

Abstract
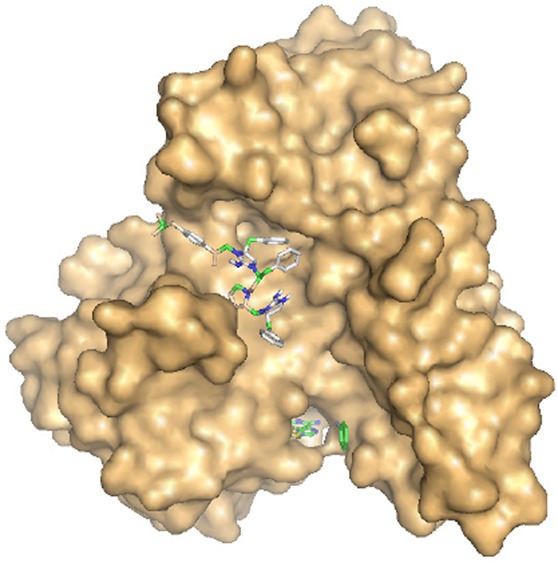
Arsenic is the most ubiquitous environmental toxin and carcinogen. Long-term exposure to arsenic is associated with human diseases including cancer, cardiovascular disease, and diabetes. Human As(III) S-adenosylmethionine (SAM) methyltransferases (hAS3MT) methylates As(III) to trivalent mono- and dimethyl species that are more toxic and potentially more carcinogenic than inorganic arsenic. Modulators of hAS3MT activity may be useful for the prevention or treatment of arsenic-related diseases. Using a newly developed high-throughput assay for hAS3MT activity, we identified 10 novel noncompetitive small molecule inhibitors. In silico docking analysis with the crystal structure of an AS3MT orthologue suggests that the inhibitors bind in a cleft between domains that is distant from either the As(III) or SAM binding sites. This suggests the presence of a possible allosteric and regulatory site in the enzyme. These inhibitors may be useful tools for future research in arsenic metabolism and are the starting-point for the development of drugs against hAS3MT.
Introduction
Arsenic is the most pervasive environmental toxin; consequently, the U.S. Environmental Protection Agency (EPA) and Agency for Toxic Substances and Disease Registry (ATSDR) rank arsenic first on the U.S. Priority List of Hazardous Substances (http://www.atsdr.cdc.gov/SPL/index.html). The EPA asserts that it pervades our drinking water,1 and the U.S. Food and Drug Administration is concerned about arsenic endangering the safety of our food supply (http://www.fda.gov/Food/FoodborneIllnessContaminants/Metals/ucm319870.htm). For example, arsenic in rice has been associated with increased risk of cancer in Chinese populations.2 The effects of arsenic exposure is not limited to cancer; there is a strong correlation between dietary arsenic and cardiovascular disease,3 diabetes,4,5 and other human disorders.6
In humans, arsenic is primarily metabolized by the enzyme As(III) SAM methyltransferase (hAS3MT), a member of the large superfamily of SAM methyltransferases (SAM MTs). Methylation was originally considered as a detoxification process but is now thought to transform arsenic into more toxic species.7 AS3MT biotransforms of inorganic arsenic (As(III)) into trivalent methylated species methylarsenite (MAs(III)) and dimethylarsenite (DMAs(III)) using SAM as the methyl donor.8,9 The trivalent methylarsenicals are excreted in urine, where they oxidize to pentavalent methylarsenate (MAs(V)) and dimethylarsenate (DMAs(V)).10−12 hAS3MT is an example of a “Phase II” enzyme that is simultaneously protective but also activates arsenic into more toxic and potentially carcinogenic organoarsenicals.13
How would more or less hAS3MT activity affect susceptibility to arsenic-related diseases? On the one hand, it seems reasonable to consider that lower rates of methylation would be protective. If so, drugs that inhibit AS3MT would be useful for prevention or treatment of arsenic-related diseases. However, it is not known whether humans require AS3MT, or what the physiological consequences of its absence might be. AS3MT knockout mice have reduced arsenic methylation14 and are sensitive to high levels of arsenic.15 However, mice do not develop arsenic-related bladder cancer, so the mouse may not be the best model for arsenic carcinogenesis in humans.16
On the other hand, more rapid production of methylated species can lead to faster urinary excretion, and higher clearance rates might be protective. In that case, drugs that activate AS3MT might be valuable. Individuals who excrete more DMAs in urine have lower rates of bladder cancer and other arsenic-related diseases.17 Conversely, higher DMAs is also associated with increased incidence of diabetes5 and is considered to be a carcinogen in rats at high concentrations.18,19 Thus, it is not clear whether drug development should be directed toward inhibitors or activators of AS3MT.
To date, only nonspecific inhibitors of AS3MT have been identified.20 The objective of this study was to identify small molecules that bind to hAS3MT and modulate its activity. In this report, we adapted a commercial methyltransferase method for use as a rapid and sensitive microplate assay to identify inhibitors of AS3MT activity. The assay utilizes time-resolved Förster (fluorescence) resonance energy transfer (TR-FRET) to directly measure conversion of SAM to S-adenosylhomocysteine (SAH). This assay uses terbium FRET with an anti-SAH antibody labeled with a Tb(III) cryptate and a SAH-dye acceptor. We screened the Torrey Pines Institute for Molecular Studies (TPIMS) scaffold ranking compound libraries that consist of more than 30 million small molecules. The TPIMS compounds were designed around 70 molecular scaffolds systematically arranged in positional scanning and scaffold ranking formats.21,22 This approach has been successfully used to isolate compounds selective for their targets, for example, selective α4β2 nAChR antagonists.23 We identified 10 compounds that inhibit hAS3MT with IC50 values of the order of 30–50 μM. None of the compounds inhibit the binding of either As(III) or MAs(III), and they do not inhibit catachol o-methyltransferase (COMT), a nonarsenic SAM MT.24 Thus, they appear to be noncompetitive inhibitors selective for AS3MT. In silico docking analysis of the inhibitors to the crystal structure of an AS3MT orthologue25 indicates that the TPIMS molecules bind in a cleft in the enzyme located distant from the As(III) and SAM binding sites. The inhibitor binding location may be an allosteric regulatory site, which implies the existence of physiological regulators of arsenic methylation. These are the first identified selective small molecule inhibitors of hAS3MT. They provide a scaffold for future rational design of clinically useful drugs that modulate hAS3MT activity and may also be useful probes for further research into AS3MT function.
