

INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common human malignancies world wide[1,2], and is closely associated with infection of HBV and HCV and contamination of aflatoxin B1[3-6]. Although the molecular mechanisms of hepatocarcinogenesis remain poorly understood, an increasing number of genetic abnormalities have been recognized[7-10], for example, the p16 gene[11,12] the p53 gene[13-18], the E-cadherin gene[19], and the c-myc gene[20]. In relevance to HCC, inactivation of a tumor suppressor gene p16, which is important in the regulation of cell cycling, has recently been described in a significant proportion of patients with HCC[11]. An important mechanism of gene inactivation is aberrant methylation in the promoter region of the gene[11,21,22]. Evidence of p16 hypermethylation has also been documented in several human cancers [21-26]. However, to our knowledge, there were few studies on the p16 gene in HCC. 5-Aza-2’-deoxycytidine (5-Aza-cdR) is thought to act by inhibiting DNA methyltransferase that methylates cytosine residues in eukaryotic DNA[27]. Cedar et al[28] even took it as an experimental tool for demethylation. This drug appears to incorporate into the cellular DNA where it acts as a noncompetitive inhibitor of the maintenance methylase[29]. Methel moieties are thus passively removed when the DNA undergoes replication, and indeed, numerous inactive cellular and viral genes can be turned on following exposure to 5-Aza-cdR[30]. In order to determine whether p16 hypermethylation is involved in hepatocarcinogenesis, we determined the p16 gene protein, mRNA expression and methylation status in HCC cell line HepG2 before and after treatment with 5-Aza-cdR, and observed the growth change.
MATHERIALS AND METHODS
Cell line culture
Human hepatocellular carcinoma cell line HepG2 was obtained from the Laboratory of Gastroenterology, Southwest Hospital. It was cultured in RPMI 1640 supplemented with 100 mL/L fetal bovine serum, 100 KU/L penicillin/ streptomycin and 5% sodium pyruvate at 37 °C in humidified atmosphere of 50 mL/L CO2.
5-Aza-CdR treatment
Cells (approximately 1 × 105/mL) were plated in the vial, and treated with 5 × 10-7M 5-Aza-cdR (Sigma Co.) for 24 h. The medium was changed after 24 h drug treatment and cultured for 9 d, to ensure complete recovery from the immediate toxic effects of 5-Aza-cdR[31]. Untreated cells were analyzed under similar conditions as a control.
Determination of cell cycle profile
Cells (2 × 106/100 mL vial) were plated and treated with 5×10-7 M 5-Aza-cdR. Cells were fixed after 7 d with 700 mL/L ethanol, and stained with propidium iodide (50 mg/L: Crze Co.). DNA content at each cell cycle stage was determined via flow cytometry.
Effects of 5-Aza-CdR on tumorigenicity
HepG2 cells (2 × 106) were injected into the right flank of 4-week-old Balb/c male nude mice 11 d after treatment with 5-Aza-cdR. Untreated cells were injected into the left flank under the same condition as a control. Tumor sizes were measured 4 weeks after injection.
Immunohistochemistry
Cells (1 × 106/3 cm dish) were cultured onto acid-washed 0.8 × 0.8 (mm) coverslips. The cells were fixed in 4% parafomaldehyde for 30 min at room temperature. The slides were washed in phosphate-buffered saline (PBS) three times for 3 minutes. The endogenous peroxidase activity was quenched by 10 minute incubation in methanol with 3% hydrogen peroxide (Zhongshan Chemical Co.). The fixation slides were then cleared with PBS. Nonspecific binding was blocked by applying normal goat serum in humidity chamber in a dilution of 1:10 for 30 minutes. Slides were blotted, and the anti-entire-human p16 antibody (Zhongshan Chemical Co.) (ready to use) as a primary antibody was applied for 1 hour at 37 °C in a humidity chamber. Secondary antibody (rabbit anti-mouse IgG) (Zhongshan Chemical Co.) diluted with PBS and normal human serum (40 μL rabbit anti-mouse antibody, 50 μL normal human serum, and 910 μL PBS) was applied for 1 hour at 37 °C in a humidity chamber. Finally, peroxidase-antiperoxidase conjugate diluted 1:100 in PBS was applied for 1 hour at 37 °C after washing with PBS. Slides were kept in diaminobenzidine tetrahydrochloride for 10 minutes (50 mg in 200 mL PBS with 25 μL of 30% H2O2) (DAB; Zhongshan Chemical Co.). The manufacturer provides the presence (+) and absence (-) controls. The expression of p16 gene was observed under the light microscope and analyzed by the figure analysis method.
