

INTRODUCTION
p16 gene (also known as MTS-1, INK4a, CDKN2A), located on chromosome 9p21, is a G1-specific cell-cycle regulatory gene. It is composed of three exons, which encode 156 amino acids[1]. The gene is frequently inactivated in many human cancers. Unlike other tumor-suppressor genes that are commonly inactivated by point mutations, small homozygous deletions and methylation of the promoter represent the major mechanism of p16 gene inactivation[2,3]. In the Western countries, colorectal carcinoma ranks first among malignant tumors. The mortality from colorectal carcinoma has been rapidly growing in China in the last two to three decades. The genetic alterations involved in this tumor are still unclear. Rare mutation and infrequent deletion of p16 gene in primary colorectal car cinoma has been widely reported[4]. There were a few papers on p16 gene methylation in colorectal carcinoma, but the results were contradictory[5,6]. In this study, we have examined a total of 60 samples of colorectal carcinoma and paired 60 samples of the normal colonic mucosa for methylation by means of PCR-based methylation assay. There is infrequent methylation in the promoter of p16 gene both in colorectal carcinoma and normal colonic mucosa. The methylation status in 5’ CpG island of p16 gene in colorectal carcinoma is not related to the clinical pathologic parameters of these tumors.
MATERIALS AND METHODS
Patients and specimens
The samples were provided by the First and Second Affiliated Hospitals of Zhejiang Medical University and Hangzhou Railway Central Hospital from 1996 to 1998. Colorectal carcinoma specimens (n = 60) and matched normal colorectal mucosa (n = 60) were obtained from 60 patients (38 men and 22 women) with colorectal carcinoma. Their ages ranged from 20 to 78 years with a mean age of 57. The size of the tumors ranged from 1.5 cm to 7.8 cm in diameter.
Template preparation
DNA was isolated by proteinase K digestion and phenol chloroform extraction. The DNA concentration and purity were determined on the ultraviolet ray spectrometer (Pharmacia Bioth Ultrospec 2000). All DNA templates were diluted to 1 μg/μL with TE.
PCR-based methylation assay
Genomic DNA (1 μg) was either overdigested with 20 U three methylation-sensititive restriction enzymes (Hpa II, Sac II and Sma I) or placed in the appropriate buffer without enzyme (control) overnight. A liquots of 120 ng of the digested and non- digested DNA were PCR amplified using primers 5’GAAGAAAGAGGAGGGGGTG-3’ (sense) and 5’GCGCTACCTGATTCCAATTC-3’ (antisense), which corresponded to a part of 5’ noncoding region and a part of the 3’ end of exon 1 of the p16 gene. The undigested DNA of the correspondent samples was also amplified and used as control. A total of 25 μL volume of PCR mixture contained 2 μL of 10 × buffer, 0.8 μL of MgCl2 (25 mM), 2 μL of dNTP (0.2 mM), 2 μL of primer (20 pM), 15 U of Taq polymerase and 2.5 μL of digested DNA. Amplifications were performed in a temperature cycler (MJ Research, Inc., USA) for 1 cycle at 95 °C(5 min), 30 cycles at 95 °C (30 s), 55 °C(30 s) and 72 °C (30 s), and 1 cycle at 72 °C (5 min). After PCR, the reaction mixture was electrophoresed in 1.5% agarose gel and examined to find out whether a 340 bp long product was generated. The sample which is unmethylated and cut by restriction enzyme, has no amplified product while the sample which is methylated and not cut by restriction enzyme, has amplified product.
Statistical analysis
Statistical analysis was made by Chi-square test. The difference was regarded significant when P value is less than 0.05.
RESULTS
Methylation of the p16 gene in colorectal carcinoma and normal colonic mucosa. We have investigated the biopsies of 60 cases of primary colorectal carcinoma and paired 60 cases of normal colonic mucosa for methylation of the p16 gene in Hpa-II, Sac-II and Sma-I sites. All of the undigested control of 60 cases of colorectal carcinoma and 60 cases with normal colonic mucosagot the same PCR products of 340 bp, while the digested samples achieved the different results (Figure 1). In the normal colonic mucosa, at the Hpa-II site in 5’CpG island of p16 gene, 13 (21.6%) of 60 samples showed full methylation and 7 (11.6%) samples partial methylation, at the Sac-II site 28 (46.6%) of 60 showed full methylation and 8 (13.3%) partial methylation and at the Sma-I site, 5 (8.3%) showed full methylation and 4 (6.7%) partial methylation. In the colorectal carcinoma, at the Hpa-II site, 11 (18.3%) samples showed full methylation and 10 (16.7%) partial methylation, at the Sac-II site, 22 (33.3%) full methylation and 6 (10%) partial methylation, and at the Sma-I site, 9 (15%) full methylation and 3 (5%) partial methylation. In comparison of the methylation status of colorectal carcinoma and that of normal colonic mucosa, there was no significant difference (P > 0.05).
Figure 1.