Materials and Methods
Reagents
SAM was purchased from Cayman Chemical Co. (Ann Arbor, MI). Boric acid was purchased from Fisher Scientific (Pittsburgh, PA). All other reagents were purchased from Sigma Chemical Co. (St. Louis, MO), as was porcine catechol O-methyltransferase. The EPIgeneous Methyltransferase Assay kit was purchased from Cisbio Assays (Bedford, MA). Tris(2-carboxyethyl)phosphine (TCEP) was prepared as a 0.5 M stock solution adjusted to pH 7.0. MAs(V) was reduced to MAs(III) as described previously.20,26,27
Purification of AS3MT Enzymes
hAS3MT, Chlamydomonas reinhardtii CrAS3MT, and its single-tryptophan derivative Y72W were purified by Ni-NTA chromatography from cells of Escherichia coli, as described previously.9,20
Synthesis of TPIMS Compounds
Individual TPIMS compounds TPI-1 through TPI-11 were synthesized using the general approaches described previously (Figure S1).28,29 The parallel solid phase synthesis was performed using the tea-bag method.30 The initial acylated peptides were made using standard Boc chemistry conditions with diisopropylcarbodiimide and 1-hydroxybenzotriazole hydrate as the activating reagents. Boc-Phenylalanine was used for the R1 position of TPI-3 through TPI-11, Boc-tyrosine(2-Br-Z) for TPI-1, and Boc-d-naphthylalanine for TPI-2. Boc-phenylalanine was used for the R2 and R3 positions of all 11 compounds. The following carboxylic acids were used for the R4 position: phenylacetic acid (TPI-1 and TPI-2), 3-bromophenylacetic acid (TPI-3), 4-isobutyl-alpha-methylphenylacetic acid (TPI-4), 3,4-dichlorophenylacetic acid (TPI-5), cyclohexanebutyric acid (TPI-6), cyclohexanepropionic acid (TPI-7), 4-tert-butyl-cyclohexancecarboxylic acid (TPI-8), 4-biphenylacetic acid (TPI-9), 1-adamantancecarboxylic acid (TPI-10), and isobutyric acid (TPI-11). The amide bonds were reduced using a 1.0 M borane tethahydrofuran (THF) complex solution (40-fold excess per amide) heated at 65 °C for 72 h followed by overnight treatment with piperidine at 65 °C. The guanidine cyclization was accomplished using a 5-fold excess of cyanogen bromide (CNBr) in a 0.1 M anhydrous solution of dichloromethane. After cyclization, the compounds were removed from the solid support with a solution of hydrogen fluoride (HF), 0 °C for 1 h.
TR-FRET Assay of Methyltransferase Activity
AS3MT activity was determed with an EPIgeneous Methyltransferase Assay kit by assaying the conversion of SAM to SAH according to the manufacturer’s directions. The assay was carried out using a low volume 384-well microtiter plate in a buffer consisting of 50 mM MOPS, pH 7.5, containing 0.15 M KCl, 10 μM SAM, 10 μM As(III), 20 μM TCEP, and TPIMS inhibitors, as indicated. Purified CrAS3MT or hAS3MT at 1 μM, final concentration, was added to initiate the reaction. The reactions were carried out for 5 min at either room temperature or 37 °C, as indicated. The reaction was terminated and developed by the addition of SAH-d2 and anti-SAH-Lumi4-Tb detection reagents. The plates were incubated for 1 h, and fluorescence was measured at both 665 and 620 nm with excitation at 337 nm in a Synergy H4 Hybrid Multi-Mode microplate reader. The homogeneous time-resolved fluorescence (HTRF) was calculated from the ratio of emission at 665 and 620 nm. The concentration of SAH was calculated with a calibration curve constructed with known concentrations of SAH.
ICP-MS Assay of Arsenic Methylation
Arsenic methylation was assayed as described previously31 in a buffer consisting of 50 mM NaH2PO4, pH 8.0, containing 0.3 M NaCl, 0.5 mM SAM, 10 μM As(III), and 1 mM TCEP and TPIMS inhibitors, as indicated. Purified hAS3MT was added at 2 μM, final concentration, to initiate the reaction. The reactions were carried out at 37 °C for 4 h, at which time they were terminated by the addition of H2O2 at 6%, final concentration. The assay solution was immediately passed through a 3 kDa cutoff Amicon ultrafilter (Millipore, Billerica, MA). Arsenic speciation was determined by high pressure liquid chromatography (HPLC) (PerkinElmer Series 2000) with a C18 300A reverse-phase column (Chromservis s.r.o., Brno, Czech Republic), with arsenic measured by inductively coupled plasma mass spectrometry (ICP-MS) using an ELAN 9000 (PerkinElmer, Waltham, MA).
