

Abstract
Ubiquitin ligases are critical components of the ubiquitin proteasome system (UPS), which governs fundamental processes regulating normal cellular homeostasis, metabolism, and cell cycle in response to external stress signals and DNA damage. Among multiple steps of the UPS system required to regulate protein ubiquitination and stability, UBLs define specificity, as they recognize and interact with substrates in a temporally- and spatially-regulated manner. Such interactions are required for substrate modification by ubiquitin chains, which marks proteins for recognition and degradation by the proteasome, or alters their subcellular localization or assembly into functional complexes. UBLs are often deregulated in cancer, altering substrate availability or activity in a manner that can promote cellular transformation. Such deregulation can occur at the epigenetic, genomic, or post-translational levels. Alterations in UBL can be used to predict their contributions, affecting tumor suppressors or oncogenes in select tumors. Better understanding of mechanisms underlying UBL expression and activities is expected to drive the development of next generation modulators that can serve as novel therapeutic modalities. This review summarizes our current understanding of UBL deregulation in cancer and highlights novel opportunities for therapeutic interventions.
Introduction
Clearance of functional proteins limits their availability and activity and is critical for their regulation. This process is carried out in a timely manner by the proteasome ubiquitin system (UPS), which consists of ubiquitin ligases and accessory adaptor and regulatory components, all of which act in a concerted manner to tag proteins for ubiquitination in a spatially- and temporally-regulated manner. As a result, the ubiquitinated protein is often degraded by proteasomes, multi-subunit complexes that recycle proteins to their amino acid components. This critical post-translational modification is recognized as a key regulator of every cellular process under normal homeostatic conditions or in response to stress such as DNA damage, cell cycling, altered mitochondrial dynamics or cellular metabolism. Processing of proteins through ubiquitination also governs cell fate decisions including senescence, autophagy or cell death, and controls cellular proliferation and differentiation. Thus, UPS perturbations either increase or reduce availability of cellular regulatory proteins and perturb normal cellular activity, possibly resulting in pathological conditions, including cancer.
A key regulatory step in this process is substrate recognition by ubiquitin ligases (UBLs), an interaction that determines a substrate’s fate by modifying it with one or more ubiquitin moieties. Notably, not all ubiquitin-conjugation result in substrate degradation: that outcome is determined by ubiquitin chain topology, which in some cases governs a protein’s subcellular localization or its ability to participate in a large signaling complex.
The covalent conjugation of ubiquitin occurs through the formation of an isopeptide bond between lysine residues in both ubiquitin and the substrate. Ubiquitin can be attached to substrates as a monomer (monoubiquitination) or as ubiquitin chains (polyubiquitination). The latter adopt different topologies defined based on the position of respective lysines in ubiquitin, which enable linking of one ubiquitin molecule to another to form polyubiquitin. Ubiquitin K48-linked and K63-linked chains are the best studied: the former are associated with substrate degradation by the proteasome, while the latter are implicated in formation of signaling complexes.
Ubiquitination is carried out by sequential activity of ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), and ubiquitin ligases (E3s). Specifically, E3 ubiquitin ligases play a key role in this cascade by recruiting ubiquitin-loaded E2s, recognizing specific substrates, and then facilitating or directly catalyzing ubiquitin transfer to substrate lysine residues. E3 ligases can be classified into three families, of which only one (the HECT family) exhibits intrinsic enzymatic activity. The most abundant family includes a few hundred RING domain-containing E3 ligases, which structurally display a cysteine-histidine RING motif (the name is peculiarly derived from “really interesting new gene”).
This group relies on enzymatic activity of E2s to ubiquitinate proteins bound by RING ligases. RING E3 ligase proteins act as either single-molecule E3 ligases or as part of multi-subunit ubiquitin ligase complexes. Somewhat similar to RING ligases are U-box (UFD2 homology) ubiquitin ligases, which function as a scaffold to facilitate ubiquitin transfer from E2 to target proteins. The third group consists of few dozen proteins that display a HECT domain (for “homologous to E6AP carboxyl terminus”). HECT ligases can catalyze transfer of ubiquitin to a target substrate independent of E2 catalytic activity.
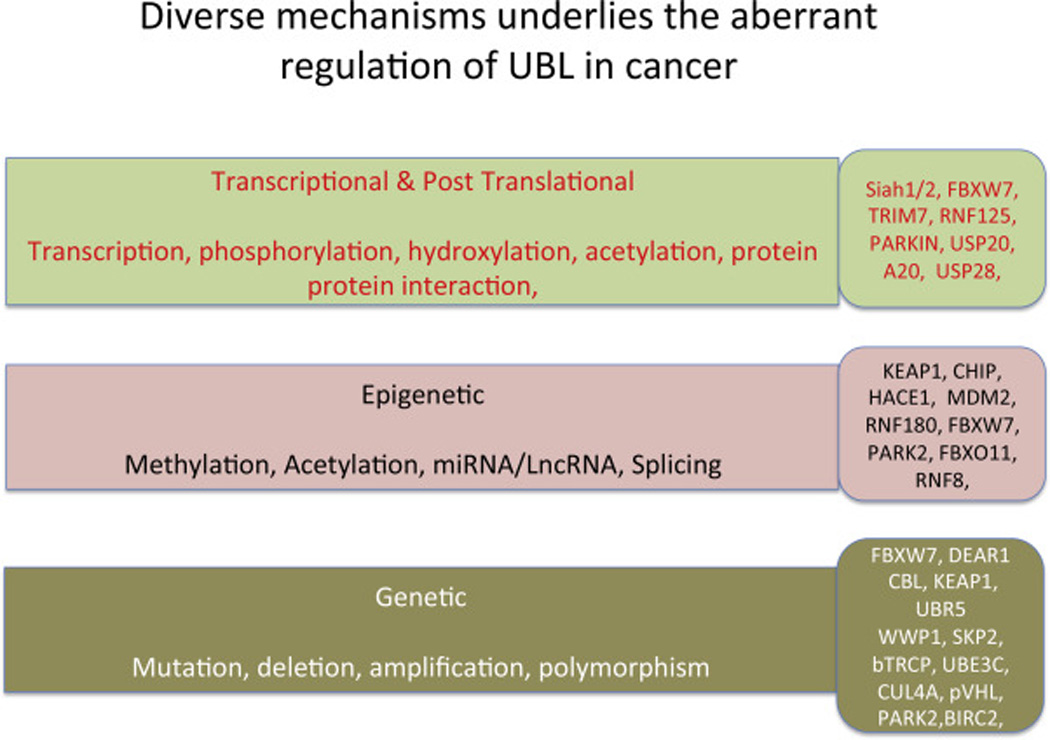
Growing evidence suggests that deregulated E3 ubiquitin ligases play a crucial role in development, progression and response to therapy of human cancers and thus could serve as promising therapeutic targets for anti-tumor drugs. Depending on the substrates they ubiquitinate and degrade, E3 ubiquitin ligases themselves can play tumor-promoting or tumor-suppressing roles. Here, we summarize key changes that underlie the genetic and epigenetic deregulation of E3 ubiquitin ligases in human cancer, including mutations, deletions, gene amplification, and altered transcription, as well as the activity of microRNAs and effects of post-translational modifications of the E3 ligases themselves (Figure 1).
Fig. 1.
Outline of key steps involved in the regulation of ubiquitin ligases (UBL) which are deregulated in cancer.
Genetic alterations: Mutations
Genetic mutations can affect ligase activity directly or indirectly. In terms of direct effects, mutations in the E3 ligases SPOP and FBXW7 (discussed below) attenuate their activity. Indirectly, mutations in upstream factors that regulate ligases (largely signal transduction components) can perturb the ability of an E3 to associate with a given substrate, which in turn alters substrate stability. Representative indirect regulators (discussed below) include the kinases GSK3 and BRAF.
Cullin-RING ubiquitin ligases (CRLs) are multi-subunit complexes that include a cullin scaffold protein, a RING domain protein (Rbx1 or Rbx2) that interacts with E2, and an adaptor protein that determines substrate specificity. Speckle-type POZ protein (SPOP) is the adaptor protein for the Cullin3/Rbx1 CRL and it selectively recruits substrates via its N-terminal MATH domain, while its BTB domain mediates dimerization and interaction with the scaffold CUL3 [1]. SPOP reportedly mediates degradation of several substrates including the steroid receptor co-activator SRC-3, androgen receptors (ARs), and estrogen receptors (ERs), among others [2,3,4]
SPOP is mutated in approximately 5–15% of patients with prostate cancer across ethnic and demographic backgrounds [5,6]. Prostate tumors with mutant SPOP show a distinct pattern of genomic alterations, suggesting that SPOP mutations could define a molecular subtype of prostate cancer [5]. SPOP mutations occur at regions encoding specific amino acid residues within the substrate-binding pocket and are predicted to attenuate substrate binding. In fact, mutated SPOP protein reportedly exhibits reduced binding to substrates such as SRC-3 [7] or AR [8]. Consistent with a proposed tumor suppressor role, expression of mutant SPOPs in prostate cancer cells promotes cell proliferation in vitro and tumor growth in immunodeficient mice [3]. SPOP is also one of the most frequently altered genes in endometrial cancer, based on somatic point mutations identified by large-scale exome sequencing [9, 10, 11, 12].
FBXW7 (F-box and WD repeat domain-containing 7) is an F-box family protein that functions as a substrate recognition component of the Skp-Cullin-F-box (SCF) E3 ubiquitin ligase [13]. It has a F-box domain, which associates with the SCF complex through interaction with Skp1 [14], and eight tandem repeats of the WD40 domain containing three critical arginine residues (R465, R479 and R505), which recognize a consensus phosphodegron motif within substrate proteins [15]. FBXW7 is generally viewed as a tumor suppressor in human cancers, and the SCF FBXW7 complex targets several well-known onco-proteins for ubiquitin-mediated, phosphorylation-dependent degradation, including c-Jun [16], c-Myc [17], Cyclin E [8], KLF5 [9], Mcl-1 [20], Notch [21] and mTOR [22]. Loss-of-function mutations in Fbw7 promote both hematopoietic and solid organ tumor formation in mice [23,24,25]. Furthermore, Fbw7 mutations have been identified in diverse human cancers, including cholangiocarcinoma [26], T cell acute lymphoblastic leukemia [27], cervical carcinoma [28], endometrial cancer [29], and colon cancer [30]. Notably, almost half of FBXW7 mutations found in cancers are missense mutations in regions encoding the arginine residues within the WD40 domain [31] critical for interaction with phosphorylated substrates. Thus, FBXW7 mutations associated with cancer appear to disrupt substrate recognition.
