

Abstract
Introduction
Resuscitation with saline is a standard initial response to hypotension or shock of almost any cause. Saline resuscitation is thought to generate an increase in cardiac output through a preload-dependent (increased end-diastolic volume) augmentation of stroke volume. We sought to confirm this to be the mechanism by which high-volume saline administration (comparable to that used in resuscitation of shock) results in improved cardiac output in normal healthy volunteers.
Methods
Using a standardized protocol, 24 healthy male (group 1) and 12 healthy mixed sex (group 2) volunteers were infused with 3 l normal (0.9%) saline over 3 hours in a prospective interventional study. Individuals were studied at baseline and following volume infusion using volumetric echocardiography (group 1) or a combination of pulmonary artery catheterization and radionuclide cineangiography (group 2).
Results
Saline infusion resulted in minor effects on heart rate and arterial pressures. Stroke volume index increased significantly (by approximately 15–25%; P < 0.0001). Biventricular end-diastolic volumes were only inconsistently increased, whereas end-systolic volumes decreased almost uniformly. Decreased end-systolic volume contributed as much as 40–90% to the stroke volume index response. Indices of ventricular contractility including ejection fraction, ventricular stroke work and peak systolic pressure/end-systolic volume index ratio all increased significantly (minimum P < 0.01).
Conclusion
The increase in stroke volume associated with high-volume saline infusion into normal individuals is not only mediated by an increase in end-diastolic volume, as standard teaching suggests, but also involves a consistent and substantial decrease in end-systolic volumes and increases in basic indices of cardiac contractility. This phenomenon may be consistent with either an increase in biventricular contractility or a decrease in afterload.
Keywords: cardiac output, resuscitation, saline, ventricular volume, volunteers
Introduction
Initial resuscitation of hypovolemic and distributive shock involves the aggressive infusion of large volumes (several liters) of intravenous fluids (colloids or crystalloids). For example, American Trauma Life Support guidelines [1] advocate rapid administration of 1–2 l crystalloid in the initial management of hypovolemic shock. Subsequently, volume infusion is frequently used in critically ill patients to challenge persistent hypotension or tachycardia. In the teaching of resuscitative physiology, clinicians are told that the role of volume infusion is to increase stroke volume (SV) through an increase in preload (an increase in end-diastolic volume [EDV]) without a change in afterload or contractility.
Few studies have examined the effect of large quantities of resuscitative fluids such as normal (0.9%) saline on cardiac function in normal healthy volunteers. In the present study, normal volunteers were infused with 3 l normal saline over 3 hours in order to assess how typical resuscitative volumes affect cardiac volumes and performance. The specific objective was to determine the extent to which increases in EDV account for augmentation of SV after large volume resuscitation. Initial studies were performed using noninvasive echocardiographic techniques. Biventricular radionuclide ventriculography and invasive hemodynamic techniques (thermodilution-capable pulmonary artery catheter [PAC] and arterial catheter) were utilized for confirmation of results and extension of findings to right ventricular function.
Normal saline was used because it is the usual crystalloid solution used in initial resuscitation.
Methods
A total of 36 individuals aged 18–40 years volunteered and gave written informed consent to participate in the study. They were within 15% of their ideal body weight, as determined using Metropolitan Life Tables. The participants were required to have a normal history, physical examination, and electrocardiogram within 2 weeks before the start of the study. Basic hematology, coagulation, biochemistry, and infectious serology assays, as well as an electrocardiogram, were found to be normal. Participants were studied after an overnight fast. Basic screening laboratory studies, including a complete blood count and electrocardiogram, were repeated.
The individuals had an 18-gauge peripheral intravenous catheter placed in each arm. Assessment of baseline hemodynamics was initially done before saline infusion after a 20-min period of stability of vital signs, including heart rate (HR). After evaluating baseline vital signs and conducting echocardiographic or radionuclide ventriculographic/invasive hemodynamic studies, normal saline infusion was begun intravenously at a rate of 1 l/hour for 3 hours.
