

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that affects motor neurons in the brain and spinal cord, resulting in paralysis of voluntary skeletal muscles and eventually death, usually within 2~3 years of symptom onset. The pathophysiology mechanism underlying ALS is not yet clearly understood. Moreover the available medication for treating ALS, riluzole, only modestly improves neurological symptoms and increases survival by a few months. Therefore, improved therapeutic strategies are urgently needed. In the present study, we investigated whether rosmarinic acid has a therapeutic potential to alleviate neurological deterioration in the G93A-SOD1 transgenic mouse model of ALS. Treatment of G93A-SOD1 transgenic mice with rosmarinic acid from 7 weeks of age at the dose of 400 mg/kg/day significantly extended survival, and relieved motor function deficits. Specifically, disease onset and symptom progression were delayed by more than one month. These symptomatic improvements were correlated with decreased oxidative stress and reduced neuronal loss in the ventral horns of G93A-SOD1 mice. These results support that rosmarinic acid is a potentially useful supplement for relieving ALS symptoms.
Keywords: Rosmarinic acid, ALS, neuroprotection, antioxidant
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the development of voluntary muscle paralysis that usually begins focally. This disease eventually leads to death, usually within 2~3 years of symptom onset [1,2]. Important pathological features of ALS include the selective neuronal loss of lower motor neurons in the spinal cord and upper motor neurons in the brain in the absence of sensory symptoms [3,4]. A subset of ALS cases have genetic causes, whereas over 90% of all ALS cases are sporadic [5,6].
Approximately 20% of all familial ALS cases are caused by mutations in the SOD1 (also called Cu/Zn SOD) gene [7,8,9]. The SOD1 enzyme is 153 amino acids long and converts the superoxide anion to hydrogen peroxide [7,8]. Transgenic mice overexpressing a mutant form of human SOD1 (G93A-SOD1) display various neurological deficits that resemble those of human ALS, including neuronal loss in the spinal cord [10]. ALS spinal cords show enhanced levels of lipid peroxidation, protein carbonylation, DNA oxidation, and peroxynitrite-mediated damage [11]. These results suggest that oxidative stress contributes to the pathophysiology of ALS. The definitive pathophysiological mechanisms underlying genetic and sporadic ALS cases are not yet clearly understood; however mitochondrial dysfunction [11,12,13,14], glutamate excitotoxicity [15,16,17], disrupted axonal transport [18,19], inflammation [5,20,21,22], and miRNA modifications and epigenetic mechanisms [23,24] have all been postulated to play a role. Currently, riluzole is the only clinically aviable drug for treating ALS; however, but riluzole only delays the progression of ALS symptoms [25,26,27,28]. Therefore, new curative and/or additive treatments for ALS are urgently needed.
Rosmarinic acid is a phenolic compound that is an ester of caffeic acid and 3,4-dihydroxyphenyllactic acid. It is found in plants, such as species of the Boraginaceae family and the subfamily Nepetoideae of the Lamiaceae family. Rosmarinic acid has a number of biological activities, including antiviral, antibacterial, anti-inflammatory and antioxidant [29]. The intraperitoneal administration of rosemary extract and of rosmarinic acid to presymptomatic G93A-SOD1 transgenic mice was recently reported to slightly delay the onset of motor dysfunction and to attenuate motor neuron degeneration [30]. However, the dose of rosmarinic acid used in their study was relatively low (0.13 mg/kg), so that it remains uncertain whether its weak therapeutic effects were due to the use of a low dose. Moreover, the temporal profiles of behavioral changes were not compared with those achieved with riluzole, making it difficult to interpret the therapeutic value of rosmarinic acid. In the present study, we compared the effects of high doses of rosmarinic acid (30 or 400 mg/kg/day) with those of riluzole on G93A-SOD1 transgenic mice with respect to behavioral, biochemical and histological parameters.
MATERIALS AND METHODS
Animals
Transgenic G93A-SOD1 mice carrying the G93A mutation in human SOD1 [10] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). G93A-SOD1 mice were maintained by crossing male G93A-SOD1 mice with female C57BL/J6×SJL F1 hybrid as previously described [31]. Mice were housed in pairs in a standard clear plastic cage with free access to food and water. The environment was temperature-controlled (23~24℃) and humidity-controlled (50~60%) and maintained under a 12 h light/dark cycle. All animals were handled in accordance with the animal care guidelines of the Ewha Womans University School of Medicine.
Rosmarinic acid and riluzole treatments
Treatments of G93A-SOD1 mice with rosmarinic acid and riluzole were carried out as previously described [31]. Drug treatments and behavioral assessments of the present study were conducted as a sister experiment of our previous study [31]. G93A-SOD1 mice were divided into four experimental groups according to their diet plans: lab chow alone (Tg-control; n=6 animals), lab chow containing rosmarinic acid (30 or 400 mg/kg/day) (Tg+RA30 and Tg+RA400; n=13 animals for each dose), and lab chow containing riluzole (35 mg/kg/day) (Tg+Rilu; n=8 animals). Lab chows containing supplements were prepared by grinding regular lab chow, mixing the lab chow powder (grindate) with rosmarinic acid or riluzole, remolding, and gamma-ray-irradiating the remodeled grindate. Control lab chow was prepared from regular lab chow by the same procedure. Lab chow mixtures and their control were prepared on a weekly basis. G93A-SOD1 mice were administered rosmarinic acid or riluzole from 7 weeks of age to 16 weeks of age. The amount of food consumed by each mouse was carefully monitored and the treatment doses of rosmarinic acid or riluzole were maintained as indicated.
