport of nanotherapies is enhanced through junctions that are altered by the presence of disease. However, while essentially all GBM patients have significant BBB disruption, the disruption is variable across the tumor and GBM patients also have regions of tumor with limited BBB permeability (16). Therefore, the development of strategies to enhance drug accumulation is important. Further, when drugs are delivered to the GBM tumor parenchyma, efflux transporters actively pump the drug out of the target cell (15, 17–19). GBM therapeutics must be administered at a high dose that can lead to severe side effects and early termination of treatment, and thus, there is an urgent need to develop nanocarriers for the treatment of GBM.
It is well known that the surface chemistry of a nanocarrier determines its pharmacokinetics (PK), biodistribution and clearance pathway (20), and PEGylation is required to avoid recognition by the reticuloendothelial system (RES) and extend the circulation time. However, in the absence of additional surface modification, PEGylated nanocarriers typically do not cross the BBB (21, 22). When the BBB is comprised by disease, passive delivery of nanotherapeutics is feasible. Passive delivery of long-circulating nanoparticles via the enhanced permeability retention (EPR) effect has been the major mechanism for nanoparticle uptake into tumors (23–27). In general, smaller nanoparticles (15 ~ 50 nm) demonstrate a greater EPR effect and intratumoral distribution than larger nanoparticles (100 ~ 300 nm) and therefore show the potential to enhance accumulation (28, 29). However, systematic studies of the effect of nanocarrier size and surface chemistry on the carrier’s ability to accumulate within GBM tumor tissue have been lacking. Previous studies have shown that the vascular permeability increases in highly angiogenic glioblastoma due to the disrupted BBB providing a conduit for the delivery of nanotherapies (30–32). However, the vascular permeability is reduced in brain tumors as compared with tumors within other organs and the size limit for nanoparticles observed to preferentially accumulate in glioblastoma (7–100 nm) is smaller than that in colorectal carcinoma, hepatoma, and sarcoma (380 – 2000 nm) (33). Once localized in the tumor, there is increasing evidence that nanocarriers need to be below a certain size to achieve significant tumor penetration (34–37).
Enhanced delivery to brain tumors with small nanoparticles has not yet been experimentally validated. Hobbs et al. demonstrated that particle permeability for orthotopic brain tumors was limited to particles with a diameter ranging from 7 to 100 nm (33); however, differences within the size range were not described. Kim et al. reported that PEGylated silica nanoparticle uptake in a U87MG mouse xenograft was greater with 100–150 nm particles as compared with larger and smaller particles (40 and > 300 nm) (38). However, in this study the tumor was implanted in the mouse shoulder, which may differ in the pore cutoff size as compared with the orthotopic brain tumor.
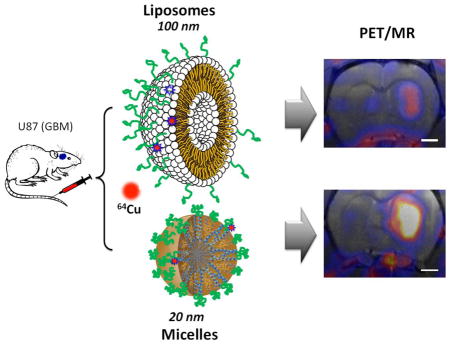
The study compares the accumulation of two 64Cu-labeled nanocarriers: a PEGylated 110-nm liposome with similar pharmacokinetics to other long-circulating liposomes (39) and recently developed 20 nm 3-helix micelles (3HM) (40). This family of highly stable, long circulating 3HM is based on a coiled-coil protein tertiary structure that is routinely used to present ligand clusters on the cell surface, where the peptide-polymer conjugate amphiphile is schematically shown in Fig. 1. The headgroup of the amphiphile consists of a peptide that self-associates to form a coiled-coil 3-helix bundle and a PEG chain (2000 Da) attached to the exterior of the 3-helix bundle at the middle position. A short PEG chain (750 Da) is also attached to one end of the peptide (C-terminus) and acts as a stealth layer on the surface of the micelle. The hydrophobic portion of the amphiphile is a double alkyl tail attached to the other end of the peptide (N-terminus). The amphiphile can be readily synthesized at high purity. Once dissolved in aqueous solution, the amphiphile self-assembles to form a 3HM that is ~20 nm by dynamic light scattering and 18 nm by small angle. x-ray scattering in size with very low polydispersity (40–44). Systematic characterization confirmed very slow subunit exchange kinetics and excellent kinetic stability of the micelle under physiological condition (40, 42). Micelles labeled with the FRET pair DiI and DiO demonstrated a trace level of cargo dissociation from the micelles over 24 hours in BSA (40).
Figure 1.

Schematic figure of the experimental procedure, which compares the accumulation of liposomes and micelles within glioblastoma multiforme in the rat brain. Green represents polyethylene glycol (PEG) on the surface of the nanoparticles (dC18: distearoyl lipid, 6-BAT: 6-aminobenzylTETA). Nanoparticles were intravenously injected through the tail vein.
We have previously developed methods to label liposomes and micelles with 64Cu using the 6-BAT chelator and have shown these labels to be stable in serum over 48 hours (39). Less than 8% of the 64Cu label disassociated from liposomes over 48 h of serum incubation at 37 °C and results with micelles were similar. In a mouse model, the circulation half-life for the 64Cu-labeled micelles was 29.5 h and ~15% of the injected dose/gram (%ID/g) remained in circulation after 48 h, as compared with 7 %ID/g for PEGylated liposomes. In vivo studies further showed that the accumulation of 3HM in the liver and spleen is substantially reduced as compared with PEGylated liposomes (40).
Using co-registered positron emission tomography (PET) and magnetic resonance (MR) images, here we report on systematic studies to investigate how the nanocarrier’s size affects the pharmacokinetics and biodistribution in rodents with GBM xenograft and evaluate the unique 3HM for treatment of GBM. The resulting data suggest that imaging of nanoparticle distribution and tumor kinetics can be used to improve the design of nanoparticles for GBM treatment and confirm that GBM delivery can be improved with small nanocarriers.
2. Materials and methods
An overview of the experimental procedures is provided in Fig. 1. HSPC, cholesterol, DSPE-PEG2k-OMe, were purchased from Avanti Polar Lipids (Alabaster, AL). Solvents and other agents were all of analytical purity and purchased from Sigma-Aldrich (Milwaukee, WI) and VWR (Brisbane, CA). 64CuCl2 was purchased from MIR Radiological Science (St. Louis, MO) under a protocol controlled by the University of California Davis. Phosphate-buffered saline (PBS) was purchased from Invitrogen Corporation (Carlsbad, CA).