RT-PCR
RNA was isolated using Tripure isolation reagent (Boehringer Mannheim Co.). RNA was reverse transcribed according to the manufacturer’s instructions of the access RT-PCR system (Promega Co.). cDNA was amplified using primers specific for both the p16 gene and GAPDH gene, the latter being used as a control. The primers used for p16 amplifications were located in exon 1 and exon 2 of the gene. Their sequences were 5’-AGCCTTCGGCTGACTGGCTGG-3’(sense) and 5’-CTGCCCATCATCATGACCTGGA-3’-(antisense). (Shanghai Sheng Gong Chemical Co., PAG pure). Standard PCR reaction was performed in total volume of 25 μL reaction mixture, including dNTP 0.2 mM, 1 mM each primer 1 (sense) and Primer 2 (antisense), 1 mM MgSO4, 0.1 U/mL of reverse transcriptase AMV, 0.1 U/mL of Taq DNA polymerase and 100 ng RNA. Conditions for p16 amplifications were 94 °C for 2 min, 40 cycles of 94 °C for 30 s; 60 °C for 45 s; and 68 °C for 90 s, followed by incubation at 68 °C for 7 min. RT-PCR amplification product is 428 base pair[bp]. Primers for the GAPDH gene were 5’-CCACCCATGGCAAATTCCATGGCA-3’ (sense), and 5’-TCTAGACGGCAGGTCAGGTCCAC-3’. (antisense) Conditions for GAPDH amplifications were 94 °C for 2 min, 40 cycles of 94 °C for 30 s, 60 °C for 40 s and 68 °C for 60 s, followed by incubation at 68 °C for 7 min. The amplification product is 598 bp. RT-PCR products were resolved on 2% agarose gel by figure analysis method.
PCR-based methylation assay
DNA was isolated with the Tripure isolation reagent. A PCR assay relying on the inability of some restriction enzymes, e.g. Hpa II or Msp I, to cut methylated sequences was used to analyze the methylation status of the second exon of the p16. DNA digests were performed according to the manufacturer’s directions (Boehringer Mannheim Co.). DNA (1 μg) was digested for 2 h with 10 units of enzyme/μg of DNA. 200 ng of the digested DNA were amplified with primers flanking the restriction sites. The primers set used for methylation a nalysis of p16 exon 2 were 5’-CTGCTTGGCGGTGAGGGGG-3’ (sense) and 5’-CCTCACCTGAGGGACCTTC-3’ (antisense). The amplifiction products was 402 bp. Conditions were: 94 °C for 3 min, 35 cycles of 94 °C for 1 min, 57 °C 30 s, and 72 °C for 40 s, followed by incubation at 72 °C for 1 min. PCR products were resolved on 2% agarose gel. To rule out the possibility of incomplete restriction, all samples were digested twice with each of the enzymes in independent experiments. PCR ampifications from each of the duplicate digests were repeated at least twice to ensure reproducibility of the results.
Statistical analysis
t tests were used for statistical analysis. Significance was defined as P < 0.05.
RESULTS
Cell cycle arrest after treatment with 5-Aza-cdR
HepG2 cells treated with 5-Aza-cdR were analyzed by flow cytometry to identify any change in its cell cycle profiles. The treated cells showed an increase in the proportion in G1,from 79.4% to 82.2% and a decrease in S, from 11.9% to 6.4%. Apoptotic rate increased from 6.5% to 14.5%. These results indicated that 5-Aza-cdR may induce cancer cell division and apoptosis.