Mortality test
At the terminal stage of symptoms, G93A-SOD1 mice become completely paralyzed and death follows within a few hours. The time of death was defined as the date on which this symptomatic sequence occurred.
Evaluation of behavioral performance
Hindlimb extension reflex test
The hindlimb extension reflex test was performed as previously described [31]. Briefly, mice were suspended by the tail, and hindlimb extension reflex deficits were scored from 0 to 2 as follows: 2, normal extension reflex in both hind limbs; 1.5, imbalanced extension in the hind limbs; 1.0, extension reflex in only one hindlimb; 0.5, the absence of any hindlimb extension; and 0, total paralysis.
Rota-rod test
The rota-rod test was performed as previously described [31]. Briefly, mice were placed individually on the rotating cylinder of a rota-rod apparatus (Dae-Jong Co. Inc., Seoul, Korea) revolving at a constant speed of 16 rpm. The motor coordination and balance of each mouse were assessed by measuring total riding time on the rotating cylinder. Each animal was given three trials, and the longest latency time before falling was recorded. An arbitrary cut-off time of 300 sec was applied.
Paw grip endurance test
The paw grip endurance (PaGE) test was performed as previously described [31]. The wire lid of the mouse cage was gently presented to a subject mouse to elicit gripping. Next, the lid was swiftly turned downwards such that the mouse held its own body against the pull of gravity by only its forelimbs and hindlimbs. The time that each mouse could successfully grip he lid was recorded. Each mouse was given up to three trials to hold onto the lid, and the longest latency time before falling was recorded. An arbitrary cut-off time of 90 sec was applied.
Grip strength test
The grip strength test was performed as previously described [31]. This test was developed as a supplemental method for the PaGE test. While the PaGE test evaluates the ability of a mouse to support its own body weight using only forelimbs and hindlimbs against gravity, body weight varies between individual mice, in part because of the heterogeneous C57BL/J6×SJL F1 hybrid genetic background. Therefore, the PaGE test is limited in its ability to accurately measure forelimb and hindlimb muscle strength of an individual mouse. To overcome this limitation, mice were presented with an irongrid with a fixed weight of 24 g. The irongrid was 5.5 cm×8 cm, and was constructed with an iron wire 1.0 mm in diameter. To elicit gripping, the irongrid was presented to each mouse. By lifting the grid up, the mice was required to hold its own body with its forelimbs and hindlimbs against the pull of gravity. The time that each mouse held onto the grid was recorded. Each mouse was given up to three trials to hold onto the grid for 10 sec, and the longest latency time was recorded.
Cell culture
The SH-SY5Y neuroblastoma cell line was cultured as previously described [31]. Briefly, SH-SY5Y cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL, Gaithersburg, MD, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco-BRL), penicillin (20 units/ml), and streptomycin (20 mg/ml) at 37℃ in a humidified incubator. Cells were maintained in a 95% air and 5% CO2 atmosphere.
The viability of SH-SY5Y against 800 µM hydrogen peroxide (H2O2) in the presence of rosmarinic acid (15 µg/ml), trolox (200 µM) or vitamin C (200 µM) in serum-free DMEM. Cell viability was determined using the WST-1 assay as previously described [31].
Histological analyses
Histological analyses of lumbar cords (L3~5) were performed as previously described [31,32]. Briefly, lumbar cords were surgically removed and post-fixed overnight in 4℃ in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Fixed cords were cut coronally into 40 µm-thick sections with a vibratome (Leica VT 1000S; Leica Instruments, Nussloch, Germany).
Cord sections were stained with 1% cresyl violet and the numbers of stained cells were determined using the TOMORO ScopeEye 3.6 system (Olympus, Japan) as previously described [31,32]. For immunohistochemistry, free-floating sections were blocked with 4% bovine serum albumin in PBST (PBS containing 0.1% Tween-20, pH 7.4) for 1 h. Sections were then incubated with anti-HNE (Alpha Diagnostic Intl. Inc., TX, USA) or anti-ChAT (Chemicon, Temecula, CA, USA) antibodies at 4℃ overnight. The sections were then washed with PBST and incubated with biotinylated secondary antibodies diluted 1:200 in PBST. Immunoreactive regions were visualized using an ABC Elite kit (Vector Laboratories; Burlingame, CA, USA). Anti-HNE stained sections were incubated with TRITC-conjugated secondary antibody (Sigma-Aldrich Inc., St. Louis, MO, USA) diluted 1:200 in PBST. After washing, the sections were mounted with VECTASHIELD® Mounting Medium (Vector Laboratories). Anti-ChAT stained sections were washed with PBST and incubated with biotinylated secondary antibody diluted 1:200 in PBST. Immunostained sections were visualized using an ABC Elite kit (Vector Laboratories, Burlingame, CA, USA). Images were captured with an Olympus BX 51 microscope equipped with a DP71 camera and DP-B software (Olympus Co., Tokyo, Japan). Fluorescence intensities, which reflect the extent of anti-HNE staining, were measured using TOMORO ScopeEye 3.6 software (Techsan Community, Seoul, Korea) as previously described [32].