Mutations in key signaling factors, which often occur in cancer, can enhance or decrease ligase binding to substrates. For example, Glycogen Synthase Kinase 3 (GSK3) is an enzyme functioning in inhibitory phosphorylation of proteins regulated by FBXW7. Hence, mutation of a GSK3 phosphorylation site in c-Myc is seen in lymphoma [32], and mutation of a phosphorylation site in v-JUN priming it for GSK3 phosphorylation allows v-JUN to escape FBXW7 recognition in cancer [16], resulting in greater oncogenic activity. In addition to mutations in the above E3 ligases or their regulators, other ubiquitin ligases have been found mutated in cancer. They are summarized in Table 1.
Table 1.
Mutations
| RNF43 | somatic mutation in 18% (35/185) of colorectal adenocarcinomas and endometrial carcinomas 201 somatic mutation in 14% (8/57) of intraductal papillary mucinous neoplasms of the pancreas 202 Somatin mutation in 4.8% of MSS (microsatellite stable) and 54.6% of MSI (microsatellite instable) group of gastric cancers 203 mutation in 4% (1 out of 23) pancreatic carcinomas 204 mutated in 13.3% (2 out of 15) mucinous ovarian carcinoma 205; mutation in 9% (2/22) mucinous ovarian borderline tumors and 21% (6/29) mucinous ovarian carcinomas 206 mutation in 9.4% (5 out of 54) liver fluke–associated cholangiocarcinomas 207 |
| CBL | mutation in 10% of myeloid neoplasms 208, 209 mutation in Juvenile myelomonocytic leukemia 210 |
| BIRC2/BIRC3 | BIRC2/BIRC3 mutation in multiple myeloma 211 BIRC3 mutation in the splenic marginal zone lymphoma (SMZL) 212 BIRC3 mutation in the mantle cell lymphoma (MCL) 213 |
| PARK2 | mutation in the familial lung cancers 187 mutation in glioblastoma, lung cancer 42 |
| ZNRF3 | deletion or mutation in adrenocortical carcinoma 214, 215 |
| UBE3C | mutation in hepatocellular carcinoma 216 |
| KEAP1 | mutation in 12% lung squamous cell carcinoma 217, 218 mutation in clear-cell renal cell carcinoma 37 mutation in hepatocellular carcinoma 219 mutation in non-small cell lung carcinoma 220 mutation in 60% pulmonary papillary adenocarcinoma 221 mutation in 19% non-small cell lung carcinoma 222 |
| UBR5 | mutated in 18% mantle cell lymphoma 223 |
Genetic deletion
The von Hippel–Lindau protein (pVHL) forms a multiprotein ubiquitin ligase complex with elongins B and C, Cullin 2 and Rbx-1 (known collectively as the VBC-CR complex), and pVHL serves as the substrate recognition subunit in the complex. The best-known targets of ubiquitination by pVHL are the hypoxia-inducible factors (HIFs), the master transcription factors regulating the hypoxia response. HIF stabilization due to VHL loss or mutation is a key oncogenic event for VHL disease, a dominantly inherited familial cancer syndrome characterized by the development of hemangioblastomas, clear cell renal cell carcinoma (ccRCC), pheochromocytoma and pancreatic neuroendocrine tumors. Increased HIF stability also underlies development of certain sporadic tumors, including ccRCC and hemangioblastomas [33].
Patients with VHL disease or sporadic ccRCC harbor a single mutant allele of the VHL gene, and inactivation or loss of the wild-type allele is required for tumor development [34,35]. ccRCC is characterized by a very high frequency of biallelic VHL inactivation with a reported incidence up to 91% [33]. Over 90% of ccRCC cases show loss of heterozygosity (LOH) on chromosome 3p, where VHL is located [36, 37]. In one study of ccRCC, LOH at 3p was found in 94% of 240 tumor specimens [37]. ccRCC patients with LOH at 3p usually exhibit inactivation of the remaining VHL allele due to somatic mutations or promoter methylation. Among the somatic VHL mutations found in sporadic ccRCC, over 50% are frameshift or nonsense mutations [33] predicted to promote loss of function.
PARK2 encodes the ubiquitin ligase PARKIN, which consists of a C-terminal ubiquitin-like (UBL) domain and an N-terminal RING-IBR-RING motif [38]. PARK2 genetic alterations are common across many human cancers as well as in hereditary Parkinson’s disease [39, 40, 41]. PARK2 is mutated or deleted in cancers, with copy number loss being the primary genetic alteration [39, 40, 42]. PARK2 is a potential tumor suppressor gene at chromosome 6q25-q27 and is often lost in human cancers [43, 44, 45]. Analysis of approximately 5,000 tumor genomes shows that PARK2 deletions were the fourth most significant deletion among 70 significantly recurrent deleted regions across the entire data set [40]. Focal PARK2 deletions have been identified in 11% of tumors across all lineages, and loss of the entire chromosome arm occurred in 19% of samples, resulting in an overall 30% PARK2 deletion [40]. Specifically, PARK2 deletion occurs in various cancers, including serous ovarian, bladder and breast cancer [40], colorectal cancer [46], glioblastoma [42], lung cancer [47], and ovarian cancer 45. Other ubiquitin ligases reportedly deleted in various cancers include FBXW7, KEAP1, CBL, BIRC2/3 and DEAR1 and are listed in Table 2.
Table 2.
Deletions
| BIRC2/BIRC3 | deletion in multiple myeloma 211 |
| DEAR1 | deletion in breast cancer 224 |
| ZNRF3 | deletion in adrenocortical carcinoma 214, 215 |
| KEAP1 | deletion in non-small cell lung carcinoma 220 |
| CBL | deletion in non-small cell lung cancer 225 |
| FBXW7 | deletion in uterine serous carcinoma 12 deletion in 4% Will’s tumor 226 deletion in esophageal squamous cell carcinoma 227 deletion in gastric cancer 228 deletion in colorectal cancer 229 |
Promoter hypermethylation
DNA methylation of promoter regions is a key epigenetic event that underlies transcriptional deregulation of cancer-associated genes. Examples include deregulation of BRCA1, which is associated with susceptibility to breast and ovarian cancers. BRCA1 plays a key role in the DNA damage response, DNA repair, cell cycle regulation, chromatin remodeling, and transcriptional regulation. BRCA1-deficient cells display significant genomic instability and sensitivity to genotoxic agents, which are believed to underlie tumor development enhanced by BRCA1 deficiency.
BRCA1 is a RING finger ubiquitin ligase that forms a functional complex with BRCA1-associated RING domain protein 1 (BARD1) 48. The BRCA1/BARD1 heterodimer can ubiquitinate substrates such as H2A, H2B, H3, H4, H2AX, CtIP, estrogen receptor alpha, RNAPII and TFIIE 49, 50, 51, 52, 53, 54. How BRCA1’s function as a tumor suppressor is linked to its ubiquitin ligase activity remains controversial. Cancer-predisposing mutations within its RING domain were shown to abolish its E3 ligase activity, and these mutants cannot rescue cell cycle checkpoint and γ-radiation hypersensitivity of the BRCA1-null human breast cancer line HCC1937 55, suggesting a requirement for BRCA1 ubiquitin ligase activity in γ-radiation protection. In a mouse breast cancer model, mice harboring a known clinical mutant (C61G) of BRCA1 that disrupts its E3 activity and interaction with BARD1 showed the genomic instability and tumor development similar to the BRCA1-null mice, suggesting the essential role of BRCA1 ubiquitin ligase activity in tumor suppression 56. In contrast, mice harboring a synthetic BRCA1 mutation (I26A) that abrogates E3 ligase activity do not develop tumors to a similar degree as do wild-type mice 57, suggesting that BRCA1 ligase activity is not required for tumor suppression in these tumor models. In addition to ubiquitin ligase activity, BRCA1 has been shown to function as a scaffold for multiple protein complexes that regulate diverse activities including DNA damage signaling, homologous recombination, cell cycle checkpoint control and transcriptional regulation. It is likely that both the protein-protein interaction and ubiquitin ligase activity of BRCA1 function in tumor suppression.
BRCA1 and BRCA2 are distinct tumor suppressor genes: both function in DNA damage repair, but they are structurally unrelated and BRCA2 has no RING domain. Carriers of BRCA1 or BRCA2 mutations have a significantly increased risk of developing breast cancer (up to 81%) and ovarian cancer (up to 39%) 58, 59, 60. BRCA1 and BRCA2 mutations are seen in 20% of inherited breast cancer but are rare in sporadic breast and ovarian cancers. BRCA1 can also be lost by gene deletion or promoter hypermethylation 61. The incidence of BRCA1 promoter methylation in breast cancer tissues is significantly higher (up to 82.1%) than in non-cancerous tissues 62, 63, 64, 65, and gene methylation status is inversely correlated with BRCA1 mRNA expression 63, 65, 66. BRCA1 promoter methylation is also correlated with decreased overall survival and disease-free survival for triple-negative and basal-like breast cancer 62, 66. Methylation of promoters of upstream UBLs or cooperating factors governs expression of genes including HACE1, RNF180, CHIP, KEAP1 and PARK1. These are listed in Table 3.
Table 3.
Promoter hypermethylation
| KEAP1 | promoter methylation in papillary thyroid carcinoma 230 promoter methylation in non-small cell lung carcinoma 220 |
| RNF180 | promoter methylation in gastric cancer 231, 232 |
| CHIP | promoter methylation in colorectal cancer 233 |
| HACE1 | promoter methylation in hepatocellular carcinoma 234 promoter methylation in sporadic Wilms’ tumor 235 promoter methylation in colorectal cancer 236 promoter methylation in gastric cancer 237 |
| PARK2 | promoter methylation in leukemia 238 |
Gene amplification
While methylation often silences genes that control expression or activity of oncogenes, gene amplification is often associated with ligases or ligase regulatory factors that regulate tumor suppressor genes, thereby limiting their availability.
One such case is relevant to MDM2 (murine double minute 2), a RING finger E3 originally discovered at a genomic locus amplified on double minute chromosomes in transformed mouse NIH-3T3 fibroblasts 67. MDM2 consists of an N-terminal p53-binding domain, a central region that contains a nuclear localization sequence (NLS), a nuclear export sequence (NES), an acidic domain and zinc finger, followed by a C-terminal RING finger domain. MDM2 has a number of substrates, of which the best characterized is the tumor suppressor p53. MDM2 negatively regulates p53 degradation, affects its nuclear export, transcriptional activity or translation 68, 69, 70, 71, 72.
MDM2 overexpression is observed in a variety of human tumors of distinct tissue origin, such as sarcoma, glioma, leukemia, melanoma, lung cancer and breast cancer 73, 74. High MDM2 expression levels decrease p53 protein levels and activity, thereby increasing cancer initiation and progression. The fact that MDM2 overexpression and p53 mutation in human cancers are usually mutually exclusive 74 highlights distinct mechanisms to limit p53 activity. MDM2 may also have oncogenic functions independent of p53 75.