The 3HM amphiphile can be readily synthesized using solid phase peptide synthesizer. Detailed chemistry and purification procedure have been documented in detail (40). Briefly, the amphiphile is based on a 3-helix bundle forming peptide, 1COI (EVEALEKKVAALECKVQALEKKVEALEHGW)(45, 46). The peptide was synthesized on a Prelude solid phase peptide synthesizer (Protein Technologies, Az) using standard 9-fluorenylmethyl carbamate (Fmoc) chemistry. For the synthesis of amphiphilic subunits, the alkyl chains were conjugated through reaction of stearic acid (C18) with deprotected Fmoc-Lys(Fmoc)-OH to generate a branched alkyl tail at the N-terminus. Modification of the C-terminus was achieved through an orthogonal Fmoc-Lys(Alloc)-OH protection strategy employed in Fmoc-SPPS. The resulting free amino groups of lysine were utilized for conjugating carboxy-terminated PEG (750 Da) using HBTU/DIPEA chemistry. Cleavage was carried out using a cocktail of 90:8:2 TFA/TIS/water for 3 h. Crude peptides were precipitated in cold ether, isolated, and dried for the conjugation of polymers. To conjugate PEG, a cysteine at position 14 facilitates the site-specific coupling of maleimide-functionalized PEG of molecular weight 2000 g/mol to the middle of the peptide sequence. The conjugation reaction was carried out in phosphate buffer (pH = 7.4) overnight with a reaction ratio of PEG to peptide at 5:1. Cysteine at the C-terminus of dC18-1COI(P2k) allows for the conjugation of 6-BAT-maleimide onto the peptides for PET imaging.
2.1. Preparation of 64Cu-labeled liposomes and micelles
Preparation of liposomes and micelles followed our previously reported methods (39, 40). To facilitate post-labeling for PET, a custom lipid-chelator conjugate containing the BAT chelator is incorporated into liposomes and micelles. Synthesis of the lipid-chelator conjugates was previously described (39, 40). In brief, for liposome preparation: in a glass test tube, the dried lipid film (20 mg, HSPC:6-BAT-lipid:DSPE-PEG2k-OMe:cholesterol= 55.5:0.5:5:39, mole percent) was suspended in 0.1 M ammonium citrate buffer (pH 5.5, 0.5 mL) and the solution was incubated for 30 min at 60 °C. The lipid mixture was then extruded 21 times through mini-extruder with a 100-nm membrane filter under 60–65 °C heating. After cooling, the solution was kept at room temperature until 64Cu labeling was complete. For micelle preparation: dC18-1COI(P2k)-P750 with 2 mol% dC18-1COI(P2k)-6-BAT(10 mg) was dissolved in double-distilled water (0.5 mL) and spontaneously self-assembled into micelles with incubation at 70 °C until the solution became clear (approximately 1 h). Particle size and zeta-potential were measured via dynamic light scattering (DLS) with a Zetasizer Nano (Malvern Instruments Inc., Westborough, MA).
Liposomes (0.2 mL of 40 mg/mL solution) and micelles (0.4 mL of a 20 mg/mL solution) were added to 64CuCl2 (Washington University, MO) buffered in 0.1 M ammonium citrate (pH 5.5, 0.1 mL) and incubated for 50 minutes. 0.1 M EDTA (20 mL) in double-distilled water was added in order to remove the non-specifically bound 64Cu from the particles. Completion of 64Cu labeling was monitored by instant thin-layer chromatography (ITLC) eluted by a 0.1 M ammonium citrate solution (pH 5.5). The chemical purity of isolated 64Cu-liposomes and 64Cu-micelles after size-exclusion column chromatography (Sephadex-G75 superfine, 6 mL bed volume, DPBS) was determined by ITLC.
2.2. 3HM and characterization
After dissolving the lyophilized amphiphile powder into aqueous solution, dynamic light scattering (DLS) reveals a hydrodynamic diameter of ~20 nm and a fairly uniform size distribution of micelles. We further performed solution small angle x-ray scattering studies to verify the particle size and the outer PEG layer thickness. The surface property of the micelle has significant effects on the in vivo behavior of nanocarrier. Although previous in vivo studies confirmed the effective stealth PEG layer on the 3HM surface, it is important to determine the PEG 750 conformation and the PEG brush layer density.
Small-angle X-ray scattering of 3HM
Small-angle x-ray scattering (SAXS) experiments were carried out at the Advanced Light Source (ALS) at the Lawrence Berkeley National Lab, Berkeley, California at the SAXS/WAXS/GISAXS beamline 7.3.3. The instrument was operated using an X-ray energy of 10 keV and a sample–detector length of 1.2 m and a 1 M Pilatus detector. Samples were contained in standard boron–quartz capillaries situated in a homemade sample holder. Using this setup, background subtraction could be performed quantitatively. Samples were dissolved in phosphate buffer (25 mM, pH 7.4) at a concentration of ~5 mg/ml, annealed at 70°C for 1 hour and allowed to equilibrate at room temperature overnight before SAXS measurements were performed.
2.3. Animal model
All animal experiments were conducted under a protocol approved by the University of California, Davis, Animal Use and Care Committee (Davis, CA). Eleven male athymic nude rats were purchased from Harlan Laboratories (Hayward, CA) and weighed ~250 g upon arrival. U87MG cells at 3 × 106 cells/10 μL were intracranially inoculated through a small burr hole in the skull into the right striatum of each rat. Imaging studies were completed at nine (n = 6) and sixteen days (n = 5) post-surgery; at this time the age ranged from 82 to 93 days and the average body weight was 294 ± 35 g.
2.4. Positron Emission Tomography/Magnetic Resonance (PET/MR) imaging
Radioactivity was handled under a university-approved radiation use authorization (Davis, CA). Glioblastoma-bearing rats were administered 200 μL of 64Cu-liposomes (690 ± 325 μCi, 4.15 ± 0.75 mg, n = 6) and 64Cu-micelles (284 ± 97 μCi, 4.22 ± 0.99 mg, n = 5) via the tail vein under 1.5% isoflurane anesthesia. The critical micelle concentration (CMC) tested in this experiment was ~4 μM (~0.03 mg/mL). Thus, the micelle concentration (0.23 mg/mL) (calculated by dividing the average dose (4.22 ± 0.99 mg) of micelles by estimated blood volume (18.4 mL) (47)) was seven times higher than the CMC.