Effects of 5-Aza-cdR on tumorigenicity
Tumors induced via the injection of untreated HepG2 cells were approximately 1.32 cm3 in size, whereas the tumors induced by HepG2 cells treated with 5-Aza-cdR averaged 0.98 cm3 in size. These demonstrated that the growth-inhibitory effects of 5-Aza-cdR treatment in vivo were still recognizable.
Effect of 5-Aza-cdR on P16 protein expression
The positive 5-Aza-cdR-treated cells showed intense homogenous brown staining of their nucler. After treatment with 5 × 10-7 M 5-A2a-cdR, HepG2 showed a higher expression of P16 protein than untreated cells. We observed under light microscope 5 fields in every slide at random, measured the staining degree of every 124 positive cells by the figure analysis method, IOD was 212.81 ± 62.56 and 495.89 ± 52.12 respectively in the untreated group and the treated group. This showed a significant difference (P < 0.05). Although P16 protein expression alternations are very complicated and still undefined, our data showed that P16 protein expression could be silenced by hyper methylation of p16 gene (Figure 1, Figure 2).
Figure 1.

Immunohistochemical demonstration of p16 protein expression in HepG2 cell line before treatment with 5-Aza-cdR. SABC × 200
Figure 2.
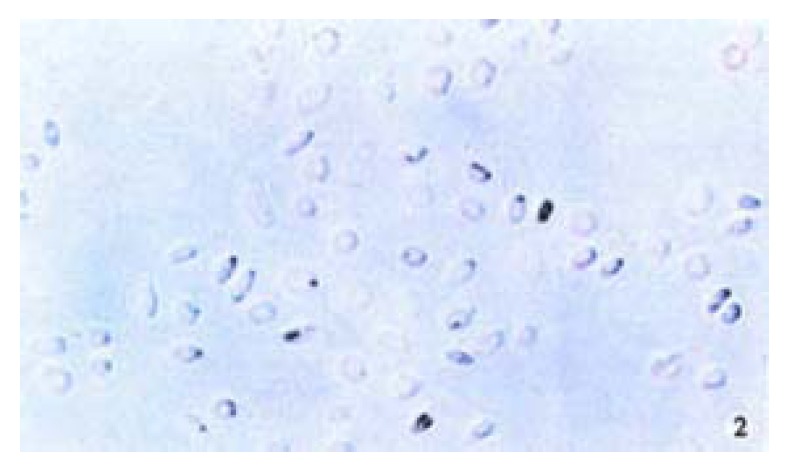
Immunohistochemical demonstration of p16 protein expression in HepG2 cell line after treatment with 5-Aza-cdR. SABC × 200
Effect of 5-Aza-CdR on p16 mRNA expression
Figure 3 shows that expression level of p16 mRNA in HepG2 cells was increased after treated with 5 × 10-7 M 5-aza-cdR. The p16 gene, was selected for this study, because it was known to frequently undergo de novo methylation during tumorigenesis and because 5-Aza-cdR might be highly effective in inducing the expression of the gene inappropriately silenced by de novo methylation[18]. These results demonstrated a strong correlation between hypermethylation of the p16 and transcriptional silencing of this gene.
Figure 3.
