

Summary
The current classification system for breast cancer is based on expression of prognostic and predictive biomarkers. As an alternative, we propose a hypothesis-based ontological breast cancer classification modeled after the taxonomy of species in evolutionary biology. This approach uses normal breast epithelial cell types and differentiation lineages as the gold standard to classify tumors. We show that there are at least eleven previously undefined normal cell types in human breast epithelium and that each breast carcinoma is related to one of these normal cell types. We find that triple negative breast cancers do not have a ‘basal-like’ phenotype. Normal breast epithelial cells conform to four novel hormonal differentiation states and almost all human breast tumors duplicate one of these hormonal differentiation states which have significant survival differences. This ontological classification scheme provides actionable treatment strategies and provides an alternative approach for understanding tumor biology with wide-ranging implications for tumor taxonomy.
Keywords: breast cancer diagnostics, Nurses’ Health Study, pathology, biomarkers, heterogeneity, -omics
Traditionally, the development of a taxonomy of a disease entity has been based upon an understanding of the underlying pathogenesis of a particular disease. Once a disease is defined as a single and pathophysiologically uniform entity, various clinical and molecular prognostic features are then used to define the severity of the disease.
This paradigm has been difficult to follow for classification of cancer due to our lack of understanding of the underlying mechanisms. In the case of breast cancer an empirical system has been developed over the past three decades without a clear underlying organizing principle. The widely accepted paradigm for the classification of human breast cancers has been to group tumors into three categories based on the presence of estrogen receptor (ER+), progesterone receptor (PR+), and human epidermal growth factor receptor 2 (HER2+), or by their absence in triple-negative breast cancers (ER/PR/HER2-,TNBC). These categories are based on the expression of molecular targets that predict response to different types of treatment such as with the ER-antagonist Tamoxifen, the selective estrogen receptor down-regulator Fulvestrant and the anti-HER2 monoclonal antibody Herceptin. Though pragmatic for dictating clinical treatment, such an ad-hoc classification scheme does not provide insights about the pathogenesis or about the true phylogeny of breast cancer.
In recent years, purely prognostic molecular classification schemes have been proposed to replace the above described empirical classification system for breast cancer Several high-throughput molecular tools and associated statistical methods such as mRNA expression profiles have been used to define several prognostic subgroups of breast cancer: Luminal A, Luminal B, Basal-like, Claudin-low and Her2-like (1, 2). Likewise, DNA methylation patterns have been used to identify five distinct groups (3) and ten different breast cancer subtypes have been identified based on a DNA copy number based genetic classification system (4, 5).
However, while prognostic categories subdivide diagnostic categories into distinct outcome groups, they cannot be the sole basis of a comprehensive classification approach. The principle reason for this is that in a purely prognostic approach the only criterion that distinguishes two entities is their difference in clinical outcome. Hence, two different entities with the similar outcome but with different underlying mechanisms of pathogenesis cannot be distinguished with this approach; such as heart attacks vs. strokes. This is not a trivial issue since differences in pathophysiology may reasonably require very different treatment approaches. In addition, a purely prognostic approach may end up categorizing two different stages of a single disease as different entities; such as three vessel coronary artery disease vs. one vessel disease.
Consequently, purely molecular prognostic approaches have not yet lead to a comprehensive classification system. Furthermore, there has been little overlap among the mRNA expression, DNA copy number and methylation based prognostic groups, because they are not based on a common pathophysiology (6). As a result, a breast cancer task force recently concluded that, at the moment, molecular tools do not provide sufficiently robust information beyond histological type, grade, and ER, PR, and HER2 status (7) and these molecular tests are therefore not routinely performed for diagnostic purposes at most institutions (8).
We set out to provide a pathophysiological framework that could provide a biological setting in which prognostic categories could be discovered (9). Notably, the phylogeny of normal cell types have been successfully used as a reference point to classify lymphomas and leukemias (10). The discovery of morphologic and molecular similarities between the various subtypes of leukemias and lymphomas with normal hematopoietic cell types was very important in this process and has been an important factor in the successful classification and treatment of many hematopoietic malignancies.
In solid tissues, an in-depth characterization of the normal cell subtypes has been very difficult. Until recently only two cell types - luminal and myoepithelial cells - had been described in the human breast (11). This limited understanding of the cell types that comprise normal breast tissue has precluded a normal cell type-based classification system for breast cancer. Inspired by the classification of hematopoietic malignancies, we hypothesized that a more detailed description of normal cell types in the human breast may be important for the effective classification of human breast tumors.