All participants were continuously monitored with electrocardiography, pulse oximetry, and either automatic sphygmomanometry (Dinamap Pro 300®, GE Medical Systems, Tampa, FL, USA) in participants who were studied using echocardiography or radial arterial catheter in those participants who were studied using radionuclide ventriculography/PAC. A nurse took vital signs (temperature, blood pressure, HR, and respiratory rate) every 15 min for the 4–6 hour duration of the study. At least one physician was present during the entire period of study. Repeat echocardiograms or radionuclide ventriculography/invasive hemodynamic data, as well as a repeat complete blood count, were again obtained after infusion of 3 l saline at the end of the study period. Subjects were supine throughout the study period.
Participants with PACs and arterial catheters placed had them removed after the study, and all were discharged 1–2 hours after the final assessment.
Echocardiography
Twenty-four males were recruited for the echocardiographic portion of the study (group 1). Standard views of the parasternal long and short axes, apical four chamber and two chamber, and Doppler outflow tract across the aortic valve were taken using a Hewlett Packard 5500 ultrasound device (Hewlett-Packard, Palo Alto, CA, USA).
SV was determined using the measured left ventricular outflow (aortic valve) diameter from the parasternal long-axis view, and an outflow tract velocity measured at the aortic valve with a Doppler probe from the apical five-chamber view [2,3]. SV at each point was determined as the mean of five consecutive measurements obtained at end-expiration. Cardiac output (CO) was calculated by multiplying this SV by the simultaneous HR. Left ventricular volumes were obtained using Simpson's Rule (method of disks), utilizing the average of volumes from apical four-chamber and two-chamber views [4]. Ejection fraction (EF) was obtained using the following equation: (EDV - end-systolic volume [ESV])/EDV. The total peripheral resistance (TPR) was determined using this calculated CO and the measured mean arterial pressure (MAP) from the Dinamap®, using the following equation: TPR (dyne·s/cm5) = (79.9 × MAP)/CO. The central venous pressure (CVP) was omitted in this calculation because of the negligible effect that the right atrial pressure exerts on this calculation in normal individuals.
Echocardiograms were read by a single, highly experienced echocardiographer who was blinded to the individual and study sequence. Intra-observer and inter-observer variability for 10 discrete measurements of SV were 6 ± 3% and 7 ± 3%, respectively. Accuracy of ventricular volumes was internally validated by comparing SVs derived from integration of the flow velocity across the aortic valve and subtraction of the ESV from the EDV. Previous studies have demonstrated that mean changes of greater than 2% in EDV, 5% in ESV, and 2% in left ventricular ejection fraction (LVEF) in groups of comparable size to that in the present study represent clinically significant alterations [5].
Pulmonary artery and radial artery catheterization
Twelve subjects (mixed sex) were recruited for the invasive catheterization/radionuclide ventriculography portion of the study (group 2). After informed consent was obtained, a 9-Fr introducer (Arrow Co., Reading, PA, USA) and a 110 cm 7.5-Fr PAC Swan–Ganz catheter (PA-Edwards Life Sciences, Irvine, CA, USA) were placed in the right femoral vein using minimal local anesthesia (lidocaine 2%), followed by ultrasound and brief fluoroscopic guidance. In addition, a 20 g, 1.5-inch Quick-Flash radial artery catheter (Arrow Co.) was placed in either the right or left arm. Placement of all invasive devices was performed by an experienced invasive cardiologist. Participants were rested for 45 min before initiation of the study initiation to allow any residual sympathetic stimulation to settle. Fluid loading was begun only when vital signs, including HR, had returned to prestudy values for at least 20 min.
Thermodilution COs were measured by three successive injections of 10 ml cold (6–10°C) dextrose 5% in water at end-expiration as per standard protocol. The recorded value was the mean of the three individual values. Recorded values for pulmonary artery pressure, pulmonary artery wedge pressure (PWP) and CVP were also obtained at end-expiration from graphic recordings. Arterial pressure was simultaneously recorded from the arterial catheter.