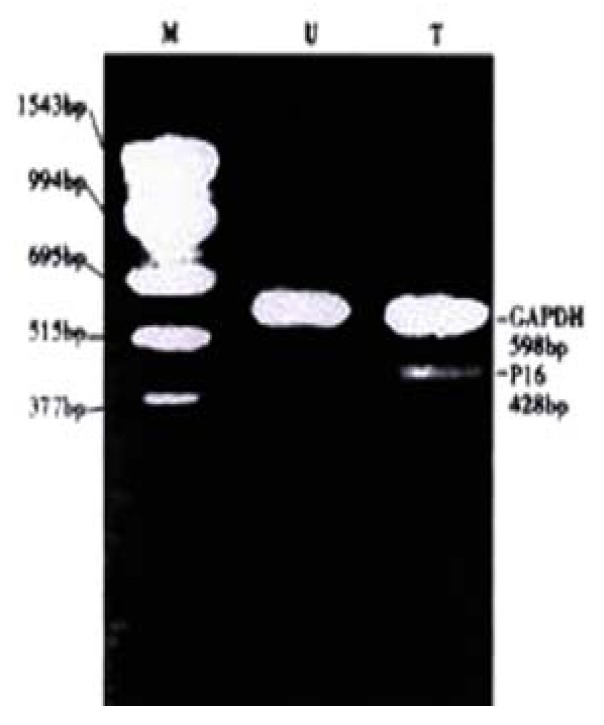
Effects of 5-Aza-cdR on the p16 mRNA expression in HepG2 cell line. Cells were treated with 5 × 10-7 M 5-Aza-cdR for 24 hours; after culturing for 9 d, p16 mRNA were determined by RT-PCR. U: untreated T: treated with 5-Aza-CdR
Effect of 5-Aza-CdR on p16 gene methylation status
Before 5-aza-cdR treatment the p16 exon2 showed hypermethylation, and therefore could not be deaved by loth HpaII and Msp I. It has been known that only the DNA indigested with Hpa II or Msp I can be amplified. After treated with 5-aza-cdR, the p16 gene became demethylated, then could be deaved by loth of the enzymes, and this failed to be emplified. Our experiment demonstrated that after hypermethylation in the p16 exon2 and 5-aza-cdR could enhance demethylation (Figure 4).
Figure 4.
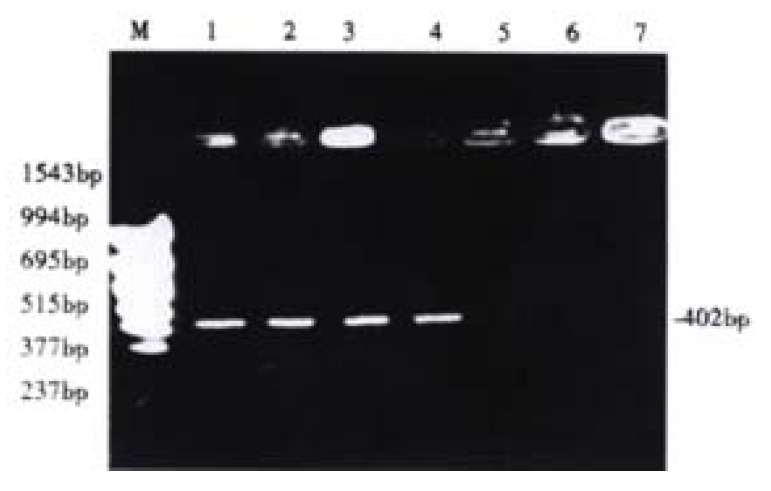
Detection of aberrant methylation of the p16 gene in HCC cell line HepG2. Lane M: molecular weight marker; Lane 1: Neither treated with 5-aza-cdR nor digested with Hpa II or Msp I; Lane 2: Untreated with 5-aza-cdR, but digested with Hpa II; Lane 3: Untreated with 5-Aza-cdR, but digested with Msp I; Lane 4: Treated with 5-aza-cdR, but not digested with Hpa II or Msp I; Lane 5: Treated with 5-aza-cdR, and digested with Hpa II; Lane 6: Treated with 5-aza-cdR, and digested with Msp I.
