

Abstract
Background
A new class of non-coding RNAs, known as long non-coding RNAs (lncRNAs), has been recently described. These lncRNAs are implicated to play pivotal roles in various molecular processes, including development and oncogenesis. Gene expression profiling of human B-ALL samples showed differential lncRNA expression in samples with particular cytogenetic abnormalities. One of the most promising lncRNAs identified, designated B-ALL associated long RNA-6 (BALR-6), had the highest expression in patient samples carrying the MLL rearrangement, and is the focus of this study.
Results
Here, we performed a series of experiments to define the function of BALR-6, including several novel splice forms that we identified. Functionally, siRNA-mediated knockdown of BALR-6 in human B-ALL cell lines caused reduced cell proliferation and increased cell death. Conversely, overexpression of BALR-6 isoforms in both human and mouse cell lines caused increased proliferation and decreased apoptosis. Overexpression of BALR-6 in murine bone marrow transplantation experiments caused a significant increase in early hematopoietic progenitor populations, suggesting that its dysregulation may cause developmental changes. Notably, the knockdown of BALR-6 resulted in global dysregulation of gene expression. The gene set was enriched for leukemia-associated genes, as well as for the transcriptome regulated by Specificity Protein 1 (SP1). We confirmed changes in the expression of SP1, as well as its known interactor and downstream target CREB1. Luciferase reporter assays demonstrated an enhancement of SP1-mediated transcription in the presence of BALR-6. These data provide a putative mechanism for regulation by BALR-6 in B-ALL.
Conclusions
Our findings support a role for the novel lncRNA BALR-6 in promoting cell survival in B-ALL. Furthermore, this lncRNA influences gene expression in B-ALL in a manner consistent with a function in transcriptional regulation. Specifically, our findings suggest that BALR-6 expression regulates the transcriptome downstream of SP1, and that this may underlie the function of BALR-6 in B-ALL.
Electronic supplementary material
The online version of this article (doi:10.1186/s12943-015-0485-z) contains supplementary material, which is available to authorized users.
Keywords: lncRNA, B-ALL, MLL, SP1, Microarray, Leukemia, RNA, Non-coding RNA
Background
The human genome produces thousands of non-coding transcripts [1]. These include the recently described class of long non-coding RNAs (lncRNAs), which have distinct chromatin signatures and epigenetic marks, designating them as unique structures that are conserved in mammals [2, 3]. More recently, comparison of lncRNA expression in zebrafish to that of mammals has suggested that although these structures retain limited overall sequence conservation among vertebrates, they show strong conservation of short stretches of sequence, chromosomal synteny and functional conservation [4]. Prior studies have shown that lncRNAs play a variety of roles in the regulation of transcription, splicing and miRNA function [5–7]. This may not be an exhaustive description of the functions of lncRNAs, as new functions are being discovered in other cellular processes [8, 9]. As might be expected, considering their roles in critical cellular functions, lncRNAs have been found to be dysregulated in cancer, with functional roles in oncogenesis described for a handful of lncRNAs so far [10–13].
B-lymphoblastic leukemia (B-acute lymphoblastic leukemia, B-ALL) is a malignancy of precursor B-cells harboring mutations and translocations that result in dysregulated gene expression [14, 15]. We have recently completed a comprehensive description of lncRNAs in B-ALL and analyzed the association of lncRNA expression with clinicopathologic parameters. Our study showed differential lncRNA expression in samples with particular cytogenetic abnormalities [16]. One of the lncRNAs from our study, designated B-ALL associated long RNA-6 (BALR-6), was significantly upregulated in all subsets of patient samples when compared to normal CD19+ cells. Interestingly, the highest expression of BALR-6 was seen in patient samples carrying the MLL rearrangement [16]. MLL rearranged B-ALL cases have a very poor prognosis and occur in infants, making them particularly hard to treat [17].
Located on chromosome 3p24.3 in humans, BALR-6 exists in a syntenic gene block with neighboring genes SATB1 and TBC1D5 that is conserved in several vertebrate species (Fig. 1a, b and d) [16]. Analysis of publically available data from the Broad Institute/ENCODE shows H3K4m3 and H3K36m3 modifications along the promoter and gene body at LOC339862, where BALR-6 resides, indicating that it is a transcriptional element (Fig. 1a) [4, 16, 18–20]. Alternative splicing analysis by the Swiss Institute of Bioinformatics predicted multiple transcripts expressed at this gene locus (Additional file 1: Figure S1A) [21]. Moreover, 100 Vertebrate PhastCons analyses of the BALR-6 locus demonstrated significant conservation of the gene body, suggesting a functional transcript (Fig. 1b) [22].
Fig. 1.
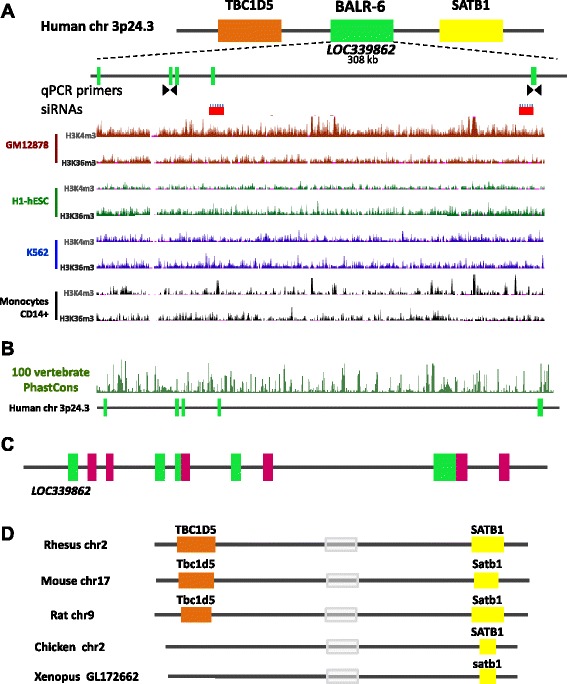
Molecular characterization of BALR-6. a Top: Chromosomal location of BALR-6 in the human genome, surrounding genes, qPCR primers, siRNAs, known annotated exons (green boxes), known introns (black lines) are shown. Bottom: Chip-Seq histone modification map from the Broad institute/ENCODE, taken from UCSC genome browser, shows H3K4m3 and H3K36m3 patterns at LOC339862 in four different cell types indicating active transcription of the lncRNA. b The 100 Vertebrate PhastCons plot from the UCSC whole-genome shows conserved regions among 98 vertebrates including mice and zebrafish throughout the locus. c RACE discovered unannotated exons (magenta) depicted with known annotated exons (green) at LOC339862. d Schematic depicting genomic conservation of the syntenic block among multiple vertebrates, as analyzed by BLAT. Grey box indicates location of homology to BALR-6
To further study this lncRNA we undertook loss-of-function analyses in B-ALL cell lines and gain-of-function analyses in vivo. We found that BALR-6 is a pro-survival factor for B-ALL cell lines, and that its knockdown led to decreased growth and increased apoptosis of these cells. In vivo, overexpression of BALR-6 led to an alteration of hematopoiesis with a shift to more immature progenitor populations. Gene expression analyses of knockdown cell lines showed a differentially expressed gene set in BALR-6 knockdown cells, with enrichment for SP1 transcriptional targets and leukemogenic genes. Finally, luciferase assays demonstrated an increase in transcriptional activity when SP1 and BALR-6 were co-expressed. Together, these findings point to a role for BALR-6 in cellular survival, leukemogenesis and highlight the role of novel elements of gene regulation in B-ALL.