PET images were acquired with a Focus 120 scanner (Siemens Medical Solutions Inc., USA) over 30 minutes at 0, 3.5, 7, and 21 h after injection of nanoparticles. After PET scanning at 21 hours, MR imaging was immediately performed with a Bruker Biospec 7 Tesla (7T) small-animal scanner (Bruker BioSpin MRI, Ettlingen, Germany). A 72-mm internal diameter linear resonator was used for RF transmission and a four-channel rat brain phased array surface coil was used for signal reception. Rat brains were imaged coronally with a fast-spin echo sequence (“RARE”; axial: TE/TR = 8 ms/750 ms; FOV = 40 × 40 mm2; MTX = 256 × 256; ST/SI = 1 mm/1 mm; ETL = 4. Coronal: TE/TR = 9ms/1200ms; FOV = 50 × 30 mm2; MTX = 320 × 192; ST/SI = 1 mm/1 mm; ETL = 4.). Data were acquired and reconstructed with ParaVision 5.1 software (Bruker BioSpin MRI). PET/MR images were co-registered on Inveon Research Workspace 4.2 (Siemens Medical Solutions Inc., USA)
2.5. Biodistribution
After PET/MR imaging, animals were immediately euthanized with Euthasol (Western Medical Supply, Arcadia, CA). Blood was collected by syringe from the left ventricle and perfused from the body with Dulbecco’s Modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA). Heart, lungs, stomach, intestine, muscle, bone, liver, kidneys, spleen and brain were harvested and placed in a gamma counter (Perkin-Elmer life Sciences). Values are presented as the %ID/g.
2.6. Image analysis for pharmacokinetics
All PET images were reconstructed with the maximum a posteriori (MAP) reconstruction algorithm and analyzed with AsiPro software (Onccorde Microsystems Inc., Knoxville, TN) and Inveon Research Workspace 4.2 (Siemens Medical Solutions Inc., USA). Regions of interest (ROIs) within the glioblastoma and contralateral left brain (striatum) were drawn on co-registered PET/MR images with a volume ranging from 38 to 201 mm3 and 33 to 138 mm3, respectively. ROIs in the contralateral brain (striatum) were of a similar size and location to those applied in the tumor. The radioactivity within the blood pool was obtained using ROIs in the heart chamber from the PET images.
Time-activity curves (TAC) of blood radioactivity subtracted 64Cu-liposomes and 64Cu-micelles uptake in glioblastoma were obtained by equation 1 and values are given in Table S2,
| Eq. 1 |
where RGBM,ROI(t) is the tumor radioactivity (%ID/cc) at any given time point (t), %VBGBM(t0) is the percent blood volume in glioblastoma measured at the 0 h time point (t0) and RBlood,ROI(t) is the radioactivity (%ID/cc) of blood at each time point.
The percent tumor blood volume (TBV) and left brain blood volume (LBV) was calculated by equation 2. TBV and LBV are presented as percent vascular volume in glioblastoma and left brain (striatum)
| Eq. 2 |
where RGBM,ROI(t0) and RLB,ROI(t0) are the radioactivity (%ID/cc) in the glioblastoma and left brain (striatum) at the 0 h time point (t0), RBlood,ROI(t0) is the radioactivity (%ID/cc) of blood at the 0 h time point (t0).
To determine the circulation half-life of the 64Cu-liposomes and 64Cu-micelles, the %ID/cc obtained from the ROI image was fitted to a one-phase decay curve using Prism 6 for Mac OS X software (La Jolla, CA). Data are presented as percent injected dose per cubic centimeter (%ID/cc).
2.7. Autoradiography
At necropsy, the sample was placed in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA), frozen down in a mixture of isopropanol and dry ice and placed in the cryostat (Leica Microsystems Inc, Buffalo Grove, IL) to equalize the temperature. The sample was then mounted on the cutting stage with O.C.T. and 10–20 μm slices were taken in succession from the front of the brain to the rear of the brain. Slices were adhered onto glass slides (Fisher Scientific, Waltham, MA) and once dry were placed on an autoradiography cassette and exposed to the Storage Phosphor Screen (Molecular Dynamics, Sunnyvale, CA) for 24 h before analysis on a Phosphor Imager STORM 860 (Amersham Biosciences, NJ). Contrast of the autoradiography image was normalized with the injected dose to compare the intensity of particle accumulation within the brain tumor.
2.8. H&E and Immunohistochemistry (IHC)
Tissues for microscopic analysis were fixed overnight in 4% buffered formalin and transferred to 70% ethanol the next day. A Tissue-Tek VIP autoprocessor (Sakura, Torrance, CA) was used to process samples for paraffin-embedding. Tissue blocks were then sectioned to 4 μm sections mounted on glass slides then stained with ayer’s hematoxylin and eosin. Samples were processed for immunohistochemistry (IHC) with a goat anti-mouse PECAM-1 (CD31) primary antibody (1:1600; SC-1506, Santa Cruz Biotechnology, Santa Cruz, CA). All IHC was performed manually. Antigen retrieval was performed in a Decloaking Chamber (Biocare Medical, Concord, CA) with citrate buffer at pH 6.0, 125°C and pressure of 15 psi within 45 min. Incubation with the primary antibody was performed at room temperature overnight in a humidified chamber and normal horse serum was used for blocking. Biotinylated horse anti-goat (1:1000; Vector Labs, Burlingame, CA) was the secondary antibody used with a Vectastain ABC Kit Elite and a Peroxidase Substrate Kit DAB (both from Vector Labs), which were used for amplification and visualization of the signal, respectively. Tissues known to contain the assessed antigen were used as positive controls.
2.9. Statistical Methods
Values are presented as means ± S.E.M. Statistical analyses were conducted using GraphPad Prism (v6). For the statistical analysis of tumor accumulation of liposomes and micelles (Fig. 5a, 5c, 6a and 6b), two-way ANOVA corrected by Sidak’s multiple comparisons was performed. Other values were analyzed using unpaired t-test (two-tailed with Welch’s correction. A corrected P value of * <0.05 was considered significant.
Figure 5.

Blood clearance of 64Cu-liposomes (black circle) and 64Cu-micelles (gray circle) obtained from ROI analyses at 0, 3.5, 7 and 21 h post-injection. Curve was fit with a one phase decay (Yliposomes= 6.104exp−0.04206× (R2 = 0.8330) and Ymicelles= 6.432exp−0.04461× (R2 = 0.8167). (b) Radioactivity (%ID/g) of liposomes (black bar) and micelles (gray bar) in blood at 22 h post-injection.
Figure 6.

Quantification of liposomes (n = 6) and micelles (n = 5) in glioblastoma, obtained from ROI analysis (glioblastoma) of PET/MR images. (a) Tumor blood volume (TBV) and contralateral left brain blood volume (LBV) calculated by ROI analysis of glioblastoma (right brain) and contralateral striatum (left brain) from 64Cu-liposome (black bar) and -micelle (gray bar) injected rats. (b) Comparison of % vascular volume between two groups with different size of glioblastoma. (c) Blood radioactivity subtracted time activity curves of liposomes (round with dashed line) and micelles (square with dotted line). Data points represent 0, 3.5, 7, and 21 h post-injection. (d) Glioblastoma-to-background (BG) ratio of liposomes (black) and micelles (gray) in glioblastoma over contralateral left striatum, obtained from PET/MR images with blood radioactivity at 21 h (Statistical significance of (a) and (c) was determined by two-way ANOVA analysis corrected by Sidak’s multiple comparison test and that of (d was determined by an unpaired t test with Welch’s correction significance: * = P < 0.05 ** = P < 0.01 *** = P < 0.001, ****= P < 0.0001).