Radionuclide ventriculography
Sequential measurement of biventricular EF in group 2 participants was performed by repeat first-pass radionuclide ventriculograms using 99mTc-DPTA, which was injected as a tight bolus into the central veins using the PAC introducer. In the study the baseline radionuclide tracer dose was 3 mCi and the follow-up dose was 7 mCi. The study was performed in a 30° right anterior oblique projection with a slant hole collimator fitted onto a small field γ camera interfaced with a dedicated computer system (ICON, Siemens, Gammasonic, Hoffman Estates, IL, USA). The data were acquired in frame mode with 440 frames, each of 60 ms duration. The first transit cardiac data were reformatted into a multigated study using the participant's electrocardiogram recorded with the first pass data. This method provides independent cinematic display of both right and left ventricles. EFs are calculated from the reformatted gated first-pass studies using standard dual region of interest and background correction [6,7].
For group 2 participants, SV was derived by dividing the thermodilution CO by the concomitant HR. EDV was obtained by dividing SV by EF, and ESV was calculated as EDV - SV. Systemic vascular resistance index was calculated as (79.9 × [MAP - CVP])/cardiac index (CI), and pulmonary vascular resistance index as (79.9 × [mean pulmonary artery pressure - PWP])/CI. Left ventricular stroke work index was calculated as 0.0136 × (MAP - CVP) × SVI, and right ventricular stroke work index as 0.0136 × (mean pulmonary artery pressure - PWP) × stroke volume index (SVI).
The study received ethics approval from the Institutional Review Board at Rush-Presbyterian-St. Luke's Medical Center, in compliance with the Helsinki declaration.
Statistical analysis
Hemodynamic values at baseline and completion of the 3-hour saline infusion were pooled to derive means and standard errors of the mean. Hemodynamic values after saline infusion were compared with baseline values using two-tailed paired t-test analysis. P < 0.05 was considered statistically significant. Values are expressed as means ± standard error.
Results
Group 1 findings: echocardiography study
Age, weight, height, and body surface area were 27.8 ± 1.8 years, 76.6 ± 2.8 kg, 173.3 ± 2.5 cm, and 1.91 ± 0.04 m2, respectively.
High-volume saline infusion exerted significant although modest hemodynamic effects on the normal healthy volunteers in group 1 (Table 1, Fig. 1). The HR of 64.4 ± 1.8 beats/min was unchanged after infusion of 3 l saline (mean 64.1 ± 2.0 beats/min). MAP, diastolic blood pressure, and systolic blood pressure (SBP) were also unchanged throughout the study.
Table 1.
Hemodynamic response to volume loading in echocardiographically studied group (group 1)
| Parameter | Before saline infusion | After saline infusion | Percentage change | P |
| HR (beats/min) | 64.4 ± 1.8 | 64.1 ± 2.0 | -0.02 ± 1.9 | NS |
| SBP (mmHg) | 115.4 ± 2.2 | 115.2 ± 2.7 | -0.1 ± 1.9 | NS |
| DBP (mmHg) | 59.3 ± 1.8 | 57.3 ± 1.8 | -3.5 ± 1.5 | NS |
| MAP (mmHg) | 80.7 ± 1.8 | 79.6 ± 1.9 | -1.2 ± 1.3 | NS |
| TPR (dyne·s/cm5) | 599 ± 25 | 491 ± 27 | -17.8 ± 3.8 | <0.0001 |
| CI (l/min per m2) | 2.90 ± 0.07 | 3.32 ± 0.09 | 14.7 ± 2.4 | <0.0001 |
| SVI (ml/m2) | 47.5 ± 1.0 | 54.3 ± 1.2 | 14.6 ± 1.4 | <0.0001 |
| LVEDVI (ml/m2) | 72.2 ± 1.8 | 72.7 ± 1.7 | 1.0 ± 1.3 | NS |
| LVESVI (ml/m2) | 24.6 ± 1.3 | 17.9 ± 1.0 | -26.4 ± 2.4 | <0.0001 |
| LVEF (%) | 65.5 ± 1.2 | 74.5 ± 1.0 | 14.0 ± 1.4 | <0.0001 |
| SBP/LVESVI (mmHg/ml per m2) | 5.00 ± 0.31 | 6.88 ± 0.41 | 40.2 ± 6.4 | <0.0001 |
CI, cardiac index; HR, heart rate; DBP, diastolic blood pressure; LVEDVI, left ventricular end-diastolic volume index; LVEF, left ventricular ejection fraction; LVESVI, left ventricular end-systolic volume index; MAP, mean arterial pressure; SBP, systolic blood pressure; SVI, stroke volume index; TPR, total peripheral resistance.