Results
BALR-6 knockdown inhibits proliferation of human B-ALL cell lines
To comprehensively study the function for this novel lncRNA, we first characterized the transcripts originating at the genomic locus corresponding to BALR-6. Using RS4;11 cell line mRNA, Rapid Amplification of cDNA Ends (RACE) uncovered multiple isoforms; from these, three were cloned and sequenced corresponding to the genomic locus as shown (Additional file 1: Figure S1A-B). Northern Blot analysis of RS4;11 DNAse treated RNA revealed the expression of two isoforms containing exon 3 and exon 5 sequences, one sized at ~3.8 Kb and the other at ~1.2 Kb (Additional file 1: Figure S1C). The annotated mRNA and new alternative splice forms, including unannotated exons, were confirmed as depicted in Fig. 1c. Isoform 1 contains several small open reading frames (ORFs), however no Kozak sequences are found in their initial ATG region, and the predicted ORFs do not resemble any known functional proteins or peptide [23]. Isoforms 2 and 3 lacked open reading frames and translation initiation sites as evaluated by EMBOSS Transeq, predicting them to be non-coding transcripts (Additional file 1: Figure S1D).
To map the murine homologous transcript, we carried out 5′RACE and 3′RACE using mRNA extracted from murine pre B-ALL cell line 70Z/3. The sequences uncovered matched the human BALR-6 sequence, confirming that there is a murine transcript originating from this same locus (Additional file 1: Figure S1E). Further analysis by BLAT showed genomic conservation of syntenic blocks in a variety of vertebrates, including Xenopus tropicalis (Fig. 1d). Together, these data demonstrate a highly conserved, functional and complex gene locus that expresses multiple non-coding transcripts, some yet to be discovered. During normal B cell development, BALR-6 is dynamically expressed, with high expression in pre-B cells and subsequent downregulation (Fig. 2a). This suggests that the high expression of BALR-6 in B-ALL could represent a stage-specific expression pattern in leukemia derived from early stages of B-cell development. To elucidate a cellular function for BALR-6, we first evaluated the expression levels of the transcripts in human B-ALL cell lines. BALR-6 expression was highest in RS4;11 cells and MV(411) cells, which carry the MLL-AF4 rearrangement, when compared to other lines (Fig. 2b). Additionally, RS4;11 cells treated with bromodomain and extra-terminal (BET) motif binding protein inhibitor I-BET151 [24] showed decreased levels of BALR-6 in a dose-dependent manner (Fig. 2c). Given that I-BET151 has previously been shown to inhibit transcription downstream of MLL, we propose that BALR-6 expression is induced by MLL, although this effect may not be entirely specific to MLL-AF4.
Fig. 2.
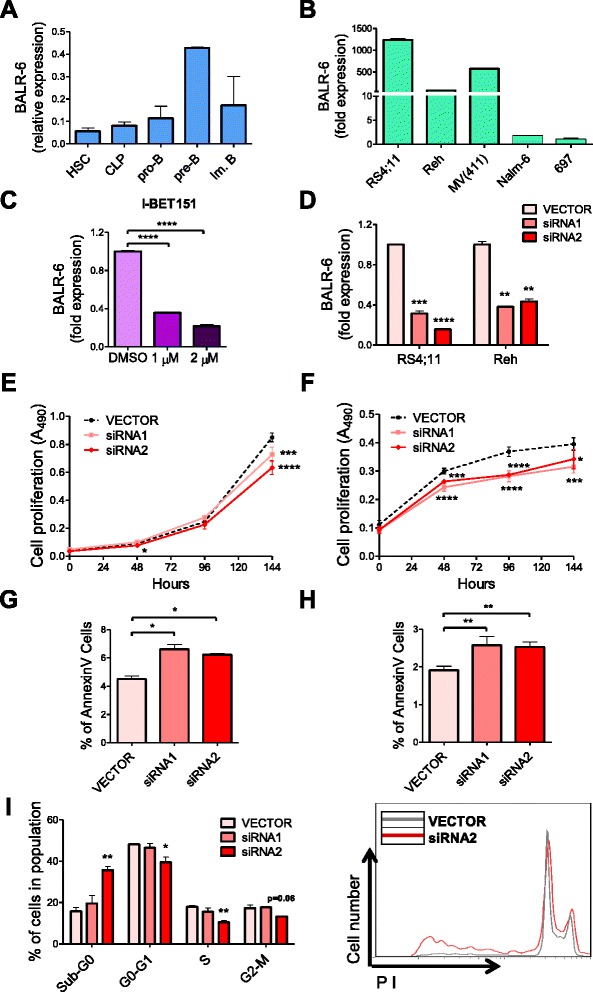
BALR-6 knockdown reduces cell proliferation and increases apoptosis in human B-ALL cells. a BALR-6 expression in human bone marrow B-cell subsets by qRT-PCR. Normalized to ACTIN. b Quantitation of BALR-6 expression in human B-ALL cell lines by qRT-PCR confirming elevated levels in MLL translocated cell lines RS4;11, and MV(411). Normalized to ACTIN. c RS4;11 cell lines treated with 1 μM, and 2 μM of I-BET151 inhibitor for 36 h, presented a decrease in BALR-6 expression levels. Normalized to ACTIN. d qRT-PCR quantification of BALR-6 in RS4;11 and Reh cell lines transduced with vector control, siRNA1 or siRNA2. Normalized to ACTIN. e, f Decreased cell proliferation, upon siRNA mediated knockdown of BALR-6 in RS4;11 cells e, and Reh cells f as measured by MTS. g, h AnnexinV staining showed that siRNA mediated knockdown of BALR-6 in RS4;11 cells g, and Reh cells h resulted in an increase of apoptosis. i Propidium iodide staining of RS4;11 knockdown cell lines showed an increase in Sub-G0 and a decrease in G0-G1, S and G2-M cells. Representative histogram of (i) confirms cell cycle changes by siRNA2, shown to the right. HSC, hematopoietic stem cell; CLP, common lymphoid progenitor; pro-B, progenitor B; pre-B, precursor B; DMSO, dimethyl sulfoxide. Evaluations were made using a two-tailed T-test, p < 0.05 (*); p < 0.005 (**); p < 0.0005 (***); p < 0.0001 (****)
Using the approach described previously, siRNAs against the splice junctions between exons of BALR-6 were cloned into a mmu-miR-155 expression cassette (Additional file 1: Figure S2A) [4, 16, 25, 26]. We observed knockdown of all the identified transcripts in multiple B-ALL cell lines (Fig. 2d and Additional file 1: Figure S2B). Transduced B-ALL cells showed a reduction in proliferation as early as 48 h after plating, with consistent reduction in proliferation observed over the full duration of the assay (up to 144 h) (Fig. 2e, f and Additional file 1: Figure S2C). siRNA-transduced B-ALL cells had significantly higher levels of apoptosis, as measured by AnnexinV, when compared with vector-transduced lines (Fig. 2g, h and Additional file 1: Figure S2D). Flow cytometry demonstrated that the siRNA2-transduced RS4;11 cell lines had an increase in Sub-G0 cells and a decrease in all other cell stages, consistent with increased apoptosis and decreased flux through the cell cycle (Fig. 2i). Together, these findings suggest a modest yet conserved role for BALR-6 in the regulation of B-ALL cell survival and proliferation.
Constitutively expressed BALR-6 supports cell survival and proliferation
To examine the effects of BALR-6 gain of function, we overexpressed the previously identified isoforms in the human B-ALL cell line Nalm-6, which has relatively low endogenous levels of the transcript (Figs. 2b and 3a). Gene transfer was conducted via a lentiviral expression system that has proven successful in our previous studies (Additional file 1: Figure S2E) [16]. Constitutive overexpression of BALR-6 Isoforms 2 and 3 led to a significant increase in proliferation as measured by MTS (Fig. 3c). In addition to an observed increase in overall growth rate, BALR-6 Isoforms 2 and 3 caused an increase in S phase cells and G2-M cells (Fig. 3d). Furthermore, AnnexinV staining showed significantly lower numbers of apoptotic cells under basal growth conditions in cell lines overexpressing any of the BALR-6 isoforms (Additional file 1: Figure S2G).