Increased MDM2 expression in human tumors is caused primarily by gene amplification. The human MDM2 gene is located on chromosome 12 (12q14–15), and its amplification is observed in colon cancer 76, 77, gastric cancer 78, 79, leukemia, breast cancer 80, glioblastoma 81, neuroblastoma 82, leukemia 83, and sarcoma 84. Other UBLs subject to genetic amplification include SKP2, CUL-4A, SMURF1, WWP1 and are summarized in Table 4.
Table 4.
Gene amplification
| WWP1 | WWP1 gene had copy number gain in 15 of 34 (44%) xenografts and cell lines from prostate cancer and 15 of 49 (31%) clinical prostate cancer samples 239 A copy number gain of WWP1 was found in 41% (17/41) of primary breast tumors. In a panel of cDNA from primary breast tumors and normal tissues, expression of WWP1 in tumors is significantly higher than that in normal tissues 240 |
| SMURF1 | amplification in 4.2% pancreatic cancer 241 amplification in pancreatic adenocarcinoma 242 |
| CUL-4A | amplification in breast cancer 243 amplification in hepatocellular carcinoma 244 amplification in pleural mesothelioma 245 amplification in esophageal squamous cell carcinoma 246 |
| SKP2 | amplification of SKP2 in 38% (31/82) of myxofibrosarcoma 122 amplification of SKP2 gene in 23 of 50 (46%) esophageal squamous cell carcinoma (ESCC) tumors 121 amplification in non-small cell lung cancer (NSCLC) 120 amplification in glioblastoma 247 |
Gene polymorphisms
A common single nucleotide polymorphism (SNP) located in the second intronic promoter (P2) of MDM2 constitutes a T to G transversion, termed SNP309 (SNP309T>G; rs2279744) due to its position 309 bps downstream of MDM2 exon 1. The G allele (SNP309G) extends a binding site for the transcription factor Sp1, enhancing MDM2 transcription 85. Transgenic mice carrying the SNP309G/G allele are more prone to tumor development than those carrying the SNP309T/T allele 86. In addition, the SNP309G allele is associated with susceptibility to a variety of human cancers 87, 88, 89, 90.
A less common MDM2 polymorphism (SNP285G>C; rs117039649) is located 24 bps upstream of SNP309 in the same MDM2 intronic promoter (P2). Like SNP309, SNP285 is located within a predicted Sp1 binding site; however, the presence of the SNP285C-allele reduces Sp1 binding affinity for the MDM2 promoter, as compared to the SNP285G allele, and is thus associated with reduced cancer risk 91, 92.
The well-studied F-box protein β-TrCP (beta-transducin repeat-containing protein) contains an N-terminal F-box motif and C-terminal substrate-binding WD-40 repeats, which recognize substrates phosphorylated within the consensus DSGXXS degron 93. β-TrCP targets degradation of many substrates including the tumor suppressor protein IkappaB 94, FOXO3 95, and REST 96. Targeted β-TrCP overexpression in mouse mammary gland promotes breast tumor development 97. β-TrCP is upregulated in several types of cancers including colorectal cancer 98, pancreatic cancer 99 and hepatoblastoma 100. A 9-bp (AACAGTGGA) ins/del polymorphism (rs16405) in the 3’-untranslated region (UTR) of β-TrCP is associated 101 with altered β-TrCP mRNA expression: β-TrCP transcript levels in hepatocellular carcinoma (HCC) tissues harboring homozygous 9N ins/ins were 4-fold and 7-fold higher, respectively, than tissues with heterozygous 9N ins/del and homozygous 9N del/del. The presence of the 9-bp insertion allele is proposed to disrupt miR-920 binding to the β-TrCP 3’-UTR, increasing β-TrCP mRNA levels and conferring susceptibility to HCC. However, the β-TrCP 9N ins/del polymorphism is not associated with susceptibility to ovarian 102 or breast cancer 103.
The rs6788895 SNP is located in an intronic region of the ubiquitin ligase SIAH2 and associated with estrogen receptor-positive breast cancer in Chinese 104 or Japanese 105 patients. SNPs rs2714805 and rs2255137 in FBXW7 intron 2 are associated with breast cancer risk 106. It remains to be determined whether or how polymorphisms of SIAH2 or FBXW7 alter their expression or activity.
Transcriptional regulation
In addition to the above mechanisms, UBLs are also subject to transcriptional regulation. S-phase kinase-associated protein 2 (Skp2) belongs to the F-box protein family and is the substrate-recognizing subunit of the SCF Skp2 E3 ligase, which consists of Skp1, Cul-1 (Cullin-1), F-box protein Skp2, and Rbx1. The cell cycle inhibitor p27 is the most well-characterized Skp2 substrate and is a physiological Skp2 target 107, 108. Other Skp2 substrates include p57 109, and p21 110, FOXO1 109, among others. Skp2 is overexpressed in a variety of human cancers, including lymphomas, prostate cancer 111, colorectal cancer 112, melanoma 113, non-small cell lung cancer 114, gastric cancer 115, pancreatic cancer 116, and breast cancer 117. Targeted overexpression of SKP2 in the mouse prostate induces hyperplasia, dysplasia, and low-grade carcinoma 118, whereas Skp2 knockout inhibits tumor development in a PTEN/P53 mouse prostate cancer model 119. Thus, SKP2 is believed to function as an oncogenic protein.
Amplification of the Skp3 locus on chromosome 5p13 is reported in several tumor types including non-small cell lung cancer (NSCLC) 120, esophageal squamous cell carcinoma (ESCC) 121, myxofibrosarcoma 122, melanoma 113, and glioblastoma 123. At a different regulatory layer, Skp2 transcription is regulated by other oncogenic factors including E2F1 124, Sp1, Elk-1 125, NF-κB 126, Myc 127, 128, and STAT3 129. Skp2 expression is also upregulated by phosphoinositol 3-kinase (PI3K)/Akt signaling 130, 131.
Transcriptional silencing of the gene that encodes the ubiquitin ligase RNF125 is associated with resistance of melanoma to vemurafenib 132, one of the lead BRAF inhibitors used in the clinic. MITF and SOX10 have been identified as upstream regulators of RNF125 transcription, and downregulation of both in drug resistant tumors decreases RNF125 expression, in turn upregulating JAK1, a RNF125 substrate, and promoting concomitant upregulation of receptor tyrosine kinases. The latter include EGFR and AXL, which are also implicated in melanoma resistance to vemurafenib. Notably, RNF125 is deregulated genetically and transcriptionally in other tumor types, including pancreatic and colorectal cancers, indicating that diverse mechanisms govern its regulation and function in cancer.
Regulation by microRNAs
It is now appreciated that miRNAs play an important role in regulating gene expression 133. miRNAs bind to the 3’UTR of mRNAs, destabilizing them or inhibiting their translation. FBXW7 expression is regulated by multiple miRNAs including miR-27a 134, 135, miR-223 136, 137, 138, miR-25 139, miR-92a 140, miR-182 and miR-503 141. An inverse correlation of miR-223 and FBXW7 expression is observed in several cancers including T-cell acute lymphoblastic leukemia 142, gastric cancer 138 and esophageal squamous cell carcinoma 137. miR-223 overexpression downregulates FBXW7 expression 136, 137, 138, 143, whereas inhibition of miR-223 upregulates FBXW7 protein levels 136. miR-223 represses activity of a luciferase reporter construct containing the FBXW7 3-UTR; in contrast, mutation of predicted miR-223 binding site within the FBXW7 3’-UTR relieves repression of reporter activity 136. These results support direct targeting of the FBXW3 3’-UTR by miR-223.
Several microRNAs reportedly control UBL expression and activity. For example, Mir21 regulates expression of the F-box protein FBXO11 144. RNF8 is a RING-finger E3 ligase recruited to DNA damage sites and required for assembly of repair proteins 145, 146. Mir214 regulates RNF8, and is therefore expected to impact chromosomal stability in ovarian cancer 147. Notably, UBLs also regulate microRNA expression, as demonstrated for the ligase TIM65, which regulates miRs by ubiquitinating the protein known as trinucleotide repeat-containing 6 (TNRC6), a component of the RISC complex 148.
Regulation by alternative splicing
Alternative splicing includes or excludes specific exons from a gene transcript and generates multiple mRNAs and proteins from a single gene 149. Like other proteins, ubiquitin ligases can also undergo alternative splicing to generate splice variants with diverse activities. For example, MDM2 splice variants have been detected with high frequency in several types of human cancers including invasive breast cancer 150, pediatric rhabdomyosarcoma 151, soft tissue sarcoma 152, ovarian carcinoma and bladder cancer 152. MDM2 splice variants are associated with progression of glioma, ovarian and bladder cancer 152, 153. Ectopic overexpression of some MDMD2 splice variants can transform NIH3T3 cells 152 and promote lymphomagenesis by HSC cells isolated from Eµ-myc mice 154. Overexpression of MDM2-B (the most commonly detected MDM2 splice variant in human cancer) in transgenic mice induced formation of sarcoma and lymphoma 155. More than 40 MDM2 splice variants have been identified, and many exhibit loss of the p53 binding domain or regions required to regulate p53 nuclear activity 156. Therefore, most MDM2 splice variants cannot directly bind p53 or regulate its expression and activity, and it remains unclear how these variants promote tumorigenesis. It is now recognized that many tumor-derived, mutant forms of p53 protein not only lose wild-type tumor suppressor function but gain oncogenic function 157. The MDM2-B variant reportedly interacts with full-length MDM2 to inhibit the latter’s ability to degrade mutant p53, leading to accumulation of oncogenic forms of mutant p53 and tumorigenesis 158, and MDM2-B overexpression is correlated with mutant p53 accumulation in human tumors. A MDM2 splice variant similar to human MDM2-B is overexpressed in tumors of mice harboring knock-in mutant (R172H) p53 (equivalent to human R175H) and is correlated with accumulation of mutant p53 in these tumors 158. Furthermore, like full-length MDM2, MDM2 splice variants may also have p53-independent functions in tumorigenesis 155, 159.
FBXW7 can also undergo alternative splicing. For example, three FBXW7 splice variants (α, β, and γ) use distinct first exons, resulting in proteins with different N-terminal domains 160. Differential localization of these variants has been observed: FBXW7-α, - β and - γ are largely localized to the nucleus, cytoplasm and nucleolus, respectively 161, 162. Nucleolar localization of FBXW7-γ reportedly regulates expression of nucleolar c-Myc and cell size 161. In addition, several forms of FBXW-α alternatively spliced at the 5’-UTR show significant differences in translational efficiency 163. Transcript levels of splice variants with high translational efficiency are specifically lower in more than 80% of breast cancer cell lines and in more than 50% of various human cancers, suggesting that differential expression of FBXW7-α variants constitutes a novel mechanism underlying FBXW7 downregulation in human cancer 163.
