

TEXT
Duodenal ulcer (DU) can be developed via several different mechanisms. Hypersecretion of gastric acid is, however, a common denominator. A massive hypersec retion of acid can by itself evoke a DU, e.g. in the Zollinger-Ellison syndrome. Irrespective of the mechanism behind the development of a DU, powerful antisecretory treatment will heal the ulcer and prevent recurrence.
The hypersecretion of acid in DU patients is well characterized (Table 1). The maximal acid secretory capacity is increased in about half of the patients[1], probably due to an enhanced trophic effect of gastrin on the oxyntic mucosa. Several mechanisms normally inhibiting gastric acid secretion, has been found defective in DU patients[2-4], i.a. resulting in an incre ased release of gastrin[2]. The final result is a raised and prolonged acid response to every meal[5].
Table 1.
Gastric acid secretory characteristics of duodenal patients (all of which might be Helicobacter pylori induced)
| Increased maximal acid secretory capacity |
| Increased parietal cell mass (trophic effect of gastrin?) |
| Increased basal acid secretion |
| Increased basal release of gastrin |
| Increased and prolonged acid response to meals |
| Increased release of gastrin |
| Defective inhibitory mechanisms |
| Antral acidification |
| Antral distension |
| Fat in the duodenum (H. pylori induced?) |
The most common cause of DU is Helicobacter pylori (H. pylori) infection. It is, however, only a minority (10%-15%) of all H. pylori infected subjects, who will develop DU. The sequence of events leading to DU includes hypersecretion of acid, development of gastric metaplasia in the proximal duodenum with colonization of H. pylori in the duodenal bulb, progress towards a high density of virulent H. pylori bacteria in the bulb with a marked active and chronic inflammation, and a profoundly reduced bicarbonate secretion in the bulb. The overview is a short presentation of evidence supporting this concept.
H. PYLORI EFFECTS ON ACID SECRETION
A pronounced H. pylori induced inflammation of the antral mucosa in the presence of an intact oxyntic mucosa will result in acid hypersecretion, due to a blockade of mechanisms normally inhibiting gastric acid secretion[6,7]. It should be emphasized that this acid hypersecretion does exist in all subjects with a H. pylori infection predominantly localized to the antrum, and is thus not a characteristic only for DU patients.
If, however, a pronounced H. pylori induced inflammation also includes the oxyntic mucosa, the acid secretion will instead be reduced due to inhibition induced by H. pylori inflammation on parietal cell level and a subsequent devel opment of atrophic gastritis[8].
Maximal acid secretory capacity
DU patients have a higher maximal acid secretory capacity than subjects without the ulcer disease, but there is a considerable overlapping between the two groups. H. pylori infection of the antrum results in a moderate increase of the release of gastrin from the antrum. Gastrin has a trophic effect on the acid secreting mucosa, that can produce a markedly increased maximal acid secretory capacity, e.g. in patients with the Zollinger-Ellison syndrome. Also moderately increased serum gastrin levels seem to be able to enhance the maximal acid secretory capacity[9,10]. For instance, eradication of H. pylori has been followed by a reduced maximal acid secretory capacity[9]. This conclusion is supported by the finding that resection of the antrum with retention of the whole acid secreting part of the stomach in DU patients markedly reduced the maximal acid secretory capacity[11].
The substantial overlapping of the maximal acid secretory capacity between DU patients and subjects without ulcer can be explained partly by the fact that many subjects without ulcer have H. pylori infection and consequently an increased release of gastrin, and partly by the possibility that the balance between the gastrins having acid stimulatory effect and those having only trophic effect[12], might vary from subject to subject.