Statistical analysis
Data were analyzed using GraphPad Prism 4 (GraphPad Software, San Diego, CA, USA). Two-sample comparisons were carried out using Student's t-test, while multiple comparisons were made using one-way ANOVA followed by the Newman-Keuls multiple range test. All data are presented as means±SEM.
RESULTS
Rosmarinic acid increases survival of G93A-SOD transgenic mice
The G93A-SOD1 transgenic mice (G93A-SOD1 mice) exhibited various neurological symptoms from 7 weeks of age. Some of untreated G93A-SOD1 mice died as expected at around 12 weeks of age. In contrast, both groups (30 or 400 mg/kg/day from 7 weeks of age) of rosmarinic acid-treated G93A-SOD1 mice survived longer and began to die at 13 and 15 weeks, respectively (Fig. 1A). At 16 weeks of age, the survival rate of untreated G93A-SOD1 mice was 0%, while the survival rates of rosmarinic acid-treated G93A-SOD1 mice (30 and 400 mg/kg/day) were 57% and 66%, respectively. None of riluzole-treated G93A-SOD1 mice had died by this stage. Thus, the survival of G93A-SOD1 mice was moderately extended by rosmarinic acid administration; however, it was lower than that of the riluzole-treated group. During the period of rosmarinic acid treatment (from 7 weeks to 16 weeks of age), the weekly body weight measurements of the G93A-SOD1 mice were slightly lower than those of the non-transgenic controls, whereas the body weights of rosmarinic acid-treated G93A-SOD1 mice were not significantly different from those of the untreated G93A-SOD1 mice or the riluzole-treated G93A-SOD1 mice (Fig. 1B).
Fig. 1. Rosmarinic acid extends the survival of G93A-SOD1 transgenic mice. (A) G93A-SOD1 transgenic mice treated with rosmarinic acid (30 or 400 mg/kg/day) from 7 weeks of age exhibited extended survival compared with untreated G93A-SOD1 mice. (B) Body weight changes of G93A-SOD1 mice during the administration of rosmarinic acid. Body weights were monitored weekly and are presented as percentages of mean body weight of each group at 7 weeks of age. Rosmarinic acid was administrated at 30 or 400 mg/kg/day to G93A-SOD1 mice from 7 weeks of age. Non-Tg, non-transgenic WT control; Tg-Con, G93A-SOD1 transgenic control; Tg+RA30 and Tg+RA400, G93A-SOD1 transgenic mice treated with rosmarinic acid of 30 mg/kg/day and 400 mg/kg/day, respectively; Tg+Rilu, G93A-SOD1 transgenic mice treated with 35 mg/kg/day of riluzole. Data are presented as means±SEM (n=6~13). **denotes differences from the Tg-control at 16 weeks at p<0.01. Two-way ANOVA and Bonferroni post hoc test were used.

Rosmarinic acid relieves motor function deficits in G93A-SOD1 mice
We next examined whether high doses of rosmarinic acid relieve motor function deficits in G93A-SOD1 mice. G93A-SOD1 mice were treated with rosmarinic acid (30 or 400 mg/kg/day) from 7 weeks of age to 16 weeks of age. Beginning at 8 weeks of age, a set of behavioral tests were carried out twice a week in the following order: rota-rod test, PaGE test, grip test, and hindlimb extension reflex test.
G93A-SOD1 control mice showed behavioral deficits in the hindlimb extension reflex test when beginning from 8 weeks of age; these symptoms progressively deteriorated over time. In contrast, G93A-SOD1 mice treated with rosmarinic acid (30 or 400 mg/kg/day) showed a delay of 4~5 weeks in the onset and progression of neurological symptoms compared with the G93A-SOD1 control mice (Fig. 2A). Rosmarinic acid was more effective at 400 mg/kg than at 30 mg/kg throughout the whole test period until 16 weeks of age, but it was slightly less effective than riluzole.
Fig. 2. Rosmarinic acid improves motor function deficits in G93A-SOD1 transgenic mice. (A~D) Motor function levels in the limb extension reflex test (A), rota-rod test (B), PaGE test (C), and grip strength test (D). Behavioral tests were performed from 7 weeks of age, twice a week described in Materials and Methods. Tests were performed in the sequence listed above. Non-Tg, non-transgenic WT control; Tg-Con, G93A-SOD1 transgenic control; Tg+RA30 and Tg+RA400, G93A-SOD1 transgenic mice treated with 30 mg/kg/day and 400 mg/kg/day rosmarinic acid, respectively; Tg+Rilu, G93A-SOD1 transgenic mice treated with 35 mg/kg/day riluzole. Data are presented as means±SEM (n=6~13). * and ** denote differences from the control at the indicated points at p<0.05 or p<0.01, respectively. Two-way ANOVA and Bonferroni post hoc test were used.