With this goal in mind, we recently analyzed more than 15,000 normal breast cells and described the normal phylogeny of cell subtypes in the luminal layer of human breast (9). We identified molecules that have bimodal patterns of expression (i.e. ‘on’ or ‘off’) in the luminal and myoepithelial layers of the breast. We first started with intermediate filament markers such as cytokeratins which we found to be particularly useful, especially CKs 5, 7, 8, 14, 17,18 and 19. This characterization showed that CKs 7 and 18 and Claudin-4 are expressed in all luminal layer cells but that they are not expressed in the myoepithelial layer. Conversely, CD10, SMA and p63 are expressed in all of the myoepithelial cells but not in the cells comprising the luminal layer of normal breast. Of note, this analysis revealed important insights into the expression of cytokeratins such as CKs 5 and 14 that had previously been considered as “basal’ keratins. CK5 and 14 were presumed to have expression restricted to the normal myopepithelial cells and this misconception was the basis for defining CK5 and CK14 positive breast cancers as ‘basal-like’ (a subset of triple negative breast cancers). Our observations and those of others (12–14) support that CK5 and 14 have been mistakenly referred to as 'basal keratins' and that they are clearly expressed in luminal cells in the lobules of normal human breast. Moreover, our analysis of tumors shows that the name "basal-like' is not an appropriate description of the differentiation state or the cell-of-origin of CK5 and 14 expressing breast cancers as this differentiation state is similar to the subset of normal luminal cells of the breast that express CK5 and 14 – a distinct normal luminal cell population that does not express ER or PR.
While characterizing additional protein expression patterns in the set of over 15,000 normal breast cells we noted the bimodal expression of the estrogen receptor (ER), the androgen receptor (AR) and the vitamin D receptor (VDR) in normal luminal cells. A comprehensive assessment of these cells using double and triple immunofluorescence analyses and a novel multiplexed immunostaining technology platform (15) showed that the luminal cells conform to four hormone receptor differentiation groups based on estrogen, androgen and vitamin D receptor (ER, AR and VDR) expression in normal human breast cells – HR0 cells expressing none of these receptors, HR1 cells expressing only one of these three receptors, HR2 cells expressing any two of these receptors and HR3 cells expressing all three of the receptors. In summary, our results indicate that the composition of normal breast epithelium is much more complex than previously appreciated – our breast taxonomy comprises at least 11 cellular differentiation states in normal human breast lobules, which can be divided into four hormone receptor groups (HR0, 1, 2, and 3; Table 1 and Figure 1).
Table 1. Normal human breast cell types.
Immunostaining of normal breast sections from multiple donors (n=36) with fourteen different markers identified multiple normal breast cell subtypes. We grouped the 11 differentiation states in the luminal layer of human breast lobules (L1–11) into hormone receptor negative (HR0; L1–3), single hormone receptor positive (HR1+: ER, AR or VDR; L4–L6), double hormone receptor positive (HR2+: ER/AR, ER/VDR, or AR/VDR; L7–L10) and triple hormone receptor positive (HR3+: ER/AR/VDR; L11) states. All luminal cells expressed K7/18 and Cld-4. In the myoepithelial layer, all cells expressed SMA, CD10 and p63, with two subtypes that were either K5/14/17[−] or K5/14/17[+] (M1 and M2).
| Cellular Differentiation States in Normal Human Breast Lobules | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CELL TYPE | ER | AR | VDR | K5 K14 K17 |
Ki67 | Cld-4 | K7 K8 K18 |
CD10 SMA p63 |
|||
| L1 | HR0 | Ki67+ | Luminal | − | − | − | − | + | + | + | − |
| L2 | HR0 | K18+ | Luminal | − | − | − | − | − | + | + | − |
| L3 | HR0 | K5+ | Luminal | − | − | − | + | − | + | + | − |
| L4 | HR1 | ER+ | Luminal | + | − | − | − | − | + | + | − |
| L5 | HR1 | AR+ | Luminal | − | + | − | − | − | + | + | − |
| L6 | HR1 | VDR+ | Luminal | − | − | + | − | − | + | + | − |
| L7 | HR1 | K5/VDR+ | Luminal | − | − | + | + | − | + | + | − |
| L8 | HR2 | ER/AR+ | Luminal | + | + | − | − | − | + | + | − |
| L9 | HR2 | ER/VDR+ | Luminal | + | − | + | − | − | + | + | − |
| L10 | HR2 | AR/VDR+ | Luminal | − | + | + | − | − | + | + | − |
| L11 | HR3 | ER/AR/VDR+ | Luminal | + | + | + | − | − | + | + | − |
| M1 | CD10+ | Myoepithelial | − | − | − | − | − | − | − | + | |
| M2 | K5+ | Myoepithelial | − | − | − | + | − | − | − | + | |
Figure 1. Putative differentiation lineage hierarchy of normal human breast.