Defective inhibitory mechanisms
In subjects with a predominantly antral H. pylori infection the gastrin release is increased in the fasting condition, during meals, and by experimental administration of gastrin releasing peptide (GRP)[6,13-17], which probably is a consequence of a reduction of the somatostatin in the antrum by the H. pylori infection[18,19]. Antral somatostatin acts as a physiological inhibitor of the gastrin release. After eradication of H. pylori the gastrin release is normalized. During i.v. infusion of GRP the gastrin release and acid secretion was significantly higher in H. pylori infected subjects, and these responses were nor malized after eradication of H. pylori[20]. Interestingly the acid response to GRP in H. pylori infected DU patients was twice that in H. pylori infected subjects without ulcer despite similar gastrin responses in both groups. Whether this experim ental situation reflects physiological conditions is open to question for several reasons, but the results indicate that DU patients may have a more pronounced hypersecretion of acid than H. pylori infected subjects without ulcer.
Antral H. pylori infection gives rise also to a blockade of an inhibitory nervous reflex from the antrum to the acid secreting mucosa[7]. In subjects without H. pylori infection distension of the antrum provokes an in hibition of acid secretion via a reflex pathway, and this inhibition is completely absent in subjects with H. pylori infection. The blockade of the inhibitory reflex is probably a result of the inflammatory process in the H. pylori infected antrum, since the inhibitory reflex seems to turn up again only when the inflammatory reaction has ceased after eradication of H. pylori.
The defective inhibitory mechanisms caused by antral H. pylori infection results in acid hypersecretion under physiological conditions. The gastrin release during meals is increased in H. pylori infected subjects[6,13-15], and contributes to an increased and prolonged acid response to meals[15]. The well-known inhibition of gastrin release by acidification of the antrum is markedly impaired in H. pylori infected subjects[14,15], contributing to the acid hypersecretion of the infected subjects. The hypersecretion of acid obviously results in an increased acid load on the duodenal bulb[15] both in H. pylori infected subjects without ulcer and DU patients. Thus, it seems reasonable that the explanation for the fact that only a minority of all H. pylori infected subjects will develop DU has to be searched for in the duodenal bulb, but with acid hypersecretion as necessary prerequisite.
GASTRIC METAPLASIA
Gastric metaplasia (GM) is islands of gastric mucosa in the duodenal bulb. GM develops as a result of an increased acid load on the bulb, and is e.g. rather extensive in patients with the Zollinger-Ellison syndrome[21]. GM has been found in 90% of H. pylori infected DU patients, and in about 60% of H. pylori infected subjects without ulcer[22,23], in accordance with the increased duodenal acid load in both these groups. GM is a prerequisite for colonization of H. pylori in the duodenal bulb. It is possible that the extension of GM is facilitated by the H. pylori induced inflammatory process in the bulb, since a combination of H. pylori eradication and antisecretory treatment reduced the GM area more effectively than either treatment alone[25,26].
Hypersecretion of acid and development of GM exists in both DU patients and H. pylori infected subjects without ulcer, albeit somewhat more pronounced in DU patients. It therefore seems reasonable to assume that a critical factor in the development of DU could be a large number of H. pylori and/or particularly virulent H. pylori in the duodenal bulb of DU patients. The density and virulence of H. pylori in DU patients has previously been determined in several studies, but in biopsies taken from the antrum. In these studies of antral H. pylori density and virulence, only a marginal difference was found between DU patients and H. pylori infected subjects without ulcer. However, findings regarding H. pylori in the antrum does not necessarily reflect the situation in the duodenal bulb.
GM in the duodenal bulb is found in patches that cannot be visualized at or dinary gastroduodenoscopy. In a recent study[24] the extent of GM in the duodenal bulb was therefore determined more systematically by taking 2 biopsies from each quadrant of the bulb. The area of GM in the multiple biopsies was 4 times larger in the DU patients than in the H. pylori infected subjects without ulcer[24]. These biopsies were also used to determine the prevalence and density of virulent H. pylori as well as the type (active and chronic) and degree of the inflammatory reaction in both the antrum and the duodenal bulb of DU patients (n = 20) and H. pylori infected subjects without ulcer (n = 21).