Fig. 3.
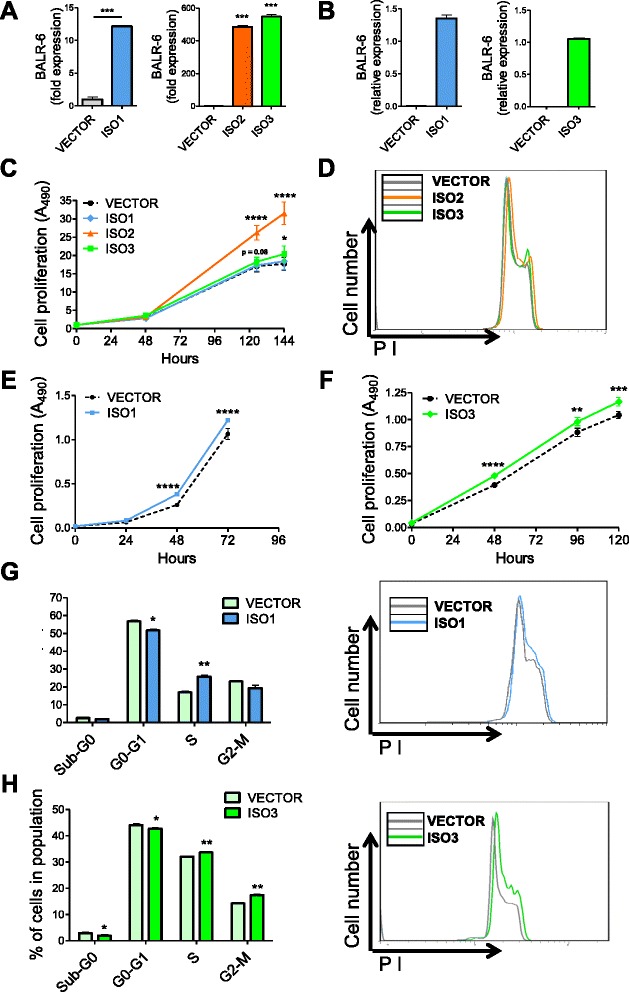
BALR-6 overexpression increases proliferation in human Nalm-6 and murine 70Z/3 cells. a qRT-PCR quantitation of BALR-6 isoform expression in Nalm-6 human pre B-ALL cell line. Normalized to ACTIN. b qRT-PCR quantification of BALR-6 isoforms in 70Z/3 mouse pre B-ALL cell line. Normalized to Actin (ISO1) or L32 (ISO3). c Increased cell proliferation in BALR-6 overexpressing Nalm-6 cell lines, as measured by MTS. d Representative histogram of Nalm-6 overexpression lines, stained with propidium iodide, shows an increase in S phase cells and G2-M cells. e, f Increased cell proliferation in BALR-6 Isoform 1 (e) and Isoform 3 (f) overexpressing 70Z/3 cell lines, as measured by MTS. g, h Propidium iodide staining of 70Z/3 cells overexpressing BALR-6 Isoform 1 (g) and Isoform 3 (h), shows a consistent increase in G2-M cells, and a decrease in Sub-G0 cells. Representative histogram of figures (g-h) confirmed the increase in cells in the G2-M when compared to the empty vector, shown to the right. ISO1, Isoform 1; ISO2, Isoform 2; ISO3, Isoform 3. Evaluations were made using a two-tailed T-test, p < 0.05 (*); p < 0.005 (**); p < 0.0005 (***); p < 0.0001 (****)
To overexpress BALR-6 in mouse cells, we constructed a set of MSCV-based bicistronic vectors (Fig. 3b, Additional file 1: Figure S2F). Successful overexpression of these constructs in murine pre B-ALL 70Z/3 cells led to a modest increase in proliferation (Fig. 3e and f). Cell cycle analysis of these lines showed an increase of S phase cells, G2-M cells (in Isoform 3 overexpressing lines) and a reduction in Sub-G0 cells, similar to the effects in Nalm-6 cells (Fig. 3g and h). Analysis by AnnexinV staining confirmed the lower number of apoptotic cells in Isoform 3 expressing cell lines (Additional file 1: Figure S2H). Moreover, these 70Z/3 Isoform 3 overexpression lines were less vulnerable to prednisolone-induced apoptosis (Additional file 1: Figure S2I). Conversely, siRNA-transduced RS4;11 cells were more prone to prednisolone-induced apoptosis (Additional file 1: Figure S2I). Therefore, knockdown and overexpression of BALR-6 had opposing phenotypes in B-ALL cell lines, and gain-of-function phenotypes were conserved in both human and mouse cells.
Enforced BALR-6 expression promotes expansion of hematopoietic progenitor populations in vivo
Since BALR-6 is highly expressed in B-ALL, we tested the effects of constitutive expression in an in vivo model [16]. 5-FU enriched bone marrow was transduced with retrovirus expressing the BALR-6 Isoform 3 and transplanted into lethally irradiated hosts (Fig. 3 and Additional file 1: Figure S2F, 2H). Mice were followed with peripheral bleeds for 16 weeks and then sacrificed for analysis. Peripheral white blood cell counts were not statistically different between the control and experimental groups. However, mice with enforced expression of BALR-6 showed a trend towards lower red blood cell counts, hematocrit and platelet counts (Additional file 1: Figure S3A). Flow cytometry revealed a lower percentage of CD11b + myeloid cells and a higher percentage of B220+ B-cells, but no difference in CD3ε + T-cell percentage in the eGFP+ population of experimental mice (Additional file 1: Figure S3B-C).
Mice were sacrificed following 4 months of reconstitution. Gross analysis showed no changes in the thymus, spleen, livers or kidneys. Microscopic inspection of hematoxylin and eosin – stained tissues did not reveal any differences (Additional file 1: Figure S3D). In the bone marrow, qRT-PCR confirmed successful overexpression of BALR-6 (Additional file 1: Figure S4A-B). Analysis by flow cytometry revealed an increase in precursor cell populations in the eGFP+ population of the experimental mice, when compared to the control group (Fig. 4a and b, Additional file 1: Figure S5C). After exclusion of differentiated cells in the bone marrow, we observed increased relative proportion of Lin-Sca1 + c-Kit + (LSK) cells, hematopoietic stem cells (HSCs) and lymphoid-primed multipotent progenitors (LMPPs) in mice overexpressing BALR-6 (Fig. 4a and b). An increase in the relative population of Lin-Sca1loc-Kitlo cells and a trend towards increased relative population of common lymphoid progenitors (CLPs) was also observed (Additional file 1: Figure S4C). The developmental pathway of B-cells in the bone marrow was investigated by the method of Hardy et al. [27]. Once again, trends towards higher relative proportions of these B-cell developmental stages were observed (fractions A-F, Additional file 1: Figure S4D). Taken together, these results suggest that BALR-6 overexpression leads to an enrichment of early developmental stage cells in murine bone marrow, indicating that its expression confers a survival advantage or increased proliferation for cells in these earlier stages.
Fig. 4.