Figure 1.
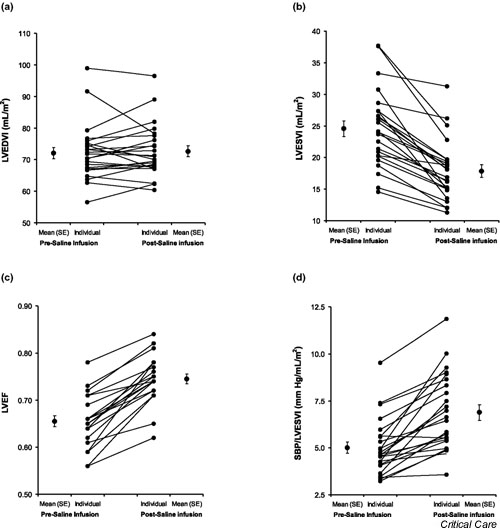
Individual and mean (± standard error) response to 3 l volume loading as measured echocardiographically. (a) Left ventricular end-diastolic volume index (LVEDVI), (b) left ventricular end-systolic volume index (LVESVI), (c) left ventricular ejection fraction (LVEF), (d) peak systolic blood pressure/end systolic volume index (PSP/ESVI; left ventricular contractility).
Given the absence of any change in HR, the increase in CI (14.7 ± 2.4%; P < 0.0001) was almost exactly matched by the increase in SVI (14.6 ± 1.4%; P < 0.0001). SVI and CI increased in all 24 participants. The approximately 15% increase in SVI was generated almost entirely through a mean 26.4 ± 2.4% decrease in left ventricular end-systolic volume index (LVESVI; P < 0.0001). Again, every participant in the study exhibited a fall in LVESVI with saline loading. The decrease in LVESVI was responsible for more than 90% of the increase in SVI in this group. Virtually no increase in mean left ventricular end-diastolic volume index (LVEDVI) was noted (1.0 ± 1.5%; not significant) after infusion of 3 l saline. Overall, 13 participants exhibited increased LVEDVI, whereas the other 11 had decreased LVEDVI.
Parameters of contractility were significantly increased with saline infusion. The baseline LVEF increased by 14.0 ± 1.4% (from 65.5 ± 1.2% to 74.5 ± 1.0% absolute value; P < 0.0001) after infusion of 3 l saline. All participants exhibited an increase in LVEF. Similarly, SBP/LVESVI [8], a relatively load-independent measure of contractility, exhibited a significant increase (40.2 ± 6.4%; P < 0.0001) from baseline after high-volume saline infusion. Again, this was a highly uniform response, with 23 out of 24 participants exhibiting a clear increase in this parameter.
Given that CO was increased and that MAP was unchanged by saline infusion, calculated afterload decreased. TPR fell 17.8 ± 3.8% after infusion of 3 l saline (P < 0.0001).
Group 2 findings: radionuclide cineangiography/pulmonary artery catheterization
Twelve subjects (8 male, 4 female) were recruited for the invasive monitoring portion of the study. Age, height, and weight were 30.9 ± 2.8 years, 173.3 ± 2.5 cm, and 86.3 ± 5.1 kg, respectively.