BACTERIAL DENSITY AND cagA STATUS IN THE DUODENAL BULB
H. pylori colonization of GM in the duodenal bulb is very common. It was found in 95% of DU patients and in 80% of infected subjects without ulcer[24], determined by quantitative culture[27]. Rigorous precautions were taken at the endoscopy to avoid contamination of the duodenal biopsies with H. pylori from the stomach. Gastric epithelial cells express the blood group antigen Lewis-b, and H. pylori have the ability to bind to these antigens, thereby facilitating the colonization of the mucosa[28]. H. pylori also has the ability to express various Lewis antigens, that might imply greater opportunity to escape the immune response of the host. It was recently shown[29] that H. pylori expressing various Lewis antigens exist in a considerably higher frequency in the duodenal bulb of DU patients (90%) than in the bulb of infected subjects without ulcer (42%).
The density of H. pylori was much lower in the duodenal bulb than in the an trum, even after correction for the area of GM[24], suggesting that the environment in the duodenal bulb is less favourable for colonization of H. pylori. The mean H. pylori density in the bulb was about 20 times higherin DU patients than in infected subjects without ulcer, despite the fact that the bacterial density in the antrum was similar for both groups[24]. There was, however, a substantial overlap of the bacterial density in the bulb between DU patients and infected subjects without ulcer.
The clinically relevant virulence factors of H. pylori have not yet been identified. The presence of the cagA gene (cytotoxin associated gene A) is so far the best marker of virulence. H. pylori bacteria that are cagA positive can induce a more powerful release of proinflammatory cytokines with a subsequent potential of mucosal damage[30]. The proportion of cagA positive bacteria might therefore be a significant factor determining the local mucosal damaging effect of colonized H. pylori. DU patients had a much higher prevalence of cagA positive bacteria in the duodenal bulb (81%) than H. pylori infected subjects without ulcer (30%), despite the fact that the prevalence of cagA positive bacteria in the antrum was of the same order for both groups (86% and 75%, respectively)[24]. Furthermore, subjects with a predominance of cagA positive bacteria in the bulb had about 10 times higher bacterial density than subjects with cagA negative bacteria[24].
H. PYLORI INDUCED INFLAMMATION IN THE DUODENAL BULB
The H. pylori bacteria are colonizing the GM in the duodenal bulb, and consequently the inflammatory cells are found in connection with the GM. The chronic duodenitis, determined by the degree of infiltration of lymphocytes, was found more marked in DU patients than in H. pylori infected subjects without ulcer, but again with overlapping between the two groups[24]. Active duodenitis, defined by the presence of neutrophil leucocytes, is an established and common finding in DU patients. Active duodenitis was found only in DU patients and almost exclusively in DU patients with cagA positive H. pylori in the bulb[24]. Active duodenitis is thus a characteristic for DU patients. Activation of neutrophil leucocytes can result in tissue damage by release of proteolytic enzymes[31] and by induction of reactive oxygen metabolites in the gastric epithelial cells[32].
BICARBONATE SECRETION IN THE DUODENAL BULB
Hypersecretion of gastric acid with increased duodenal acid load can result in mucosal damage and development of ulcer. Under normal circumstances the acid ification of the duodenal bulb will immediately activate bicarbonate secretion from the duodenal mucosa resulting in neutralization of the acid with the formati on of carbon dioxide and water. The carbon dioxide will then stimulate the nitric oxide (NO) producing enzyme NO-synthase[33], that has been demonstrated in the duodenal mucosa[34]. NO will finally activate the bicarbonate secretion[35].
DU patients have a markedly reduced bicarbonate secretion in response to acidification of the duodenal bulb[36]. It has been shown more recently that the bicarbonate secretion is normalized after eradication of H. pylori[37], implying that the reduced bicarbonate response to acidification of the bulb is a result of the H. pylori infection. Interestingly, the bicarbonate secretion was found normal in H. pylori infected subjects without ulcer[37]. The H. pylori dependent reduction of the bicarbonate secretion is thus another characteristic of DU patients.