Assay for Catechol O-Methyl Transferase Activity
Catechol O-methyltransferase was assayed as described.24 The assay was carried out in 30 μL of a buffer consisting of 40 mM TES, pH 7.6, containing 0.1 mM dihydroxyacetone phosphate, 1.2 mM MgCl2, 4 mM DTT, and 0.6 units of COMT. TPI compounds or sinefungin were added as indicated. The reaction was initiated by the addition of 1 mM SAM, final concentration. After 2 h at 37 °C, the reaction was terminated by the addition of 0.2 M sodium borate, pH 10.0, final concentration. Total methylated products were estimated from the absorption at 344 nm using a Synergy H4 Hybrid Multi-Mode microplate reader.
Assays of Substrate-Dependent Quenching of Tryptophan Fluorescence
Measurement of protein fluorescence of purified CrAS3MT Y72W were assayed in 384 well microtiter plates with a total volume of 15 μL of a buffer consisting of 50 mM MOPS-KOH, pH 7.5, containing 0.15 M KCl, and 5 μM Y72W CrAS3MT, final concentration. Substrates were added to the assay buffer to initiate the reaction. Protein fluorescence was determined with a Synergy H4 Hybrid Multi-Mode microplate reader, with excitation and emission wavelengths of 295 and 345 nm, respectively.
In Silico Docking Analysis
The TPIMS molecules were converted to 3D structures using the Open Babel program (http://openbabel.org/) and saved in Protein Data Bank (PDB) format. Further energy minimization was performed by employing the Dundee PRODRG server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg). Graphical user interface program AutoDockTools 1.5.632 was used to merge nonpolar hydrogens, add Gasteiger charges, and set rotatable bonds. Finally, all ligands were saved in PDBQT file format for use by AutoDock Vina (ADVina).33 The CmArsM (4FS8) and COMT (3BWM) structures were prepared for docking analysis by removing the water molecules and cofactors/ligands from the PDB files, assigning polar hydrogens, adding Gasteiger charges, and finally selecting the proteins as rigid models using AutoDockTools 1.5.6. The TPIMS inhibitors were docked with the two proteins using the open source docking program ADVina. A docking grid search space volume was employed with a size of 30 Å × 30 Å × 30 Å for a grid spacing of 1 Å, and the grid center was fixed at the dimensions (X, Y, Z) 6.683, −2.966, Z = 23.928 (for 4FS8), and (X, Y, Z) −8.839, −5.628, −9.694 (for 3BWM), respectively. The exhaustiveness parameter in ADVina was set at 9. All automated docking evaluations by Vina employed iteration with local search global optimization. The docked score results are given as most favorable free energy of binding (ΔG) and are clustered together within the 1 Å positional root-mean squared deviation (RMSD). Predicted molar dissociation constants (Kd) were calculated from the ΔG values using the relationship Kd = exp(ΔG/RT), where R is 1.9872, and T is 298.15 K. Molecular models were generated using PyMol.34
Results
Screening of Compound Scaffold Ranking Libraries
Two AS3MT orthologues were used in this study. Human hAS3MT was purified by the expression of a synthetic gene that produces a highly active form of the enzyme.9 The second CrAS3MT (also called CrArsM) is an AS3MT orthologue from the eukaryotic alga Chlamydomonas reinhardtii.35 Homology models of hAS3MT and CrAS3MT based on the crystal structure of the orthologue from the alga Cyanidioschyzon merolae (termed CmArsM or CmAS3MT)25 shows that the two structures are overall superimposible.9 Initial screening was performed with CrAS3MT because it has robust activity at room temperature, while hAS3MT has a temperature optimum of 37 °C.
A newly developed TR-FRET assay for AS3MT activity that is both rapid and highly sensitive was used for high throughput screening of potential AS3MT inhibitors.20 Utilizing the TPIMS Scaffold Ranking Library, which contained over 30 million synthetic compounds systematically arranged into 70 samples,22,23 the core bisguanidine pyrrolide scaffold (Figure S2) was identified as the scaffold most likely to provide individual inhibitory compounds. From these initial results, a set of individual compounds all containing the bisguanidine pyrrolide core with differing R groups was screened. Ten compounds, designated TPI-1 to TPI-10, inhibited CrAS3MT methylation activity by at least 75% (Figures 1 and 2). The effect of the putative inhibitors on hAS3MT activity was examined. Each of the 10 TPIMS compounds inhibited hAS3MT with IC50 values in the range of 30 to 50 μM. As examples, compounds TPI-2, TPI-4, TPI-5, and TPI-6 showed IC50 values of 38, 51, 31, and 38 μM, respectively (Figure 2). Eight of the active inhibitors differ only in R4. One compound, TPI-11, that did not inhibit also has the same R1, R2, and R3 groups as the inhibitors TPI-3 through TPI-10. The only difference between these 8 inhibitors and TPI-11 occurs in the substitution at the R4 position. TPI-11 contains an isobutyl group at the R4 position which is significantly less bulky than any of the other functional groups contained in the inhibitors suggesting that the size of the functional group at this position may contribute to the compound’s ability to inhibit.
Figure 1.

Inhibition of AS3MT activity by TPIMS inhibitors. Methyltranserase activity was assayed with CrAS3MT using the TR-FRET method, as described under Materials and Methods. As(III) was added at 10 μM, SAM was added at 20 μM, and small molecule compounds were added at 50 μM, final concentrations. The reaction was initiated by the addition of enzyme at 1 μM, final concentration. The reaction was terminated after 5 min at room temperature, and SAH production analyzed. The data are the mean ± SE (n = 3). The dotted line indicates 75% inhibition.
Figure 2.