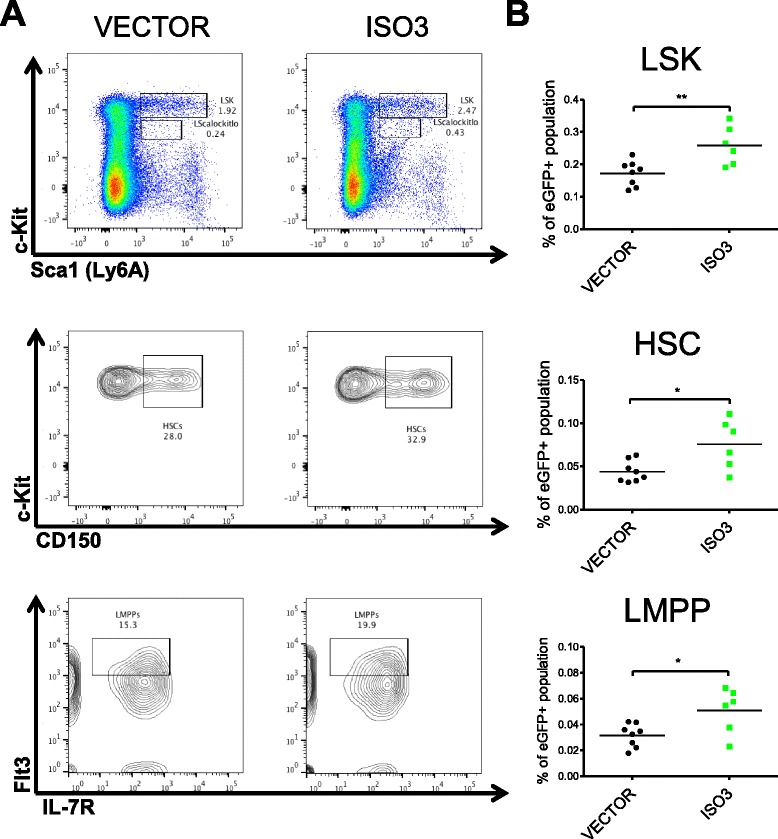
BALR-6 overexpression causes an increase in hematopoietic precursor cells in vivo. a Representative FACS plots of hematopoietic progenitor populations LSK, HSC and LMPP in bone marrow transfer mice. b Quantitation of progenitor populations showing a significant increase in experimental mice when compared to control. Number of mice used in this analysis: VECTOR, n = 8; ISO3, n = 6. ISO3, Isoform 3; HSC, hematopoietic stem cell; LMPP, lymphoid primed multipotent progenitor; LSK, lineage- Sca1+ c-Kit+. Evaluations made using a two-tailed T-test, p < 0.05 (*); p < 0.005 (**)
BALR-6 regulates expression of genes involved in multiple biological processes
At the molecular level, several studies have demonstrated that many lncRNAs act as transcriptional regulators [5, 11, 23, 28, 29]. To explore whether or not BALR-6 regulates gene expression, RNA isolated from knockdown cell lines was analyzed by microarray [30, 31]. Upon siRNA mediated knockdown of BALR-6, 2499 probes showed differential expression. Of these, 1862 unambiguously mapped to 1608 unique Entrez Gene IDs. Unsupervised hierarchical clustering analysis identified differentially expressed genes in the siRNA-expressing cell lines (Fig. 5a).
Fig. 5.
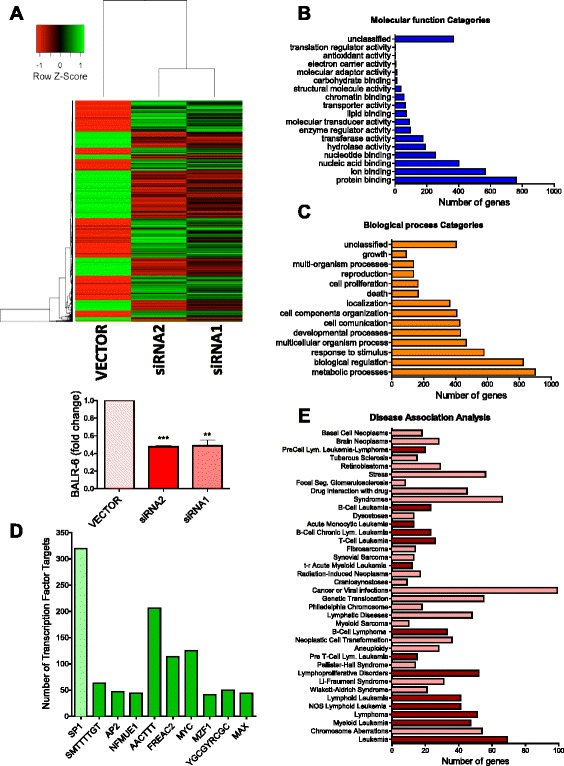
BALR-6 knockdown leads to global differential expression of genes. a Unsupervised hierarchical gene clustering of differentially expressed genes upon BALR-6 siRNA mediated knockdown in RS4;11 cells (PPDE ≥ 95 %, fold change ≥ 1.5). qRT-PCR confirmation of BALR-6 knockdown shown below. Normalized to ACTIN. b, c Bar graphs of GO Slim classification enrichment analysis of differentially expressed genes by molecular function (b), and biological processes (c). d Enrichment analysis of transcription factor targets. Top ten transcription factors with a p-value ≤ 0.0001 are shown. SP1 had the highest number if dysregulated targets, highlighted in light green. For unknown transcription factors, binding site sequence is shown. e Disease association analysis by GLAD4U, revealing enrichment of genes associated to various malignancies, in particular, hematological malignancies (dark red). Diseases with a p-value ≤ 0.05 are shown.; PPDE, posterior probability of differential expression. Evaluations were made using a two-tailed T-test, p < 0.005 (**); p < 0.0005 (***)
Further data analysis was carried out using WebGESTALT [32, 33]. Gene Ontology (GO) slim classification of differentially expressed genes by molecular function was utilized to provide insight into the pathways in which BALR-6 is involved, with protein binding function category having the most dysregulated genes (Fig. 5b). A number of biological processes, as annotated in the GO database, were significantly dysregulated in BALR-6 knockdown cell lines, including cell death and cell proliferation (Fig. 5c). Disease associated enrichment analysis, which was inferred using GLAD4U, showed an enrichment of genes known to be dysregulated in various disease states (Fig. 5e). Of the 38 significantly associated disease states, 14 were of leukemic origin. Transcription factor enrichment analysis showed a significant enrichment of genes that are predicted targets of SP1, among other transcription factors (Fig. 5d). Taken together, these data revealed the biological importance of BALR-6. A detailed description of the microarray analyses can be found in the methods.
SP1 transcriptome is modulated by BALR-6
As indicated by the transcription factor enrichment analysis, we confirmed that the expression SP1 and CREB1, a target and interactor of SP1, were dysregulated upon BALR-6 knockdown (Fig. 6a). The strongest phenotype was seen in the siRNA2-mediated knockdown, which also showed the strongest cellular phenotypes in the majority of pre B-ALL cell lines (Figs. 2d, i and 6a, Additional file 1: Figure S2B-D and Figure S5A-B). Conversely, increased levels of SP1 and CREB1 correlated with overexpression of BALR-6 isoforms in both human and murine cell lines (Nalm-6 and 70Z/3) (Fig. 6b).
Fig. 6.