The mechanism by which the H. pylori infection is reducing the bicarbonate secretion has been studied recently. The bicarbonate secretion in response to acidification of the duodenal bulb in rats could be inhibited by local administration of a water extract of H. pylori[38]. In these experiments a substantial increase of the NO synthase inhibitor asymmetrical dimethyl arginine (ADMA) was demonstrated in the duodenal mucosa. Separate administration of ADMA markedly reduced the bicarbonate response to acidification of the bulb. The presence of ADMA has not yet been determined in the bulb mucosa of humans. In the antrum, however, the ADMA concentration was 65 times higher in H. pylori infected subjects than in non-infected subjects[39]. It is possible that H. pylori delivers peptides that are degraded by proteolysis in the antrum-bulb region to for instance ADMA. It seems reasonable to assume that the high density of cagA positive H. pylori bacteria in the bulb of DU patients can produce high enough ADMA concentrations to explain the markedly reduced bicarbonate secretion that is a characteristic of DU patients.
WHY DOES ONLY A MINORITY OF H. PYLORI INFECTED SUBJECTS DEVELOP DUODENAL ULCER?
A special series of events seem to result in the development of DU in H. pylori infected subjects. The first part of this series of events should be regarded more as a prequisite for the development of DU, and is common for all subjects with an antrum predominant H. pylori gastritis (Figure 1). The antrum gastritis results in hypersecretion of acid, the increased acid load to the duodenal bulb gives rise to formation of GM, and these islands of gastric mucosa can be colonized by H. pylori.
Figure 1.

Successive prerequisites for the development of Helicobacter pylori induced duodenal ulcer. Reprinted from Best Practice & Research in Clinical Gastroenterology, 14 (1), L Olbe et al, Conceivable mechanisms by which Helicobacter pylori provokes duodenal ulcer disease, 1-12, 2000, by permission of the publisher Bailliere Tindall.
The most important factor for the development of DU seems to be a high density of virulent H. pylori bacteria in the duodenal bulb. When the density of virulent strains / with cagA positivity as a marker of virulence / reaches a high level, two effects are triggered that are characteristic for DU patients. Firstly, a high density of virulent H. pylori induces a pronounced release of proinflammatory cytokines, leading to a substantial chronic inflammation and an active duodenitis with its tissue damaging potential. Secondly, a high density of virulent H. pylori seems to produce enough amounts of NO synthase inhibitors to significantly reduce the bicarbonate secretion in response to acidification of the duodenal bulb. This entails a high acidity in the bulb leading to a potential increase of GM and tissue damage.
In summary the pivotal components in the conceptual development of DU are hypersecretion of acid and colonization of the duodenal bulb with a high density of virulent H. pylori strains resulting in an active duodenitis and reduced bicarbonate secretion (Figure 2).
Figure 2.
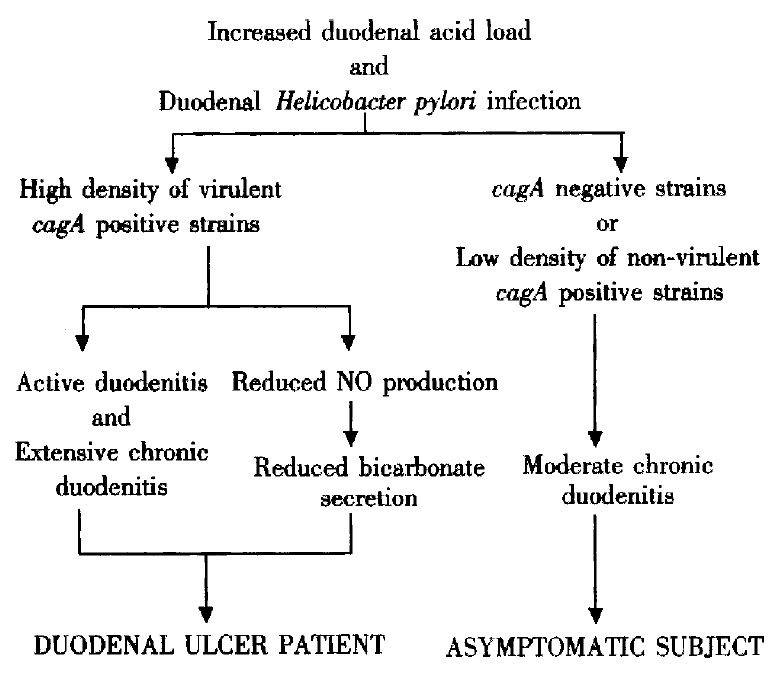
Conceivable sequence of events in the development of Helicobacter pylori induced duodenal ulcer. Reprinted from Best Practice & Research in Clinical Gastroenterology, 14 (1), L Olbe et al, Conceivable mechanisms by which Helicobacter pylori provokes duodenal ulcer disease, 1-12, 2000, by permission of the publisher Bailliere Tindall.
