

TEXT
H. pylori infects half of the world population and the prevalence varies widely in different parts of the world with average rates of 40%-50% in western countries, rising to more than 90% in the developing world[1,2]. Compelling evidence from epidemiological and histopathological studies has linked H. pylori infection to the subsequent development of gastric carcinogenesis[3]. Furthermore, Watanabe and colleagues recently induced gastric adenocarcinoma in 37% of orally infected Mongolian gerbils, which were preceded with a series of premalignant changes in gastric mucosa of these gerbils[4]. However, in spite of the established causal relationship between H. pylori infection and gastric carcinogenesis, the underlying mechanisms remain unknown.
Disturbances in cell turnover in the gastrointestinal tract are believed to predispose to cancer development, and until recently, these changes were considered to be a marker of increased cancer risk[5]. It is clear that this organism is the main cause of chronic gastritis and capable of modifying epithelial cell turnover within gastric glands and in culture gastric epithelial cells, by influencing the balance between cell proliferation and apoptosis[6-9]. We and others have studied the effect of H. pylori infection on gastric epithelial cell turnover and found that patients infected with H. pylori had significantly higher proliferation rates as compared with uninfected controls[6,8-12]. The density of H. pylori may be one of the important determinants as we found that H. pylori at low inocula, stimulates cell proliferation, but at higher inocula (bacterium to cell ratio > 100), it causes a time- and concentration-dependent reduction of cell number and a marked increase in apoptosis and cell cycle arrest at G1 phase[8].
Antioxidant vitamins C, E and β-carotene affect cell growth in various human cells directly or through their a ntioxidant properties[13,14]. Our in vitro studies showed that the above vitamins can also significantly inhibit gastric cancer cell proliferation and induce apoptosis[15]. Investigations on gastric vitamins in patients with and without H. pylori infection suggest that H. pylori infection affects the concentrations of vitamin C, E and β-carotene in the stomach and the CagA+ strains have an even greater ability to reduce gastric juice vitamin C levels[16,17].
Although many factors may be related to H. pylori associated gastric carcinogenesis, the underlying molecular mechanisms are still unknown. However, many mediators and signal transduction pathways are involved in the regulation of gastric epithelial cell homeostasis, some of which may determine the final outcome of H. pylori infection. Understanding the molecular basis of H. pylori associated gastric carcinogenesis is important for determining prognosis, prevention and treatment of H. pylori infection. This review examines the possible molecular mechanisms responsible for H. pylori associated gastric carcinogenes is.
BACTERIAL VIRULENCE FACTORS
Although many factors contribute to H. pylori virulence, few have been directly related to gastric carcinogenesis[18]. Most strains of H. pylori from patients with intestinal-type gastric cancer or with atrophic gastritis are type I and secrete vacuolating cytotoxin (VacA) and carry CagA, a gene that encodes an immunodominant protein of unknown function, whereas many of the strains from asymptomatically infected persons lack this gene[19-21]. It has been shown that patients harbouring CagA+ strains have significantly higher gastric epithelial proliferation rates than patients infected with CagA- strains, but the apoptotic index in patients infected with CagA+ strains are lower than in patients infected with CagA- strains. Increased cell proliferation in the absence of a corresponding increase in apoptosis may explain the increased gastric cancer risk associated with infection by CagA+ strains[10]. Although there is no direct evidence suggesting that the CagA or VacA protein is carcinogenic, the enhanced inflammation and marked reduction of gastric juice vitamin C levels in CagA+ strain infected patients may ply a role in H. pylori associated carcinogenesis[16].
H. pylori expresses a powerful urease enzyme, which catalyses the conversion of urea to ammonia. Individuals with H. pylori infection have higher ammonia concentrations in gastric juice than uninfected controls[22]. A series of studies have demonstrated that concentrations of ammonia comparable to those found in gastric juice of infected individuals can cause gastric atrophy in rats, increase epithelial cell proliferation, and act as a promoter in the methyl-N’nitro-N-nitrosoguanidine (MNNG) rat model of gastric cancer[23-28].
Furthermore, H. pylori phospholipases, proteases and oxidases have been shown to cause degradation of many molecules, such as phospholipids in bio-membranes, transforming growth factor-β (TGF-β) and vitamin C, which are important in preventing carcinogenesis[16,29-31].
OXIDATIVE DNA DAMAGE AND p53
Mutation of the p53 tumor suppressor gene is the most common alteration found in a variety of human tumor cells and is considered to be one of the steps leading to the neoplastic state. The importance of p53 protein may be due to its effect on cell cycle progression, including cell proliferation and apoptosis, in response to cell DNA damage[32]. The activated p53 protein can affect the expression of a number of genes, including the cyclin-dependent kinase inhibitor (CKI) p21, which regulates G1 cell cycle check point and bax, a gene involved in apoptosis. Therefore, the protein can either prevent the cell from entering the S phase until the DNA damage is repaired and/or can turn on the apoptotic pathways to destroy an abnormal cell[33].