Beginning from 9 weeks of age, G93A-SOD1 control mice showed impaired performance while running at 16 rpm in the rota-rod test; moreover their performance progressively worsened over time. The administration of rosmarinic acid (30 or 400 mg/kg/day) to G93A-SOD1 mice delayed the onset and progression of neurological symptoms by 3~5 weeks compared with G93A-SOD1 control mice throughout the test period (Fig. 2B). Rosmarinic acid was more effective at 400 mg/kg than at 30 mg/kg until 14 weeks of age. However, no significant differences were observed between the two doses at 15~16 weeks of age; both doses were less effective than riluzole.
The paw grip endurance (PaGE) test requires mice to hold their own bodies against the pull of gravity with their hindlimbs by grasping a metal lid. In this test, G93A-SOD1 mice were impaired in their ability to hang onto the lid beginning from 9 weeks of age. Moreover, their performance progressively deteriorated over time. Administration of rosmarinic acid (400 mg/kg/day) delayed the onset and progression of neurological symptoms by 3~5 weeks during the test period, although its effects were weaker than those by riluzole (Fig. 2C). Moreover, 30 mg/kg rosmarinic acid was far less effective in improving behavioral deficits compared with 400 mg/kg.
In the grip strength test, mice were required to hold an iron grid (5.5 cm×8 cm) with a fixed weight of 24 g using forelimbs and hindlimbs, G93A-SOD1 mice showed an impaired ability to hold the grid, beginning at 10 weeks of age, with the holding time gradually decreasing over time. At 15 weeks, G93A-SOD1 mice were not able to properly hold the iron grid. Administration of rosmarinic acid (400 mg/kg/day) delayed the onset and progression of neurological symptoms by 5 weeks, although the effects of rosmarinic acid were slightly weaker than those of riluzole (Fig. 2D). The effects of 30 mg/kg rosmarinic acid were much less dramatic compared to the effects of 400 mg/kg.
Rosmarinic acid suppresses oxidative stress in the spinal cords of G93A-SOD1 mice
We next tested whether rosmarinic acid exerts neuroprotective effects against oxidative stress-induced cell death. SH-SY5Y cells treated with 800 µM H2O2 for 24 h were only 30% viabile, whereas the viabilities of H2O2-treated cells were improved to 38% and 57% in the presence of 15 µg/ml and 30 µg/ml of rosmarinic acid, respectively (Fig. 3A).
Fig. 3. Rosmarinic acid treatment suppresses oxidative stress in vitro and in the lumbar cords of G93A-SOD1 transgenic mice. (A) Rosmarinic acid treatment suppressed H2O2 (800 µM)-induced cytotoxicity in SH-SY5Y neuroblastoma cells. Cells were treated with 15 or 30 µg/ml rosmarinic acid (RA). Vitamin C (VitC) and trolox (TRX) were used at 200 µM. Cell viability was measured using the WST-1 assay after 24 h of treatment. Data points represent means±SEM (n=6). **denotes difference compared to untreated control at p<0.01. ##denotes difference compared to H2O2-treated cells at p<0.01. One-way ANOVA and Newman-Keuls post hoc test were used. (B~F) Photomicrographs showing anti-HNE-stained ventral horns of the lumbar cord of non-transgenic control mice (Non-Tg; B), G93A-SOD1 transgenic control mice (Tg-Con; C), G93A-SOD1 transgenic mice treated with 400 mg/kg/day rosmarinic acid (Tg+RA; D), and G93A-SOD1 mice treated with 35 mg/kg/day riluzole (Tg+Rilu; E). (F) Fluorescence levels in the ventral horns [circled area in (B), 600 µm in diameter] of the lumbar cord of each group were quantified. All animals were examined at 16 weeks of age. Scale bar; 500 µm. Data are presented as means±SEM (n=8~13). * and ** denote differences compared to non-transgenic control (NonTg) at p<0.05 and p<0.01, respectively. # and ## denote difference compared to Tg-CON at p<0.05 and p<0.01, respectively. One-way ANOVA and Newman-Keuls post hoc test were used.