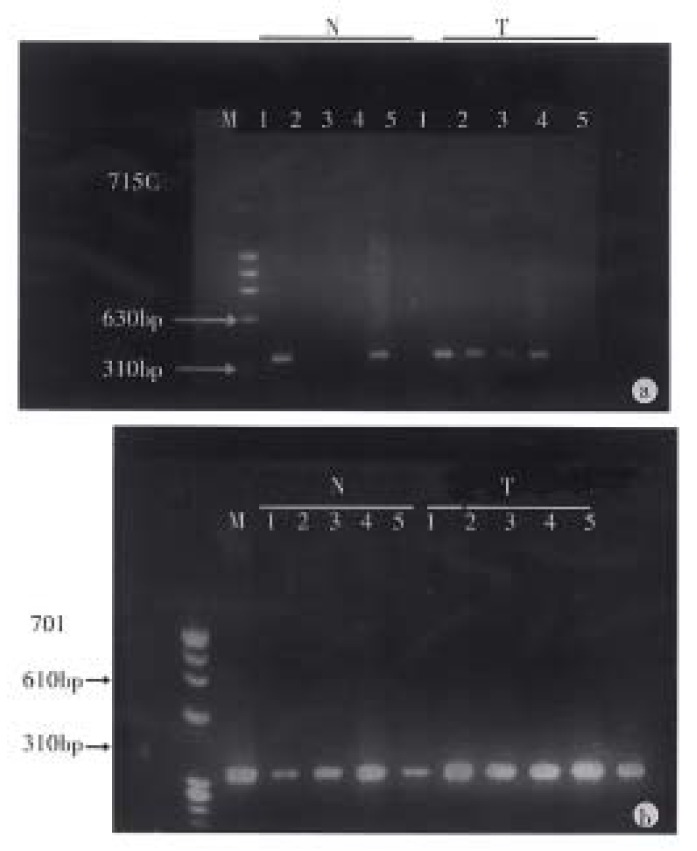
PCR-based detection of 5’ CpG island methylation. N: normal colonic mucosa; T: colorectal carcinoma. A 5’ CpG island sequence (340 bp) of the p16 gene was PCR amplified after sufficient digestion with a methylation sensitive enzyme Hpa-II (2), Sac-II (3) and Sma-I (5) and undige sted control (1, 4). A: case 715c, in the normal colonic mucosa, Hpa-II and Sma-I sites unmethylation (N2, N5) and Sac-II sites partial methylation (N3); in the colorectal carcinoma, Hpa-II and Sac-II site partial methylati on (T2, T3) and Sma-I site unmethylation (T5). B: case 701, in the normal co lonic mucosa, Sac-II site full methylation (N3) and Sma-I and Hpa-II sites partial methylation (N5, N2); in the colorectal carcinoma, Hpa-II, Sac-II and Sma-I sites full methylation (T2, T3, T5).
Statistical analysis revealed a marked significant difference in the methylation status of Hpa-II, Sac-II and Sma-I sites of p16 gene in normal colonic mucosa (χ2 = 28.6, P < 0.01), and the methylation of pro moter in p16 gene tended to be associated with aging (χ2 = 5.64, 0.1 > P > 0.05) (Table 1).
Table 1.
Correlation between methylation of p16 gene and age, enzyme cutting site of normal clonic mucosa
| Number of site | Full methylation | Partial methylation | Un-methylation | |
| Age (yrs) | ||||
| ≤ 40 | 33 | 4 | 3 | 26a |
| 41-60 | 60 | 19 | 8 | 33 |
| > 60 | 87 | 23 | 8 | 56 |
| Site | ||||
| Hpa-II | 60 | 13 | 7 | 40b |
| Sac-II | 60 | 28 | 8 | 24 |
| Sma-I | 60 | 5 | 4 | 51 |
0.1 > P > 0.05,
P < 0.005.
The 60 cases of colorectal carcinoma were divided into groups according to the pathological types and size of cancer and the lymph node metastasis. It was found that the methylation of 5’ CpG island of the p16 gene was not related to the clinicopathologic parameters (Table 2).
Table 2.
Correlation between methylation of p16 gene and clinicopathologic parameters of colorectal carcinoma
| Numbers of site | Full methylation | Partial methylation | Un-methylation | |
| Histological type | ||||
| (adenocacinoma) | ||||
| Well-differentiated | 18 | 6 | 3 | 9a |
| Moderately-differentiated 117 | 26 | 14 | 77 | |
| Poorly-differentiated | 15 | 3 | 2 | 10 |
| Mucinous | 21 | 6 | 0 | 15 |
| Size (cm) | ||||
| ≤ 2.5 | 27 | 5 | 4 | 18a |
| 2.6-4 | 54 | 15 | 5 | 34 |
| >4 | 87 | 21 | 10 | 56 |
| Lymph node metastases | ||||
| With LN metastases | 78 | 20 | 5 | 53a |
| Without LN metastases | 102 | 26 | 14 | 62 |
P > 0.05.