3. Results
3.1. Preparation of 64Cu-liposomes and -micelles
To facilitate post-labeling, a custom lipid-PEG-chelator conjugate is incorporated into the self-assembled liposomes and micelles. As illustrated in Fig. 1, liposomes with 0.5 mol% 6-BAT lipid and micelles with 2 mol% of dC18-1COI(P2k)-6-BAT were successfully prepared in 0.1 M ammonium citrate buffer (pH 5.5) and deionized water, respectively. The average mean diameter of the liposomes and micelles was 111.9 ± 5.7 and 19.6 ± 7.4 nm, respectively (Table 1). The Z-average particle size of the liposomes was about 6-fold greater than that of the micelles (Fig. 1). The zeta-potential of the liposomes and micelles was −15.6 ± 3.5 and −13.6 ± 1.4 mV under physiological pH, where the negative charge of micelles and liposomes results from PEG on the surface. 64Cu was efficiently incorporated into the 6-BAT chelator on both particles resulting in an 80 ± 19% radiolabeling yield, which is comparable to the previous reports (39, 40). The radiochemical purities of the liposomes and micelles measured by ITLC were above 98% after size-exclusion chromatography. The specific activities of the liposomes and micelles were 159 ± 50.1 μCi/mg (115.6 ± 36.4 μCi/μmollipid) and 75.3 ± 40.7 μCi/mg (559.8 ± 303.1 μCi/μmollipid), respectively. The specific activity of both particles was sufficient to evaluate the pharmacokinetics within the glioblastoma model.
Table 1.
Characterization of liposomes and micelles with particle size and zeta potential
| Liposomes | Micelles | |
|---|---|---|
| Z-average size (mean ± SD, nm)a | 111.9 ± 5.7 | 19.6 ± 7.4 |
| Zeta-potential (mean ± SD, mV)a | −15.6 ± 3.5 | −13.6 ±1.4 |
Average mean and standard deviation is calculated from two means of particles used for two in vivo experiments under physiological pH (7.3–7.5).
3.2. Physiochemical characterization of the 3HM
3HM have been thoroughly characterized using transmission electron microscopy (TEM) and DLS as reported previously (40). To extract the PEG shell thickness on the outer layer of 3HM, solution SAXS experiments were performed. Fig. 2 shows the solution SAXS profiles of 3HM with and without PEG750 attached to the micelle surface. A core-shell form factor model was used to fit the SAXS data and the parameters of best fit are listed in Table 2. Based on these data, the PEG750 chains form an outer layer with a thickness of ~0.8 nm. The DLS and SAXS gave slightly different values for particle diameter. Since the liposome diameter was measured using DLS, we cite the DLS value for comparison. The micelle diameter based on SAXS was ~18 nm.
Figure 2.

Solution SAXS results comparing micelles with and without PEG750 layer on the exterior of the micelle. The lines indicate best fit to the core-shell model. Data for dC18-1CW(P2k) have been offset vertically for clarity.
Table 2.
Core-shell parameters from model fitting of SAXS data in Fig. 2.
| Core radius (nm) | Shell thickness (nm) | |
|---|---|---|
| w/o P750 | 3.0 | 5.2 |
| with P750 | 3.0 | 6.0 |
3.3 In vivo PET/MR imaging
T1w MRI contrast (without injection of an exogenous contrast agent) was sufficient to visualize the glioblastoma lesion in the right brain (Fig. 3, lower row) and large blood vessels (white arrows in Figs. 3 & 4) in the tumor center. MR images in Fig. S1 demonstrated that intracranial injection of U87MG cells in the right brain resulted in a highly localized GBM within the right brain. The average tumor volume at 9 days after surgery (n = 6) was < 100 mm3 (50 ± 15 mm3) and 16 days after surgery (n = 5) was > 100 mm3 (154 ± 36 mm3) (Fig. S2).
Figure 3.

Coregistered PET/MR images (upper) and MR only images (lower) of the rat brain at 21 h post-injection of 64Cu-liposomes and 64Cu-micelles. Arrows indicate developed blood vessels in glioblastoma. Maximum and minimum color scale from PET images represent 1 and 0 %ID/cc, respectively and the size of the white scale bars in image is 2 mm.
Figure 4.

Coregistered PET/MR images of rat brain post injection of 64Cu-liposomes (upper row) and 64Cu-micelles (lower row). (a) From left to right, PET/MR images are acquired at 0, 3.5, 7.5 and 21 h after injection. (b) PET/MR images acquired at 21 hours after injection, from left (posterior) to right (anterior). Each image represents a 1 mm thick slice image of the glioblastoma lesion. Arrows indicate blood vessels. Maximum and minimum values of the color scale are 1.0 and 0.3 %ID/cc, respectively.
Co-registered PET/MR images obtained 21 h after injection of 64Cu–liposomes and –micelles depict the enhanced accumulation of both particles within the tumor as compared with the adjacent striatum in the left brain (Fig. 3). Accumulation of particles increased gradually from 0.5 to 21 h, with evident accumulation of micelles from the 3.5 h time point (Fig. 4a). The accumulation of 20-nm 64Cu-micelles was substantially greater than that observed for 110-nm 64Cu-liposomes.
Radioactivity associated with both of the 64Cu-labeled nanoparticles was first observed in the center of the tumor (3.5 h vs 21 h, Fig. 4a), reaching the periphery at later time points. 64Cu-liposomes were also observed to localize around large vessels within the tumors (white arrow, upper row in Fig. 4b). At 21 hours after injection, serial brain slices of the PET/MR images from posterior to anterior (Fig. 4b) also demonstrate that the liposome and micelle concentration remained greater in the tumor center than in the periphery. No significant differences in the relative intratumoral distribution were observed (Fig. S3).
3.4 Pharmacokinetics and biodistribution of liposomes and micelles in blood
The pharmacokinetics of liposomes and micelles in blood were measured from the ROI analysis of radioactivity in the cardiac chambers. The clearance of 64Cu-liposomes and 64Cu-micelles in the blood pool was fit by a one-phase decay curve. The half-clearance time of liposomes and micelles was 16.5 and 15.5 h, respectively (Fig. 5a). Radioactivity quantified for 64Cu-liposomes (2.36 ± 0.47 %ID/g, n = 6) and -micelles (2.29 ± 0.50 %ID/g, n = 5) from blood collected at 22 h after injection (Fig. 5b) was similar to the image-derived values (liposomes: 2.64 ± 0.16 %ID/cc, micelles: 2.74 ± 0.35 %ID/cc). The slightly lower values calculated for the image-derived estimates are expected due to partial volume effects.