Effects of infusion of 3 l saline on left ventricular performance in group 2, studied using radionuclide cineangiography/ PACs, were generally similar to those noted echocardiographically in group 1 (Table 2, Fig. 2f,2g,2h,2i,2j). A modest increase in HR by 5.7 ± 3.5% did not achieve statistical significance, whereas small increases in SBP (5.9 ± 1.8%; P = 0.0075), diastolic blood pressure (9.6 ± 3.0%; P = 0.0081) and MAP (7.8 ± 2.2%; P = 0.004) did. Similarly, there were significant but more substantial relative increases in pulmonary artery pressures. Not surprisingly, PWP almost doubled (77.8 ± 20.6% increase; P = 0.0128) from baseline. Systemic vascular resistance index and pulmonary vascular resistance index both fell significantly (by approximately 17% and 28%, respectively). In all cases, at least 10 of the 12 participants exhibited changes from baseline consistent with the mean change in the parameter (i.e. in the same direction).
Table 2.
Hemodynamic response to volume loading in pulmonary artery catheter/radionuclide cineangiogram studied group
| Parameter | Before saline infusion | After saline infusion | Percentage change | P |
| HR (beats/min) | 68.4 ± 3.4 | 72.2 ± 4.1 | 5.7 ± 3.5 | NS |
| SBP (mmHg) | 126.0 ± 4.7 | 132.4 ± 4.2 | 5.9 ± 1.8 | 0.0075 |
| DBP (mmHg) | 69.4 ± 2.8 | 75.7 ± 2.8 | 9.6 ± 3.0 | 0.0081 |
| MAP (mmHg) | 88.1 ± 3.3 | 94.6 ± 3.0 | 7.8 ± 2.2 | 0.004 |
| CVP (mmHg) | 9.4 ± 0.7 | 12.4 ± 0.9 | 41.5 ± 15.2 | 0.028 |
| SPAP (mmHg) | 22.5 ± 1.0 | 29.2 ± 1.4 | 30.6 ± 5.6 | 0.0001 |
| DPAP (mmHg) | 11.3 ± 0.8 | 15.4 ± 0.7 | 44.1 ± 13.4 | 0.0065 |
| MPAP (mmHg) | 15.1 ± 0.7 | 20.0 ± 0.9 | 36.2 ± 9.2 | 0.0019 |
| PWP (mmHg) | 9.7 ± 0.9 | 15.3 ± 0.8 | 77.8 ± 26.4 | 0.0128 |
| CI (l/min per m2) | 2.96 ± 0.12 | 3.87 ± 0.29 | 30.0 ± 6.5 | 0.0006 |
| SVI (ml/m2) | 44.0 ± 1.9 | 54.1 ± 3.0 | 23.1 ± 4.7 | 0.0005 |
| LVEDVI (ml/m2) | 70.6 ± 2.2 | 76.3 ± 4.0 | 7.7 ± 2.7 | 0.0138 |
| LVESVI (ml/m2) | 26.6 ± 0.8 | 22.3 ± 0.6 | -17.1 ± 5.1 | 0.0131 |
| LVEF (%) | 62 ± 1 | 69 ± 1 | 11.2 ± 2.2 | 0.0003 |
| LVSWI (g/beats per m2) | 46.6 ± 2.5 | 58.0 ± 3.6 | 25.1 ± 5.1 | 0.0004 |
| SBP/LVESVI (mmHg/ml per m2) | 4.77 ± 0.25 | 5.71 ± 0.40 | 19.9 ± 6.1 | 0.0071 |
| SVRI (dyne·s per cm5 per m2) | 2140 ± 100 | 1779 ± 114 | -16.5 ± 4.6 | 0.0039 |
| RVEDVI (ml/m2) | 81.8 ± 4.2 | 89.6 ± 5.8 | 9.5 ± 3.5 | 0.019 |
| RVESVI (ml/m2) | 37.8 ± 2.7 | 34.0 ± 2.9 | -6.5 ± 2.7 | 0.0335 |
| RVEF (%) | 54 ± 1 | 61 ± 2 | 12.9 ± 1.6 | <0.0001 |
| RVSWI (g/beat per m2) | 3.4 ± 0.5 | 5.8 ± 0.9 | 88.7 ± 24.8 | 0.0038 |
| SPAP/RVESVI (mmHg/ml per m2) | 0.62 ± 0.04 | 0.94 ± 0.13 | 48.2 ± 14.6 | 0.0063 |
| PVRI (dyne·s per cm5 per m2) | 147 ± 17 | 100 ± 16 | -27.6 ± 10.9 | 0.0264 |