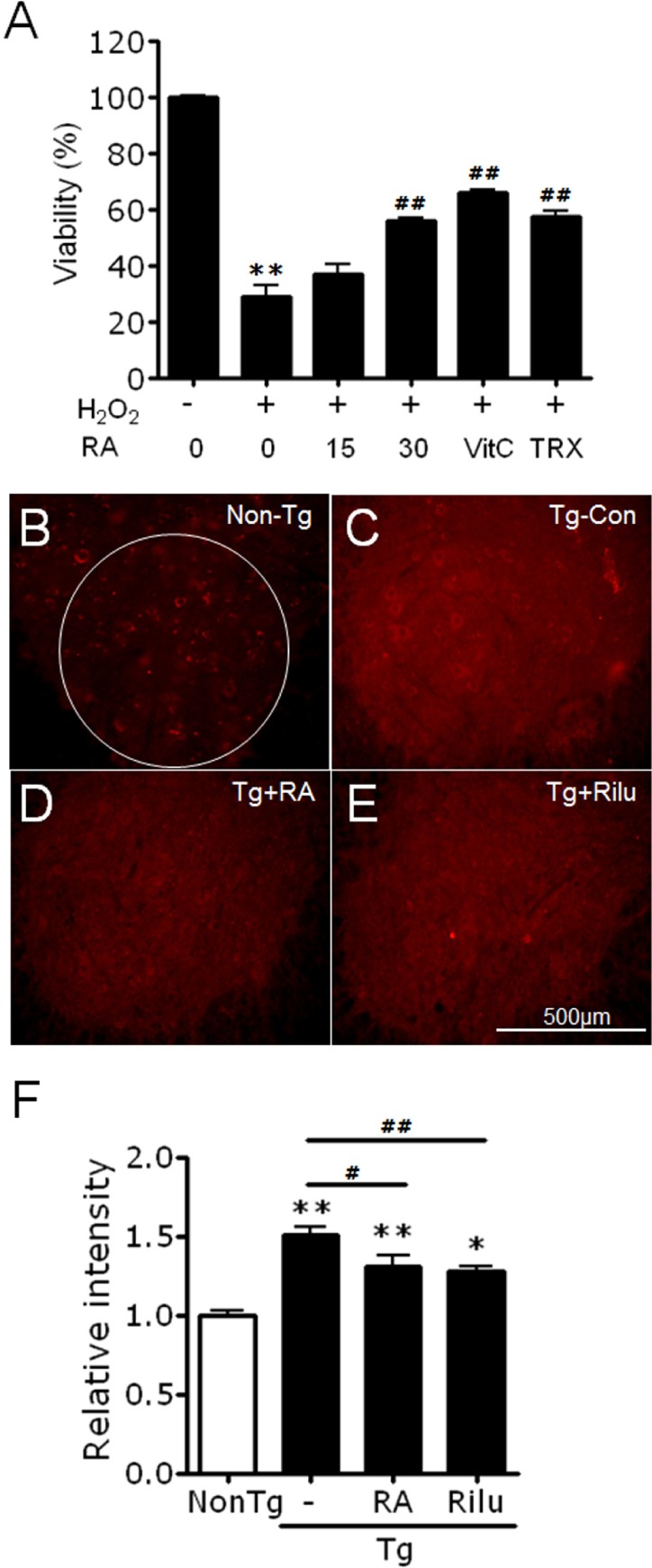
We next examined whether rosmarinic acid suppresses the accumulation of oxidative stress in the spinal cords of G93A-SOD1 mice. Spinal cord sections of untreated G93A-SOD1 mice that survived to 16 weeks of age were also prepared. Immunohistochemical staining with anti-HNE antibody showed that the levels of HNE (4-hydroxy-2-nonenal), a product of lipid peroxidation, were significantly reduced in the ventral horns of the spinal cords (L3~L5) of rosmarinic acid-treated (400 mg/kg/day) G93A-SOD1 mice compared with control G93A-SOD1 mice (Fig. 3B-F).
Rosmarinic acid reduces neuronal loss in the spinal cords of G93A-SOD1 mice
We examined whether rosmarinic acid protects against neuronal loss in the spinal cords of G93A-SOD1 mice. Cresyl violet-staining revealed that G93A-SOD1 mice at 16 weeks of age had only 28% of the number of cresyl violet-stained cells in the ventral horns of the spinal cords (L3~L5) compared with non-transgenic control mice. In contrast, rosmarinic acid-treated (400 mg/kg/day) G93A-SOD1 mice had an increased number of cresyl violet-stained cells compared with non-transgenic control mice, 55.7% of that that in non-transgenic control mice (Fig. 4A~D, and 4I).
Fig. 4. Rosmarinic acid increases the survival of neuronal cells in the lumbar cord of G93A-SOD1 transgenic mice. (A~H) Representative photomicrographs showing cresyl violet-stained lumbar L3~5 cords (A~D) or anti-ChAT-stained lumbar L3~5 cords (E~H) of non-transgenic control mice (Non-Tg; A, E), G93A-SOD1 transgenic control (Tg-Con; B, F), G93A-SOD1 mice treated with 400 mg/kg/day rosmarinic acid (Tg+RA; C, G), and G93A-SOD1 mice treated with 35 mg/kg/day riluzole (Tg+Rilu; D, H). (I, J) Quantification of the numbers of cresyl violet-stained cells (I) or anti-ChAT-stained cells (J) in the ventral horns of non-transgenic control mice (Non-Tg), G93A-SOD1 transgenic control (Tg-Con), G93A-SOD1 mice treated with 400 mg/kg/day rosmarinic acid (Tg+RA), and G93A-SOD1 mice treated with 35 mg/kg/day riluzole (Tg+Rilu). Cresyl violet-stained cells larger than 5 µm in diameter within the circled area in (A) (600 µm in diameter) were counted in both ventral horns of each section using TOMORO ScopeEye 3.6, as described in Materials and Methods. Anti-ChAT-stained cells were counted according to the same procedure. All animals were examined at 16 weeks of age. Scale bar; 500 µm. Data are presented as means±SEM (n= 8~13). **denotes difference compared to non-transgenic control (NonTg) at p<0.01. ##denotes difference compared to Tg-CON at p<0.01. One-way ANOVA and Newman-Keuls post hoc test were used.