3.5 Calculation of tumor blood volume (TBV) and left brain (striatum) blood volume (LBV)
It has been reported that tumor blood volume changes with tumor grade (48, 49). Indeed, cerebral blood volume (CBV) of C6 gliomas measured by a previous MR study increased by 15% compared to control brain tissue (50). Here, we segmented the study into liposomes and micelles. At early time points after intravenous administration, long-circulating nanoparticles were only detected in the blood pool and provided a tool for evaluating the TBV by dividing the tumor radioactivity by the blood radioactivity at the 0 h time point (51).
Within this study, the average TBV and LBV measured by liposomes and micelles were not significantly different (Fig. 6a). However, the blood volume in the contralateral left brain (striatum, LBV) was significantly lower than that obtained from the glioblastoma (TBV) irrespective of particle size (Fig. 6a). Finally, in the two tumor size groups, the percent vascular volume within the glioblastoma was similar (<100 mm3: 4.785 ± 1.385%, >100 mm3: 5.462 ± 1.085%, P = 0.3867) as shown in Fig. 6b.
3.6 Image analysis of glioblastoma and contralateral left brain
The size-dependent accumulation of the nanoparticles in glioblastoma is summarized in the time-activity curves (%ID/cc) (Fig. 6c). Blood-pool radioactivity within the tumor was subtracted from the total local radioactivity by equation 1. At 30 min after injection of the nanoparticles, the radioactivity of the liposomes (0.082 ± 0.018 %ID/cc) and micelles (0.217 ± 0.115 %ID/cc) within the tumor was not significantly different. Based on the image data, glioblastoma accumulation of the 64Cu-micelles was significantly higher than that of 64Cu-liposomes at 7 and 21 h after injection reaching a ratio of 1.9 times greater (Fig. 6c). The contralateral left brain (striatum) was used to estimate the background radioactivity, and the tumor to background ratio of the 64Cu-micelles (5.12 ± 1.54 fold) was significantly higher than that of 64Cu-liposomes (2.78 ± 1.38, Fig. 6d).
3.7 Biodistribution of liposomes and micelles
The biodistribution of the liposomes and micelles was then measured after perfusion of animals with Dulbecco modified eagle medium (DMEM) which was used to eliminate the remaining radioactivity contributed by the circulating nanoparticles (~2 %ID/g). The radioactivity within the glioblastoma-bearing right brain and left brain was gamma-counted without tumor dissection.
The increased accumulation of micelles within the right brain (containing the glioblastoma), as compared with liposomes, was validated by biodistribution. In Fig. 7a, the accumulation of micelles and liposomes was 0.0924 ± 0.0012 %ID/g (n = 3) and 0.0372 ± 0.012 %ID/g (n = 3, p = 0.0048), respectively, in the right brain bearing a small tumor. This compares with (0.261± 0.015 %ID/g, n = 3) and (0.140 ± 0.029 %ID/g, n = 2, p = 0.0086) for micelles and liposomes, respectively, in large xenograft. In addition, the accumulation of both liposomes (p = 0.0143) and micelles (p = 0.0075) was greater in larger xenografts relative to smaller.
Figure 7.

Biodistribution of 64Cu-liposomes (black bar) and -micelles (gray bar) in (a) right and (b) left brain. Right brain bears glioblastoma. Percent injected dose per gram (%ID/g) was obtained after perfusion of blood at 22 h post-injection of 64Cu-liposomes (n=6) and -micelles (n=5). Right bar graphs are differentiated by tumor size. (c) Biodistribution of 64Cu-liposomes (black bar, n = 6) and 64Cu-micelles (gray bar, n = 5) at 22 h post-injection. (Statistical significance of (a) and (b) was determined by two-way ANOVA analysis corrected by Sidak’s multiple comparison test and that of (c was determined by unpaired t test with Welch’s correction, significance: * = P < 0.05, ** = P < 0.01, *** = P < 0.001, ****= P < 0.0001).
Surprisingly, in the contralateral left brain, accumulation of the micelles was also increased relative to that of the liposomes and the accumulation further increased in advanced xenograft peaking at 0.0304 ± 0.00041 %ID/g (Fig. 7b).
The uptake of both nanoparticles in other organs (heart, lung, stomach, intestines, muscle, bone, liver and kidneys) was similar 22 h after injection; however, splenic uptake of 64Cu-micelles (1.39 ± 0.70 %ID/g, n = 5) was significantly lower in comparison to the 64Cu-liposomes (14.8 ± 2.5 %ID/g, n = 6, p < 0.0001, Fig. 7c). Intestinal radioactivity after the injection of liposomes was significantly higher, although the difference was only ~1%ID/g.
3.7. Autoradiography and Immunohistochemistry
Optical images (upper row, Fig. S5) and autoradiography from the same slides (lower row, Fig. S5) confirmed the finding of enhanced tumor radioactivity within the co-registered PET/MR images (Fig. 4a). Histological examination (Fig. 8a,b) with H&E (upper row) and with CD31 (lower row) depicts scattered large blood vessels apparent within the glioblastoma with greater frequency as compared with the surrounding tissue. No evidence of tumor was observed in the contralateral (left) brain.
Figure 8.

Immunohistochemistry of (a) glioblastoma bearing right brain and (b) contralateral left brain. Upper and lower images are from H&E and CD31, respectively. (CC: cerebral cortex, STR: striatum, T: tumor, and V: vessel)
4. Discussion
Applying methods for the synthesis of stable particles and PET labeling demonstrated in previous studies, here, we explore the accumulation of long-circulating liposomes and 3HM in glioblastoma using 64Cu-labeled drug carriers and the combination of PET and MRI. The PEGylation on the surface of the carriers provided a similar charge and facilitated studies of the enhanced permeability and retention of nanoparticles based on differences in their diameters. Although previous studies have demonstrated that vascular permeability is reduced in brain tumors compared to tumors within other organs, enhanced delivery to brain tumors with small nanoparticles has not been clearly demonstrated. Here, we demonstrate that the uptake of 20-nm 3HM is significantly greater than 110-nm liposomes in glioblastoma 7 hours after injection (Fig. 6c). Importantly, we observed that the micelles continued to accumulate over the period studied here, and therefore these small particles were not clearing from the lesion, even in the absence of a targeting moiety. The micelles were well distributed throughout the tumor, potentially providing an opportunity to effectively treat disease when a drug or radiotherapy is attached.
The average fold increase for liposome and 3HM accumulation in glioblastoma compared to background (left striatum) was 2.78 and 5.12, respectively (Fig. 6d). Although those values were lower than those measured for human glioblastoma, which has 13~19-fold higher accumulation of stealth liposomes vs. normal brain (52), the overall results demonstrate that liposomes and micelles enhanced accumulation in glioblastoma.