DISCUSSION
HCC is one of the most frequent human malignances worldwide. A number of tumor suppressor genic abnormalities are being recognized involving in the development of HCC[7-20]. The tumor-suppressor gene, p16, coding for an inhibitor of cyclin-dependant kinase (cdk) 4, was isolated and mapped to chromosome 9p21[32,33], a locus frequently lost in diverse malignancies[34]. The p16 gene product p16 protein was originally isolated as a protein that was associated with cdk complexes[35] and thought to exert a negative type of control on cell proliferation through its binding to cdk4, thereby preventing cdk4 from forming an active complex with cyclin D protein[36]. As one of cancer-associated genes, the p16 gene abnormalities is arousing more and more attention. Germ-line mutations of the p16 gene have been identified in hereditary melanomas[37]. In addition, the gene is homozygously deleted or mutated in uncultured tumors of diverse origin[38], including pancreatic adenocarcinomas[39], melanomas[37], esophageal carcinomas[40,41], gliomas [42] lung cancer[43] and hepatocellular carcinoma[44]. Recently, it has been reported that inactivation of the p16 gene is frequently associated with aberrant DNA met hylation in all common human cancers[26]. However, there have been relatively few studies on expression of p16 in HCC.
Sun et al[45] found that DNA methyltransferase (MTase) mRNA levels were significantly higher in HCC and HCC cell lines (including HepG2 cell line), but they did not study the related genic methylation. Wong et al[46] demonstrated that aberrant methylation of the p16 gene occurred in 73% of HCC tissues. Hui et al[44] studied six HCC cell lines and found no p16 mRNA and protein in only one of them. After ruling out mutation and homozygous deletion of the p16 gene, they guessed that the silence of p16 gene might be associated with enhanced methylation of p16 gene, although the methylation status was not tested. However, p16 mRNA was detected in the other five HCC cell lines, and they thought that the lack of p16 gene transcription was not a frequent event in HCC cell line. The absence of p16 protein and presence of p16 mRNA expression strongly suggested that inactivation of p16 gene function could occur at the posttranscriptional level, but, the presence of expressions of both p16 mRNA and protein in HepG2, could not be explained. Boyes et al[47] discovered that the inhibition of gene transcription correlated with density of DNA methylation and the length of the promoter region. Hsieh[48] also found that the expression of gene transcription depended on CpG island methylation density; lower levels of methylation yielded a 67% to 90% inhibition of gene expression, higher levels of methylation extinguished gene expression completely. Numerous investigations described reactivation of genes by 5-Aza-cdR by acting on various loci of the inactive X chromosome[49], the VHL gene[50], the E-cadherin gene[51], the estrogen receptor gene[52] and the p16 gene[31]. In order to detect similar methylation changes in HepG2 cell line, we also used 5-Aza-cdR.
In this study, HepG2 cells treated with 5-Aza-cdR, showed demethylation of the p16 gene. The p16 mRNA and protein were all increased dramatically, cell cycle was arrested in G1, apoptototic rate increased and implanted tumor grew more slowly. According to our results, we thought that: p16 hypermethyla tion is associated with HCC cell line; MTase is increased in HepG2 cell line; 5’CpG island of p16 promoter was not methylated completely in this HCC cell line; p16 gene can be reactivated by inhibiting the activities of MTase; p16 methylation may be a common event during the establishment of the cell line in vitro; and p16 gene encodes a cell cycle regulatory protein that belongs to the cyclin-dependent kinase inhibitory protein family and regulates the G1/S phase cell cycle transition, and therefore, reactivated p16 may affect the growth of the HCC cell line. Xiao et al[53,54] found that 17p13.3 CpG island, PYN22.1, Rb gene all showed hypermethylation, we guess that 5-Aza-cdR may reactivate those genes.
The induction of p16 activation by 5-Aza-cdR in vivo and the decreased tumorigenicity in animal experiment suggest the chemotherapeutic potential of 5-Aza-cdR in the management of cancer.