Regulation by post-translational modifications
A central requirement for control of protein ubiquitination is the ability of an ubiquitin ligase to associate with its substrate. It is important to note that in all cases, substrate recognition by ubiquitin ligases depends on post-translational modifications of the substrate. Although less studied, post-translational modifications of the ligase also regulate its subcellular localization and activity. Below are examples of key post-translational modifications that control stability of both ligases and substrates.
The Jun oncogene is subject to several layers of regulation, which are altered by mutations observed in cancer. Mutations range from deletion of the N-terminal domain to mutations that increase Jun stability by interfering with its association with the UBL FBXW7. C-Jun association with its upstream stress kinase JNK reportedly limits its availability under non stress conditions, independent of JNK kinase activity, a mechanism required to limit availability of a several JNK substrates, including ATF2 and p53, under non-stress conditions 164, 165, 166. Correspondingly, JNK upregulation is often seen in cancer and thought to contribute to increased c-Jun stability. Ras/MAPK signaling is also linked to c-Jun stability through the UBL Trim7 167. Tumor cells harboring increased RAS-MAPK signaling show increased TRIM7 phosphorylation and consequent activation, resulting in K63 ubiquitination and stabilization of RACO, a co-factor required for c-Jun activity. Although indirect, RACO control by Trim7 determines levels of c-Jun protein. It is noteworthy that FBXW7 mutations are also seen in tumors harboring Ras mutations, although it remains to be determined whether stabilization of c-Jun by Ras/TRIM7/RACO is mutually exclusive with FBXW7 mutations.
BRAF mutations, which are seen in over 40% of human melanoma cases, result in amplification of MAPK signaling, with notable increases in ERK1/2 activity. Thus, expectedly, both substrates and UBLs that depend on ERK activity are subject to enhanced ubiquitination-dependent degradation. One example of this is BRAF-dependent regulation and consequent loss of the ligase TRIM7 167, which enhances oncogenesis 167.
Increasing evidence suggests that post-translational regulation of deubiquitinating enzymes (DUBs) has a significant effect on the UPS system, as these factors determine the stability or activity of an ubiquitinated substrate. PKA-dependent phosphorylation of the DUB USP20 was recently shown to perturb post-endocytic trafficking of the β-andregenic receptor under cellular stress conditions. USP20 is implicated in control of the DNA damage response, cell cycling, and NF-κB activity 168, 169, and its regulation by PKA links this kinase to control of DNA damage and NF-κB activity.
The UBL Mdm2 is also regulated by post-translational phosphorylation by ATM. This activity was previously shown to affect Mdm2 localization and, consequently, its ability to associate with and ubiquitinate p53 170. Conversely, MDM2 phosphorylation by CAbl results in p53 activation and stabilization 171. A more complex regulatory mechanism was recently reported by Batuello and colleagues who demonstrated that MDM2 phosphorylation by Src increased its stability and enhanced its association with UBC12, an E2 conjugating enzyme for NEDD8, enabling enhanced neddylation of p53, an activity that blocks p53 ability to activate transcriptional targets 172.
Control of stability of proteasome subunits regulates activity and function. Growing evidence suggests that diverse mechanisms, including post-translational modifications, underlie these activities. For example, phosphorylation of the proteasome subunit PSMA7 by cAbl kinase impacts its regulation by BRCA1, which controls PSMA7 ubiquitination and stability 173. Both BRCA1 and cAbl are deregulated in cancer, suggesting that deregulated PSMA7 activity may function in oncogenesis.
Post-translational modifications of the ubiquitin ligase Siah2 ubiquitin ligase determine its activity in several ways. First, Siah2 phosphorylation by p38 kinase reportedly alters its activity by promoting its nuclear localization 174. Furthermore, Siah2 regulation by the DUB USP13 limits its activity despite the fact that Siah2 expression levels increase 175. Notably, USP13 also regulates PTEN activity 176. Different forms of stress modulate these activities: hypoxia reduces USP13 expression enabling increased Siah2 activity, while cellular stress induces p38 activity and alters Siah localization, affecting its recognition and targeting of substrates 174. Siah regulation by HIPK2 kinase also modulates its activity under hypoxia, rendering it more active and resulting in HIPK2 ubiquitination and degradation 177, 178. These activities affect global transcriptional regulation mechanisms governed by HIPK2 under hypoxia.
Different post-translational modifications also control UBL function. HIF-1α, for example, is subject to prolyl hydroxylation by one of three prolyl hydroxylases (PHD1–3). Such modification is required for HIF1a association with pVHL and subsequent HIF-1α ubiquitination and degradation 179. Notably, PHD1 and PHD3 proteins themselves are regulated by the UBLs Siah1/2 180, exemplifying multilayered regulation by UBLs.
Acetylation of the protein RASSF5 reportedly restricts its interaction with the UBL Itch, thereby increasing levels of the RASS5 protein. This activity affects the G1 transition in the cell cycle and induction of apoptosis 181.
Some UBLs are also glycosylated, among them A20, a UBL that also possesses DUB activity. Specifically, A20, which is implicated in control of NF-kB signaling, is regulated by post-translational O-glucosamine-N-acetylation (O-GlcNAcylation), which is required for A20 ubiquitination and subsequent proteasomal degradation 182.
ER stress activities and the UPR maintain protein homeostasis in normal cells and are perturbed in pathological conditions, including cancer. UPR deregulation is a common occurrence and a possible causative factor in numerous diseases, and numerous pathways, from metabolism to autophagy and cell death, are affected by its deregulation. UPR components are implicated in transcriptional regulation of several ubiquitin ligases. Among them are Siah1/2, which are controlled by ATF4 and sXBP1, two of the three major transducers of the UPR sensors PERK and URE1, respectively 183. Accumulation of misfolded proteins, a common occurrence under deregulated UPR conditions or in tumor cells subjected to chemotherapies (such as taxanes), activates ER-associated degradation (ERAD), engaging ERAD-resident UBLs. This mechanism reportedly underlies the switch to glutamine metabolism seen in paclitaxel-treated breast cancer cells prompted by degradation of misfolded glutamine carrier proteins SLC1A5 and SLC38A2 by the ER resident UBL RNF5 184.
Ubiquitin phosphorylation is a recent addition to layers of post-translational control of UBL signaling. PINK kinase is the first identified ubiquitin kinase, and it reportedly regulates activity of PARKIN ubiquitin ligase 185, 186. Parkin activity is enhanced upon ubiquitin phosphorylation, a mechanism implicated in mitophagy. PARKIN, which was initially associated primarily with Parkinson’s disease, is now known to be deregulated by genetic and epigenetic mechanisms in several cancers, including lung cancer 187 and glioma 188. It is expected that ubiquitin phosphorylation will not be limited to single kinase and could affect activity of other ubiquitin ligases and their substrates.
Ubiquitin ligase expression or activity is also regulated by protein-protein interactions. For example, MDM2 ligase activity can be regulated by MDMX, an MDM2 homologue with no intrinsic E3 ubiquitin ligase activity despite sequence homology of MDMX and MDM2 RING finger domains 189. MDMX can heterodimerize with MDM2 to inhibit MDM2 self-ubiquitination and enhance MDM2-dependent ubiquitination and degradation of p53 190, 191. Consistently, knock-in mice harboring a form of MDMX with a point mutation in the MDM2 binding site show embryonic lethality and elevated p53 levels; that embryonic lethality phenotype is fully rescued by concomitant p53 deletion 192.
The ubiquitin ligase Siah2 promotes transcriptional activity of the androgen receptor (AR) gene in prostate cancer cells by regulating the turnover of AR on select promoters 193, enabling recycling of 15% of AR regulated genes associated with lipid and sterol metabolism. Among those, AKR1C3, a steroidogenic enzyme functioning in resistance of prostate cancer cells to androgen-deprivation therapy, can directly bind Siah2 and inhibit its self-ubiquitinaiton/degradation, thereby increasing its levels and ubiquitin ligase activity towards substrates such as AR or NCOR1 194. Consistently, Siah2 and AKR1C3 protein levels are positively correlated in human prostate cancer tissues 194.
Epilogue
Several mechanisms underlie regulation and activity of ubiquitin ligases; in all cases, alterations in these activities promote deleterious phenotypes. We have summarized major regulatory nodes that perturb UBL activity and are often deregulated in cancer, from genomic mutations at the DNA level, to transcriptional control and post-translational protein modifications. UBLs themselves are subject to modifications, and those activities are altered in cancer, although this area requires further study. Whether deregulation of a UBL occurs through multiple mechanisms is a question that merits further analysis. Lastly, as tumor cell plasticity enables survival of harsh environmental conditions, as exemplified by resistance to therapy or metastatic potential, it is expected that spatial and temporal deregulation of UBL activity will remain highly relevant to the development of therapies that antagonize these responses.
The critical role UBL play in fundamental processes and the fact that in cancer they are deregulated by a number of genetic, epigenetic including transcription, translation and post-translational modifications, points to opportunities for their targeting, as novel therapeutic modalities. Depending on their substrates, UBL can elicit tumor suppressor or tumor promoter, in a context-dependent manner. Thus, targeting UBL will require deep understanding of its activity in a tissue and tumor-dependent manner. For a tumor-suppressive UBL, the targeting compounds could include those that elevate its expression level or promote substrate recognition. In contrast, targeting a tumor-promoting UBL is likely to include modulators that can repress its expression, alter its subcellular localization, inhibit its interaction with substrate or its assembly in multi-subunit complex containing UBL. Developing modulators for UBL is not trivial given the need to modulate protein-protein interactions. Ongoing efforts include means to alter activity (ubiquitination capacity), alter conformation (based on structure or biophysical properties) or localization (based on temporal and spatial activities in a given tissue type). Currently, this remains among the more challenging aspects in drug development. Notably, advances are made and the notion of targeting UBL is gaining traction, exemplified by a growing number of examples 195, 196, 197, 198, 199, 200. Knowledge of structural organization and defined modifications underlying spatial and temporal activity of UBL will likely guide future development of biologics and small molecule inhibitors for this class of proteins.
Acknowledgments
Support by NCI grants CA154888 (to J.Q.) and CA128814 and CA111515 (to Z.R.). This work was also supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program under Award No.W81XWH-14-1-0551 (Z.R.). Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense.”
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jianfei Qi, Email: jqi@som.umaryland.edu.
Ze’ev A. Ronai, Email: zeev@ronailab.net.