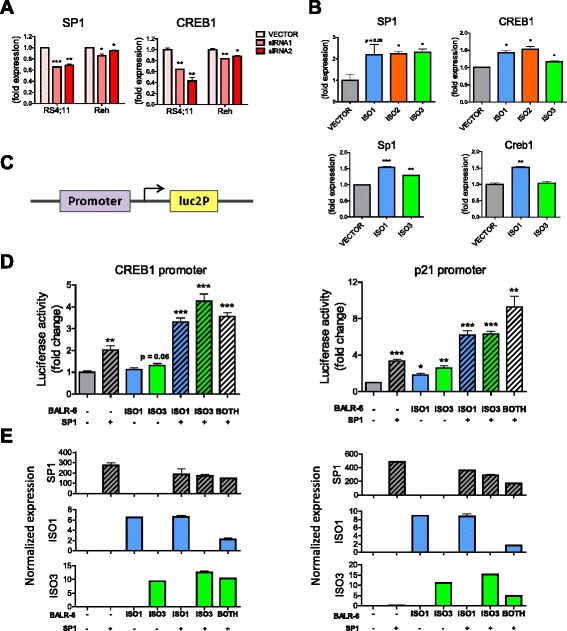
SP1 transcriptome is modulated by BALR-6. a Confirmation of SP1 and CREB1 expression in RS4;11 microarray samples, as well as Reh knockdown cell lines. Normalized to ACTIN. (b) SP1 and CREB1 transcript level increase correlates with overexpression of BALR-6 in Nalm-6 cells (top) and 70Z/3 cells (bottom). Quantitation by qRT-PCR, normalized to ACTIN (Nalm-6 cells) or L32 (70Z/3 cells). c Schematic depicting location of cloned promoter sequences in the pGL4.11 vector system for luciferase assays. d Transcriptional activity at CREB1 (left) and p21 (right) promoter regions upon SP1 and/or BALR-6 overexpression, as measured by luciferase activity. e Quantitation of overexpression in luciferase assays (as seen in (d)) by qRT-PCR of respective transcripts, normalized to ACTIN. Evaluations were made using a two-tailed T-test, p < 0.05 (*); p < 0.005 (**); p < 0.0005 (***). luc2p, synthetic firefly luciferase
To confirm our findings, a second microarray analysis was carried out with technical duplicates of RS4;11 cell lines transduced with empty vector or siRNA2. 2756 probes showed differential expression. Of these, 2280 unambiguously mapped to 2128 Entrez Gene IDs and were analyzed by hierarchical clustering (Additional file 1: Figure S6A). Enrichment analysis in WebGESTALT revealed similar GO slim classifications (Additional file 1: Figure S6B-C), and transcription factor target enrichment analysis confirmed the significant enrichment of SP1 targets seen previously (Fig. 5d, Additional file 1: Figure S6D). Additionally, enrichment of CREB1 targets was significant (Additional file 1: Figure S6D). Notably, leukemic diseases were the only ones significantly enriched in the disease association analysis (Additional file 1: Figure S6E). Together, these findings indicated a consistent change in the transcriptome, particularly downstream of SP1, upon knockdown of BALR-6 in MLL rearranged B-ALL.
To further understand the relationship of BALR-6 and SP1, we examined promoter regions of known SP1 targets (CREB1 and p21) and cloned these sequences into the luciferase reporter vector, pGL4.11 (Fig. 6c). The CREB1 promoter contained 7 putative SP1 binding sites, while the p21 promoter contained 6 such sites (Additional file 1: Figure S5C-D). Luciferase reporter assays in HEK 293 T cells with constitutive expression of SP1, Isoform 1, Isoform 3 or a combination of these vectors, revealed increased luciferase activity in both promoters (Fig. 6d and e). Notably, when SP1 and BALR-6 were co-overexpressed, we noted a strong increase in transcriptional activity with both the CREB1 and p21 promoter.
Discussion
The discovery of lncRNAs has revolutionized how we think about gene expression. The genomic organization of many lncRNAs is indeed complex. Some are found in regions overlapping with protein coding genes, while others that are exclusively intergenic [2, 4]. Some lncRNAs contain microRNAs within either their exonic or intronic sequence [34, 35]. Here, we have characterized several isoforms of a lncRNA that is overexpressed in leukemia and shows dynamic expression in hematopoietic development [16]. Expressed from a locus adjacent to genes important in lymphocyte development, BALR-6 itself is dynamically regulated during human B-cell development [36–38]. Our work significantly adds to the known repertoire of RNA molecules that are expressed from this locus, and several of these appear to be functional within a cellular context.
In this manuscript, we describe the cellular function of a second lncRNA that was discovered as being overexpressed in MLL-translocated B-ALL. In some ways, BALR-6 shows some similarities with the other lncRNA we studied, BALR-2 [16]. Indeed, knockdown of both lncRNAs led to decreased cell growth and increased apoptosis, and overexpression led to increased growth and a partial resistance to prednisolone treatment. These findings are not altogether surprising given that these lncRNAs may be contributing to the poor clinical behavior of an aggressive cytogenetic subtype of B-ALL [17]. However, there are important differences between these lncRNAs—the genomic locus for BALR-6 is more complex, there are multiple isoforms and no comparable murine transcript is described in publically available databases. Nonetheless, we have obtained fragments of a low-expression transcript from murine hematopoietic cell lines that encoded portions homologous to human BALR-6. Further characterization of the murine transcripts will be the goal of future studies.
Significantly, our study is amongst the few characterizations of lncRNA dysregulation in the hematopoietic system [16, 39–41]. LncRNAs have been ascribed functions in lymphopoiesis, myelopoiesis and erythropoiesis [42–45]. Additionally, their differential expression has been described in peripheral T-cell subsets [46]. Here, we discovered the effect of BALR-6 overexpression on early hematopoietic progenitors in the marrow, including LSK cells, HSCs and LMPPs. Constitutive expression of BALR-6 isoforms led to increased survival or proliferation of normally transient bone marrow progenitor cells. Furthermore, Hardy fractions showed a trend towards being increased when compared to control, particularly those that developmentally precede the large pre B-cell stage (fraction C’, early pre-B). The relative percentages of more mature B-lineage cells downstream of these developmental stages are largely normal. Despite increased proportions of early progenitor cells, passage through a checkpoint (such as the pre-BCR checkpoint) may reduce cell numbers back to baseline. This suggests that the function of BALR-6 in vivo may be in directing differentiation and adequate lymphoid cell development. The upregulation of this lncRNA causes a survival or proliferative advantage, a hallmark of leukemogenesis. Coupling BALR-6 overexpression with an appropriate oncogenic co-stimulus may lead to full-blown leukemogenesis or enhancement thereof, and this is currently an active area of investigation in the laboratory.
In line with a function in promoting the survival of early hematopoietic progenitors, BALR-6 clearly affects proliferation in cell line experiments. Upon siRNA mediated knockdown, we saw reduced cell proliferation and increased cell death. We observed the opposite effect when we constitutively expressed BALR-6 in human and murine B-ALL cell lines. Moreover, similar mechanisms may be operant in B-ALL with MLL translocations, and loss-of-function experiments in primary patient samples and mouse models of MLL-driven leukemia are areas for further investigation.
Given prior reports of lncRNAs serving to regulate transcriptional complexes, our finding that BALR-6 knockdown causes changes in the SP1 transcriptome is compelling. SP1 is a transcriptional regulator that is associated with dysregulated cell cycle arrest in multiple myeloma [47–49]. CREB1 is a well-known proto-oncogene that promotes cellular proliferation in hematopoietic cells [50, 51]. Here we demonstrate that SP1-mediated transcription at the CREB1 and p21 promoters are positively regulated by BALR-6, providing a putative mechanism for our observations of BALR-6′s role in B-ALL.
Conclusions
In this study, we demonstrate that the MLL-AF4-dysregulated lncRNA, BALR-6, plays a role in cell survival and regulates hematopoietic progenitors. At the molecular level, BALR-6 regulates the transcriptome of B-ALL cell lines, likely through regulating SP1-mediated transcription. In summary, our study has several novel and unique findings that help uncover a role for a poorly understood class of molecules in a pathogenetic process. This will undoubtedly have impacts on our understanding of molecular biology within cancer cells.