DISCUSSION
There are two major mechanisms of gene inactivation. One is the genetic mechanism, i.e. the aberration of DNA structure such as homozygous deletion or intragenic mutation resulting in the gene inactivation. The other is the epigenetic mechanism, i.e., the methylation of the position 5 of cytosine (C) leading to the lack of gene expression, while the structure and the product of the gene remained unchanged. In higher order eukaryotes, DNA is methylated only at cytosines located 5’ to guanosine in the CpG dinucleotide. This modification has important regulatory effects on gene expression, especially when involving CpG-rich areas known as CpG islands, located in the promoter region of many genes. While almost all gene-associated islands are protected from methylation on autosomal chromosomes, extensive methylation of CpG islands has been associated with transcriptional inactivation of selected imprinted genes and the genes on inactivated X-chromosome of females. Aberrant methylation of normally unmethylated CpG islands has been associated with transcription inactivation of gene[7].
The exon 1 coding sequences of the p16 gene resides within 5’ CpG islands. This area is not methylated in most normal tissues but methylated in many human cancers. The rates of homozygous deletion and intragenic mutation of p16 gene in colorectal carcinoma are very low, but how about the methylation of 5’ CpG islands of p16 gene in this tumor? We examined the methylation status of 340 bp sequence within 5’ CpG islands of p16 gene in colorectal carcinoma and normal colonic mucosa using the PCR-based methylation assay. Our data suggest that there was no significant difference in the methylation status of p16 gene 5’ CpG islands between the colorectal carcinoma and normal colonic mucosa. We further studied the correlation between methylation of p16 gene and clinicopathologic parameters of colorectal carcinoma, which also showed that the methylation of p16 gene did not play an important role in the progress of colorectal carcinoma. Recently, some scholars suggest that the cell proliferation in colorectal carcinoma, unlike in most other tumor types, could bypass p16ink4a-mediated growth arrest[8]. Our experiment, which combined with the reports of low rates of homozygous deletion and intragenic mutation of p16 gene in colorectal carcinoma, support s this opinion.
There are contradictory results on the methylation of the 5’ CpG island of p16 in colorectal carcinoma and normal colonic mucosa reported by Herman et al and Gonzalez-Zulueta et al[5,6]. We found the methylation of the 5’ CpG island of p16 gene in colonic mucosa was very complicated. The full methylation rates of Hpa-II and Sma-I sites in 5’ CpG island of p16 gene were low (21.6% and 8.3%), while that of Sac-II site was high (46.6%). There were significant differences in methylation status of three different sites (P < 0.01. It suggested that using different methylation-sensititive restriction enzymes in different experiments without self control might lead to contradictory results.
The existence of partial methylation status has also been verified. We could see the weak bands (1/2-1/8 of control) after the samples were digested by methylation-sensititive restriction enzymes in some cases. It was confirmed by repeated digestion. This could be explained by the presence of distinct cell subpopulation. The other explanation is the methylation of one allele. As PCR-based technique facilitates the detection of low numbers of methylated alleles, the DNA of stroma may influence the results.
DNA methylation plays an important role in the development, imprinting and aging. It has been reported that the methylation of estrogen receptor gene CpG island links with aging in human colon[9]. In this experiment, the rates of the methylation of promoter in p16 gene increased with aging in colonic mucosa.
Southern hybridization is a classical method for detecting methylation status of specific sequence in specific genes, it needs not only more DNA but also radioactive element. PCR-based methylation assay has provided significant advantages of being markedly more sensitive and highly efficient, and need no radioactive element and expensive instruments.
To avoid false positive results, the key problem of this method is the sufficient digestion of DNA templates with restriction enzymes. In this experiment, we diluted all the DNA sample to 1 μg/μL, and selected the smallest numbers of template which could find clear PCR product by ladder diluted met hod. We digested the DNA with the largest amount of enzyme, prolonged the digestion time (overnight) and repeated digestion to some partial methylation cases to confirm results. We used the digested products without ethanol precipitation as template of PCR to avoid the loss of DNA and adjusted the reaction mixture in accordance with different buffers (Hpa-II, Sac-II with buffer A, Sma-I with buffer J) used in different restriction enzyme digestion. For each case, we amplified the digested template and the undigested one simultanously with the same digestion buffers as control.
Footnotes
Supported by Scientific Foundation of Ministry of Health (No.96-1-349) and Scientific Fund of Hangzhou Railway Branch.
Edited by Ma JY
References
- 1.Kamb A, Gruis NA, Weaver-Feldhaus J, Liu Q, Harshman K, Tavtigian SV, Stockert E, Day RS, Johnson BE, Skolnick MH. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 2.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 3.Lo KW, Cheung ST, Leung SF, van Hasselt A, Tsang YS, Mak KF, Chung YF, Woo JK, Lee JC, Huang DP. Hypermethylation of the p16 gene in nasopharyngeal carcinoma. Cancer Res. 1996;56:2721–2725. [PubMed] [Google Scholar]
- 4.Okamoto A, Demetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennett WP, Forrester K, Gerwin B, Serrano M, Beach DH. Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci USA. 1994;91:11045–11049. doi: 10.1073/pnas.91.23.11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 6.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. Methylation of the 5'CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 1995;55:4531–4535. [PubMed] [Google Scholar]
- 7.Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18:869–882. doi: 10.1093/carcin/18.5.869. [DOI] [PubMed] [Google Scholar]
- 8.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 9.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]