References
- 1.Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. The EMBO journal. 2013;32:2307–2320. doi: 10.1038/emboj.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theurillat JP, et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–89. doi: 10.1126/science.1250255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geng C, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer research. 2014;74:5631–5643. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang P, et al. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell death & disease. 2015;6:e1687. doi: 10.1038/cddis.2015.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barbieri CE, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature genetics. 2012;44:685–689. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blattner M, et al. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia. 2014;16:14–20. doi: 10.1593/neo.131704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geng C, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6997–7002. doi: 10.1073/pnas.1304502110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An J, Wang C, Deng Y, Yu L, Huang H. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell reports. 2014;6:657–669. doi: 10.1016/j.celrep.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Gallo M, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nature genetics. 2012;44:1310–1315. doi: 10.1038/ng.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kandoth C, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao S, et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2916–2921. doi: 10.1073/pnas.1222577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuhn E, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. Journal of the National Cancer Institute. 2012;104:1503–1513. doi: 10.1093/jnci/djs345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nature reviews Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- 14.Bai C, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 15.Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112:243–256. doi: 10.1016/s0092-8674(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 16.Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG., Jr The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer cell. 2005;8:25–33. doi: 10.1016/j.ccr.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Yada M, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. The EMBO journal. 2004;23:2116–2125. doi: 10.1038/sj.emboj.7600217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koepp DM, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 19.Zhao D, Zheng HQ, Zhou Z, Chen C. The Fbw7 tumor suppressor targets KLF5 for ubiquitin-mediated degradation and suppresses breast cell proliferation. Cancer research. 2010;70:4728–4738. doi: 10.1158/0008-5472.CAN-10-0040. [DOI] [PubMed] [Google Scholar]
- 20.Wertz IE, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–114. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 21.Tsunematsu R, et al. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. The Journal of biological chemistry. 2004;279:9417–9423. doi: 10.1074/jbc.M312337200. [DOI] [PubMed] [Google Scholar]
- 22.Mao JH, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321:1499–1502. doi: 10.1126/science.1162981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Onoyama I, et al. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. The Journal of experimental medicine. 2007;204:2875–2888. doi: 10.1084/jem.20062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babaei-Jadidi R, et al. FBXW7 influences murine intestinal homeostasis and cancer, targeting Notch, Jun, and DEK for degradation. The Journal of experimental medicine. 2011;208:295–312. doi: 10.1084/jem.20100830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mao JH, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004;432:775–779. doi: 10.1038/nature03155. [DOI] [PubMed] [Google Scholar]
- 26.Churi CR, et al. Mutation profiling in cholangiocarcinoma: prognostic and therapeutic implications. PloS one. 2014;9:e115383. doi: 10.1371/journal.pone.0115383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malyukova A, et al. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer research. 2007;67:5611–5616. doi: 10.1158/0008-5472.CAN-06-4381. [DOI] [PubMed] [Google Scholar]
- 28.Ojesina AI, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371–375. doi: 10.1038/nature12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-Dios DA, et al. High-throughput interrogation of PIK3CA, PTEN, KRAS, FBXW7 and TP53 mutations in primary endometrial carcinoma. Gynecologic oncology. 2013;128:327–334. doi: 10.1016/j.ygyno.2012.11.037. [DOI] [PubMed] [Google Scholar]
- 30.Akhoondi S, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer research. 2007;67:9006–9012. doi: 10.1158/0008-5472.CAN-07-1320. [DOI] [PubMed] [Google Scholar]
- 31.Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welcker M, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nature reviews Cancer. 2015;15:55–64. doi: 10.1038/nrc3844. [DOI] [PubMed] [Google Scholar]
- 34.Crossey PA, et al. Molecular genetic investigations of the mechanism of tumourigenesis in von Hippel-Lindau disease: analysis of allele loss in VHL tumours. Human genetics. 1994;93:53–58. doi: 10.1007/BF00218913. [DOI] [PubMed] [Google Scholar]
- 35.Zbar B, Brauch H, Talmadge C, Linehan M. Loss of alleles of loci on the short arm of chromosome 3 in renal cell carcinoma. Nature. 1987;327:721–724. doi: 10.1038/327721a0. [DOI] [PubMed] [Google Scholar]
- 36.Gnarra JR, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nature genetics. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 37.Sato Y, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nature genetics. 2013;45:860–867. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 38.Shimura H, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature genetics. 2000;25:302–305. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 39.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong Y, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nature genetics. 2014;46:588–594. doi: 10.1038/ng.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucking CB, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. The New England journal of medicine. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- 42.Veeriah S, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nature genetics. 2010;42:77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weir BA, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cesari R, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5956–5961. doi: 10.1073/pnas.0931262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Poulogiannis G, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwakawa R, Okayama H, Kohno T, Sato-Otsubo A, Ogawa S, Yokota J. Contribution of germline mutations to PARK2 gene inactivation in lung adenocarcinoma. Genes, chromosomes & cancer. 2012;51:462–472. doi: 10.1002/gcc.21933. [DOI] [PubMed] [Google Scholar]
- 48.Wu LC, et al. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nature genetics. 1996;14:430–440. doi: 10.1038/ng1296-430. [DOI] [PubMed] [Google Scholar]
- 49.Kalb R, Mallery DL, Larkin C, Huang JT, Hiom K. BRCA1 is a histone-H2A–specific ubiquitin ligase. Cell reports. 2014;8:999–1005. doi: 10.1016/j.celrep.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mallery DL, Vandenberg CJ, Hiom K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. The EMBO journal. 2002;21:6755–6762. doi: 10.1093/emboj/cdf691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes & development. 2006;20:1721–1726. doi: 10.1101/gad.1431006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horwitz AA, Affar el B, Heine GF, Shi Y, Parvin JD. A mechanism for transcriptional repression dependent on the BRCA1 E3 ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6614–6619. doi: 10.1073/pnas.0610481104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eakin CM, Maccoss MJ, Finney GL, Klevit RE. Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5794–5799. doi: 10.1073/pnas.0610887104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. The Journal of biological chemistry. 2005;280:24498–24505. doi: 10.1074/jbc.M414020200. [DOI] [PubMed] [Google Scholar]
- 55.Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM. Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5134–5139. doi: 10.1073/pnas.081068398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Drost R, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer cell. 2011;20:797–809. doi: 10.1016/j.ccr.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 57.Shakya R, et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 2011;334:525–528. doi: 10.1126/science.1209909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antoniou A, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. American journal of human genetics. 2003;72:1117–1130. doi: 10.1086/375033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ford D, et al. Genetic heterogeneity penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. American journal of human genetics. 1998;62:676–689. doi: 10.1086/301749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foulkes WD. Inherited susceptibility to common cancers. The New England journal of medicine. 2008;359:2143–2153. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 61.Welcsh PL, King MC. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Human molecular genetics. 2001;10:705–713. doi: 10.1093/hmg/10.7.705. [DOI] [PubMed] [Google Scholar]
- 62.Zhu X, et al. Hypermethylation of BRCA1 gene: implication for prognostic biomarker and therapeutic target in sporadic primary triple-negative breast cancer. Breast cancer research and treatment. 2015;150:479–486. doi: 10.1007/s10549-015-3338-y. [DOI] [PubMed] [Google Scholar]
- 63.Li Q, Wei W, Jiang YI, Yang H, Liu J. Promoter methylation and expression changes of in cancerous tissues of patients with sporadic breast cancer. Oncology letters. 2015;9:1807–1813. doi: 10.3892/ol.2015.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Truong PK, Lao TD, Doan TP, Le TA. BRCA1 promoter hypermethylation signature for early detection of breast cancer in the Vietnamese population. Asian Pacific journal of cancer prevention : APJCP. 2014;15:9607–9610. doi: 10.7314/apjcp.2014.15.22.9607. [DOI] [PubMed] [Google Scholar]
- 65.Yamashita N, et al. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clinical breast cancer. 2015 doi: 10.1016/j.clbc.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 66.Sharma P, et al. The prognostic value of promoter methylation in early stage triple negative breast cancer. Journal of cancer therapeutics & research. 2014;3:1–11. doi: 10.7243/2049-7962-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cahilly-Snyder L, Yang-Feng T, Francke U, George DL. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somatic cell and molecular genetics. 1987;13:235–244. doi: 10.1007/BF01535205. [DOI] [PubMed] [Google Scholar]
- 68.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–1975. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 69.Boyd SD, Tsai KY, Jacks T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nature cell biology. 2000;2:563–568. doi: 10.1038/35023500. [DOI] [PubMed] [Google Scholar]
- 70.Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- 71.Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- 72.Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Molecular cell. 2008;32:180–189. doi: 10.1016/j.molcel.2008.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Onel K, Cordon-Cardo C. MDM2 and prognosis. Molecular cancer research : MCR. 2004;2:1–8. [PubMed] [Google Scholar]