The p53 gene abnormalities in gastric cancer are usually point mutations or allelic deletions leading to over-expression of the protein, loss of p53 function and with resulting defects in the protective pathways of cell cycle arrest and apoptosis. Furthermore, the increased p53 expression and gene mutations have also been reported in gastric premalignant mucosa, such as dysplasia, atrophy or even the mucosa without obvious abnormality, suggesting that p53 function is affected from the early stage of gastric carcinogenesis[34].
The role of p53 in H. pylori associated carcinogenesis is still unclear, but some evidence suggests that it may be protective in this process. In p53 knockout mice, atrophic gastritis developed in 2 of 4 animals infected with H. felis within 3 months including one which developed moderate dysplasia. In contrast, these changes were not seen in any of the control animals, suggesting that lack of functional p53 accelerated carcinogenesis in experimental Helicobacter infection[35]. Data from Fox and colleagues further support the protective role of p53. They examined the effect of infection with H. felis in heterozygous mice deficient in one p53 allele[36]. One year after infection, the wild-type and p53 heterozygous mice both showed severe adenomatous and cystic hyperplasia of the surface foveolar epithelium. However, infected p53 heterozygous mice had a higher proliferative index than the infected wild-type mice.
Whether H. pylori and its associated inflammation induces p53 mutations or affects the activity of the protein is not clear, but p53 function may be defective at an early stage in H. pylori associated gastric carcinogenesis. Some studies have reported increased expression of p53 protein in gastric mucosa infected with H. pylori. However, the enhanced p53 expression failed to have any effect on gastric epithelial cell proliferation or apoptosis, and there appeared to be a positive relationship between the accelerated cell turnover and p53 over-expression. The accumulation of p53 was also not associated with expression of the CDI p21, a down-stream effector of p53[37,38]. We have recently initiated a study investigating the role of p53 in H. pylori infected gastric cell lines and found that H. pylori associated apoptosis is independent to p53 status of gastric cells[39]. These findings suggest that p53 function is defective in H. pylori infected mucosa. There are several mechanisms which may lead to the loss of p53 function. Firstly, recent studies suggest that p53 mediated apoptosis is suppressed by signals from growth factors, such as inte rleukin-6 (IL-6), interferon-γ (IFN-γ) and protein kinase C, which are shown to be up-regulated by H. pylori infection[40-42]. Secondly, it is likely that H. pylori and its associated inflammatory responses cause p53 gene mutation. H. pylori infection induces increased production of reactive oxygen metabolites (ROMs) by persistent inflammatory cell infiltration in gastric mucosa[43-45]. Many studies have indicated that ROMs can directly interact with genomic DNA and cause damage in specific genes that control cell growth and differentiation[46-48]. Furthermore, it has been reported that intact H. pylori, as well as isolated cellular components, stimulate nitric oxide (NO) synthesis[49-51]. High concentrations of NO induce wild-type p53 protein accumulation[52,53], and the NO-related deamination of DNA has been reported to cause GC-AT transitions, which are frequently found in p53 mutations in gastric cancer[23,54]. In addition, H. pylori possesses several proteases which may directly affect p53 activity though there is no direct experimental evidence of a relationship between these proteases and p53 protein[55].
BCL-2 AND OTHER APOPTOSIS RELATED MOLECULES
Bcl-2 protein is a part of a large group of proteins encoded by specific genes belonging to the bcl-2 family, which are known to play an important role in regulation of apoptosis. Some of these proteins (bcl-2 and bcl-xL) support survival, whereas others (bax, bad, bcl-x5) are apoptosis inducers[56]. Over-expression of bax and bak proteins encoded by the two pro-apoptotic members of the bcl-2 gene family has been associated with H. pylori infection and to induce apoptosis in the AGS gastric epithelial cell line and in gastric mucosal biopsies from patients colonized by H. pylori[57]. In contrast, the expression of bcl-2 protein was not affected or even suppressed by this organism[37]. However, over-expression of bcl-2 with abnormal distribution of apoptotic cells along the glands, which are usually found at the extremities of normal gastric glands, has been described in both intestinal metaplasia and gastric dysplasia, which occur as a result of long-term H. pylori infection[58,59].