Immunohistochemical analysis with anti-choline acetyltransferase (ChAT) revealed that untreated G93A-SOD1 control mice had 32.1% of the number of ChAT-positive cells in the ventral horns of the spinal cords (L3~L5) compared with non-transgenic control mice, while rosmarinic acid-treated (400 mg/kg/day) G93A-SOD1 mice exhibited an increased numbers of ChAT-positive neurons, 50.3% of that in non-transgenic control mice (Fig. 4E~H, and 4J).
DISCUSSION
We demonstrated that supplementation of rosmarinic acid inhibited neuronal loss in the ventral horns in G93A-SOD1 mice (Fig. 4). This inhibition was accompanied with improvement of neurological symptoms and extended survival of G93A-SOD1 mice (Fig. 1 and 2). Consistent with these findings, the administration of rosmarinic acid markedly reduced the accumulation of HNE in the spinal cords of G93A-SOD1 mice (Fig. 3). These results suggest that rosmarinic acid confers protective effects against neurological symptoms and neurodegeneration in G93A-SOD1 mice. This finding might be due to its ability of anti-oxidant activity. In fact, oxidative stress is an important cause of ALS [11,33,34,35,36]. However, previous trials with vitamin A [37], vitamin E [38,39], vitamin D [40,41,42], riboflavin [43], resveratrol [44], folic acid [45], N-acetylcysteine [46], and CoQ10 [47,48] have shown complicated results and yielded little or even no therapeutic benefit, antioxidants alone might be insufficient to inhibit the pathological progression of ALS.
We recently reported that SK-PC-B70M, an oleanolic-glycoside saponin fraction extracted from the root of Pulsatilla koreana [32], delays the progression of neurological deficits in G93A-SOD1 mice by 1~2 weeks [32]. In contrast, the present study showed that rosmarinic acid delayed the onsets of symptomatic mortality as well as neurologic symptoms by more than 1 month and these effects were produced by treatment with rosmarinic acid orally. Drug treatments and behavioral assessments of the present study were conducted as a sister experiment of our previous study [31], in which the survival rate of SOD1-G93A mice at 16 weeks of age was 25%, although survived SOD1-G93A mice at this stage expressed severe neurological symptoms. So it is possible that a certain portion of SOD1-G93A mice in our experimental condition may survive longer than 16 weeks of age. Even though rosmarinic acid was found to delay symptomatic onset by more than one month, rosmarinic acid was not more effective than riluzole. Nonetheless, rosmarinic acid may still have a potential as a supplement for treating ALS. Further studies will investigate whether combination treatments of rosmarinic acid and riluzole yield greater therapeutic benefits compared with riluzole treatment alone.
ACKNOWLEDGMENTS
This research was supported by a grant (2015R1A2A2A01003413) from the Ministry of Science, ICT and Future Planning, Republic of Korea.
References
- 1.Mulder DW. Clinical limits of amyotrophic lateral sclerosis. Adv Neurol. 1982;36:15–22. [PubMed] [Google Scholar]
- 2.Mitsumoto H, Chad DA, Pioro EP. Amyotrophic lateral sclerosis. New York, NY: Oxford University Press; 1998. [Google Scholar]
- 3.Pasinelli P, Houseweart MK, Brown RH, Jr, Cleveland DW. Caspase-1 and -3 are sequentially activated in motor neuron death in Cu,Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:13901–13906. doi: 10.1073/pnas.240305897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 5.Ferraiuolo L, Heath PR, Holden H, Kasher P, Kirby J, Shaw PJ. Microarray analysis of the cellular pathways involved in the adaptation to and progression of motor neuron injury in the SOD1 G93A mouse model of familial ALS. J Neurosci. 2007;27:9201–9219. doi: 10.1523/JNEUROSCI.1470-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis. 2009;4:3. doi: 10.1186/1750-1172-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radunović A, Delves HT, Robberecht W, Tilkin P, Enayat ZE, Shaw CE, Stević Z, Apostolski S, Powell JF, Leigh PN. Copper and zinc levels in familial amyotrophic lateral sclerosis patients with CuZnSOD gene mutations. Ann Neurol. 1997;42:130–131. doi: 10.1002/ana.410420124. [DOI] [PubMed] [Google Scholar]