Footnotes
Edited by Ma JY
References
- 1.COX AJ. Stomach size and its relation to chronic peptic ulcer. AMA Arch Pathol. 1952;54:407–422. [PubMed] [Google Scholar]
- 2.Walsh JH, Richardson CT, Fordtran JS. pH dependence of acid secretion and gastrin release in normal and ulcer subjects. J Clin Invest. 1975;55:462–468. doi: 10.1172/JCI107952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schöön IM, Bergegårdh S, Grötzinger U, Olbe L. Evidence for a defective inhibition of pentagastrin-stimulated gastric acid secretion by antral distension in the duodenal ulcer patient. Gastroenterology. 1978;75:363–367. [PubMed] [Google Scholar]
- 4.Kihl B, Olbe L. Inhibition of pentagastrin-stimulated gastric acid secretion by graded intraduodenal administration of oleic acid in man. Scand J Gastroenterol. 1981;16:121–128. [PubMed] [Google Scholar]
- 5.Fordtran JS, Walsh JH. Gastric acid secretion rate and buffer content of the stomach after eating. Results in normal subjects and in patients with duodenal ulcer. J Clin Invest. 1973;52:645–657. doi: 10.1172/JCI107226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levi S, Beardshall K, Haddad G, Playford R, Ghosh P, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet. 1989;1:1167–1168. doi: 10.1016/s0140-6736(89)92752-9. [DOI] [PubMed] [Google Scholar]
- 7.Olbe L, Hamlet A, Dalenbäck J, Fändriks L. A mechanism by which Helicobacter pylori infection of the antrum contributes to the development of duodenal ulcer. Gastroenterology. 1996;110:1386–1394. doi: 10.1053/gast.1996.v110.pm8613042. [DOI] [PubMed] [Google Scholar]
- 8.El-Omar EM, Oien K, El-Nujumi A, Gillen D, Wirz A, Dahill S, Williams C, Ardill JE, McColl KE. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- 9.Harris AW, Gummett PA, Misiewicz JJ, Baron JH. Eradication of Helicobacter pylori in patients with duodenal ulcer lowers basal and peak acid outputs to gastrin releasing peptide and pentagastrin. Gut. 1996;38:663–667. doi: 10.1136/gut.38.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gillen D, Wirz AA, Ardill JE, McColl KE. Rebound hypersecretion after omeprazole and its relation to on-treatment acid suppression and Helicobacter pylori status. Gastroenterology. 1999;116:239–247. doi: 10.1016/s0016-5085(99)70118-6. [DOI] [PubMed] [Google Scholar]
- 11.Bergegårdh S, Broman G, Knutson U, Palmer L, Olbe L. Gastric acid responses to graded i.v. infusion of pentagastrin and histalog in peptic ulcer patients before and after antrum-bulb resection. Scand J Gastroenterol. 1976;11:337–346. [PubMed] [Google Scholar]
- 12.Sawada M, Dickinson CJ. The G cell. Annu Rev Physiol. 1997;59:273–298. doi: 10.1146/annurev.physiol.59.1.273. [DOI] [PubMed] [Google Scholar]
- 13.Peterson WL, Barnett CC, Evans DJ, Feldman M, Carmody T, Richardson C, Walsh J, Graham DY. Acid secretion and serum gastrin in normal subjects and patients with duodenal ulcer: the role of Helicobacter pylori. Am J Gastroenterol. 1993;88:2038–2043. [PubMed] [Google Scholar]
- 14.Tarnasky PR, Kovacs TO, Sytnik B, Walsh JH. Asymptomatic H. pylori infection impairs pH inhibition of gastrin and acid secretion during second hour of peptone meal stimulation. Dig Dis Sci. 1993;38:1681–1687. doi: 10.1007/BF01303178. [DOI] [PubMed] [Google Scholar]