Several other molecules may also play a role in H. pylori associated apoptosis. Treatment of gastric cells with TNF-α and IFN-γ markedly potentiate H. pylori induced apoptosis[60]. Rudi et al[61] have recently reported that H. pylori up-regulates the expression of the CD95 (APO-1/FAS) and CD95 ligand (CD95L), which are involved in initiating apoptosis. In fact, the enhanced CD95 expression observed in this study was associated with increased rates of apoptosis in gastric epithelial cell lines and in gastric mucosa. The importance of this study is not only the demonstration of the involvement of CD95 mediated apoptotic pathway in H. pylori associated apoptos is, but also the expression of CD95L mRNA on surface epithelium and pyloric gland cells of H. pylori infected mucosa at levels comparable to those found on lamina propria lymphocytes. CD95L is normally expressed by activated T cells and recently it has been found that many tumors, including gastric adenocarcinomas express CD95L[62]. CD95L can induce apoptosis of activated immunocytes and is thought to contribute to tumor immune escape[62]. The enhanced expression of CD95L mRNA in H. pylori infected gastric mucosa may suppress normal immune responses by inducing immunocyte apoptosis, which may further potentia te genetic instability due to the defect in the DNA damage-p53 mediated protective pathway.
TELOMERASE ACTIVITY
Activity of telomerase, which synthesizes the telomeric DNA to replaces the loss that occurs at each cell division, is suppressed in most normal human somatic cells but induced in most human cancers. Normal human cells progressively lose telomere sequences due to the lack of telomerase activity. In contrast, most immortalized cell lines and malignant human tumors appear to maintain constant telomere length via the activation of telomerase[63,64]. Reactivation of telomerase is thought to be an important step in carcinogenesis.
Expression of human telomerase RNA (hTR) and telomerase activity have been studied in gastric cancer and corresponding non-cancerous mucosa[65]. Telomerase activity was detected in 23 of 26 carcinoma tissues. Although all tumor specimens and non-cancerous mucosa expressed various levels of hTR, 21 cases expressed hTR at a higher level in the tumor than that in the adjacent normal mucosa. Nine of 26 non-cancerous mucosa showed telomerase activity and all of them contained intestinal metaplasia. The incidence of telomerase-positive mucosa in grade II intestinal metaplasia was significantly higher than that in mucosa with grade I intestinal metaplasia or without intestinal metaplasia, whereas hTR over-expression was found both in mucosa with and without intestinal metaplasia regardless to their grades. The level of hTR expression and telomerase positivity was shown to increase in parallel with the degree of H. pylori infection. These results suggest that H. pylori infection may be a strong trigger for hTR over-expression in intestinal metaplasia, and this may lead to telomerase reactivation[65,66].
Recently, Chin and colleagues examined the interaction between telomere dysfunction and p53 in cells and organs of telomerase-deficient mice. Coincident with severe telomere shortening and associated genomic instability, p53 is activated, leading to growth arrest and/or apoptosis. Deletion of p53 significantly attenuated the adverse effects of telomere dysfunction, but only during the earliest stages of genetic crisis. Correspondingly, the loss of telomere function and p53 deficiency cooperated to initiate the transformation process[67]. These findings suggest that p53 deficiency and telomerase reactivation in H. pylori infected mucosa may play an important role in H. pylori associated gastric carcinogenesis.
HOST GENETIC FACTORS
Beales et al[68] reported that the frequency of DQ 5, one of the D-related human leukocyte antigen (HLA) molecules, was significantly higher in individuals with gastric atrophy or metaplasia than in those without gastric atrophy or intestinal metaplasia and in uninfected individuals. Azuma and colleagues have studied the relationship between H. pylori infection and the genotyping of another D-type HLA molecule, DQA1 in 82 gastric adenocarcinoma patients and 167 unrelated controls. They found that the allele frequency of DQA1 was significantly lower in H. pylori positive atrophic gastritis group than in H. pylori positive superficial gastritis and normal control groups. In addition, the allele frequency of DQA1 also was significantly lower in H. pylori positive intestinal type gastric adenocarcinoma group than in normal control, H. pylori positive superficial gastritis, and diffuse type gastric adenocarcinoma groups[69].
The importance of host genetic factors in determining the outcome of H. pylori infection was further demonstrated by Sakagami and colleagues. They assessed H. pylori infection in different strains of mice and found that the level of bacterial colonisation, the severity of gastritis and the development of gastric atrophy varied within these mice. For example, in infected SJL, C3H/He, DBA/2, and C57BL/6 mice, moderate to severe chronic active gastritis was observed only in the body of the stomach, which increased in severity over time with specialised cells in the body glands being replaced. As the severity of this damage in the body increased and atrophic changes were seen, the level of bacterial colonisation of the antrum decreased. In contrast, in BALB/c and CBA mice, there was only mild gastritis in the antrum, no remarkable changes were detected in the gastric body mucosa, and no atrophy was seen over time. In both these strains of mice, heavy bacterial colonisation was seen, which tended to increase over the period of the experiment[70]. These findings suggest that the host genetic factors are important in H. pylori associated gastric carcinogenesis.