- 8.Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- 9.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 10.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 11.Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48:629–641. doi: 10.1016/j.freeradbiomed.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 12.Son M, Puttaparthi K, Kawamata H, Rajendran B, Boyer PJ, Manfredi G, Elliott JL. Overexpression of CCS in G93A-SOD1 mice leads to accelerated neurological deficits with severe mitochondrial pathology. Proc Natl Acad Sci U S A. 2007;104:6072–6077. doi: 10.1073/pnas.0610923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vehviläinen P, Koistinaho J, Gundars G. Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:126. doi: 10.3389/fncel.2014.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peixoto PM, Kim HJ, Sider B, Starkov A, Horvath TL, Manfredi G. UCP2 overexpression worsens mitochondrial dysfunction and accelerates disease progression in a mouse model of amyotrophic lateral sclerosis. Mol Cell Neurosci. 2013;57:104–110. doi: 10.1016/j.mcn.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kosuge Y, Sekikawa-Nishida K, Negi H, Ishige K, Ito Y. Characterization of chronic glutamate-mediated motor neuron toxicity in organotypic spinal cord culture prepared from ALS model mice. Neurosci Lett. 2009;454:165–169. doi: 10.1016/j.neulet.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 16.Corona JC, Tovar-y-Romo LB, Tapia R. Glutamate excitotoxicity and therapeutic targets for amyotrophic lateral sclerosis. Expert Opin Ther Targets. 2007;11:1415–1428. doi: 10.1517/14728222.11.11.1415. [DOI] [PubMed] [Google Scholar]
- 17.Shaw PJ, Ince PG. Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J Neurol. 1997;244(Suppl 2):S3–S14. doi: 10.1007/BF03160574. [DOI] [PubMed] [Google Scholar]
- 18.LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascaño J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 19.De Vos KJ, Chapman AL, Tennant ME, Manser C, Tudor EL, Lau KF, Brownlees J, Ackerley S, Shaw PJ, McLoughlin DM, Shaw CE, Leigh PN, Miller CC, Grierson AJ. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum Mol Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 21.Browne SE, Yang L, DiMauro JP, Fuller SW, Licata SC, Beal MF. Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiol Dis. 2006;22:599–610. doi: 10.1016/j.nbd.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 22.Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet Neurol. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- 23.Donnelly CJ, Grima JC, Sattler R. Aberrant RNA homeostasis in amyotrophic lateral sclerosis: potential for new therapeutic targets. Neurodegener Dis Manag. 2014;4:417–437. doi: 10.2217/nmt.14.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paez-Colasante X, Figueroa-Romero C, Sakowski SA, Goutman SA, Feldman EL. Amyotrophic lateral sclerosis: mechanisms and therapeutics in the epigenomic era. Nat Rev Neurol. 2015;11:266–279. doi: 10.1038/nrneurol.2015.57. [DOI] [PubMed] [Google Scholar]
- 25.McGeer EG, McGeer PL. Pharmacologic approaches to the treatment of amyotrophic lateral sclerosis. BioDrugs. 2005;19:31–37. doi: 10.2165/00063030-200519010-00004. [DOI] [PubMed] [Google Scholar]
- 26.Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V Riluzole/ALS Study Group II. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol. 2002;249:609–615. doi: 10.1007/s004150200071. [DOI] [PubMed] [Google Scholar]
- 27.Lacomblez L, Bensimon G, Leigh PN, Debove C, Bejuit R, Truffinet P, Meininger V ALS Study Groups I and II. Long-term safety of riluzole in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:23–29. doi: 10.1080/146608202317576507. [DOI] [PubMed] [Google Scholar]
- 28.Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:191–206. [PubMed] [Google Scholar]
- 29.Petersen M, Simmonds MS. Rosmarinic acid. Phytochemistry. 2003;62:121–125. doi: 10.1016/s0031-9422(02)00513-7. [DOI] [PubMed] [Google Scholar]
- 30.Shimojo Y, Kosaka K, Noda Y, Shimizu T, Shirasawa T. Effect of rosmarinic acid in motor dysfunction and life span in a mouse model of familial amyotrophic lateral sclerosis. J Neurosci Res. 2010;88:896–904. doi: 10.1002/jnr.22242. [DOI] [PubMed] [Google Scholar]
- 31.Seo JS, Baek IS, Leem YH, Kim TK, Cho Y, Lee SM, Park YH, Han PL. SK-PC-B70M alleviates neurologic symptoms in G93A-SOD1 amyotrophic lateral sclerosis mice. Brain Res. 2011;1368:299–307. doi: 10.1016/j.brainres.2010.10.048. [DOI] [PubMed] [Google Scholar]
- 32.Seo JS, Leem YH, Lee KW, Kim SW, Lee JK, Han PL. Severe motor neuron degeneration in the spinal cord of the Tg2576 mouse model of Alzheimer disease. J Alzheimers Dis. 2010;21:263–276. doi: 10.3233/JAD-2010-091528. [DOI] [PubMed] [Google Scholar]