From the biodistribution data obtained after perfusion (Fig. 7), the greater accumulation associated with a greater EPR effect in an advanced xenograft (>100 mm3) was confirmed. Micelle accumulation was greater than that of liposomes regardless of the progression of the xenograft (Fig. 7a). Although the radioactivity in the left brain was ~10-fold lower than in the right brain (Fig. 7b), the accumulation in the normal left brain showed two significant effects associated with the adjacent disease. First, in the contralateral left brain, 3HM uptake increased with xenograft progression in the implanted right brain. The permeability of the contralateral brain could be affected by the pressure induced by the growing tumor or by cytokines and growth factors associated within tumor (53). Second, the 110-nm liposomal uptake in the left brain was similar (~ 0.008%ID/g) regardless of the glioblastoma diameter. Thus, the extravasation of 110-nm liposomes was limited by the vascular pore size cutoff but relatively small 20-nm micelles crossed the BBB.
A major advantage of the PET-MRI techniques applied here is the opportunity to simultaneously view anatomy and accumulation and estimate the PK and the local blood volume. Large discrete blood vessels were detected within the tumor center with MRI. At the time of injection, the presence of these large vessels is expected to enhance the local radioactivity due to the significant blood volume within these vessels. Accumulation of nanoparticles via the EPR effect is expected to be greater in the periphery, occurring over tens of hours and peaking at the later time points. Here, with the combination of MRI and PET, radioactivity in the tumor center was observed initially with a gradual increase at the periphery.
The extended circulation of nanoparticles in the blood is crucial for the extravasation through leaky vasculature and accumulation in tumors. In our previous PK studies of liposomes and micelles in a mouse model (40, 54), the half-life of 64Cu-liposomes and 64Cu-micelles was 18 and 25 h (one-phase decay), respectively. Here, we observed a shorter half-life for both particles in blood (t1/2 liposomes and micelles = 16.5 and 15.5 h). The observed circulation time was longer than 99mTc-labeled HYNIC-PEG liposomes previously studied in a rat model where only 52 %ID remained in the blood pool 4 h after injection (55). We assume that the reduced half-life observed here was due to differences in the vascular physiology between the two species. Here, the similar blood clearance of the nanoparticles in blood facilitated a direct comparison of the radioactivity in the tissues at the same time point.
When evaluating long-circulating nanoparticles, the blood volume can also be estimated by evaluating the radioactivity in the blood and tumor at the time of injection as calculated by a previously described radiometric method (51). Previous MR studies in the rat brain reported a relationship between blood volume and vessel size where approximately 15% of C6 gliomas demonstrated an increased cerebral blood volume as compared to gray matter, and 90% demonstrated an increased average vessel size (50). In a subsequent study, no correlation was found between blood vessel density and tumor progression in GBM (56). Here, we observed a 62 – 82% increase in the % vascular volume in the tumor as compared to the contralateral LBV (Fig. 6a) but the % vascular volume was not significantly different between small (<100 mm3) and large tumors (>100 mm3) (Fig. 6b). The vascular volume in the adjacent left brain also was not significantly changed with xenograft progression (Fig. S4). Immunohistochemistry (IHC) with a CD31 antibody demonstrated larger vessels (Fig. 8a, black arrow) in glioblastoma lesions, which were not observed in normal brain tissue (striatum, Fig. 8b, black arrow). Previous work also demonstrated large vessels in tumors larger than 4 mm (57). In addition, in our study MR images (arrows in Fig. 3 and Fig. 4a–b) resolved large vessels within glioblastoma lesions. The TBV and LBV results suggest that vascularization of glioblastoma increases the vascular volume in glioblastoma.
The biodistribution of both nanoparticles in organs such as the heart, lung, stomach, muscle, bone, liver and kidney was similar. As we observed in our previous study (40), the micelle accumulation was significantly lower in spleen than that observed with liposomes, which could ultimately reduce the treatment toxicity.
Recently, 3HM micelles were loaded with doxorubicin and prolonged drug bioavailability in circulation (42, 43), which may improve therapeutic efficacy and reduce splenic toxicity. Success in ongoing research with respect to loading or conjugating anticancer drugs to micelles could provide a promising method to treat glioblastoma (58, 59).
In conclusion, current GBM treatment includes invasive surgery, radiotherapy, and chemotherapy; however, drug delivery remains a major challenge. Here, we demonstrated that 3HM accumulate within glioblastoma to a significantly greater extent than 110-nm liposomes. PET/MR co-registration of brain images with multiple imaging modalities may facilitate the monitoring of disease progression and planning of treatment regimens.
Supplementary Material
Acknowledgements
Rat image (Fig. 1) is modified from http://zingous.com/rat-coloring-page-free-rat-color-sheet/. liposome figure is modified from images of Wikimedia Commons, the free media repository). We acknowledge the support of NIHR01CA103828, R01CA134659, R21EB016947 and the UC Davis Research Investments in Science and Engineering.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavanee WK. WHO classification of tumours of the central nervous system. 4. Lyon, France: IARC Press; 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preusser M, de Ribaupierre S, Wohrer A, Erridge SC, Hegi M, Weller M, Stupp R. Current Concepts and Management of Glioblastoma. Annals of Neurology. 2011;70(1):9–21. doi: 10.1002/ana.22425. [DOI] [PubMed] [Google Scholar]
- 3.Wen PY, Kesari S. Malignant gliomas in adults. New England Journal of Medicine. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 4.Zustovich F, Lombardi G, Della Puppa A, Rotilio A, Scienza R, Pastorelli D. A Phase II Study of Cisplatin and Temozolomide in Heavily Pre-treated Patients with Temozolomide-refractory High-grade Malignant Glioma. Anticancer Research. 2009;29(10):4275–9. [PubMed] [Google Scholar]