Methods
Cloning and cell culture
mmu-miR-155 formatted siRNAs were cloned into BamHI and XhoI sites in the pHAGE2-CMV-ZsGreen-WPRE vector using the strategy that we have previously described to generate knockdown vectors [16, 25, 26, 52]. Using the sequence information from 5′ and 3′ RACE products we cloned full length transcripts into an MSCV-based bicistronic viral vector between the BamHI and XhoI sites, as described previously and into a pHAGE6-UBC-ZsGreen-CMV-LNC (P6UZCL) variant of the third generation lentiviral vector system, between the NotI and BamHI sites [16, 52]. Primer sequences used are listed in Additional file 2: Table S1 or mentioned previously [16]. RS4;11 and MV(411), (MLL-AF4-translocated; ATCC CRL-1873 and CRL-9591), Reh (TEL-AML1-translocated; CRL-8286), 697 (E2A-PBX1-translocated), Nalm-6, 70Z/3 (ATCC TIB-158) murine pre B-cell leukemic cell line, and the HEK 293 T cell line (ATCC CRL-11268) were grown in their corresponding media at 37 °C in a 5 % CO2 incubator as previously described [16, 53].
Rapid amplification of cDNA ends (RACE)
To determine the 5′ and 3′ transcript ends of the lncRNAs, we performed RACE using First Choice RLM-RACE kit (Ambion). Using the sequence information from 5′ and 3′ RACE products, we cloned full length transcripts into P6UZCL, and into the MSCV viral vector. Primer sequences used and isoform sequences obtained are listed in Additional file 2: Table S1.
Transduction and sorting of cell lines
Lentiviruses and MSCV-based retroviruses were produced to generate knockdown constructs as previously described [16, 25, 26, 52]. In brief, 5.0 × 105 cells were spin-infected at 30 °C for 90 min in the presence polybrene (4 μg/mL). Transduced cell lines were sorted for high green expression using a BD FACSAriaII cell sorter, and analysis was performed using BD FACSDiva software.
Biological assays
For pharmaco-induced assays, cells were cultured at a concentration of 1.0 × 106 cells per mL and treated for 36 h. I-BET151 was dissolved in dimethyl sulfoxide to desired concentrations. After treatment, cells were harvested for RNA extraction. For MTS proliferation assays, cells were cultured for at least 5 days before plating. Cells were plated at a density of 2500 cells per 100 μl of media in each well of a 96 well plate. Reagents were added according to the manufacturer’s instructions (Promega CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit) and cells were incubated at 37 °C, 5 % CO2 for 4 h before absorbance was measured at 490 nm. For apoptosis assays, cells were plated at 5.0 × 105 cells/mL for 24 h with or without prednisolone treatment. Prednisolone (TCI America) was dissolved in dimethyl sulfoxide to desired concentrations. Cells were harvested after 24 h and stained with APC-tagged AnnexinV. For cell cycle analysis, cells were synchronized by serum starvation for 12 h (human cell lines) or 4 h (murine cell lines) then plated at 5.0 × 105 cells/mL and incubated at 37 °C, 5 % CO2 for 24 h. Cells were harvested, fixed with EtOH and then stained with propidium iodide. AnnexinV stained and PI stained samples were analyzed using a BD FACS HTLSRII flow cytometer and further analysis was performed using FlowJo.
Luciferase assays
Promoter sequences for CREB1 and p21 were cloned upstream of synthetic firefly luciferase (luc2p) in the pGL4.11 vector. Renilla luciferase is expressed in the pGL4.75 vector downstream of the PGK promoter. HEK 293 T cells were transfected with the pGL4.75 and pGL4.11 containing reporter vectors at a 1:20 ratio (5 ng:100 ng), along with a combination of MSCV vector (empty, Isoform-1 or Isoform-3) and pCMV3 (empty or SP1-HA, Sino Biological Inc.) vector at a 1:1 ratio (200 ng:200 ng). For the last condition SP1, Isoform1 and Isoform 3 were transfected together at a ratio of 2:1:1 (200 ng:100 ng:100 ng). Co-transfections were performed with BioT (Bioland Scientific LLC) in 24 well plates as per the manufacturer’s instructions. Cells were lysed after 32 h and supernatant lysate was collected as per manufacturer’s instructions (Promega). The dual luciferase assay kit (Promega) was used as substrates for Renilla and firefly luciferase activity. Luminescence was measured on a Glomax-Multi Jr (Promega). The ratio of firefly to Renilla luciferase activity was calculated for all samples. The luminescence for the sample co-transfected with MSCV empty vector and pCMV3 empty vector, was used as a normalization control.
qRT-PCR and PCR
RNA from cell lines was reverse transcribed using qScript (Quantas Biosciences). Real Time quantitative PCR was performed with the StepOnePlus Real-Time PCR System (Applied Biosystems) using PerfeCTa SYBR Green FastMix reagent (Quantas Biosciences). cDNA from mice samples was amplified using KOD Master Mix (EMD Millipore) and ran on a 1.2 % agarose gel stained with ethidium bromide. Primer sequences used are listed in Additional file 2: Table S1.
Northern blot
Total RNA was separated on a 1.2 % (w/v) formaldehyde agarose gel and then blotted onto Hybond N+ nylon membranes (Amersham Biosciences) by semi-dry transfer (Bio-Rad,
Trans-Blot SD Semi-Dry Transfer Cell). DNA probes were ordered from Integrated DNA Technologies (IDT, San Diego, CA) with digoxigenin incorporated at 3′end. For ACTIN we used the RNA probe provided in the DIG Northern Starter Kit (Roche). Membranes were hybridized overnight using ULTRAhyb-Oligo Buffer (Ambion) at 37 or 42 °C with probes. Visualization was done by X-Ray film using CDP-Star reagents (Roche). X-Ray film was scanned and saved as jpeg files. Brightness and contrast was increased by 20 % for ease of visualization.
Data sources
Human genome assembly GRCh37/hg19 and the mouse genome assembly GRCm38/mm10 were used. Methylation patterns for the four cell lines were obtained from Chip-Seq data available in the UCSC genome browser generated by the Broad/ENCODE group [18–20]. Peak viewing range set from 1–50 for H3K4m3 modifications, and 1–15 for H3K36m3 modifications. Alternative splice form information was obtained from the Swiss Institute of Bioinformatics, via UCSC Genome Browser [21]. Genome alignments of RefSeq transcripts from human, mouse and other vertebrates, GenBank mRNAs and ESTs, as well as PhastCons scores were obtained from the UCSC Genome Browser [22].
Microarray data analysis
Microarray data was generated from samples of 3 different transduced RS4;11 cell lines with siRNAs against BALR-6, or the control empty vector. Samples were hybridized at the UCLA Clinical Microarray Core facility using Affymetrix HG-U133_Plus_2 microarray. The Affymetrix raw data files (.cel files) were loaded into the R program for quality control analysis. Additionally, raw hybridization intensities were normalized using the MAS5 method with the affy package in R. Normalized values were sorted by detection p-value ≤ 0.05. Differential expression analysis was performed using unpaired Bayesian comparison model (CyberT Website) [30, 31]. Data was then sorted for genes with a posterior probability of deferential expression (PPDE) ≥ 95 % and a fold change ≥ 1.5. Analysis of differentially expressed genes was carried out using the WEB-based GEne SeT AnaLysis Toolkit (WebGESTALT, http://bioinfo.vanderbilt.edu/webgestalt/) [32, 33]. This online tool uses information from different public data sources for enrichment analysis, including the Gene Ontology data base, and GLAD4U. A second (validation) microarray was carried out, as described above, with technical duplicates for RS4;11 cell lines transduced with siRNA2 or the empty vector. For differential analysis the raw data files were loaded into the R environment and analyzed using the R library of Linear Models for Microarray Data (LIMMA). Pairwise comparison and eBayes fit was carried out. Data was then sorted for genes with a p-value ≤ 0.05. Further analysis was done as described above, using WebGESTALT.