Dose–response relationship of TPIMS inhibitors and hAS3MT activity. The activity of hAS3MT was assayed as described in the legend to Figure 1 in the presence of the indicated concentrations of (A) TPI-2; (B) TPI-4; (C) TPI-5; or (D) TPI-6. The inhibitor concentrations of half-maximal inhibition (IC50) were calculated as 38 μM for TPI-2, 51 μM for TPI-5, 31 μM for TPI-5, and 38 μM for TPI-6. The data were fitted using SigmaPlot. The data are the mean ± SE (n = 3).
AS3MT TPIMS Inhibitors Do Not Inhibit COMT, a Nonarsenic SAM MT
The effect of TPI-4 on the activity of porcine liver COMT was examined. No inhibition of COMT activity activity was observed (Figure S3). In contrast, singfungin, a SAM analogue, significantly inhibited COMT activity. This indicates first that TPI-4 does not inhibit SAM binding and second and more importantly that the small molecule inhibitor is selective for AS3MT.
Effects of TPIMS Inhibitors on the First and Second Methylation Steps
AS3MT methylates arsenic at least twice, As(III) → MAs(III) and MAs(III) → DMAs(III), which is rapidly oxidized to DMAs(V) in air.12,36 We determined the effect of the small molecule inhibitors individually on the first and second methylation steps. The TR-FRET assay measures primarily the first methylation step, and each of the 10 compounds inhibits the first methylation step. To examine the effect on the second methylation step, we used the conventional assay for arsenic biotransformations, separation of the species by reverse phase HPLC coupled to arsenic detection by ICP-MS after reaction times of tens of minutes to hours.37 When the substrate is As(III), DMAs(V) is the primary final compound, a combination of both the first and second methylation steps. However, when MAs(III) is used as substrate, only the second methylation step occurs. Thus, the effect of the small molecule compounds could be examined individually on each step. Each TPIMS compound inhibited As(III) methylation (Figure 3A). Five, TPI-2, TPI-4, TPI-6, TPI-8, and TPI-9, inhibited MAs(III) methylation (Figure 3B). In contrast, the other five, TPI-1, TPI-3, TPI5, TPI-7, and TPI-10, did not inhibit MAs(III) methylation at the highest available concentration (Figure 3C). These results suggest that all 10 TPIMS compounds inhibited the first methylation step (As(III) → MAs(III)), while only 5 are effective inhibitors of the second step (MAs(III) → DMAs(III)).
Figure 3.

Effect of TPIMS inhibitors on individual methylation steps. The activity of hAS3MT was assayed by HPLC-ICP-MS, as described under Materials and Methods. The assay contained TPIMS compounds at 50 μM each and 2 μM hAS3MT, final concentrations. The reactions were terminated by the addition of 6% (v/v) H2O2 after 4 h at 37 °C to oxidize all arsenic species, which were speciated by HPLC-ICP-MS. (A) Both methylation steps contribute to the methylation of 10 μM As(III) methylation. (B and C) Only the second methylation step is required for methylation of 10 μM MAs(III). (B) TPIMS compounds that inhibit MAs(III) methylation. (C) TPIMS compounds that do not inhibit MAs(III) methylation.
Effect of Small Molecule Inhibitors on As(III) Binding to hAS3MT
To examine whether inhibition of catalytic activity resulted from the inhibition of substrate binding, a fluorescent assay for As(III) binding was employed. As(III) is bound to two conserved cysteine residues in members of the AS3MT family.9,38 Adjacent to the As(III) binding site is a tyrosine residue. When this residue is mutated to a tryptophan residue, the enzyme exhibits quenching of intrinsic protein fluorescence when either As(III) or MAs(III) is bound. The corresponding CrAS3MT residue, Tyr72, was mutated to tryptophan, providing a spectroscopic probe for the binding of substrates.20 Two TPIMS compounds, TPI-2 and TPI-9, were fluorescent and could not be used in this assay. The other eight had little effect on the fluorescence of free tryptophan, and their effect on the protein fluorescence of the Y72W construct was examined. The protein was incubated with the small molecule inhibitors for 5 min and then titrated with either As(III) (Figure 4A) or MAs(III) (Figure 4B). In no case was there significant reduction of fluorescence quenching with either As(III) or MAs(III), indicating that the small molecule compounds do not competitively inhibit substrate binding.
Figure 4.

Effect of TPIMS compound binding of As(III) or MAs(III) to AS3MT. Substrate binding was estimated from the quenching of the fluorescence of the single-tryptophan CrArsM Y72W enzyme, as described under Materials and Methods. The 5 μM enzyme was preincubated with individual TPIMS compounds at 50 μM final concentration for 5 min on ice prior to the initiation of the assay with either (A) As(III) or (B) MAs(III) at the indicated concentrations. Black circle, no addition; open circle, TPI-2; inverted black triangle, TPI-4; open triangle, TPI-5; black square, TPI-6. The data are the mean ± SE (n = 3).
In Silico Docking Analysis Inhibitor Binding to AS3MT
A virtual screening approach using Autodock Vina was applied to calculate the lowest free energy binding site for the 10 small molecule inhibitors (Table 1).33 The only available crystal structure of an AS3MT, that from C. merolae, was used for the docking studies.25 From the structure, three domains have been identified, an N-terminal domain with the SAM binding site, a middle domain with the As(III) binding site, and a C-terminal domain of unknown function. Both the As(III) binding site and C-terminal domain are unique to AS3MTs and are not found in other SAM MTs. All 10 TPIMS compounds bound in a cleft between the first and third domains located on the opposite side of the protein from the As(III) and SAM binding sites, as illustrated with TPI-4 (Figure 5). The placement of the R groups in the models varied, but since in silico analysis provides predictions and not experimental results, the more important finding is that they all bound in the same cleft on the surface of the enzyme. From the calculated free energy of binding, it is clear that each of the inhibitors are predicted to bind to AS3MT with high affinity (Table 1). In contrast, the predicted affinity of binding of TPIMS compounds to COMT was several orders of magnitude lower than to AS3MT, and the predicted binding site overlaps with the catechol binding site (Figure 5).