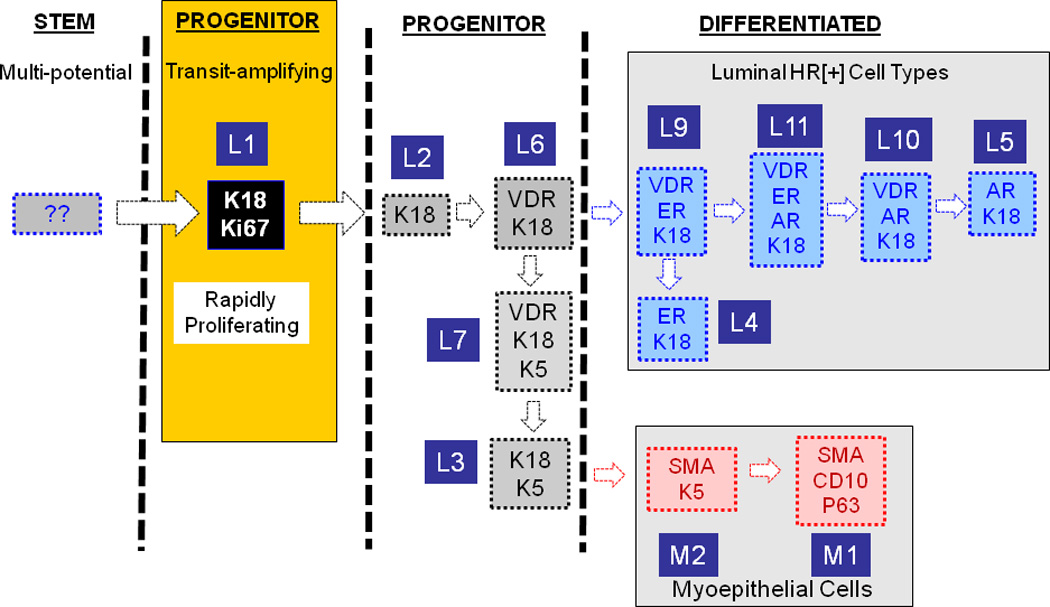
Stem cells are mostly quiescent and they rarely proliferate, but they give rise to a rapidly proliferating finite-lifespan progenitor cells with multi-lineage differentiation potential. The more differentiated cell types increasingly become mitotically less active, and finally the terminally differentiated cells become post-mitotic. Among the normal breast cell types only K18[+] cells were highly proliferative, which makes them the best candidate for the transit amplifying cells. We attempted to organize the rest of the breast cell types in a way where each differentiation step involves gain or loss of a single marker. Based on this constraint, we postulate that transit amplifying cell first loses its proliferative capacity which coincides with VDR expression giving rise to a oligo-potential progenitor (L6). This cell either maintains K18 expression and gains ER and AR expression giving rise to luminal HR+ cell types (L4–5, and L9–11), or it down-regulates K18 and up-regulate K5 (L7). When this cell downregulates VDR (L3) and upregulates SMA and p63 it generates the typical K5[+], HR[−] K18[−] myoeptihelial cell type (M2). As this cell downregulates K5 it generates the second subpopulation of K5[−] myoepithelial cells (M1).The above model depicts one possible scenario among many. Describing the interrelatedness of the breast cell types we identified and understanding their differentiation lineage hierarchy will require in vivo and functional ex vivo experiments that are not currently possible for technical reasons.
The striking heterogeneity in the molecular characteristics of individual cells in normal breast epithelium paralleled the distinct profiles of normal hematopoietic cell populations so we next assessed whether breast carcinomas resemble hematological malignancies with tumor cells maintaining cell type/differentiation specific patterns of protein expression that reflects the patterns observed in their non-neoplastic counterparts. Remarkably, when we compared the 11 normal breast cell types that we had identified with more than 3,000 human breast tumors we found that the vast majority (>95%) of patient tumors could be placed precisely in this normal cell type phylogeny as could most of over 60 cell lines that are commonly used for studying breast cancer. In addition, almost none of the breast cancers exhibit a pure basal-like phenotype as defined by the expression of true myoepithelial markers and absence of any luminal markers. Strikingly, when we classified the breast tumors from over 1,800 patients from the Nurses’ Health Study according to the HR categories we had defined in normal breast luminal cells (HR0–3), we found a very strong association between the number of receptors expressed in a breast carcinoma and the 5 year survival of the patient – with patients with HR3+ tumors having the best survival, and patients with HR0 tumors having the worst survival. We noted similar results analyzing survival based on the mRNA expression patterns of these hormone receptors from a different breast cancer cohort (16) and demonstrated effects on growth by modulating the activity of AR and VDR. In all, this data suggests that evaluating the HR status of a breast cancer could provide diagnostic, prognostic as well as predictive value.