Mice and bone marrow transplantation
Mice were housed under pathogen free conditions at the University of California, Los Angeles (UCLA). Donor mice were injected intraperitoneally with 200 mg/kg of 5-fluorouracil. After 5 days the mice were sacrificed. The bone marrow was collected under sterile conditions and plated in media enriched with IL-3, IL-6 and mSCF (Gibco). 24 h after plating, the bone marrow was spin infected twice, at 30 °C for 90 min in the presence polybrene (4 μg/mL), with retroviruses expressing the empty MSCV vector or BALR-6 Isoform 3. Recipient mice were lethally irradiated and injected with donor bone marrow 6 h after irradiation. 8 mice were used per group. One mouse in the ISO3 group died due to engraftment failure after 2 weeks post injection. These mice were bled at 8, 12 and 16 weeks post bone marrow injection. At 16 weeks the mice were sacrificed for full analysis. For statistical analysis, one mouse was excluded due to low eGFP expression. This experiment was repeated, and had similar results. All animal studies were approved by the UCLA Animal Research Committee (ARC).
Flow cytometry of samples
At 16 weeks post bone marrow transplant, blood, bone marrow, thymus and spleen were collected from the mice under sterile conditions [53]. Single cell suspensions were lysed in red blood cell lysis buffer. Fluorochrome conjugated antibodies were used for staining (antibodies were obtained from eBiosciences, and Biolegend). Cells were stained with surface marker antibodies for 30 min at 4 °C, washed twice with 1X PBS, and finally fixed with 1 % PFA. Flow cytometry was performed at the UCLA Jonsson Comprehensive Cancer Center (JCCC) and at the BROAD Stem Cell Research Flow Core. Analysis was performed using FlowJo software. The lists of antibodies used and gating schematics are provided in Additional file 2: Table S2. Normal adult human bone marrow was obtained commercially from healthy adults (All Cells, Inc.) as previously described [51]. CD34 enrichment from human bone marrow was performed using the magnetic activated cell sorting (MACS) system (Miltenyi Biotec, San Diego, CA) prior to isolation of CD34+ subsets by flow cytometry. Bone marrow CD34 selected cells were incubated with a cocktail of antibodies as well as FITC-labeled lineage depletion antibodies (Additional file 2: Table S3). CD19 was not included in the lineage depletion cocktail used for sorting the progenitor B population. The immunophenotypic definitions used to isolate progenitors from human bone marrow CD34 selected cells are described in Additional file 2: Table S3. All populations were purified using fluorescence-activated cell sorting on a FACSAria (355, 405, 488, 561 and 633 nm lasers) (BD Immunocytometry Systems).
Acknowledgements
We would like to thank the UCLA Clinical Microarray Core for performing the microarray hybridization experiments, Alejandro Balazs at Caltech for lentiviral vector backbones, and Ken Dorshkind for helpful discussions. Also, we thank Neha Goswami, Ella Waters, Jasmine Gajeton, Peter D. Pioli, Nolan Ung, Jennifer King and May Paing for their technical support. Very special thanks to Diana C. Márquez-Garbán at Dr. Richard Pietras lab (UCLA) for use of their facilities for some of the experiments. This research was supported by the Graduate Research Fellowship Program from the National Science Foundation DGE-1144087 (N.I.R.M.), the Eugene V. Cota-Robles Fellowship from UCLA (N.I.R.M., J.R.C.), Tumor Biology Training Grant T32 CA009056 from the National Institute of Health (NIH) (T.R.F.), the institutional Tumor Immunology Training Grants NIH T32CA009120 (J.R.C.) and NIH T32009056 (T.R.F.), the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA Training Program (M.O.A.), a Career Development Award K08CA133521 (D.S.R.), the Sidney Kimmel Translational Scholar Award SKF-11-013 (D.S.R), the Irving Feintech Family Foundation/Tower Cancer Research Foundation Research Grant (D.S.R), the University of California Cancer Research Coordinating Committee (D.S.R), the Stein-Oppenheimer Endowment Award (D.S.R), NIH P01 HL073104 (G.M.C.), UCLA Broad Stem Cell Research Center (BSCRC) (D.S.R., G.M.C), NIH T32 HL086345 (S.S.) and California Institute for Regenerative Medicine (CIRM) Training Grant TG2-01169 (S.S.). Flow cytometry was performed in the BSCRC FACS Core facility, and UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, the UCLA Council of Bioscience Resources, and the David Geffen School of Medicine at UCLA.
Abbreviations
- BALR
B-ALL associated long RNA
- chr
chromosome
- CLP
common lymphoid progenitor
- CMV
cytomegalovirus promoter
- DMSO
dimethyl sulfoxide
- eGFP
enhanced green fluorescent protein
- HSC
hematopoietic stem cell
- ISO1
Isoform 1
- ISO2
Isoform 2
- ISO3
Isoform 3
- LMPP
lymphoid primed multipotent progenitor
- LSK
lineage- Sca1+ c-Kit+
- LTR
long terminal repeats
- ORF
open reading frame
- PFA
paraformaldehyde
- PGK
phosphoglycerate kinase promoter
- PPDE
posterior probability of differential expression luc2p, synthetic firefly luciferase
- pre-B
precursor B
- pro-B
progenitor B
- UBC
ubiquitin C promoter
- ZsGreen
Zoanthus green fluorescent protein
Additional files
BALR-6 locus encodes numerous alternative splice forms. Figure S2. Knockdown and overexpression of full length BALR-6 isoforms in mammalian cell lines. Figure S3. Constitutive expression of BALR-6 in mice periphery. Figure S4. Elevated levels of immature B cell populations in mice with BALR-6 overexpression. Figure S5. SP1 targets in siRNA mediated knockdown cell lines. Figure S6. Confirmation of global differential expression findings seen in initial microarray. (PDF 1.19 mb)
Primers and RACE sequences for BALR-6. Table S2. Antibodies used for bone marrow transplant flow cytometry analysis, and population gating schematics. Tables S3. Antibodies used for CD34 enrichment of human bone marrow flow cytometry analysis, and population gating schematics. (PDF 125 kb)
Footnotes
Competing interests
The authors have no relevant competing interests.
Authors’ contributions
DSR designed research, analyzed data and prepared the manuscript. GMC designed research and prepared the manuscript. NIRM designed research, performed research, analyzed data and prepared manuscript. TRF, PCP, JRC, JKP, TMT, JA, MOA, KP and SS performed research, and prepared manuscript. MJD analyzed data, and prepared manuscript. All authors read and approved the final manuscript.
Contributor Information
Norma I. Rodríguez-Malavé, Email: nirodriguez@mednet.ucla.edu
Thilini R. Fernando, Email: trfernando@mednet.ucla.edu
Parth C. Patel, Email: pcpatel@mednet.ucla.edu
Jorge R. Contreras, Email: jrcontreras@mednet.ucla.edu
Jayanth Kumar Palanichamy, Email: drjayanth@aiims.edu.
Tiffany M. Tran, Email: ttran@mednet.ucla.edu
Jaime Anguiano, Email: janguiano@mednet.ucla.edu.
Michael J. Davoren, Email: mdavoren@g.ucla.edu
Michael O. Alberti, Email: malberti@mednet.ucla.edu
Kimanh T. Pioli, Email: kpioli@mednet.ucla.edu
Salemiz Sandoval, Email: salemizsandoval@mednet.ucla.edu.