SUMMARY
Many molecules are involved in H. pylori associated gastric carcinogenesis. However, how the organism and its associated inflammation, interact with these molecules in gastric mucosa to induce carcinogenesis is still unknown. Over many years, H. pylori infected mucosa may experience sequential exposure to “damage-regeneration” (Figure 1). Following the long-standing repeated damage-regenerate cycle, gastric atrophy and intestinal metaplasia gradually develop, which finally results in adenocarcinoma. During this process, the loss of p53 function may play an important role. As mentioned above, H. pylori infection may cause deficiency of p53 function and subsequently, this may not only lead to defects in the DNA damage-p53 mediated protective pathway, but the mutated p53 protein may also provide a possible selective advantage for tumor cell proliferation by attenuating apoptosis[71]. Furthermore, H. pylori infection has been shown to induce the reactivation of telomerase and to cause telomere dysfunction, which may cooperate with p53 deficiency to accelerate carcinogenesis[67]. In early stages of H. pylori infection, CD95 and bax mediated apoptosis may play an important role in eliminating damaged DNA or gene mutated cells, thereby maintaining genetic stability. However, H. pylori infection induces CD95L expression, which may suppress host immune responses by causing immunocyte apoptosis. Therefore, as shown in Figure 2, the loss of p53 function, reactivation of telomerase activity, inhibition of host immune responses, together with host genetic factors, may play important roles in the development of H. pylori associated carcinogenesis.
Figure 1.

Long-term H. pylori infection may induce a repeated “damage-regeneration” process which gradually leads to gastric atrophy, intestinal metaplasia and finally carcinoma. A. H. pylori infected gastric mucosa with inflammatory cell infiltration. B. Gastric epithelial cells are damaged, become apoptotic and then are regenerated, with enhanced inflammatory cell infiltration and disturbance of mucus layer. C. Atrophy and intestinal metaplasia develop; fibrosis and thinning of the lamina propria; finally H. pylori is lost due to inhospitable mucosa. D. Gastric carcinoma is induced.
Figure 2.
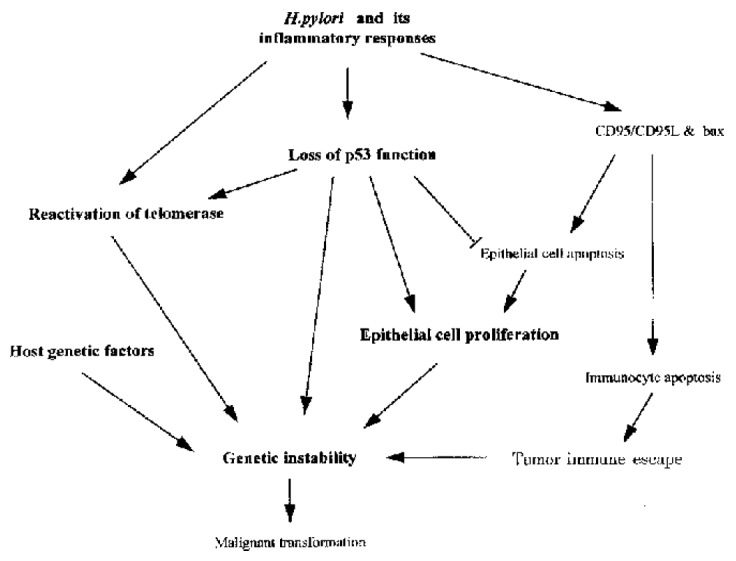
Possible molecular mechanisms of H. pylori associated carcinogenesis. H. pylori and its associated inflammatory responses cause reactivation of telomerase, the loss of p53 function and also induce CD95 and bax mediated apoptosis. CD95L produced by gastric epithelial cells m ay also cause intra-mucosal immunocyte apoptosis, which could facilitate tumor immune escape. However, mutated p53 may attenuate epithelial cell apoptosis, providing a possible selective advantage for tumor cell proliferation. Furthermore , p53 deficiency may cooperate with telomere dysfunction to accelerate carcinogenesis. All of these changes, together with host genetic factors may play important roles in the development of H. pylori associated carcinogenesis.