Footnotes
supported partly by the National Natural Science Foundation of China, No.39870344
Edited by Lu HM Proofread by Ma JY
References
- 1.Chen CJ, Yu MW, Liaw YF. Epidemiological characteristics and risk factors of hepatocellular carcinoma. J Gastroenterol Hepatol. 1997;12:S294–S308. doi: 10.1111/j.1440-1746.1997.tb00513.x. [DOI] [PubMed] [Google Scholar]
- 2.Tang ZY. Clinical research of hepatocellular carcinoma in the 21st century. China Natl J New Gastroenterol. 1995;1:2–3. [Google Scholar]
- 3.Yang JM, Wang RQ, Bu BG, Zhou ZC, Fang DC, Luo YH. Effect of HCV infection on expression of several cancer-associated gene products in HCC. World J Gastroenterol. 1999;5:25–27. doi: 10.3748/wjg.v5.i1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng DY, Chen RX, Peng Y, Zheng H, Yan YH. Effect of HCV NS(3) protein on p53 protein expression in hep atocarcinogenesis. World J Gastroenterol. 1999;5:45–46. doi: 10.3748/wjg.v5.i1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Liu H, Zhou Q, Li X. Analysis of point mutation in site 1896 of HBV precore and its detection in the tissues and serum of HCC patients. World J Gastroenterol. 2000;6:395–397. doi: 10.3748/wjg.v6.i3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng ZL, Ma Y. Aflatoxin sufferer and p53 gene mutation in hepatocellular carcinoma. World J Gastroenterol. 1998;4:28–29. doi: 10.3748/wjg.v4.i1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheu JC. Molecular mechanism of hepatocarcinogenesis. J Gastroenterol Hepatol. 1997;12:S309–S313. doi: 10.1111/j.1440-1746.1997.tb00514.x. [DOI] [PubMed] [Google Scholar]
- 8.Sun BH, Zhang J, Wang BJ, Zhao XP, Wang YK, Yu ZQ, Yang DL, Hao LJ. Analysis of in vivo patterns of caspase 3 gene expression in primary hepatocellular carcinoma and its relationship to p21(WAF1) expression and hepatic apoptosis. World J Gastroenterol. 2000;6:356–360. doi: 10.3748/wjg.v6.i3.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts LR, LaRusso NF. Potential roles of tumor suppressor genes and microsatellite instability in hepatocellular carcinogenesis in southern African blacks. World J Gastroenterol. 2000;6:37–41. doi: 10.3748/wjg.v6.i1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martins C, Kedda MA, Kew MC. Characterization of six tumor suppressor genes and microsatellite instability in hepatocellular carcinoma in southern African blacks. World J Gastroenterol. 1999;5:470–476. doi: 10.3748/wjg.v5.i6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaubert P, Gayer R, Zimmermann A, Fontolliet C, Stamm B, Bosman F, Shaw P. Germ-line mutations of the p16INK4(MTS1) gene occur in a subset of patients with hepatocellular carcinoma. Hepatology. 1997;25:1376–1381. doi: 10.1002/hep.510250613. [DOI] [PubMed] [Google Scholar]
- 12.Qin Y, Li B, Tan YS, Sun ZL, Zuo FQ, Sun ZF. Polymorphism of p16INK4a gene and rare mutation of p15INK4b gene exon2 in primary hepatocarcinoma. World J Gastroenterol. 2000;6:411–414. doi: 10.3748/wjg.v6.i3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Challen C, Lunec J, Warren W, Collier J, Bassendine MF. Analysis of the p53 tumor-suppressor gene in hepatocellular carcinomas from Britain. Hepatology. 1992;16:1362–1366. doi: 10.1002/hep.1840160610. [DOI] [PubMed] [Google Scholar]
- 14.Kress S, Jahn UR, Buchmann A, Bannasch P, Schwarz M. p53 Mutations in human hepatocellular carcinomas from Germany. Cancer Res. 1992;52:3220–3223. [PubMed] [Google Scholar]
- 15.Xiao WH, Liu WW, Lu YY, Li Z. Mutation of p53 tumor suppressor gene in hepatocellular carcinoma. Xin Xiaohuabingxue Zazhi. 1997;5:573–574. [Google Scholar]