- 74.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic acids research. 1998;26:3453–3459. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15608–15612. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Forslund A, et al. MDM2 gene amplification is correlated to tumor progression but not to the presence of SNP309 or TP53 mutational status in primary colorectal cancers. Molecular cancer research : MCR. 2008;6:205–211. doi: 10.1158/1541-7786.MCR-07-0239. [DOI] [PubMed] [Google Scholar]
- 77.Hav M, et al. MDM2 gene amplification and protein expressions in colon carcinoma: is targeting MDM2 a new therapeutic option? Virchows Archiv : an international journal of pathology. 2011;458:197–203. doi: 10.1007/s00428-010-1012-7. [DOI] [PubMed] [Google Scholar]
- 78.Lee YS, et al. Genomic profile analysis of diffuse-type gastric cancers. Genome biology. 2014;15:R55. doi: 10.1186/gb-2014-15-4-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gunther T, et al. Mdm2 gene amplification in gastric cancer correlation with expression of Mdm2 protein and p53 alterations. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2000;13:621–626. doi: 10.1038/modpathol.3880107. [DOI] [PubMed] [Google Scholar]
- 80.Choschzick M, et al. MDM2 amplification is an independent prognostic feature of node-negative, estrogen receptor-positive early-stage breast cancer. Cancer biomarkers : section A of Disease markers. 2010;8:53–60. doi: 10.3233/DMA-2011-0806. [DOI] [PubMed] [Google Scholar]
- 81.Hui AB, Lo KW, Yin XL, Poon WS, Ng HK. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Laboratory investigation; a journal of technical methods and pathology. 2001;81:717–723. doi: 10.1038/labinvest.3780280. [DOI] [PubMed] [Google Scholar]
- 82.Corvi R, et al. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene. 1995;10:1081–1086. [PubMed] [Google Scholar]
- 83.Merup M, et al. Amplification of multiple regions of chromosome 12, including 12q13-15, in chronic lymphocytic leukaemia. European journal of haematology. 1997;58:174–180. doi: 10.1111/j.1600-0609.1997.tb00944.x. [DOI] [PubMed] [Google Scholar]
- 84.Florenes VA, Maelandsmo GM, Forus A, Andreassen A, Myklebost O, Fodstad O. MDM2 gene amplification and transcript levels in human sarcomas: relationship to TP53 gene status. Journal of the National Cancer Institute. 1994;86:1297–1302. doi: 10.1093/jnci/86.17.1297. [DOI] [PubMed] [Google Scholar]
- 85.Bond GL, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 86.Post SM, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer cell. 2010;18:220–230. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moradi MT, Salehi Z, Asl SF, Aminian K, Hashtchin AR. Helicobacter pylori infection and MDM2 SNP309 association with gastric cancer susceptibility. Genetic testing and molecular biomarkers. 2013;17:794–798. doi: 10.1089/gtmb.2013.0173. [DOI] [PubMed] [Google Scholar]
- 88.Zhang Y, Bai Y, Zhang Y, Guan J, Chen L. The MDM2 309 T/G polymorphism is associated with head and neck cancer risk especially in nasopharyngeal cancer: a meta-analysis. Onkologie. 2012;35:666–670. doi: 10.1159/000343639. [DOI] [PubMed] [Google Scholar]
- 89.Bjornslett M, Knappskog S, Lonning PE, Dorum A. Effect of the MDM2 promoter polymorphisms SNP309T>G and SNP285G>C on the risk of ovarian cancer in BRCA1 mutation carriers. BMC cancer. 2012;12:454. doi: 10.1186/1471-2407-12-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xiong X, et al. Risk of MDM2 SNP309 alone or in combination with the p53 codon 72 polymorphism in acute myeloid leukemia. Leukemia research. 2009;33:1454–1458. doi: 10.1016/j.leukres.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 91.Knappskog S, et al. The MDM2 promoter SNP285C/309G haplotype diminishes Sp1 transcription factor binding and reduces risk for breast and ovarian cancer in Caucasians. Cancer cell. 2011;19:273–282. doi: 10.1016/j.ccr.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 92.Knappskog S, et al. SNP285C modulates oestrogen receptor/Sp1 binding to the MDM2 promoter and reduces the risk of endometrial but not prostatic cancer. Eur J Cancer. 2012;48:1988–1996. doi: 10.1016/j.ejca.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 93.Laney JD, Hochstrasser M. Substrate targeting in the ubiquitin system. Cell. 1999;97:427–430. doi: 10.1016/s0092-8674(00)80752-7. [DOI] [PubMed] [Google Scholar]
- 94.Winston JT, Strack P, Beer-Romero P, Chu CY, Elledge SJ, Harper JW. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes & development. 1999;13:270–283. doi: 10.1101/gad.13.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tsai WB, et al. Inhibition of FOXO3 tumor suppressor function by betaTrCP1 through ubiquitin-mediated degradation in a tumor mouse model. PloS one. 2010;5:e11171. doi: 10.1371/journal.pone.0011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Westbrook TF, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–374. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kudo Y, et al. Role of F-box protein betaTrcp1 in mammary gland development and tumorigenesis. Molecular and cellular biology. 2004;24:8184–8194. doi: 10.1128/MCB.24.18.8184-8194.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ougolkov A, et al. Associations among beta-TrCP, an E3 ubiquitin ligase receptor, beta-catenin, and NF-kappaB in colorectal cancer. Journal of the National Cancer Institute. 2004;96:1161–1170. doi: 10.1093/jnci/djh219. [DOI] [PubMed] [Google Scholar]
- 99.Muerkoster S, et al. Increased expression of the E3-ubiquitin ligase receptor subunit betaTRCP1 relates to constitutive nuclear factor-kappaB activation and chemoresistance in pancreatic carcinoma cells. Cancer research. 2005;65:1316–1324. doi: 10.1158/0008-5472.CAN-04-1626. [DOI] [PubMed] [Google Scholar]
- 100.Koch A, et al. Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:4295–4304. doi: 10.1158/1078-0432.CCR-04-1162. [DOI] [PubMed] [Google Scholar]
- 101.Chen S, et al. An insertion/deletion polymorphism in the 3’ untranslated region of beta-transducin repeat-containing protein (betaTrCP) is associated with susceptibility for hepatocellular carcinoma in Chinese. Biochemical and biophysical research communications. 2010;391:552–556. doi: 10.1016/j.bbrc.2009.11.096. [DOI] [PubMed] [Google Scholar]
- 102.Huo ZH, Zhong HJ, Zhu YS, Xing B, Tang H. Roles of functional NFKB1 and beta-TrCP insertion/deletion polymorphisms in mRNA expression and epithelial ovarian cancer susceptibility. Genetics and molecular research : GMR. 2013;12:3435–3443. doi: 10.4238/2013.March.11.6. [DOI] [PubMed] [Google Scholar]
- 103.Eskandari-Nasab E, Hashemi M, Amininia S, Ebrahimi M, Rezaei M, Hashemi SM. Effect of TP53 16-bp and beta-TrCP 9-bp INS/DEL polymorphisms in relation to risk of breast cancer. Gene. 2015;568:181–185. doi: 10.1016/j.gene.2015.05.048. [DOI] [PubMed] [Google Scholar]
- 104.Zhang B, et al. A common variant in the SIAH2 locus is associated with estrogen receptor-positive breast cancer in the Chinese Han population. PloS one. 2013;8:e79365. doi: 10.1371/journal.pone.0079365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Elgazzar S, et al. A genome-wide association study identifies a genetic variant in the SIAH2 locus associated with hormonal receptor-positive breast cancer in Japanese. Journal of human genetics. 2012;57:766–771. doi: 10.1038/jhg.2012.108. [DOI] [PubMed] [Google Scholar]
- 106.Yu JC, et al. Genetic susceptibility to the development and progression of breast cancer associated with polymorphism of cell cycle and ubiquitin ligase genes. Carcinogenesis. 2009;30:1562–1570. doi: 10.1093/carcin/bgp173. [DOI] [PubMed] [Google Scholar]
- 107.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nature cell biology. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 108.Nakayama K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. The EMBO journal. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kamura T, et al. Degradation of p57Kip2 mediated by SCFSkp2-dependent ubiquitylation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10231–10236. doi: 10.1073/pnas.1831009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11324–11329. doi: 10.1073/pnas.95.19.11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang G, et al. Elevated Skp2 protein expression in human prostate cancer: association with loss of the cyclin-dependent kinase inhibitor p27 and PTEN and with reduced recurrence-free survival. Clinical cancer research : an official journal of the American Association for Cancer Research. 2002;8:3419–3426. [PubMed] [Google Scholar]
- 112.Li JQ, et al. Correlation of Skp2 with carcinogenesis, invasion, metastasis, and prognosis in colorectal tumors. International journal of oncology. 2004;25:87–95. [PubMed] [Google Scholar]
- 113.Rose AE, et al. Clinical relevance of SKP2 alterations in metastatic melanoma. Pigment cell & melanoma research. 2011;24:197–206. doi: 10.1111/j.1755-148X.2010.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yokoi S, Yasui K, Mori M, Iizasa T, Fujisawa T, Inazawa J. Amplification and overexpression of SKP2 are associated with metastasis of non-small-cell lung cancers to lymph nodes. The American journal of pathology. 2004;165:175–180. doi: 10.1016/S0002-9440(10)63286-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Masuda TA, et al. Clinical and biological significance of S-phase kinase-associated protein 2 (Skp2) gene expression in gastric carcinoma: modulation of malignant phenotype by Skp2 overexpression, possibly via p27 proteolysis. Cancer research. 2002;62:3819–3825. [PubMed] [Google Scholar]
- 116.Einama T, et al. High-level Skp2 expression in pancreatic ductal adenocarcinoma: correlation with the extent of lymph node metastasis, higher histological grade, and poorer patient outcome. Pancreas. 2006;32:376–381. doi: 10.1097/01.mpa.0000220862.78248.c4. [DOI] [PubMed] [Google Scholar]
- 117.Traub F, Mengel M, Luck HJ, Kreipe HH, von Wasielewski R. Prognostic impact of Skp2 and p27 in human breast cancer. Breast cancer research and treatment. 2006;99:185–191. doi: 10.1007/s10549-006-9202-3. [DOI] [PubMed] [Google Scholar]
- 118.Shim EH, et al. Expression of the F-box protein SKP2 induces hyperplasia, dysplasia, and low-grade carcinoma in the mouse prostate. Cancer research. 2003;63:1583–1588. [PubMed] [Google Scholar]
- 119.Lin HK, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhu CQ, et al. Skp2 gene copy number aberrations are common in non-small cell lung carcinoma, and its overexpression in tumors with ras mutation is a poor prognostic marker. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:1984–1991. doi: 10.1158/1078-0432.ccr-03-0470. [DOI] [PubMed] [Google Scholar]
- 121.Wang XC, et al. Suppression of anoikis by SKP2 amplification and overexpression promotes metastasis of esophageal squamous cell carcinoma. Molecular cancer research : MCR. 2009;7:12–22. doi: 10.1158/1541-7786.MCR-08-0092. [DOI] [PubMed] [Google Scholar]
- 122.Li CF, et al. Characterization of gene amplification-driven SKP2 overexpression in myxofibrosarcoma: potential implications in tumor progression and therapeutics. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:1598–1610. doi: 10.1158/1078-0432.CCR-11-3077. [DOI] [PubMed] [Google Scholar]
- 123.Fimiani V, Minniti D. Anti-tumor properties of the organometallic complex cis-dimethylbis[sulfinylbis[methane]-S]platinum(II) Anti-cancer drugs. 1992;3:9–15. doi: 10.1097/00001813-199202000-00002. [DOI] [PubMed] [Google Scholar]
- 124.Zhang L, Wang C. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 2006;25:2615–2627. doi: 10.1038/sj.onc.1209286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Appleman LJ, Chernova I, Li L, Boussiotis VA. CD28 costimulation mediates transcription of SKP2 and CKS1, the substrate recognition components of SCFSkp2 ubiquitin ligase that leads p27kip1 to degradation. Cell Cycle. 2006;5:2123–2129. doi: 10.4161/cc.5.18.3139. [DOI] [PubMed] [Google Scholar]