Footnotes
Edited by Ma JY
References
- 1.Farthing MJ. Helicobacter pylori infection: an overview. Br Med Bull. 1998;54:1–6. doi: 10.1093/oxfordjournals.bmb.a011661. [DOI] [PubMed] [Google Scholar]
- 2.Zhang ZW, Xing YY, Li SQ, Huang ZH. Seroepidemiological investigation of Helicobacter pylori infection in a Beijing population (in Chinese) Beijing Yixue. 1992;23:250–252. [Google Scholar]
- 3.Zhang ZW Schistosomes. liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in mongolian gerbils. Gastroenterology. 1998;115:642–648. doi: 10.1016/s0016-5085(98)70143-x. [DOI] [PubMed] [Google Scholar]
- 5.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 6.Zhang ZW, Patchett SE, Farthing MJG. H. pylori associated reduc-tion in gastric cell number is associated with increased apoptosis rate, but not cell DN A synthesis rate (abstract) Gastroenterology. 1999;116:G1759. [Google Scholar]
- 7.Correa P, Miller MJ. Carcinogenesis, apoptosis and cell proliferation. Br Med Bull. 1998;54:151–162. doi: 10.1093/oxfordjournals.bmb.a011665. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZW, Patchett SE, Farthing MJ. Role of Helicobacter pylori and p53 in regulation of gastric epithelial cell cycle phase progression. Dig Dis Sci. 2002;47:987–995. doi: 10.1023/a:1015069519610. [DOI] [PubMed] [Google Scholar]
- 9.Patchett SE, Katelaris PH, Zhang ZW, Alstead EM, Domizio P, Farthing MJ. Ornithine decarboxylase activity is a marker of premalignancy in longstanding Helicobacter pylori infection. Gut. 1996;39:807–810. doi: 10.1136/gut.39.6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peek RM, Moss SF, Tham KT, Pérez-Pérez GI, Wang S, Miller GG, Atherton JC, Holt PR, Blaser MJ. Helicobacter pylori cagA+ strains and dissociation of gastric epithelial cell proliferation from apoptosis. J Natl Cancer Inst. 1997;89:863–868. doi: 10.1093/jnci/89.12.863. [DOI] [PubMed] [Google Scholar]
- 11.Cahill RJ, Xia H, Kilgallen C, Beattie S, Hamilton H, O'Morain C. Effect of eradication of Helicobacter pylori infection on gastric epithelial cell proliferation. Dig Dis Sci. 1995;40:1627–1631. doi: 10.1007/BF02212681. [DOI] [PubMed] [Google Scholar]
- 12.Lynch DA, Mapstone NP, Clarke AM, Sobala GM, Jackson P, Morrison L, Dixon MF, Quirke P, Axon AT. Cell proliferation in Helicobacter pylori associated gastritis and the effect of eradication therapy. Gut. 1995;36:346–350. doi: 10.1136/gut.36.3.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Poppel G, van den Berg H. Vitamins and cancer. Cancer Lett. 1997;114:195–202. doi: 10.1016/s0304-3835(97)04662-4. [DOI] [PubMed] [Google Scholar]
- 14.Brigelius-Flohé R, Flohé L. Ascorbic acid, cell proliferation, and cell differentiation in culture. Subcell Biochem. 1996;25:83–107. doi: 10.1007/978-1-4613-0325-1_5. [DOI] [PubMed] [Google Scholar]
- 15.Zhang ZW, Wilks S, Davies D, Carnaby S, Allen P, Poschet JF, Patchett SE, Farthing MJG. Comparative effects of Antioxidant vitamins C, E and β -carot ene on cell proliferation and apoptosis in a gastric epithelial cell line (abstr act). Gastroenterology 1998, 114: A709 [Google Scholar]
- 16.Zhang ZW, Patchett SE, Perrett D, Katelaris PH, Domizio P, Far-thing MJ. The relation between gastric vitamin C concentrations, mucosal histology, and CagA seropositivity in the human stomach. Gut. 1998;43:322–326. doi: 10.1136/gut.43.3.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang ZW, Patchett SE, Perrett D, Domizio P, Farthing MJG. The influen ce of chronic Helicobacter pylori infection on gastric mucosal and luminal β-carotene concentrations (abstract) Gastroenterology. 1996;110:A619. [Google Scholar]
- 18.Covacci A, Telford JL, Del Giudice G, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 19.Basso D, Navaglia F, Brigato L, Piva MG, Toma A, Greco E, Di Mario F, Galeotti F, Roveroni G, Corsini A, et al. Analysis of Helicobacter pylori vacA and cagA genotypes and serum antibody profile in benign and malignant gastroduodenal diseases. Gut. 1998;43:182–186. doi: 10.1136/gut.43.2.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301. doi: 10.1136/gut.40.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudi J, Kolb C, Maiwald M, Zuna I, von Herbay A, Galle PR, Stremmel W. Serum antibodies against Helicobacter pylori proteins VacA and CagA are associated with increased risk for gastric adenocarcinoma. Dig Dis Sci. 1997;42:1652–1659. doi: 10.1023/a:1018849112533. [DOI] [PubMed] [Google Scholar]