- 16.Lin GY, Chen ZL, Lu CM, Li Y, Ping XJ, Huang R. Immunohistochemical study on p53, H-rasp21, c-erbB-2 protein and PCNA expression in HCC tissues of Han and minority ethnic patients. World J Gastroenterol. 2000;6:234–238. doi: 10.3748/wjg.v6.i2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Shi JQ. Overexpression and mutations of tumor suppressor gene p53 in hepatocellular carcinoma. China Natl J New Gastroenterol. 1996;2:161–164. [Google Scholar]
- 18.Peng XM, Peng WW, Yao JL. Codon 249 mutations of p53 gene in development of hepatocellular carcinoma. World J Gastroenterol. 1998;4:125–127. doi: 10.3748/wjg.v4.i2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, Hirohashi S. The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer. 1997;71:355–359. doi: 10.1002/(sici)1097-0215(19970502)71:3<355::aid-ijc8>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 20.Simile MM, Pascale R, De Miglio MR, Nufris A, Daino L, Seddaiu MA, Gaspa L, Feo F. Correlation between S-adenosyl-L-methionine content and production of c-myc, c-Ha-ras, and c-Ki-ras mRNA transcripts in the early stages of rat liver carcinogenesis. Cancer Lett. 1994;79:9–16. doi: 10.1016/0304-3835(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 21.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Lai MD, Chen J. Methylation status of p16 gene in colorectal carcinoma and normal colonic mucosa. World J Gastroenterol. 1999;5:451–454. doi: 10.3748/wjg.v5.i5.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 1995;55:4531–4535. [PubMed] [Google Scholar]
- 24.Costello JF, Berger MS, Huang HS, Cavenee WK. Silencing of p16/CDKN2 expression in human gliomas by methylation and chromatin condensation. Cancer Res. 1996;56:2405–2410. [PubMed] [Google Scholar]
- 25.Otterson GA, Khleif SN, Chen W, Coxon AB, Kaye FJ. CDKN2 gene silencing in lung cancer by DNA hypermethylation and kinetics of p16INK4 protein induction by 5-aza 2'deoxycytidine. Oncogene. 1995;11:1211–1216. [PubMed] [Google Scholar]
- 26.Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 27.Pinto A, Zagonel V. 5-Aza-2'-deoxycytidine (Decitabine) and 5-azacytidine in the treatment of acute myeloid leukemias and myelodysplastic syndromes: past, present and future trends. Leukemia. 1993;7 Suppl 1:51–60. [PubMed] [Google Scholar]
- 28.Cedar H, Razin A. DNA methylation and development. Biochim Biophys Acta. 1990;1049:1–8. doi: 10.1016/0167-4781(90)90076-e. [DOI] [PubMed] [Google Scholar]
- 29.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 30.Jones PA. Altering gene expression with 5-azacytidine. Cell. 1985;40:485–486. doi: 10.1016/0092-8674(85)90192-8. [DOI] [PubMed] [Google Scholar]
- 31.Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2'-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. [PubMed] [Google Scholar]
- 32.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 33.Nobori T, Miura K, Wu DJ, Lois A, Takabayashi K, Carson DA. Deletions of the cyclin-dependent kinase-4 inhibitor gene in multiple human cancers. Nature. 1994;368:753–756. doi: 10.1038/368753a0. [DOI] [PubMed] [Google Scholar]
- 34.Cannon-Albright LA, Goldgar DE, Meyer LJ, Lewis CM, Anderson DE, Fountain JW, Hegi ME, Wiseman RW, Petty EM, Bale AE. Assignment of a locus for familial melanoma, MLM, to chromosome 9p13-p22. Science. 1992;258:1148–1152. doi: 10.1126/science.1439824. [DOI] [PubMed] [Google Scholar]
- 35.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7:1572–1583. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 36.Serrano M, Gómez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science. 1995;267:249–252. doi: 10.1126/science.7809631. [DOI] [PubMed] [Google Scholar]