- 15.Hamlet A, Olbe L. The influence of Helicobacter pylori infection on postprandial duodenal acid load and duodenal bulb pH in humans. Gastroenterology. 1996;111:391–400. doi: 10.1053/gast.1996.v111.pm8690204. [DOI] [PubMed] [Google Scholar]
- 16.Beardshall K, Moss S, Gill J, Levi S, Ghosh P, Playford RJ, Calam J. Suppression of Helicobacter pylori reduces gastrin releasing peptide stimulated gastrin release in duodenal ulcer patients. Gut. 1992;33:601–603. doi: 10.1136/gut.33.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.el-Omar E, Penman I, Dorrian CA, Ardill JE, McColl KE. Eradicating Helicobacter pylori infection lowers gastrin mediated acid secretion by two thirds in patients with duodenal ulcer. Gut. 1993;34:1060–1065. doi: 10.1136/gut.34.8.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moss SF, Legon S, Bishop AE, Polak JM, Calam J. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet. 1992;340:930–932. doi: 10.1016/0140-6736(92)92816-x. [DOI] [PubMed] [Google Scholar]
- 19.Gibbons AH, Legon S, Walker MM, Ghatei M, Calam J. The effect of gastrin-releasing peptide on gastrin and somatostatin messenger RNAs in humans infected with Helicobacter pylori. Gastroenterology. 1997;112:1940–1947. doi: 10.1053/gast.1997.v112.pm9178686. [DOI] [PubMed] [Google Scholar]
- 20.el-Omar EM, Penman ID, Ardill JE, Chittajallu RS, Howie C, McColl KE. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology. 1995;109:681–691. doi: 10.1016/0016-5085(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 21.Parrish JA, Rawlins DC. Intestinal mucosa in the Zollinger-Ellison syndrome. Gut. 1965;6:286–289. doi: 10.1136/gut.6.3.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noach LA, Rolf TM, Bosma NB, Schwartz MP, Oosting J, Rauws EA, Tytgat GN. Gastric metaplasia and Helicobacter pylori infection. Gut. 1993;34:1510–1514. doi: 10.1136/gut.34.11.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris AW, Gummett PA, Walker MM, Misiewicz JJ, Baron JH. Relation between gastric acid output, Helicobacter pylori, and gastric metaplasia in the duodenal bulb. Gut. 1996;39:513–520. doi: 10.1136/gut.39.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamlet A, Thoreson AC, Nilsson O, Svennerholm AM, Olbe L. Duodenal Helicobacter pylori infection differs in cagA genotype between asymptomatic subjects and patients with duodenal ulcers. Gastroenterology. 1999;116:259–268. doi: 10.1016/s0016-5085(99)70121-6. [DOI] [PubMed] [Google Scholar]
- 25.Wyatt JI, Rathbone BJ, Sobala GM, Shallcross T, Heatley RV, Axon AT, Dixon MF. Gastric epithelium in the duodenum: its association with Helicobacter pylori and inflammation. J Clin Pathol. 1990;43:981–986. doi: 10.1136/jcp.43.12.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khulusi S, Badve S, Patel P, Lloyd R, Marrero JM, Finlayson C, Mendall MA, Northfield TC. Pathogenesis of gastric metaplasia of the human duodenum: role of Helicobacter pylori, gastric acid, and ulceration. Gastroenterology. 1996;110:452–458. doi: 10.1053/gast.1996.v110.pm8566592. [DOI] [PubMed] [Google Scholar]