Table 1. Comparison of Binding Properties of TPI Compounds Calculated Using Autodock Vina33.
| compound | AS3MT binding affinity (kcal/mol) | AS3MT Kd (μM) | COMT binding affinity (kcal/mol) | COMT Kd (μM) |
|---|---|---|---|---|
| TPI-1 | –7.3 | 4.5 | –4.3 | 704 |
| TPI-2 | –8.4 | 0.7 | –5.6 | 79 |
| TPI-3 | –9.3 | 0.2 | –4.6 | 424 |
| TPI-4 | –8.0 | 1.3 | –3.6 | 229 |
| TPI-5 | –9.4 | 0.1 | –1.4 | 9410 |
| TPI-6 | –7.2 | 5.3 | –4.7 | 358 |
| TPI-7 | –8.5 | 0.6 | –5.0 | 216 |
| TPI-8 | –9.2 | 0.2 | –2.2 | 2440 |
| TPI-9 | –8.8 | 0.5 | –0.7 | 306800 |
| TPI-10 | –9.0 | 0.3 | –5.5 | 93 |
Figure 5.
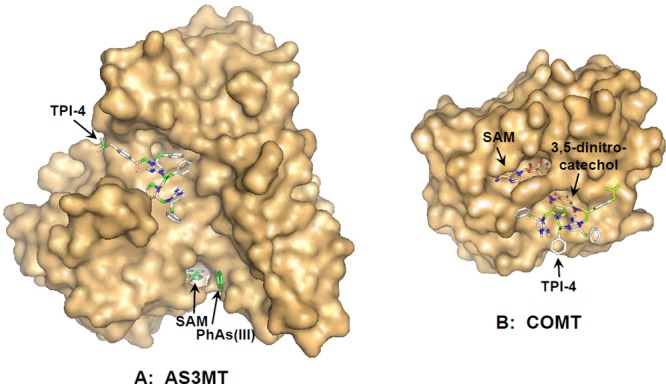
In silico binding analysis of TPI-4 to AS3MT and COMT. TPI-4 was docked with the crystal structure of (A) the AS3MT orthologue CmArsM (4FS8) and (B) COMT (3BWM) using AutoDock Vina (ADVina)33 as described under Materials and Methods. Surface plots of the two proteins are shown with TPI-4, SAM, PhAs(III), and 3,5-dinitrochatechol represented in stick form. Molecular models were generated using PyMol.34
Discussion
AS3MT catalyzes the formation of highly toxic trivalent methylated products9,39,40 that may also be more carcinogenic than inorganic arsenite.41,42 In microbes, AS3MT-catalyzed methylation of As(III) is clearly a detoxification process.43 In humans and other animals, arsenic methylation is paradoxically a detoxification process, but, in the long term, AS3MT activates arsenic into a more toxic and carcinogen species.41,44 Could modulation of human hAS3MT prevent or alleviate arsenic-related diseases? There are two possibilities: (1) inhibitors of hAS3MT could prevent the formation of MAs(III) and DMAs(III), or (2) activators of hAS3MT could increase clearance of arsenic in urine. Inhibition of hAS3MT might increase acute toxicity, as observed in knockout mice,15 but humans are rarely exposed to levels of arsenic that produce acute toxicity. Activation of hAS3MT might reduce the overall body burden, but it would not prevent the generation of intracellular MAs(III) and DMAs(III) and would not prevent intracellular events such as generation of reactive oxidative species or DNA damage. Which, if either, alternative would provide beneficial outcomes cannot be resolved without experimental evidence.
To date, no specific inhibitors of AS3MT have been identified. Our approach was to identify small molecules that bind to hAS3MT and either inhibit or activate. The objective of this study was to identify small molecules that bind to hAS3MT and affect enzymatic activity. These small molecules would be the starting point for future development of drugs to prevent arsenic-related disease. The small molecules should be selective for As(III) methyltransferases, and preferably, binding should not be competitive with As(III) or SAM. This required development of a high-throughput assay that could be used to screen small molecule libraries.20 This TR-FRET assay was adapted to screen for inhibitors, and modifications might allow it to screen for activators in future experiments.
We used this assay to screen synthetic mixture based libraries that have been used successfully in the identification of small molecule inhibitors over the last two decades.21,22 From a mixture based scaffold ranking library of over 30 million small molecule compounds, we identified 10 compounds that inhibit hAS3MT. The TPIMS inhibitors did not prevent binding of As(III), indicating noncompetitive binding at a site distinct from the substrate binding site. All 10 inhibit As(III) methylation, but only 5 inhibit MAs(III) methylation, indicating differences in their mechanism of action. We proposed that AS3MT undergoes different conformational changes during the two methylation steps that may account for the differential action of the inhibitors.9,45 Importantly, the latter observation means that at least those five do not inhibit SAM binding, or they would have inhibited all methyltransferase activity. TPI-4 also does not inhibit COMT activity, demonstrating selectivity for AS3MT. COMT is one of the few SAM MTs that is commercially available and has a convenient assay. In future experiments, the effect of the inhibitors on other members of the superfamily should be examined.