- 126.Schneider G, Saur D, Siveke JT, Fritsch R, Greten FR, Schmid RM. IKKalpha controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. The EMBO journal. 2006;25:3801–3812. doi: 10.1038/sj.emboj.7601259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Evans L, et al. SKP2 is a direct transcriptional target of MYCN and a potential therapeutic target in neuroblastoma. Cancer letters. 2015;363:37–45. doi: 10.1016/j.canlet.2015.03.044. [DOI] [PubMed] [Google Scholar]
- 128.Bretones G, et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. The Journal of biological chemistry. 2011;286:9815–9825. doi: 10.1074/jbc.M110.165977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang H, Zhao W, Yang D. Stat3 induces oncogenic Skp2 expression in human cervical carcinoma cells. Biochemical and biophysical research communications. 2012;418:186–190. doi: 10.1016/j.bbrc.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 130.Reichert M, Saur D, Hamacher R, Schmid RM, Schneider G. Phosphoinositide-3-kinase signaling controls S-phase kinase-associated protein 2 transcription via E2F1 in pancreatic ductal adenocarcinoma cells. Cancer research. 2007;67:4149–4156. doi: 10.1158/0008-5472.CAN-06-4484. [DOI] [PubMed] [Google Scholar]
- 131.Andreu EJ, et al. BCR-ABL induces the expression of Skp2 through the PI3K pathway to promote p27Kip1 degradation and proliferation of chronic myelogenous leukemia cells. Cancer research. 2005;65:3264–3272. doi: 10.1158/0008-5472.CAN-04-1357. [DOI] [PubMed] [Google Scholar]
- 132.Kim H, et al. Downregulation of the Ubiquitin Ligase RNF125 Underlies Resistance of Melanoma Cells to BRAF Inhibitors via JAK1 Deregulation. Cell reports. 2015;11:1458–1473. doi: 10.1016/j.celrep.2015.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nature reviews Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Q, et al. Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene. 2011;30:3875–3886. doi: 10.1038/onc.2011.103. [DOI] [PubMed] [Google Scholar]
- 135.Lerner M, et al. MiRNA-27a controls FBW7/hCDC4-dependent cyclin E degradation and cell cycle progression. Cell Cycle. 2011;10:2172–2183. doi: 10.4161/cc.10.13.16248. [DOI] [PubMed] [Google Scholar]
- 136.Xu Y, Sengupta T, Kukreja L, Minella AC. MicroRNA-223 regulates cyclin E activity by modulating expression of F-box and WD-40 domain protein 7. The Journal of biological chemistry. 2010;285:34439–34446. doi: 10.1074/jbc.M110.152306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kurashige J, et al. Overexpression of microRNA-223 regulates the ubiquitin ligase FBXW7 in oesophageal squamous cell carcinoma. British journal of cancer. 2012;106:182–188. doi: 10.1038/bjc.2011.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Li J, et al. MicroRNA-223 functions as an oncogene in human gastric cancer by targeting FBXW7/hCdc4. Journal of cancer research and clinical oncology. 2012;138:763–774. doi: 10.1007/s00432-012-1154-x. [DOI] [PubMed] [Google Scholar]
- 139.Lu D, et al. MiR-25 regulates Wwp2 and Fbxw7 and promotes reprogramming of mouse fibroblast cells to iPSCs. PloS one. 2012;7:e40938. doi: 10.1371/journal.pone.0040938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhou C, Shen L, Mao L, Wang B, Li Y, Yu H. miR-92a is upregulated in cervical cancer and promotes cell proliferation and invasion by targeting FBXW7. Biochemical and biophysical research communications. 2015;458:63–69. doi: 10.1016/j.bbrc.2015.01.066. [DOI] [PubMed] [Google Scholar]
- 141.Li L, et al. Sequential expression of miR-182 and miR-503 cooperatively targets FBXW7, contributing to the malignant transformation of colon adenoma to adenocarcinoma. The Journal of pathology. 2014;234:488–501. doi: 10.1002/path.4407. [DOI] [PubMed] [Google Scholar]
- 142.Kumar V, et al. Notch and NF-kB signaling pathways regulate miR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia. 2014;28:2324–2335. doi: 10.1038/leu.2014.133. [DOI] [PubMed] [Google Scholar]
- 143.Mansour MR, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. The Journal of experimental medicine. 2013;210:1545–1557. doi: 10.1084/jem.20122516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang CH, et al. The oncogenic microRNA-21 inhibits the tumor suppressive activity of FBXO11 to promote tumorigenesis. The Journal of biological chemistry. 2015;290:6037–6046. doi: 10.1074/jbc.M114.632125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Huen MS, et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–914. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mailand N, et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 147.Wang Z, et al. miR-214-mediated downregulation of RNF8 induces chromosomal instability in ovarian cancer cells. Cell Cycle. 2014;13:3519–3528. doi: 10.4161/15384101.2014.958413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li S, Wang L, Fu B, Berman MA, Diallo A, Dorf ME. TRIM65 regulates microRNA activity by ubiquitination of TNRC6. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:6970–6975. doi: 10.1073/pnas.1322545111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene. 2015;34:1–14. doi: 10.1038/onc.2013.570. [DOI] [PubMed] [Google Scholar]
- 150.Lukas J, et al. Alternative and aberrant messenger RNA splicing of the mdm2 oncogene in invasive breast cancer. Cancer research. 2001;61:3212–3219. [PubMed] [Google Scholar]
- 151.Bartel F, Taylor AC, Taubert H, Harris LC. Novel mdm2 splice variants identified in pediatric rhabdomyosarcoma tumors and cell lines. Oncology research. 2001;12:451–457. doi: 10.3727/096504001108747459. [DOI] [PubMed] [Google Scholar]
- 152.Bartel F, et al. Amplification of the MDM2 gene, but not expression of splice variants of MDM2 MRNA, is associated with prognosis in soft tissue sarcoma. International journal of cancer Journal international du cancer. 2001;95:168–175. doi: 10.1002/1097-0215(20010520)95:3<168::aid-ijc1029>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 153.Matsumoto R, Tada M, Nozaki M, Zhang CL, Sawamura Y, Abe H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer research. 1998;58:609–613. [PubMed] [Google Scholar]
- 154.Fridman JS, Hernando E, Hemann MT, de Stanchina E, Cordon-Cardo C, Lowe SW. Tumor promotion by Mdm2 splice variants unable to bind p53. Cancer research. 2003;63:5703–5706. [PubMed] [Google Scholar]
- 155.Steinman HA, et al. An alternative splice form of Mdm2 induces p53-independent cell growth and tumorigenesis. The Journal of biological chemistry. 2004;279:4877–4886. doi: 10.1074/jbc.M305966200. [DOI] [PubMed] [Google Scholar]
- 156.Harris LC. MDM2 splice variants and their therapeutic implications. Current cancer drug targets. 2005;5:21–26. doi: 10.2174/1568009053332654. [DOI] [PubMed] [Google Scholar]
- 157.Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 158.Zheng T, et al. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nature communications. 2013;4:2996. doi: 10.1038/ncomms3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Okoro DR, et al. Endogenous human MDM2-C is highly expressed in human cancers and functions as a p53-independent growth activator. PloS one. 2013;8:e77643. doi: 10.1371/journal.pone.0077643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Spruck CH, et al. hCDC4 gene mutations in endometrial cancer. Cancer research. 2002;62:4535–4539. [PubMed] [Google Scholar]
- 161.Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Current biology : CB. 2004;14:1852–1857. doi: 10.1016/j.cub.2004.09.083. [DOI] [PubMed] [Google Scholar]
- 162.Ye X, et al. Recognition of phosphodegron motifs in human cyclin E by the SCF(Fbw7) ubiquitin ligase. The Journal of biological chemistry. 2004;279:50110–50119. doi: 10.1074/jbc.M409226200. [DOI] [PubMed] [Google Scholar]
- 163.Liu Y, et al. Multiple novel alternative splicing forms of FBXW7alpha have a translational modulatory function and show specific alteration in human cancer. PloS one. 2012;7:e49453. doi: 10.1371/journal.pone.0049453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fuchs SY, Fried VA, Ronai Z. Stress-activated kinases regulate protein stability. Oncogene. 1998;17:1483–1490. doi: 10.1038/sj.onc.1202184. [DOI] [PubMed] [Google Scholar]
- 165.Fuchs SY, et al. JNK targets p53 ubiquitination and degradation in nonstressed cells. Genes & development. 1998;12:2658–2663. doi: 10.1101/gad.12.17.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fuchs SY, Dolan L, Davis RJ, Ronai Z. Phosphorylation-dependent targeting of c-Jun ubiquitination by Jun N-kinase. Oncogene. 1996;13:1531–1535. [PubMed] [Google Scholar]
- 167.Chakraborty A, Diefenbacher ME, Mylona A, Kassel O, Behrens A. The E3 ubiquitin ligase Trim7 mediates c-Jun/AP-1 activation by Ras signalling. Nature communications. 2015;6:6782. doi: 10.1038/ncomms7782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yasunaga J, Lin FC, Lu X, Jeang KT. Ubiquitin-specific peptidase 20 targets TRAF6 and human T cell leukemia virus type 1 tax to negatively regulate NF-kappaB signaling. Journal of virology. 2011;85:6212–6219. doi: 10.1128/JVI.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shanmugam I, et al. Ubiquitin-specific peptidase 20 regulates Rad17 stability, checkpoint kinase 1 phosphorylation and DNA repair by homologous recombination. The Journal of biological chemistry. 2014;289:22739–22748. doi: 10.1074/jbc.M114.550459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14973–14977. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Goldberg Z, et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. The EMBO journal. 2002;21:3715–3727. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Batuello CN, Hauck PM, Gendron JM, Lehman JA, Mayo LD. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1749–1754. doi: 10.1073/pnas.1416656112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Li D, et al. c-Abl regulates proteasome abundance by controlling the ubiquitin-proteasomal degradation of PSMA7 subunit. Cell reports. 2015;10:484–496. doi: 10.1016/j.celrep.2014.12.044. [DOI] [PubMed] [Google Scholar]
- 174.Khurana A, Nakayama K, Williams S, Davis RJ, Mustelin T, Ronai Z. Regulation of the ring finger E3 ligase Siah2 by p38 MAPK. The Journal of biological chemistry. 2006;281:35316–35326. doi: 10.1074/jbc.M606568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Scortegagna M, et al. USP13 enzyme regulates Siah2 ligase stability and activity via noncatalytic ubiquitin-binding domains. The Journal of biological chemistry. 2011;286:27333–27341. doi: 10.1074/jbc.M111.218214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Zhang J, et al. Deubiquitylation and stabilization of PTEN by USP13. Nature cell biology. 2013;15:1486–1494. doi: 10.1038/ncb2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Polonio-Vallon T, Kirkpatrick J, Krijgsveld J, Hofmann TG. Src kinase modulates the apoptotic p53 pathway by altering HIPK2 localization. Cell Cycle. 2014;13:115–125. doi: 10.4161/cc.26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Winter M, et al. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nature cell biology. 2008;10:812–824. doi: 10.1038/ncb1743. [DOI] [PubMed] [Google Scholar]
- 179.Kim W, Kaelin WG., Jr The von Hippel-Lindau tumor suppressor protein: new insights into oxygen sensing and cancer. Current opinion in genetics & development. 2003;13:55–60. doi: 10.1016/s0959-437x(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 180.Nakayama K, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 181.Suryaraja R, Anitha M, Anbarasu K, Kumari G, Mahalingam S. The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner. Cell death & disease. 2013;4:e565. doi: 10.1038/cddis.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Shrikhande GV, et al. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PloS one. 2010;5:e14240. doi: 10.1371/journal.pone.0014240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Scortegagna M, et al. Fine tuning of the UPR by the ubiquitin ligases Siah1/2. PLoS genetics. 2014;10:e1004348. doi: 10.1371/journal.pgen.1004348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jeon YJ, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer cell. 2015;27:354–369. doi: 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Durcan TM, Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes & development. 2015;29:989–999. doi: 10.1101/gad.262758.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Koyano F, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 187.Xiong D, et al. A recurrent mutation in PARK2 is associated with familial lung cancer. American journal of human genetics. 2015;96:301–308. doi: 10.1016/j.ajhg.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Viotti J, et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene. 2014;33:1764–1775. doi: 10.1038/onc.2013.124. [DOI] [PubMed] [Google Scholar]