- 27.Atherton JC, Tham KT, Peek RM, Cover TL, Blaser MJ. Density of Helicobacter pylori infection in vivo as assessed by quantitative culture and histology. J Infect Dis. 1996;174:552–556. doi: 10.1093/infdis/174.3.552. [DOI] [PubMed] [Google Scholar]
- 28.Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 29.Thoreson AC, Hamlet A, Celik J, Byström M, Nyström S, Olbe L, Svennerholm AM. Differences in surface-exposed antigen expression between Helicobacter pylori strains isolated from duodenal ulcer patients and from asymptomatic subjects. J Clin Microbiol. 2000;38:3436–3441. doi: 10.1128/jcm.38.9.3436-3441.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crabtree JE, Farmery SM, Lindley IJ, Figura N, Peichl P, Tompkins DS. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J Clin Pathol. 1994;47:945–950. doi: 10.1136/jcp.47.10.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320:365–376. doi: 10.1056/NEJM198902093200606. [DOI] [PubMed] [Google Scholar]
- 32.Davies GR, Simmonds NJ, Stevens TR, Sheaff MT, Banatvala N, Laurenson IF, Blake DR, Rampton DS. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut. 1994;35:179–185. doi: 10.1136/gut.35.2.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holm M, Johansson B, Pettersson A, Fändriks L. Carbon dioxide mediates duodenal mucosal alkaline secretion in response to luminal acidity in the anesthetized rat. Gastroenterology. 1998;115:680–685. doi: 10.1016/s0016-5085(98)70147-7. [DOI] [PubMed] [Google Scholar]
- 34.Holm M, Powell T, Casselbrant A, Johansson B, Fändriks L. Dynamic involvement of the inducible type of nitric oxide synthase in acid-induced duodenal mucosal alkaline secretion in the rat. Dig Dis Sci. 2001;46:1765–1771. doi: 10.1023/a:1010674109111. [DOI] [PubMed] [Google Scholar]
- 35.Holm M, Johansson B, Pettersson A, Fändriks L. Acid-induced duodenal mucosal nitric oxide output parallels bicarbonate secretion in the anaesthetized pig. Acta Physiol Scand. 1998;162:461–468. doi: 10.1046/j.1365-201X.1998.0307f.x. [DOI] [PubMed] [Google Scholar]
- 36.Isenberg JI, Selling JA, Hogan DL, Koss MA. Impaired proximal duodenal mucosal bicarbonate secretion in patients with duodenal ulcer. N Engl J Med. 1987;316:374–379. doi: 10.1056/NEJM198702123160704. [DOI] [PubMed] [Google Scholar]
- 37.Hogan DL, Rapier RC, Dreilinger A, Koss MA, Basuk PM, Weinstein WM, Nyberg LM, Isenberg JI. Duodenal bicarbonate secretion: eradication of Helicobacter pylori and duodenal structure and function in humans. Gastroenterology. 1996;110:705–716. doi: 10.1053/gast.1996.v110.pm8608879. [DOI] [PubMed] [Google Scholar]
- 38.Fändriks L, von Bothmer C, Johansson B, Holm M, Bölin I, Pettersson A. Water extract of Helicobacter pylori inhibits duodenal mucosal alkaline secretion in anesthetized rats. Gastroenterology. 1997;113:1570–1575. doi: 10.1053/gast.1997.v113.pm9352859. [DOI] [PubMed] [Google Scholar]
- 39.Fändriks L, von Bothmer C, Lnroth H, Olbe L, Pettersson A. Presence of NO synthase inhibitor ADMA in H. pylori infected antral mucosa. Gastroenterology. 1998;114:A118: G486. [Google Scholar]