Gay M. Crooks, Email: gcrooks@mednet.ucla.edu
Dinesh S. Rao, Email: drao@mednet.ucla.edu
References
- 1.Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–8. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 2.Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–7. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niazi F, Valadkhan S. Computational analysis of functional long noncoding RNAs reveals lack of peptide-coding capacity and parallels with 3′ UTRs. RNA. 2012;18:825–43. doi: 10.1261/rna.029520.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–50. doi: 10.1016/j.cell.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–23. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–38. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–69. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491:454–7. doi: 10.1038/nature11508. [DOI] [PubMed] [Google Scholar]
- 9.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements. Nature. 2011;470:284–8. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC, et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol. 2011;29:742–9. doi: 10.1038/nbt.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutschner T, Hammerle M, Eissmann M, Hsu J, Kim Y, Hung G, et al. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013;73:1180–9. doi: 10.1158/0008-5472.CAN-12-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isin M, Dalay N. LncRNAs and neoplasia. Clin Chim Acta. 2015;444:280–8. doi: 10.1016/j.cca.2015.02.046. [DOI] [PubMed] [Google Scholar]
- 14.Mullighan CG, Downing JR. Global genomic characterization of acute lymphoblastic leukemia. Semin Hematol. 2009;46:3–15. doi: 10.1053/j.seminhematol.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122:3407–15. doi: 10.1172/JCI61203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernando TR, Rodriguez-Malave NI, Waters EV, Yan W, Casero D, Basso G, et al. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol Cancer Res. 2015;13:839–51. doi: 10.1158/1541-7786.MCR-15-0006-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pui CH, Behm FG, Downing JR, Hancock ML, Shurtleff SA, Ribeiro RC, et al. 11q23/MLL rearrangement confers a poor prognosis in infants with acute lymphoblastic leukemia. J Clin Oncol. 1994;12:909–15. doi: 10.1200/JCO.1994.12.5.909. [DOI] [PubMed] [Google Scholar]
- 18.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–81. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ram O, Goren A, Amit I, Shoresh N, Yosef N, Ernst J, et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–39. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. GenBank: update. Nucleic Acids Res. 2004;32:D23–6. doi: 10.1093/nar/gkh045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y, Zhong Y, Wang Y, Zhang X, Batista DL, Gejman R, et al. Activation of p53 by MEG3 non-coding RNA. J Biol Chem. 2007;282:24731–42. doi: 10.1074/jbc.M702029200. [DOI] [PubMed] [Google Scholar]
- 24.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–33. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A. 2009;106:7113–8. doi: 10.1073/pnas.0902636106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao DS, O'Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D. MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity. 2010;33:48–59. doi: 10.1016/j.immuni.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 28.Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–74. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A. 2009;106:11667–72. doi: 10.1073/pnas.0904715106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17:509–19. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 31.Kayala MA, Baldi P. Cyber-T web server: differential analysis of high-throughput data. Nucleic Acids Res. 2012;40:W553–9. doi: 10.1093/nar/gks420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–8. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–83. doi: 10.1093/nar/gkt439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lerner M, Harada M, Loven J, Castro J, Davis Z, Oscier D, et al. DLEU2, frequently deleted in malignancy, functions as a critical host gene of the cell cycle inhibitory microRNAs miR-15a and miR-16-1. Exp Cell Res. 2009;315:2941–52. doi: 10.1016/j.yexcr.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 35.Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007;13:313–6. doi: 10.1261/rna.351707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dickinson LA, Joh T, Kohwi Y, Kohwi-Shigematsu T. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell. 1992;70:631–45. doi: 10.1016/0092-8674(92)90432-C. [DOI] [PubMed] [Google Scholar]
- 37.Will B, Vogler TO, Bartholdy B, Garrett-Bakelman F, Mayer J, Barreyro L, et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nat Immunol. 2013;14:437–45. doi: 10.1038/ni.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohwi-Shigematsu T, Poterlowicz K, Ordinario E, Han HJ, Botchkarev VA, Kohwi Y. Genome organizing function of SATB1 in tumor progression. Semin Cancer Biol. 2013;23:72–9. doi: 10.1016/j.semcancer.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang K, Han BW, Chen ZH, Lin KY, Zeng CW, Li XJ, et al. A distinct set of long non-coding RNAs in childhood MLL-rearranged acute lymphoblastic leukemia: biology and epigenetic target. Hum Mol Genet. 2014;23:3278–88. doi: 10.1093/hmg/ddu040. [DOI] [PubMed] [Google Scholar]
- 40.Garitano-Trojaola A, Agirre X, Prosper F, Fortes P. Long non-coding RNAs in haematological malignancies. Int J Mol Sci. 2013;14:15386–422. doi: 10.3390/ijms140815386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Dominguez JR, Hu W, Gromatzky AA, Lodish HF. Long noncoding RNAs during normal and malignant hematopoiesis. Int J Hematol. 2014;99:531–41. doi: 10.1007/s12185-014-1552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkatraman A, He XC, Thorvaldsen JL, Sugimura R, Perry JM, Tao F, et al. Maternal imprinting at the H19-Igf2 locus maintains adult haematopoietic stem cell quiescence. Nature. 2013;500:345–9. doi: 10.1038/nature12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aoki K, Harashima A, Sano M, Yokoi T, Nakamura S, Kibata M, et al. A thymus-specific noncoding RNA, Thy-ncR1, is a cytoplasmic riboregulator of MFAP4 mRNA in immature T-cell lines. BMC Mol Biol. 2010;11:99. doi: 10.1186/1471-2199-11-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X, Lian Z, Padden C, Gerstein MB, Rozowsky J, Snyder M, et al. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood. 2009;113:2526–34. doi: 10.1182/blood-2008-06-162164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev. 2011;25:2573–8. doi: 10.1101/gad.178780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xia F, Dong F, Yang Y, Huang A, Chen S, Sun D, et al. Dynamic transcription of long non-coding RNA genes during CD4+ T cell development and activation. PLoS One. 2014;9:e101588. doi: 10.1371/journal.pone.0101588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fulciniti M, Amin S, Nanjappa P, Rodig S, Prabhala R, Li C, et al. Significant biological role of sp1 transactivation in multiple myeloma. Clin Cancer Res. 2011;17:6500–9. doi: 10.1158/1078-0432.CCR-11-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amodio N, Di Martino MT, Foresta U, Leone E, Lionetti M, Leotta M, et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012;3:e436. doi: 10.1038/cddis.2012.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Yan Z, Fulciniti M, Li Y, Gkotzamanidou M, Amin SB, et al. Transcription factor-pathway coexpression analysis reveals cooperation between SP1 and ESR1 on dysregulating cell cycle arrest in non-hyperdiploid multiple myeloma. Leukemia. 2014;28:894–903. doi: 10.1038/leu.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A, et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–62. doi: 10.1016/j.ccr.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 51.Esparza SD, Chang J, Shankar DB, Zhang B, Nelson SF, Sakamoto KM. CREB regulates Meis1 expression in normal and malignant hematopoietic cells. Leukemia. 2008;22:665–7. doi: 10.1038/sj.leu.2404933. [DOI] [PubMed] [Google Scholar]
- 52.O'Connell RM, Balazs AB, Rao DS, Kivork C, Yang L, Baltimore D. Lentiviral vector delivery of human interleukin-7 (hIL-7) to human immune system (HIS) mice expands T lymphocyte populations. PLoS One. 2010;5:e12009. doi: 10.1371/journal.pone.0012009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Contreras JR, Palanichamy JK, Tran TM, Fernando TR, Rodriguez-Malave NI, Goswami N. MicroRNA-146a modulates B-cell oncogenesis by regulating Egr1. Oncotarget. 2015;6:11023–37. doi: 10.18632/oncotarget.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]