Crystallography has been instrumental in rational structure-based drug design.46 In silico docking of the TPIMS compounds was conducted using the crystal structure of an hAS3MT orthologue.25 There were no constraints imposed on the docking, just that the final solution should have the lowest free energy. It is significant that the lowest free energy site for all 10 inhibitors were at approximately the same location on the surface of the enzyme. While the differences in the binding affinity and inhibitory properties among the 10 cannot be deduced from the docking analysis, the fact that they are all predicted to bind at a similar region of the enzyme suggests that this is a site worthy of further analysis. Although AS3MT is a member of the large superfamily of SAM methyltransferases, the structure of the enzyme at the inhibitor binding site is unique in AS3MT and not found in other members of the superfamily.25 As noncompetitive inhibitors, their binding to a site distant from the substrate binding site implies a conformational change that may be regulatory and not linear with substrate binding. Noncompetitive inhibitors often bind to allosteric sites in enzymes. For example, a similar docking analysis of binding of 6-hydroxyflavon, a noncompetitive inhibitor of cytochrome P-450 2C9, indicates that it binds to the same allosteric regulatory site as warfarin,47 an important drug for treatment of blood clotting.48 An allosteric site on AS3MT implies that cells regulate its activity. A search for physiological regulators may be productive. In addition, a regulatory site could be a target for future development of modulators of hAS3MT activity. Future studies will be directed at cocrystallization of hAS3MT and orthologues with inhibitors and design of new inhibitors with high affinity as candidates of drug development, as well as understanding of the arsenic methylation mechanism. Meanwhile, these inhibitors will serve as useful reagents for research on the molecular mechanism of AS3MT.
Conclusions
New high-throughput assays and crystal structures have allowed the identification of synthetic small molecule inhibitors of AS3MT, the enzyme that activates arsenic into more highly toxic and potentially carcinogen species. The inhibitors bind to a potential allosteric site on the surface of AS3MT, suggesting physiological regulation of arsenic methylation. These small molecule inhibitors are the starting point for the development of drugs to prevent arsenic-related diseases.
Glossary
Abbreviations
- SAM
S-adenosylmethionine
- SAH
S-adenosylhomocysteine
- AS3MT
As(III) SAM methyltransferase
- SAM MT
SAM methyltransferase
- TR–FRET
time-resolved Förster resonance energy transfer
- As(III)
arsenite
- MAs(III)
methylarsenite
- MAs(V)
methyl arsenate
- DMAs(III)
dimethylarsenite
- DMAs(V)
dimethylarsenate
- TCEP
tris(2-carboxyethyl)phosphine
- COMT
catechol O-methyltransferase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.5b00432.
Scheme for the synthesis of TPI compounds; chemical structure of TIPMS compounds; effect of compound TPI-4 on COMT activity (PDF)
This work was supported by grants to B.P.R. from the National Institutes of Health (R01 ES023779) and the Bankhead-Coley Program (Grant # 4BF01), State of Florida Department of Health. Inhibitor development was supported in part by funding to the Torrey Pines Institute for Molecular Studies from the Florida Drug Discovery Acceleration Program, State of Florida Department of Health. M.M. was supported by a Raman Postdoctoral Fellowship from the University Grants Commission, Government of India.
The authors declare no competing financial interest.
Supplementary Material
References
- Council, National Research (2001) Arsenic in Drinking Water: 2001 Update. http://www.nap.edu/books/0309076293/html/.
- Li G.; Sun G. X.; Williams P. N.; Nunes L.; Zhu Y. G. (2011) Inorganic arsenic in Chinese food and its cancer risk. Environ. Int. 37, 1219–1225 10.1016/j.envint.2011.05.007. [DOI] [PubMed] [Google Scholar]
- States J. C.; Srivastava S.; Chen Y.; Barchowsky A. (2009) Arsenic and cardiovascular disease. Toxicol. Sci. 107, 312–323 10.1093/toxsci/kfn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. C.; Howard B. V.; Umans J. G.; Gribble M. O.; Best L. G.; Francesconi K. A.; Goessler W.; Lee E.; Guallar E.; Navas-Acien A. (2015) Arsenic exposure, arsenic metabolism, and incident diabetes in the strong heart study. Diabetes Care 38, 620–627 10.2337/dc14-1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobna Z.; Del Razo L. M.; Garcia-Vargas G. G.; Sanchez-Pena L. C.; Barrera-Hernandez A.; Styblo M.; Loomis D. (2013) Environmental exposure to arsenic, AS3MT polymorphism and prevalence of diabetes in Mexico. J. Exposure Sci. Environ. Epidemiol. 23, 151–155 10.1038/jes.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K.; Jenisova Z.; Feszterova M.; Baros S.; Liska J.; Hudecova D.; Rhodes C. J.; Valko M. (2011) Arsenic: toxicity, oxidative stress and human disease. J. Appl. Toxicol. 31, 95–107 10.1002/jat.1649. [DOI] [PubMed] [Google Scholar]