- 5.Brandes AA, Basso U, Reni M, Vastola F, Tosoni A, Cavallo G, Scopece L, Ferreri AJ, Panucci MG, Monfardini S, et al. First-line chemotherapy with cisplatin plus fractionated temozolomide in recurrent glioblastoma multiforme: A phase II study of the Gruppo Italiano Cooperativo di Neuro-Oncologia. Journal of Clinical Oncology. 2004;22(9):1598–604. doi: 10.1200/JCO.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 6.Zhang GB, Huang SY, Wang ZC. A meta-analysis of bevacizumab alone and in combination with irinotecan in the treatment of patients with recurrent glioblastoma multiforme. Journal of Clinical Neuroscience. 2012;19(12):1636–40. doi: 10.1016/j.jocn.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 7.Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS. Experience with irinotecan for the treatment of malignant glioma. Neuro-Oncology. 2009;11(1):80–91. doi: 10.1215/15228517-2008-075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WKA, Paleologos N, Nicholas MK, Jensen R, et al. Bevacizumab Alone and in Combination With Irinotecan in Recurrent Glioblastoma. Journal of Clinical Oncology. 2009;27(28):4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 9.Marx GM, Pavlakis N, McCowatt S, Boyle FM, Levi JA, Bell DR, Cook R, Biggs M, Little N, Wheeler HR. Phase II study of Thalidomide in the treatment of recurrent glioblastoma multiforme. Journal of Neuro-Oncology. 2001;54(1):31–8. doi: 10.1023/a:1012554328801. [DOI] [PubMed] [Google Scholar]
- 10.Puduvalli VK, Giglio P, Groves MD, Hess KR, Gilbert MR, Mahankali S, Jackson EF, Levin VA, Conrad CA, Hsu SH, et al. Phase II trial of irinotecan and thalidomide in adults with recurrent glioblastoma multiforme. Neuro-Oncology. 2008;10(2):216–22. doi: 10.1215/15228517-2007-060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. New England Journal of Medicine. 2014;370(8):699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, et al. Bevacizumab plus Radiotherapy-Temozolomide for Newly Diagnosed Glioblastoma. New England Journal of Medicine. 2014;370(8):709–22. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]
- 13.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncology. 2009;10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 14.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 15.Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 16.Oberoi RK, Parrish KE, Sio TT, Mittapalli RK, Elmquist WF, Sarkaria JN. Strategies to improve delivery of anticancer drugs across the blood–brain barrier to treat glioblastoma. Neuro-Oncology. 2015 doi: 10.1093/neuonc/nov164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhaskar S, Tian F, Stoeger T, Kreyling W, de la Fuente JM, Grazu V, Borm P, Estrada G, Ntziachristos V, Razansky D. Multifunctional Nanocarriers for diagnostics, drug delivery and targeted treatment across blood-brain barrier: perspectives on tracking and neuroimaging. Particle and Fibre Toxicology. 2010:7. doi: 10.1186/1743-8977-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilhelm I, Fazakas C, Krizbai IA. In vitro models of the blood-brain barrier. Acta Neurobiologiae Experimentalis. 2011;71(1):113–28. doi: 10.55782/ane-2011-1828. [DOI] [PubMed] [Google Scholar]
- 19.Yang H. Nanoparticle-Mediated Brain-Specific Drug Delivery, Imaging, and Diagnosis. Pharmaceutical Research. 2010;27(9):1759–71. doi: 10.1007/s11095-010-0141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu XY, Zhang XF, Wang XH, Li YX, Jing XB. Comparative study of paclitaxel physically encapsulated in and chemically conjugated with PEG-PLA. Polymers for Advanced Technologies. 2009;20(11):843–8. [Google Scholar]
- 21.Zensi A, Begley D, Pontikis C, Legros C, Mihoreanu L, Buechel C, Kreuter J. Human serum albumin nanoparticles modified with apolipoprotein A-I cross the blood-brain barrier and enter the rodent brain. Journal of Drug Targeting. 2010;18(10):842–8. doi: 10.3109/1061186X.2010.513712. [DOI] [PubMed] [Google Scholar]
- 22.Zensi A, Begley D, Pontikis C, Legros C, Mihoreanu L, Wagner S, Buechel C, von Briesen H, Kreuter J. Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. Journal of Controlled Release. 2009;137(1):78–86. doi: 10.1016/j.jconrel.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Miladi I, Le Duc G, Kryza D, Berniard A, Mowat P, Roux S, Taleb J, Bonazza P, Perriat P, Lux F, et al. Biodistribution of ultra small gadolinium-based nanoparticles as theranostic agent: Application to brain tumors. Journal of Biomaterials Applications. 2013;28(3):385–94. doi: 10.1177/0885328212454315. [DOI] [PubMed] [Google Scholar]
- 24.Huynh NT, Morille M, Bejaud J, Legras P, Vessieres A, Jaouen G, Benoit JP, Passirani C. Treatment of 9L Gliosarcoma in Rats by Ferrociphenol-Loaded Lipid Nanocapsules Based on a Passive Targeting Strategy via the EPR Effect. Pharmaceutical Research. 2011;28(12):3189–98. doi: 10.1007/s11095-011-0501-y. [DOI] [PubMed] [Google Scholar]
- 25.Steiniger SCJ, Kreuter J, Khalansky AS, Skidan IN, Bobruskin AI, Smirnova ZS, Severin SE, Uhl R, Kock M, Geiger KD, et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. International Journal of Cancer. 2004;109(5):759–67. doi: 10.1002/ijc.20048. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita Y, Saito R, Krauze MT, Kawaguchi T, Noble C, Drummond DC, Kirpotin DB, Park JW, Berger MS, Bankiewicz KS. Convection-enhanced delivery of liposomal doxorubicin in intracranial brain tumor xenografts. Targeted Oncology. 2006;1(2):79–85. [Google Scholar]
- 27.Brigger I, Morizet J, Aubert G, Chacun H, Terrier-Lacombe MJ, Couvreur P, Vassal G. Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. Journal of Pharmacology and Experimental Therapeutics. 2002;303(3):928–36. doi: 10.1124/jpet.102.039669. [DOI] [PubMed] [Google Scholar]
- 28.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nature Nanotechnology. 2011;6(12):815–23. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 29.Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3(5):703–17. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horowitz M, Blasberg R, Molnar P, Strong J, Kornblith P, Pleasants R, Fenstermacher J. Regional [Misonidazole-C-14 Distribution in Experimental Rt-9-Brain Tumors. Cancer Research. 1983;43(8):3800–7. [PubMed] [Google Scholar]
- 31.Levin VA, Freemandove M, Landahl HD. Permeability Characteristics of Brain Adjacent to Tumors in Rats. Archives of Neurology. 1975;32(12):785–91. doi: 10.1001/archneur.1975.00490540029003. [DOI] [PubMed] [Google Scholar]