Hence, our efforts offer a different approach for tumor classification that differs from efforts that are focused on developing a comprehensive molecular analysis based exclusively on tumor genomic information. While such genomic efforts are clearly revealing new targetable lesions for treating some cancers, these efforts may ultimately not provide a rational classification system – particularly in tumors which have very complex molecular genetic aberrations, where each individual has a tumor with a nearly unique set of genetic changes. Likely, these '-omics’ approaches have not yielded the anticipated results because they have low morphologic resolution, lack objective points of reference and, most importantly, they are not hypothesis driven. These shortcomings can result in a loss of tumor lineage information, can lead to redundant classification schemes and can split tumors into smaller and smaller arbitrary groups. Instead, we propose a very different approach: the use normal cell types as a gold standard to classify tumors and offer an approach for assessing risk on information garnered from analysis of cells (i.e. cell-based risk assessment rather than on expression based on analyses of homogenized cells). In normal tissues, each cell subtype is designed to perform a specific function. Since these functions are defined and finite, the maximum number of biologically important normal cell types are limited, unchanging and can be precisely defined. Tumors are similarly restricted. Therefore, our method objectively constrains the arbitrary splitting of tumors into endless subclasses, and provides a durable context within which molecular data may be accurately interpreted.
What we propose here is a stepwise classification system that places tumors into lineage based diagnostic categories based on their distinct tissue of origin, cell-of-origin and differentiation lineage. Upon defining uniform lineage based classes, we propose to use molecular and genetic classifiers to distinguish prognostic subsets within each lineage.
Footnotes
Financial and competing interests disclosure
T Ince was a scientific advisor to 30M Inc. (2007–2012). S Santagata was cofounder of and scientific advisor to Bayesian Diagnostics. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Contributor Information
Sandro Santagata, Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Department of Cancer Biology, Dana-Farber Cancer Institute, 77 Avenue Louis Pasteur, Boston, Massachusetts 02115, Ph: 617-525-5686, Fax: 617-975-0944, ssantagata@partners.org.
Tan A. Ince, Sylvester Comprehensive Cancer Center, Braman Family Breast Cancer Institute and Interdisciplinary Stem Cell Institute, University of Miami Miller School of Medicine, Biomedical Research Building (BRB) Room 907, Miami, FL, 33136, Ph: 305-243-1782, Fax: 305-243-5929, TInce@med.miami.edu.
REFERENCES
- 1.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer research : BCR. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2010;5:5–23. doi: 10.1016/j.molonc.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson SJ, Rueda OM, Aparicio S, Caldas C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013;32:617–628. doi: 10.1038/emboj.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaffe MB. The scientific drunk and the lamppost: massive sequencing efforts in cancer discovery and treatment. Sci Signal. 2013;6:pe13. doi: 10.1126/scisignal.2003684. [DOI] [PubMed] [Google Scholar]
- 7.Guiu S, Michiels S, Andre F, Cortes J, Denkert C, Di Leo A, et al. Molecular subclasses of breast cancer: how do we define them? The IMPAKT 2012 Working Group Statement. Ann Oncol. 2012;23:2997–3006. doi: 10.1093/annonc/mds586. [DOI] [PubMed] [Google Scholar]
- 8.Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23(Suppl 2):S60–S64. doi: 10.1038/modpathol.2010.33. [DOI] [PubMed] [Google Scholar]
- 9.Santagata S, Thakkar A, Ergonul A, Wang B, Woo T, Hu R, et al. Taxonomy of breast cancer based on normal cell phenotype predicts outcome. The Journal of clinical investigation. 2014;124:859–870. doi: 10.1172/JCI70941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swerdlow S, Campo E, Harris NL, Jaffe E, Pileri S, Stein H. WHO classification of Tumours of Haematopoietic and Lymphoid Tissues (International Agency for Research on Cancer, Lyon, France) 2008 [Google Scholar]
- 11.Jones C, Mackay A, Grigoriadis A, Cossu A, Reis-Filho JS, Fulford L, et al. Expression Profiling of Purified Normal Human Luminal and Myoepithelial Breast Cells Identification of Novel Prognostic Markers for Breast Cancer. Cancer research. 2004;64:3037–3045. doi: 10.1158/0008-5472.can-03-2028. [DOI] [PubMed] [Google Scholar]
- 12.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–417. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Gusterson B. Do 'basal-like' breast cancers really exist? Nat Rev Cancer. 2009;9:128–134. doi: 10.1038/nrc2571. [DOI] [PubMed] [Google Scholar]
- 14.Gusterson BA, Ross DT, Heath VJ, Stein T. Basal cytokeratins and their relationship to the cellular origin and functional classification of breast cancer. Breast cancer research : BCR. 2005;7:143–148. doi: 10.1186/bcr1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harrell JC, Prat A, Parker JS, Fan C, He X, Carey L, et al. Genomic analysis identifies unique signatures predictive of brain, lung, and liver relapse. Breast Cancer Res Treat. 2012;132:523–535. doi: 10.1007/s10549-011-1619-7. [DOI] [PMC free article] [PubMed] [Google Scholar]