- Styblo M.; Del Razo L. M.; Vega L.; Germolec D. R.; LeCluyse E. L.; Hamilton G. A.; Reed W.; Wang C.; Cullen W. R.; Thomas D. J. (2000) Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch. Toxicol. 74, 289–299 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- Drobna Z.; Xing W.; Thomas D. J.; Styblo M. (2006) shRNA silencing of AS3MT expression minimizes arsenic methylation capacity of HepG2 cells. Chem. Res. Toxicol. 19, 894–898 10.1021/tx060076u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dheeman D. S.; Packianathan C.; Pillai J. K.; Rosen B. P. (2014) Pathway of human AS3MT arsenic methylation. Chem. Res. Toxicol. 27, 1979–1989 10.1021/tx500313k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes M. F.; Devesa V.; Adair B. M.; Conklin S. D.; Creed J. T.; Styblo M.; Kenyon E. M.; Thomas D. J. (2008) Tissue dosimetry, metabolism and excretion of pentavalent and trivalent dimethylated arsenic in mice after oral administration. Toxicol. Appl. Pharmacol. 227, 26–35 10.1016/j.taap.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le X. C.; Lu X.; Ma M.; Cullen W. R.; Aposhian H. V.; Zheng B. (2000) Speciation of key arsenic metabolic intermediates in human urine. Anal. Chem. 72, 5172–5177 10.1021/ac000527u. [DOI] [PubMed] [Google Scholar]
- Gong Z.; Lu X.; Cullen W. R.; Chris Le X. (2001) Unstable trivalent arsenic metabolites, monomethylarsonous acid and dimethylarsinous acid. J. Anal. At. Spectrom. 16, 1409–1413 10.1039/b105834g. [DOI] [Google Scholar]
- Guengerich F. P. (2000) Metabolism of chemical carcinogens. Carcinogenesis 21, 345–351 10.1093/carcin/21.3.345. [DOI] [PubMed] [Google Scholar]
- Drobna Z.; Naranmandura H.; Kubachka K. M.; Edwards B. C.; Herbin-Davis K.; Styblo M.; Le X. C.; Creed J. T.; Maeda N.; Hughes M. F.; Thomas D. J. (2009) Disruption of the arsenic (+3 oxidation state) methyltransferase gene in the mouse alters the phenotype for methylation of arsenic and affects distribution and retention of orally administered arsenate. Chem. Res. Toxicol. 22, 1713–1720 10.1021/tx900179r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokohira M.; Arnold L. L.; Pennington K. L.; Suzuki S.; Kakiuchi-Kiyota S.; Herbin-Davis K.; Thomas D. J.; Cohen S. M. (2010) Severe systemic toxicity and urinary bladder cytotoxicity and regenerative hyperplasia induced by arsenite in arsenic (+3 oxidation state) methyltransferase knockout mice. A preliminary report. Toxicol. Appl. Pharmacol. 246, 1–7 10.1016/j.taap.2010.04.013. [DOI] [PubMed] [Google Scholar]
- Cohen S. M.; Ohnishi T.; Arnold L. L.; Le X. C. (2007) Arsenic-induced bladder cancer in an animal model. Toxicol. Appl. Pharmacol. 222, 258–263 10.1016/j.taap.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Chung C. J.; Huang Y. L.; Huang Y. K.; Wu M. M.; Chen S. Y.; Hsueh Y. M.; Chen C. J. (2013) Urinary arsenic profiles and the risks of cancer mortality: A population-based 20-year follow-up study in arseniasis-endemic areas in Taiwan. Environ. Res. 122, 25–30 10.1016/j.envres.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Kenyon E. M.; Hughes M. F. (2001) A concise review of the toxicity and carcinogenicity of dimethylarsinic acid. Toxicology 160, 227–236 10.1016/S0300-483X(00)00458-3. [DOI] [PubMed] [Google Scholar]
- Yamanaka K.; Kato K.; Mizoi M.; An Y.; Takabayashi F.; Nakano M.; Hoshino M.; Okada S. (2004) The role of active arsenic species produced by metabolic reduction of dimethylarsinic acid in genotoxicity and tumorigenesis. Toxicol. Appl. Pharmacol. 198, 385–393 10.1016/j.taap.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Dong H.; Xu W.; Pillai J. K.; Packianathan C.; Rosen B. P. (2015) High-throughput screening-compatible assays of As(III) S-adenosylmethionine methyltransferase activity. Anal. Biochem. 480, 67–73 10.1016/j.ab.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinilla C.; Appel J. R.; Borras E.; Houghten R. A. (2003) Advances in the use of synthetic combinatorial chemistry: mixture-based libraries. Nat. Med. 9, 118–122 10.1038/nm0103-118. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Giulianotti M. A.; Appel J. R.; Dooley C. T.; Nefzi A.; Ostresh J. M.; Yu Y.; Maggiora G. M.; Medina-Franco J. L.; Brunner D.; Schneider J. (2008) Strategies for the use of mixture-based synthetic combinatorial libraries: scaffold ranking, direct testing in vivo, and enhanced deconvolution by computational methods. J. Comb. Chem. 10, 3–19 10.1021/cc7001205. [DOI] [PubMed] [Google Scholar]
- Wu J.; Zhang Y.; Maida L. E.; Santos R. G.; Welmaker G. S.; LaVoi T. M.; Nefzi A.; Yu Y.; Houghten R. A.; Toll L.; Giulianotti M. A. (2013) Scaffold ranking and positional scanning utilized in the discovery of nAChR-selective compounds suitable for optimization studies. J. Med. Chem. 56, 10103–10117 10.1021/jm401543h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchardt R. T. (1974) A rapid spectrophotometric assay for catechol-O-methyltransferase. Anal. Biochem. 58, 382–389 10.1016/0003-2697(74)90206-1. [DOI] [PubMed] [Google Scholar]
- Ajees A. A.; Marapakala K.; Packianathan C.; Sankaran B.; Rosen B. P. (2012) Structure of an As(III) S-adenosylmethionine methyltransferase: insights into the mechanism of arsenic biotransformation. Biochemistry 51, 5476–5485 10.1021/bi3004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reay P. F.; Asher C. J. (1977) Preparation and purification of 74As-labeled arsenate and arsenite for use in biological experiments. Anal. Biochem. 78, 557–560 10.1016/0003-2697(77)90117-8. [DOI] [PubMed] [Google Scholar]