- 33.Boillée S, Cleveland DW. Revisiting oxidative damage in ALS: microglia, Nox, and mutant SOD1. J Clin Invest. 2008;118:474–478. doi: 10.1172/JCI34613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Apolloni S, Parisi C, Pesaresi MG, Rossi S, Carrì MT, Cozzolino M, Volonté C, D'Ambrosi N. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol. 2013;190:5187–5195. doi: 10.4049/jimmunol.1203262. [DOI] [PubMed] [Google Scholar]
- 36.Marrali G, Casale F, Salamone P, Fuda G, Caorsi C, Amoroso A, Brunetti M, Restagno G, Barberis M, Bertuzzo D, Canosa A, Moglia C, Calvo A, Chiò A. NADPH oxidase (NOX2) activity is a modifier of survival in ALS. J Neurol. 2014;261:2178–2183. doi: 10.1007/s00415-014-7470-0. [DOI] [PubMed] [Google Scholar]
- 37.Crochemore C, Virgili M, Bonamassa B, Canistro D, Pena-Altamira E, Paolini M, Contestabile A. Long-term dietary administration of valproic acid does not affect, while retinoic acid decreases, the lifespan of G93A mice, a model for amyotrophic lateral sclerosis. Muscle Nerve. 2009;39:548–552. doi: 10.1002/mus.21260. [DOI] [PubMed] [Google Scholar]
- 38.Ascherio A, Weisskopf MG, O'reilly EJ, Jacobs EJ, McCullough ML, Calle EE, Cudkowicz M, Thun MJ. Vitamin E intake and risk of amyotrophic lateral sclerosis. Ann Neurol. 2005;57:104–110. doi: 10.1002/ana.20316. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, O'Reilly ÉJ, Weisskopf MG, Logroscino G, McCullough ML, Schatzkin A, Kolonel LN, Ascherio A. Vitamin E intake and risk of amyotrophic lateral sclerosis: a pooled analysis of data from 5 prospective cohort studies. Am J Epidemiol. 2011;173:595–602. doi: 10.1093/aje/kwq416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long Kv, Nguyê˜n LT. Roles of vitamin D in amyotrophic lateral sclerosis: possible genetic and cellular signaling mechanisms. Mol Brain. 2013;6:16. doi: 10.1186/1756-6606-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gianforcaro A, Hamadeh MJ. Vitamin D as a potential therapy in amyotrophic lateral sclerosis. CNS Neurosci Ther. 2014;20:101–111. doi: 10.1111/cns.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camu W, Tremblier B, Plassot C, Alphandery S, Salsac C, Pageot N, Juntas-Morales R, Scamps F, Daures JP, Raoul C. Vitamin D confers protection to motoneurons and is a prognostic factor of amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35:1198–1205. doi: 10.1016/j.neurobiolaging.2013.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Moges H, Vasconcelos OM, Campbell WW, Borke RC, McCoy JA, Kaczmarczyk L, Feng J, Anders JJ. Light therapy and supplementary Riboflavin in the SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis (FALS) Lasers Surg Med. 2009;41:52–59. doi: 10.1002/lsm.20732. [DOI] [PubMed] [Google Scholar]
- 44.Mancuso R, del Valle J, Modol L, Martinez A, Granado-Serrano AB, Ramirez-Núñez O, Pallás M, Portero-Otin M, Osta R, Navarro X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics. 2014;11:419–432. doi: 10.1007/s13311-013-0253-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Chen S, Li L, Wang Q, Le W. Folic acid protects motor neurons against the increased homocysteine, inflammation and apoptosis in SOD1 G93A transgenic mice. Neuropharmacology. 2008;54:1112–1119. doi: 10.1016/j.neuropharm.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 46.Louwerse ES, Weverling GJ, Bossuyt PM, Meyjes FE, de Jong JM. Randomized, double-blind, controlled trial of acetylcysteine in amyotrophic lateral sclerosis. Arch Neurol. 1995;52:559–564. doi: 10.1001/archneur.1995.00540300031009. [DOI] [PubMed] [Google Scholar]
- 47.Molina JA, de Bustos F, Jiménez-Jiménez FJ, Gómez-Escalonilla C, García-Redondo A, Esteban J, Guerrero-Sola A, del Hoyo P, Martínez-Salio A, Ramírez-Ramos C, Indurain GR, Arenas J. Serum levels of coenzyme Q10 in patients with amyotrophic lateral sclerosis. J Neural Transm. 2000;107:1021–1026. doi: 10.1007/s007020070050. [DOI] [PubMed] [Google Scholar]
- 48.Kaufmann P, Thompson JL, Levy G, Buchsbaum R, Shefner J, Krivickas LS, Katz J, Rollins Y, Barohn RJ, Jackson CE, Tiryaki E, Lomen-Hoerth C, Armon C, Tandan R, Rudnicki SA, Rezania K, Sufit R, Pestronk A, Novella SP, Heiman-Patterson T, Kasarskis EJ, Pioro EP, Montes J, Arbing R, Vecchio D, Barsdorf A, Mitsumoto H, Levin B QALS Study Group. Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann Neurol. 2009;66:235–244. doi: 10.1002/ana.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]