- 22.Marshall BJ, Langton SR. Urea hydrolysis in patients with Campylobacter pyloridis infection. Lancet. 1986;1:965–966. doi: 10.1016/s0140-6736(86)91060-3. [DOI] [PubMed] [Google Scholar]
- 23.Tsuji S, Tsujii M, Sun WH, Gunawan ES, Murata H, Kawano S, Hori M. Helicobacter pylori and gastric carcinogenesis. J Clin Gastroenterol. 1997;25 Suppl 1:S186–S197. doi: 10.1097/00004836-199700001-00030. [DOI] [PubMed] [Google Scholar]
- 24.Tsujii M, Kawano S, Tsuji S, Ito T, Nagano K, Sasaki Y, Hayashi N, Fusamoto H, Kamada T. Cell kinetics of mucosal atrophy in rat stomach induced by long-term administration of ammonia. Gastroenterology. 1993;104:796–801. doi: 10.1016/0016-5085(93)91015-a. [DOI] [PubMed] [Google Scholar]
- 25.Tsujii M, Kawano S, Tsuji S, Takei Y, Tamura K, Fusamoto H, Kamada T. Mechanism for ammonia-induced promotion of gastric carcinogenesis in rats. Carcinogenesis. 1995;16:563–566. doi: 10.1093/carcin/16.3.563. [DOI] [PubMed] [Google Scholar]
- 26.Tsujii M, Kawano S, Tsuji S, Nagano K, Ito T, Hayashi N, Fusamoto H, Kamada T, Tamura K. Ammonia: a possible promotor in Helicobacter pylori-related gastric carcinogenesis. Cancer Lett. 1992;65:15–18. doi: 10.1016/0304-3835(92)90207-c. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki H, Seto K, Mori M, Suzuki M, Miura S, Ishii H. Monochloramine induced DNA fragmentation in gastric cell line MKN45. Am J Physiol. 1998;275:G712–G716. doi: 10.1152/ajpgi.1998.275.4.G712. [DOI] [PubMed] [Google Scholar]
- 28.Matsui T, Matsukawa Y, Sakai T, Nakamura K, Aoike A, Kawai K. Ammonia inhibits proliferation and cell cycle progression at S-phase in human gastric cells. Dig Dis Sci. 1997;42:1394–1399. doi: 10.1023/a:1018837920769. [DOI] [PubMed] [Google Scholar]
- 29.Dorrell N, Martino MC, Stabler RA, Ward SJ, Zhang ZW, McColm AA, Farthing MJ, Wren BW. Characterization of Helicobacter pylori PldA, a phospholipase with a role in colonization of the gastric mucosa. Gastroenterology. 1999;117:1098–1104. doi: 10.1016/s0016-5085(99)70394-x. [DOI] [PubMed] [Google Scholar]
- 30.Piotrowski J, Slomiany A, Slomiany BL. Suppression of Helicobacter pylori protease activity towards growth factors by sulglycotide. J Physiol Pharmacol. 1997;48:345–351. [PubMed] [Google Scholar]
- 31.Odum L, Andersen LP. Investigation of Helicobacter pylori ascorbic acid oxidating activity. FEMS Immunol Med Microbiol. 1995;10:289–294. doi: 10.1111/j.1574-695X.1995.tb00046.x. [DOI] [PubMed] [Google Scholar]
- 32.Oren M. The involvement of oncogenes and tumor suppressor genes in the control of apoptosis. Cancer Metastasis Rev. 1992;11:141–148. doi: 10.1007/BF00048060. [DOI] [PubMed] [Google Scholar]
- 33.Kastan MB, Kuerbitz SJ. Control of G1 arrest after DNA damage. Environ Health Perspect. 1993;101 Suppl 5:55–58. doi: 10.1289/ehp.93101s555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moss SF. Review article: Cellular markers in the gastric precancerous process. Aliment Pharmacol Ther. 1998;12 Suppl 1:91–109. doi: 10.1111/j.1365-2036.1998.00002.x. [DOI] [PubMed] [Google Scholar]
- 35.Dunn BE, Phadnis SH, Henderson J, Choi H. Induction of gastric dysplas ia by H. felis in p53 deficient mice (abstract) Gut. 1995;37:A40. [Google Scholar]
- 36.Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R, Wang TC. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996;110:155–166. doi: 10.1053/gast.1996.v110.pm8536852. [DOI] [PubMed] [Google Scholar]
- 37.Hibi K, Mitomi H, Koizumi W, Tanabe S, Saigenji K, Okayasu I. Enhanced cellular proliferation and p53 accumulation in gastric mucosa chronically infected with Helicobacter pylori. Am J Clin Pathol. 1997;108:26–34. [PubMed] [Google Scholar]