- Chen J.; Sun S.; Li C. Z.; Zhu Y. G.; Rosen B. P. (2014) Biosensor for organoarsenical herbicides and growth promoters. Environ. Sci. Technol. 48, 1141–1147 10.1021/es4038319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley C. T.; Ny P.; Bidlack J. M.; Houghten R. A. (1998) Selective ligands for the mu, delta, and kappa opioid receptors identified from a single mixture based tetrapeptide positional scanning combinatorial library. J. Biol. Chem. 273, 18848–18856 10.1074/jbc.273.30.18848. [DOI] [PubMed] [Google Scholar]
- Nefzi A.; Dooley C.; Ostresh J. M.; Houghten R. A. (1998) Combinatorial chemistry: from peptides and peptidomimetics to small organic and heterocyclic compounds. Bioorg. Med. Chem. Lett. 8, 2273–2278 10.1016/S0960-894X(98)00412-0. [DOI] [PubMed] [Google Scholar]
- Houghten R. A. (1985) General method for the rapid solid-phase synthesis of large numbers of peptides: specificity of antigen-antibody interaction at the level of individual amino acids. Proc. Natl. Acad. Sci. U. S. A. 82, 5131–5135 10.1073/pnas.82.15.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J.; Rosen B. P.; Zhang Y.; Wang G.; Franke S.; Rensing C. (2006) Arsenic detoxification and evolution of trimethylarsine gas by a microbial arsenite S-adenosylmethionine methyltransferase. Proc. Natl. Acad. Sci. U. S. A. 103, 2075–2080 10.1073/pnas.0506836103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. (2005) A component-based software environment for visualizing large macromolecular assemblies. Structure 13, 447–462 10.1016/j.str.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. (2009) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. (2001) The PyMOL User’s Manual, DeLano Scientific, San Carlos, CA. [Google Scholar]
- Chen J.; Qin J.; Zhu Y. G.; de Lorenzo V.; Rosen B. P. (2013) Engineering the soil bacterium Pseudomonas putida for arsenic methylation. Appl. Environ. Microbiol. 79, 4493–4495 10.1128/AEM.01133-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Razo L. M.; Garcia-Vargas G. G.; Valenzuela O. L.; Castellanos E. H.; Sanchez-Pena L. C.; Currier J. M.; Drobna Z.; Loomis D.; Styblo M. (2011) Exposure to arsenic in drinking water is associated with increased prevalence of diabetes: a cross-sectional study in the Zimapan and Lagunera regions in Mexico. Environ. Health 10, 73. 10.1186/1476-069X-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- B’Hymer C.; Caruso J. A. (2004) Arsenic and its speciation analysis using high-performance liquid chromatography and inductively coupled plasma mass spectrometry. J. Chromatogr. A 1045, 1–13 10.1016/j.chroma.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Marapakala K.; Qin J.; Rosen B. P. (2012) Identification of catalytic residues in the As(III) S-adenosylmethionine methyltransferase. Biochemistry 51, 944–951 10.1021/bi201500c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Shi Q.; Nix F. B.; Styblo M.; Beck M. A.; Herbin-Davis K. M.; Hall L. L.; Simeonsson J. B.; Thomas D. J. (2002) A novel S-adenosyl-L-methionine:arsenic(III) methyltransferase from rat liver cytosol. J. Biol. Chem. 277, 10795–10803 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- Drobna Z.; Waters S. B.; Devesa V.; Harmon A. W.; Thomas D. J.; Styblo M. (2005) Metabolism and toxicity of arsenic in human urothelial cells expressing rat arsenic (+3 oxidation state)-methyltransferase. Toxicol. Appl. Pharmacol. 207, 147–159 10.1016/j.taap.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styblo M.; Drobna Z.; Jaspers I.; Lin S.; Thomas D. J. (2002) The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ. Health Perspect. 110(Suppl 5), 767–771 10.1289/ehp.02110s5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobna Z., Jaspers I., Thomas D. J., and Styblo M. (2002) Activation of AP-1 in Urotsa Cells by Methylated Trivalent Arsenicals, in EPA Science Inventory (Agency U. S. E. P., Ed.), Environmental Protection Agency, Washington, DC. [Google Scholar]
- Ye J.; Rensing C.; Rosen B. P.; Zhu Y. G. (2012) Arsenic biomethylation by photosynthetic organisms. Trends Plant Sci. 17, 155–162 10.1016/j.tplants.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D. J., and Rosen B. P. (2013) Arsenic Methyltransferases, in Encyclopedia of Metalloproteins (Kretsinger R. H., Uversky V. N., and Permyakov E. A., Eds.) pp 138–143, Springer, New York, NY. [Google Scholar]
- Marapakala K.; Packianathan C.; Ajees A. A.; Dheeman D. S.; Sankaran B.; Kandavelu P.; Rosen B. P. (2015) A disulfide-bond cascade mechanism for As(III) S-adenosylmethionine methyltransferase. Acta Crystallogr., Sect. D: Biol. Crystallogr. 71, 505–515 10.1107/S1399004714027552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschamps J. R. (2005) The role of crystallography in drug design. AAPS J. 7, E813–819 10.1208/aapsj070478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si D.; Wang Y.; Zhou Y. H.; Guo Y.; Wang J.; Zhou H.; Li Z. S.; Fawcett J. P. (2009) Mechanism of CYP2C9 inhibition by flavones and flavonols. Drug Metab. Dispos. 37, 629–634 10.1124/dmd.108.023416. [DOI] [PubMed] [Google Scholar]
- Roden D. M. (2015) Cardiovascular pharmacogenomics: current status and future directions. J. Hum. Genet. 10.1038/jhg.2015.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.