- 37.Hussussian CJ, Struewing JP, Goldstein AM, Higgins PA, Ally DS, Sheahan MD, Clark WH, Tucker MA, Dracopoli NC. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 38.Spruck CH, Gonzalez-Zulueta M, Shibata A, Simoneau AR, Lin MF, Gonzales F, Tsai YC, Jones PA. p16 gene in uncultured tumours. Nature. 1994;370:183–184. doi: 10.1038/370183a0. [DOI] [PubMed] [Google Scholar]
- 39.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- 40.Mori T, Miura K, Aoki T, Nishihira T, Mori S, Nakamura Y. Frequent somatic mutation of the MTS1/CDK4I (multiple tumor suppressor/cyclin-dependent kinase 4 inhibitor) gene in esophageal squamous cell carcinoma. Cancer Res. 1994;54:3396–3397. [PubMed] [Google Scholar]
- 41.Zhou X, Tarmin L, Yin J, Jiang HY, Suzuki H, Rhyu MG, Abraham JM, Meltzer SJ. The MTS1 gene is frequently mutated in primary human esophageal tumors. Oncogene. 1994;9:3737–3741. [PubMed] [Google Scholar]
- 42.Giani C, Finocchiaro G. Mutation rate of the CDKN2 gene in malignant gliomas. Cancer Res. 1994;54:6338–6339. [PubMed] [Google Scholar]
- 43.Okamoto A, Demetrick DJ, Spillare EA, Hagiwara K, Hussain SP, Bennett WP, Forrester K, Gerwin B, Serrano M, Beach DH. Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci USA. 1994;91:11045–11049. doi: 10.1073/pnas.91.23.11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui AM, Sakamoto M, Kanai Y, Ino Y, Gotoh M, Yokota J, Hirohashi S. Inactivation of p16INK4 in hepatocellular carcinoma. Hepatology. 1996;24:575–579. doi: 10.1002/hep.510240319. [DOI] [PubMed] [Google Scholar]
- 45.Sun L, Hui AM, Kanai Y, Sakamoto M, Hirohashi S. Increased DNA methyltransferase expression is associated with an early stage of human hepatocarcinogenesis. Jpn J Cancer Res. 1997;88:1165–1170. doi: 10.1111/j.1349-7006.1997.tb00345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wong IH, Lo YM, Zhang J, Liew CT, Ng MH, Wong N, Lai PB, Lau WY, Hjelm NM, Johnson PJ. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999;59:71–73. [PubMed] [Google Scholar]
- 47.Boyes J, Bird A. Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. EMBO J. 1992;11:327–333. doi: 10.1002/j.1460-2075.1992.tb05055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hsieh CL. Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994;14:5487–5494. doi: 10.1128/mcb.14.8.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones PA, Taylor SM, Mohandas T, Shapiro LJ. Cell cycle-specific reactivation of an inactive X-chromosome locus by 5-azadeoxycytidine. Proc Natl Acad Sci USA. 1982;79:1215–1219. doi: 10.1073/pnas.79.4.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700–9704. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshiura K, Kanai Y, Ochiai A, Shimoyama Y, Sugimura T, Hirohashi S. Silencing of the E-cadherin invasion-suppressor gene by CpG methylation in human carcinomas. Proc Natl Acad Sci USA. 1995;92:7416–7419. doi: 10.1073/pnas.92.16.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ottaviano YL, Issa JP, Parl FF, Smith HS, Baylin SB, Davidson NE. Methylation of the estrogen receptor gene CpG island marks loss of estrogen receptor expression in human breast cancer cells. Cancer Res. 1994;54:2552–2555. [PubMed] [Google Scholar]
- 53.Xiao WH, Liu WW, Lu YY, Gao CF, Chui JT, Lu R. Analysis of methylation states of CpG island at locus 17p13.3 and its significance in hepatocellular carcinoma. Zhonghua Yixue Yichuanxue Zazhi. 1997;14:297–299. [Google Scholar]
- 54.Xiao WH, Liu WW, Lu YY, Gao CF. Study the mechanisms of many suppresses genes inactivation. Disan Junyi Daxue Xuebao. 1997;19:324–327. [Google Scholar]