- 38.Jones NL, Shannon PT, Cutz E, Yeger H, Sherman PM. Increase in proliferation and apoptosis of gastric epithelial cells early in the natural history of Helicobacter pylori infection. Am J Pathol. 1997;151:1695–1703. [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang ZW, Patchett SE, Farthing MJG. P53 signal transduction pathway is not essential for H. pylori associated apoptosis and cell growth inhibition in gastric cells (abstract) Gastroenterology. 1999;116:G0688. [Google Scholar]
- 40.Hong M, Lai MD, Lin YS, Lai MZ. Antagonism of p53-dependent apoptosis by mitogen signals. Cancer Res. 1999;59:2847–2852. [PubMed] [Google Scholar]
- 41.Lindholm C, Quiding-Järbrink M, Lönroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. doi: 10.1128/iai.66.12.5964-5971.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terres AM, Pajares JM, Hopkins AM, Murphy A, Moran A, Baird AW, Kelleh er D. Helicobacter pylori disrupts epithelial barrier function in a process inhibited by protein kinase C activators. Infect Immun. 1998;66:2943–2950. doi: 10.1128/iai.66.6.2943-2950.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hahm KB, Lee KJ, Kim JH, Cho SW, Chung MH. Helicobacter pylori infection, oxidative DNA damage, gastric carcinogenesis, and reversibility by rebamipide. Dig Dis Sci. 1998;43:72S–77S. [PubMed] [Google Scholar]
- 44.Farinati F, Cardin R, Degan P, Rugge M, Mario FD, Bonvicini P, Naccarato R. Oxidative DNA damage accumulation in gastric carcinogenesis. Gut. 1998;42:351–356. doi: 10.1136/gut.42.3.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bagchi D, Bhattacharya G, Stohs SJ. Production of reactive oxygen species by gastric cells in association with Helicobacter pylori. Free Radic Res. 1996;24:439–450. doi: 10.3109/10715769609088043. [DOI] [PubMed] [Google Scholar]
- 46.Wei YH, Lu CY, Lee HC, Pang CY, Ma YS. Oxidative damage and mutation to mitochondrial DNA and age-dependent decline of mitochondrial respiratory function. Ann N Y Acad Sci. 1998;854:155–170. doi: 10.1111/j.1749-6632.1998.tb09899.x. [DOI] [PubMed] [Google Scholar]
- 47.Barnett YA, Barnett CR. DNA damage and mutation: contributors to the age-related alterations in T cell-mediated immune responses? Mech Ageing Dev. 1998;102:165–175. doi: 10.1016/s0047-6374(98)00018-9. [DOI] [PubMed] [Google Scholar]
- 48.Olinski R, Jaruga P, Zastawny TH. Oxidative DNA base modifications as factors in carcinogenesis. Acta Biochim Pol. 1998;45:561–572. [PubMed] [Google Scholar]
- 49.Brzozowski T, Konturek PC, Sliwowski Z, Drozdowicz D, Pajdo R, Stachura J, Hahn EG, Konturek SJ. Lipopolysaccharide of Helicobacter pylori protects gastric mucosa via generation of nitric oxide. J Physiol Pharmacol. 1997;48:699–717. [PubMed] [Google Scholar]
- 50.Nagata K, Yu H, Nishikawa M, Kashiba M, Nakamura A, Sato EF, Tamura T, Inoue M. Helicobacter pylori generates superoxide radicals and modulates nitric oxide metabolism. J Biol Chem. 1998;273:14071–14073. doi: 10.1074/jbc.273.23.14071. [DOI] [PubMed] [Google Scholar]
- 51.Shapiro KB, Hotchkiss JH. Induction of nitric oxide synthesis in murine macrophages by Helicobacter pylori. Cancer Lett. 1996;102:49–56. doi: 10.1016/0304-3835(96)04154-7. [DOI] [PubMed] [Google Scholar]
- 52.Ambs S, Hussain SP, Harris CC. Interactive effects of nitric oxide and the p53 tumor suppressor gene in carcinogenesis and tumor progression. FASEB J. 1997;11:443–448. doi: 10.1096/fasebj.11.6.9194524. [DOI] [PubMed] [Google Scholar]
- 53.Forrester K, Ambs S, Lupold SE, Kapust RB, Spillare EA, Weinberg WC, F elley-Bosco E, Wang XW, Geller DA, Tzeng E, et al. Nitric oxide -induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci USA. 1996;93:2442–2447. doi: 10.1073/pnas.93.6.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsuji S, Kawano S, Tsujii M, Takei Y, Tanaka M, Sawaoka H, Nagano K, F usamoto H, Kamada T. Helicobacter pylori extract stimulates inflammatory nitric oxide production. Cancer Lett. 1996;108:195–200. doi: 10.1016/s0304-3835(96)04410-2. [DOI] [PubMed] [Google Scholar]
- 55.Nilius M, Malfertheiner P. Helicobacter pylori enzymes. Aliment Pharmacol Ther. 1996;10 Suppl 1:65–71. doi: 10.1046/j.1365-2036.1996.22164007.x. [DOI] [PubMed] [Google Scholar]