- 32.Oberoi RK, Mittapalli RK, Elmquist WF. Pharmacokinetic Assessment of Efflux Transport in Sunitinib Distribution to the Brain. Journal of Pharmacology and Experimental Therapeutics. 2013;347(3):755–64. doi: 10.1124/jpet.113.208959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(8):4607–12. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nature Reviews Cancer. 2006;6(8):583–92. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 35.Popovic Z, Liu WH, Chauhan VP, Lee J, Wong C, Greytak AB, Insin N, Nocera DG, Fukumura D, Jain RK, et al. A Nanoparticle Size Series for In Vivo Fluorescence Imaging. Angewandte Chemie-International Edition. 2010;49(46):8649–52. doi: 10.1002/anie.201003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fox ME, Szoka FC, Frechet JMJ. Soluble Polymer Carriers for the Treatment of Cancer: The Importance of Molecular Architecture. Accounts of Chemical Research. 2009;42(8):1141–51. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nasongkla N, Chen B, Macaraeg N, Fox ME, Frechet JMJ, Szoka FC. Dependence of Pharmacokinetics and Biodistribution on Polymer Architecture: Effect of Cyclic versus Linear Polymers. Journal of the American Chemical Society. 2009;131(11):3842. doi: 10.1021/ja900062u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim HL, Lee SB, Jeong HJ, Kim DW. Enhanced tumor targetability of PEGylated mesoporous silica nanoparticles on in vivo optical imaging according to their size. Rsc Advances. 2014;4(59):31318–22. [Google Scholar]
- 39.Seo JW, Zhang H, Kukis DL, Meares CF, Ferrara KW. A Novel Method to Label Preformed Liposomes with 64Cu Positron Emission Tomography (PET) Imaging. Bioconjugate Chemistry. 2008;19(12):2577–84. doi: 10.1021/bc8002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong H, Dube N, Shu JY, Seo JW, Mahakian LM, Ferrara KW, Xu T. Long-Circulating 15 nm Micelles Based on Amphiphilic 3-Helix Peptide-PEG Conjugates. Acs Nano. 2012;6(6):5320–9. doi: 10.1021/nn301142r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong H, Shu JY, Dube N, Ma Y, Tirrell MV, Downing KH, Xu T. 3-Helix Micelles Stabilized by Polymer Springs. Journal of the American Chemical Society. 2012;134(28):11807–14. doi: 10.1021/ja3048128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dube N, Shu JY, Dong H, Seo JW, Ingham E, Kheirolomoom A, Chen P-Y, Forsayeth J, Bankiewicz K, Ferrara KW, et al. Evaluation of Doxorubicin-Loaded 3-Helix Micelles as Nanocarriers. Biomacromolecules. 2013;14(10):3697–705. doi: 10.1021/bm4010518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dube N, Seo JW, Dong H, Shu JY, Lund R, Mahakian LM, Ferrara KW, Xu T. Effect of Alkyl Length of Peptide-Polymer Amphiphile on Cargo Encapsulation Stability and Pharmacokinetics of 3-Helix Micelles. Biomacromolecules. 2014;15(8):2963–70. doi: 10.1021/bm5005788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong H, Lund R, Xu T. Micelle Stabilization via Entropic Repulsion: Balance of Force Directionality and Geometric Packing of Subunit. Biomacromolecules. 2015;16(3):743–7. doi: 10.1021/bm501659w. [DOI] [PubMed] [Google Scholar]
- 45.Ogihara NL, Weiss MS, Eisenberg D, Degrado WF. The crystal structure of the designed trimeric coiled coil coil-VaLd: Implications for engineering crystals and supramolecular assemblies. Protein Science. 1997;6(1):80–8. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shu JY, Tan C, DeGrado WF, Xu T. New design of helix bundle peptide-polymer conjugates. Biomacromolecules. 2008;9(8):2111–7. doi: 10.1021/bm800113g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee HB, Blaufox MD. Blood-Volume in the Rat. Journal of Nuclear Medicine. 1985;26(1):72–6. [PubMed] [Google Scholar]
- 48.Aronen HJ, Gazit IE, Louis DN, Buchbinder BR, Pardo FS, Weisskoff RM, Harsh GR, Cosgrove GR, Halpern EF, Hochberg FH, et al. Cerebral Blood-Volume Maps of Gliomas - Comparison with Tumor Grade and Histologic-Findings. Radiology. 1994;191(1):41–51. doi: 10.1148/radiology.191.1.8134596. [DOI] [PubMed] [Google Scholar]
- 49.vanDijke CF, Brasch RC, Roberts TPL, Weidner N, Mathur A, Shames DM, Mann JS, Demsar F, Lang P, Schwickert HC. Mammary carcinoma model: Correlation of macromolecular contrast-enhanced MR imaging characterizations of tumor microvasculature and histologic capillary density. Radiology. 1996;198(3):813–8. doi: 10.1148/radiology.198.3.8628876. [DOI] [PubMed] [Google Scholar]
- 50.Dennie J, Mandeville JB, Boxerman JL, Packard SD, Rosen BR, Weisskoff RM. NMR imaging of changes in vascular morphology due to tumor angiogenesis. Magnetic Resonance in Medicine. 1998;40(6):793–9. doi: 10.1002/mrm.1910400602. [DOI] [PubMed] [Google Scholar]
- 51.Engvall C, Ryding E, Wirestam R, Holtas S, Ljunggren K, Ohlsson T, Reinstrup P. Human cerebral blood volume (CBV) measured by dynamic susceptibility contrast MRI and Tc-99m-RBC SPECT. Journal of Neurosurgical Anesthesiology. 2008;20(1):41–4. doi: 10.1097/ANA.0b013e31815d4c70. [DOI] [PubMed] [Google Scholar]
- 52.Koukourakis MI, Koukouraki S, Fezoulidis I, Kelekis N, Kyrias G, Archimandritis S, Karkavitsas N. High intratumoural accumulation of stealth (R) liposomal doxorubicin (Caelyx (R)) in glioblastomas and in metastatic brain tumours. British Journal of Cancer. 2000;83(10):1281–6. doi: 10.1054/bjoc.2000.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Senger DR, Brown LF, Claffey KP, Dvorak HF. Vascular-Permeability Factor, Tumor Angiogenesis and Stroma Generation. Invasion & Metastasis. 1994;14(1–6):385–94. [PubMed] [Google Scholar]
- 54.Seo JW, Mahakian LM, Kheirolomoom A, Zhang H, Meares CF, Ferdani R, Anderson CJ, Ferrara KW. Liposomal Cu-64 labeling method using bifunctional chelators: poly(ethylene glycol) spacer and chelator effects. Bioconjugate Chemistry. 2010;21(7):1206–15. doi: 10.1021/bc100018n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dams ETM, Laverman P, Oyen WJG, Storm G, Scherphof GL, Van der Meer JWM, Corstens FHM, Boerman OC. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. Journal of Pharmacology and Experimental Therapeutics. 2000;292(3):1071–9. [PubMed] [Google Scholar]
- 56.Ricard C, Stanchi F, Rodriguez T, Amoureux MC, Rougon G, Debarbieux F. Dynamic Quantitative Intravital Imaging of Glioblastoma Progression Reveals a Lack of Correlation between Tumor Growth and Blood Vessel Density. Plos One. 2013;8(9):15. doi: 10.1371/journal.pone.0072655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deane BR, Lantos PL. The Vasculature of Experimental Brain-Tumors. 1. A Sequential Light and Electron-Microscope Study of Angiogenesis. Journal of the Neurological Sciences. 1981;49(1):55–66. doi: 10.1016/0022-510x(81)90188-x. [DOI] [PubMed] [Google Scholar]
- 58.Nghi N, Montagnese J, Rogers LR, Sher A, Wolansky L. Positron Emission Tomography-Magnetic Resonance Imaging in the Evaluation of Brain Tumors: Current Status and Future Prospects. Seminars in Roentgenology. 2014;49(3):275–89. doi: 10.1053/j.ro.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 59.Ertl-Wagner B, Ingrisch M, Niyazi M, Schnell O, Jansen N, Forster S, la Fougere C. PET-MR in patients with glioblastoma multiforme. Radiologe. 2013;53(8):682–90. doi: 10.1007/s00117-013-2500-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.