- 189.Badciong JC, Haas AL. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. The Journal of biological chemistry. 2002;277:49668–49675. doi: 10.1074/jbc.M208593200. [DOI] [PubMed] [Google Scholar]
- 190.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12009–12014. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wang X, Wang J, Jiang X. MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. The Journal of biological chemistry. 2011;286:23725–23734. doi: 10.1074/jbc.M110.213868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Huang L, et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12001–12006. doi: 10.1073/pnas.1102309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Qi J, et al. The E3 ubiquitin ligase Siah2 contributes to castration-resistant prostate cancer by regulation of androgen receptor transcriptional activity. Cancer cell. 2013;23:332–346. doi: 10.1016/j.ccr.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fan L, et al. The steroidogenic enzyme AKR1C3 regulates stability of the ubiquitin ligase Siah2 in prostate cancer cells. The Journal of biological chemistry. 2015 doi: 10.1074/jbc.M115.662155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu J, et al. Targeting the ubiquitin pathway for cancer treatment. Biochimica et biophysica acta. 2015;1855:50–60. doi: 10.1016/j.bbcan.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nature reviews Drug discovery. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer cell. 2014;26:455–464. doi: 10.1016/j.ccell.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Landre V, Rotblat B, Melino S, Bernassola F, Melino G. Screening for E3-ubiquitin ligase inhibitors: challenges and opportunities. Oncotarget. 2014;5:7988–8013. doi: 10.18632/oncotarget.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pal A, Young MA, Donato NJ. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer research. 2014;74:4955–4966. doi: 10.1158/0008-5472.CAN-14-1211. [DOI] [PubMed] [Google Scholar]
- 200.Zhao Y, Sun Y. Cullin-RING Ligases as attractive anti-cancer targets. Current pharmaceutical design. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Giannakis M, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nature genetics. 2014;46:1264–1266. doi: 10.1038/ng.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sakamoto H, et al. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2015;28:261–267. doi: 10.1038/modpathol.2014.98. [DOI] [PubMed] [Google Scholar]
- 203.Wang K, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nature genetics. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 204.Jiao Y, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. The Journal of pathology. 2014;232:428–435. doi: 10.1002/path.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zou Y, et al. RNF43 mutations are recurrent in Chinese patients with mucinous ovarian carcinoma but absent in other subtypes of ovarian cancer. Gene. 2013;531:112–116. doi: 10.1016/j.gene.2013.08.054. [DOI] [PubMed] [Google Scholar]
- 206.Ryland GL, et al. RNF43 is a tumour suppressor gene mutated in mucinous tumours of the ovary. The Journal of pathology. 2013;229:469–476. doi: 10.1002/path.4134. [DOI] [PubMed] [Google Scholar]
- 207.Ong CK, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nature genetics. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 208.Katzav S, Schmitz ML. Mutations of c-Cbl in myeloid malignancies. Oncotarget. 2015;6:10689–10696. doi: 10.18632/oncotarget.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sanada M, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009;460:904–908. doi: 10.1038/nature08240. [DOI] [PubMed] [Google Scholar]
- 210.Niemeyer CM, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nature genetics. 2010;42:794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Keats JJ, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Rossi D, et al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118:4930–4934. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- 213.Bea S, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18250–18255. doi: 10.1073/pnas.1314608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Assie G, et al. Integrated genomic characterization of adrenocortical carcinoma. Nature genetics. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 215.Juhlin CC, et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. The Journal of clinical endocrinology and metabolism. 2015;100:E493–502. doi: 10.1210/jc.2014-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Jiang JH, et al. Clinical significance of the ubiquitin ligase UBE3C in hepatocellular carcinoma revealed by exome sequencing. Hepatology. 2014;59:2216–2227. doi: 10.1002/hep.27012. [DOI] [PubMed] [Google Scholar]
- 217.Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kim Y, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:121–128. doi: 10.1200/JCO.2013.50.8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cleary SP, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–1702. doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Muscarella LA, et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics. 2011;6:710–719. doi: 10.4161/epi.6.6.15773. [DOI] [PubMed] [Google Scholar]
- 221.Li QK, Singh A, Biswal S, Askin F, Gabrielson E. KEAP1 gene mutations and NRF2 activation are common in pulmonary papillary adenocarcinoma. Journal of human genetics. 2011;56:230–234. doi: 10.1038/jhg.2010.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Singh A, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS medicine. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Meissner B, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121:3161–3164. doi: 10.1182/blood-2013-01-478834. [DOI] [PubMed] [Google Scholar]
- 224.Lott ST, et al. DEAR1 is a dominant regulator of acinar morphogenesis and an independent predictor of local recurrence-free survival in early-onset breast cancer. PLoS medicine. 2009;6:e1000068. doi: 10.1371/journal.pmed.1000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tan YH, et al. CBL is frequently altered in lung cancers: its relationship to mutations in MET and EGFR tyrosine kinases. PloS one. 2010;5:e8972. doi: 10.1371/journal.pone.0008972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Williams RD, et al. Subtype-specific FBXW7 mutation and MYCN copy number gain in Wilms’ tumor. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:2036–2045. doi: 10.1158/1078-0432.CCR-09-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Yokobori T, et al. Copy number loss of FBXW7 is related to gene expression and poor prognosis in esophageal squamous cell carcinoma. International journal of oncology. 2012;41:253–259. doi: 10.3892/ijo.2012.1436. [DOI] [PubMed] [Google Scholar]
- 228.Milne AN, et al. Loss of CDC4/FBXW7 in gastric carcinoma. Cellular oncology : the official journal of the International Society for Cellular Oncology. 2010;32:347–359. doi: 10.3233/CLO-2010-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Iwatsuki M, et al. Loss of FBXW7, a cell cycle regulating gene, in colorectal cancer: clinical significance. International journal of cancer Journal international du cancer. 2010;126:1828–1837. doi: 10.1002/ijc.24879. [DOI] [PubMed] [Google Scholar]
- 230.Martinez VD, Vucic EA, Pikor LA, Thu KL, Hubaux R, Lam WL. Frequent concerted genetic mechanisms disrupt multiple components of the NRF2 inhibitor KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex in thyroid cancer. Molecular cancer. 2013;12:124. doi: 10.1186/1476-4598-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Xie X, et al. Evaluating the clinical feasibility: The direct bisulfite genomic sequencing for examination of methylated status of E3 ubiquitin ligase RNF180 DNA promoter to predict the survival of gastric cancer. Cancer biomarkers : section A of Disease markers. 2015 doi: 10.3233/CBM-150466. [DOI] [PubMed] [Google Scholar]
- 232.Deng J, et al. Methylation of CpG sites in RNF180 DNA promoter prediction poor survival of gastric cancer. Oncotarget. 2014;5:3173–3183. doi: 10.18632/oncotarget.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wang Y, et al. CHIP/Stub1 functions as a tumor suppressor and represses NF-kappaB-mediated signaling in colorectal cancer. Carcinogenesis. 2014;35:983–991. doi: 10.1093/carcin/bgt393. [DOI] [PubMed] [Google Scholar]
- 234.Sakata M, et al. Methylation of the HACE1 gene is frequently detected in hepatocellular carcinoma. Hepato-gastroenterology. 2013;60:781–783. doi: 10.5754/hge10439. [DOI] [PubMed] [Google Scholar]
- 235.Anglesio MS, et al. Differential expression of a novel ankyrin containing E3 ubiquitin-protein ligase, Hace1, in sporadic Wilms’ tumor versus normal kidney. Human molecular genetics. 2004;13:2061–2074. doi: 10.1093/hmg/ddh215. [DOI] [PubMed] [Google Scholar]
- 236.Hibi K, et al. Aberrant methylation of the HACE1 gene is frequently detected in advanced colorectal cancer. Anticancer research. 2008;28:1581–1584. [PubMed] [Google Scholar]
- 237.Sakata M, et al. Methylation of HACE1 in gastric carcinoma. Anticancer research. 2009;29:2231–2233. [PubMed] [Google Scholar]
- 238.Agirre X, et al. Abnormal methylation of the common PARK2 and PACRG promoter is associated with downregulation of gene expression in acute lymphoblastic leukemia and chronic myeloid leukemia. International journal of cancer Journal international du cancer. 2006;118:1945–1953. doi: 10.1002/ijc.21584. [DOI] [PubMed] [Google Scholar]
- 239.Chen C, et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene. 2007;26:2386–2394. doi: 10.1038/sj.onc.1210021. [DOI] [PubMed] [Google Scholar]
- 240.Chen C, Zhou Z, Ross JS, Zhou W, Dong JT. The amplified WWP1 gene is a potential molecular target in breast cancer. International journal of cancer Journal international du cancer. 2007;121:80–87. doi: 10.1002/ijc.22653. [DOI] [PubMed] [Google Scholar]
- 241.Kwei KA, et al. SMURF1 amplification promotes invasiveness in pancreatic cancer. PloS one. 2011;6:e23924. doi: 10.1371/journal.pone.0023924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Birnbaum DJ, et al. Genome profiling of pancreatic adenocarcinoma. Genes, chromosomes & cancer. 2011;50:456–465. doi: 10.1002/gcc.20870. [DOI] [PubMed] [Google Scholar]
- 243.Chen LC, et al. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer research. 1998;58:3677–3683. [PubMed] [Google Scholar]
- 244.Yasui K, et al. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology. 2002;35:1476–1484. doi: 10.1053/jhep.2002.33683. [DOI] [PubMed] [Google Scholar]
- 245.Hung MS, et al. Cul4A is an oncogene in malignant pleural mesothelioma. Journal of cellular and molecular medicine. 2011;15:350–358. doi: 10.1111/j.1582-4934.2009.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Shinomiya T, et al. Comparative genomic hybridization of squamous cell carcinoma of the esophagus: the possible involvement of the DPI gene in the 13q34 amplicon. Genes, chromosomes & cancer. 1999;24:337–344. [PubMed] [Google Scholar]
- 247.Saigusa K, et al. Overexpressed Skp2 within 5p amplification detected by array-based comparative genomic hybridization is associated with poor prognosis of glioblastomas. Cancer science. 2005;96:676–683. doi: 10.1111/j.1349-7006.2005.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]