- 56.O'Connor L, Strasser A. The Bcl-2 protein family. Results Probl Cell Differ. 1999;23:173–207. doi: 10.1007/978-3-540-69184-6_9. [DOI] [PubMed] [Google Scholar]
- 57.Chen G, Sordillo EM, Ramey WG, Reidy J, Holt PR, Krajewski S, Reed JC, Blaser MJ, Moss SF. Apoptosis in gastric epithelial cells is induced by Helicobacter pylori and accompanied by increased expression of BAK. Biochem Biophys Res Commun. 1997;239:626–632. doi: 10.1006/bbrc.1997.7485. [DOI] [PubMed] [Google Scholar]
- 58.Clarke MR, Safatle-Ribeiro AV, Ribeiro U, Sakai P, Reynolds JC. bcl-2 protein expression in gastric remnant mucosa and gastric cancer 15 or more years after partial gastrectomy. Mod Pathol. 1997;10:1021–1027. [PubMed] [Google Scholar]
- 59.Lauwers GY, Scott GV, Hendricks J. Immunohistochemical evidence of aberrant bcl-2 protein expression in gastric epithelial dysplasia. Cancer. 1994;73:2900–2904. doi: 10.1002/1097-0142(19940615)73:12<2900::aid-cncr2820731205>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 60.Wagner S, Beil W, Westermann J, Logan RP, Bock CT, Trautwein C, Bleck JS, Manns MP. Regulation of gastric epithelial cell growth by Helicobacter pylori: offdence for a major role of apoptosis. Gastroenterology. 1997;113:1836–1847. doi: 10.1016/s0016-5085(97)70003-9. [DOI] [PubMed] [Google Scholar]
- 61.Rudi J, Kuck D, Strand S, von Herbay A, Mariani SM, Krammer PH, Galle PR, Stremmel W. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest. 1998;102:1506–1514. doi: 10.1172/JCI2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bennett MW, O'connell J, O'sullivan GC, Roche D, Brady C, Kelly J, Collins JK, Shanahan F. Expression of Fas ligand by human gastric adenocarcinomas: a potential mechanism of immune escape in stomach cancer. Gut. 1999;44:156–162. doi: 10.1136/gut.44.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reddel RR. Genes involved in the control of cellular proliferative potential. Ann N Y Acad Sci. 1998;854:8–19. doi: 10.1111/j.1749-6632.1998.tb09887.x. [DOI] [PubMed] [Google Scholar]
- 64.Meyerson M. Telomerase enzyme activation and human cell immortalization. Toxicol Lett. 1998;102-103:41–45. doi: 10.1016/s0378-4274(98)00278-1. [DOI] [PubMed] [Google Scholar]
- 65.Kuniyasu H, Domen T, Hamamoto T, Yokozaki H, Yasui W, Tahara H, Tahara E. Expression of human telomerase RNA is an early event of stomach carcinogenesis. Jpn J Cancer Res. 1997;88:103–107. doi: 10.1111/j.1349-7006.1997.tb00353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kameshima H, Yagihashi A, Yajima T, Watanabe N, Ikeda Y. Helicobacter pylori infection induces telomerase activity in premalignant lesions. Am J Gastroenterol. 1999;94:547–548. doi: 10.1111/j.1572-0241.1999.00547.x. [DOI] [PubMed] [Google Scholar]
- 67.Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 68.Beales IL, Davey NJ, Pusey CD, Lechler RI, Calam J. Long-term sequelae of Helicobacter pylori gastritis. Lancet. 1995;346:381–382. doi: 10.1016/s0140-6736(95)92263-6. [DOI] [PubMed] [Google Scholar]
- 69.Azuma T, Ito S, Sato F, Yamazaki Y, Miyaji H, Ito Y, Suto H, Kuriyama M, Kato T, Kohli Y. The role of the HLA-DQA1 gene in resistance to atrophic gastritis and gastric adenocarcinoma induced by Helicobacter pylori infection. Cancer. 1998;82:1013–1018. doi: 10.1002/(sici)1097-0142(19980315)82:6<1013::aid-cncr2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 70.Sakagami T, Dixon M, O'Rourke J, Howlett R, Alderuccio F, Vella J, Shimoyama T, Lee A. Atrophic gastric changes in both Helicobacter felis and Helicobacter pylori infected mice are host dependent and separate from antral gastritis. Gut. 1996;39:639–648. doi: 10.1136/gut.39.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishida M, Gomyo Y, Ohfuji S, Ikeda M, Kawasaki H, Ito H. Evidence that expression of a mutated p53 gene attenuates apoptotic cell death in human gastric intestinal-type carcinomas in vivo. Jpn J Cancer Res. 1997;88:468–475. doi: 10.1111/j.1349-7006.1997.tb00405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]