CI, cardiac index; CVP, central venous pressure; DBP, diastolic blood pressure; DPAP, diastolic pulmonary artery pressure; HR, heart rate; LVEDVI, left ventricular end-diastolic volume index; LVEF, left ventricular ejection fraction; LVESVI, left ventricular end-systolic volume index; LVSWI, left ventricular stroke work index; MAP, mean arterial pressure; MPAP, mean pulmonary artery pressure; PAC, pulmonary artery catheter; PSP, peak systolic blood pressure; PVRI, pulmonary vascular resistance index; PWP, pulmonary artery wedge pressure; RVEDVI, right ventricular end-diastolic volume index; RVEF, right ventricular ejection fraction; RVESVI, right ventricular end-systolic volume index; RVSWI, right ventricular stroke work index; SBP, systolic blood pressure; SPAP, systolic pulmonary artery pressure; SVI, stroke volume index; SVRI, systemic vascular resistance index.
Figure 2.
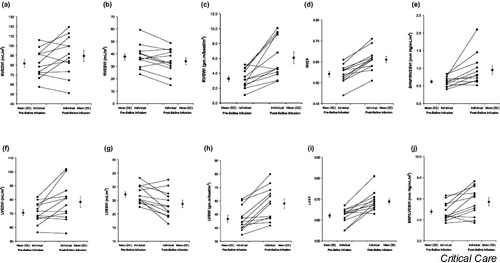
Individual and mean (± standard error) to 3 l volume loading as measured using radionuclide cineangiography and invasive hemodynamic monitoring. (a) Right ventricular end-diastolic volume index (RVEDVI), (b) right ventricular end-systolic volume index (RVEDVI), (c) right ventricular stroke work index (RVSWI), (d) right ventricular ejection fraction (RVEF), (e) peak systolic pulmonary artery pressure/right ventricular end-systolic volume index (right ventricular contractility), (f) left ventricular end-diastolic volume index (LVEDVI), (g) left ventricular end-systolic volume index (LVESVI), (h) left ventricular stroke work index (LVSWI), (i) left ventricular ejection fraction (LVEF), (j) peak systolic blood pressure/left ventricular end-systolic volume index (PSP/LVESVI).
CI rose almost 30% (P = 0.0006) with SVI increasing by 23% (P < 0.0005) from baseline. All participants exhibited an increase in these parameters. The remainder of the increase in CI was accounted for by the modest and nonsignificant increase in HR. Unlike in group 1, LVEDVI increased modestly (approximately 8%; P = 0.0138) but inconsistently (8/12 participants), whereas LVESVI fell to a greater extent (approximately -17%, P = 0.0131; LVESVI decreased in 10/12 participants). The decrease in LVESVI accounted for 43% of the increase in SVI. All parameters of left ventricular contractility (LVEF, left ventricular left ventricular stroke work index, SBP/ LVESVI) were increased by 11–25%. All but one participant exhibited increases in these parameters with volume loading.
Right ventricular responses substantially paralleled the left (Table 2; Fig. 2a,2b,2c,2d,2e). Right ventricular end-diastolic volume index increased modestly with saline infusion (approximately 10%; P = 0.019) whereas right ventricular end-systolic volume index (RVESVI) decreased by a smaller but still significant amount (approximately 7%; P = 0.0335). Eight out of 12 participants exhibited this increase in right ventricular end-diastolic volume index, and 8 out of 12 also demonstrated a decrease in RVESVI with volume loading. The decrease in RVESVI represented 38% of the measured increased in SVI. Right ventricular contractility parameters (right ventricular EF, right ventricular stroke work index, systolic pulmonary artery pressure/RVESVI), like those of the left, were consistently elevated by 13–88%. Again, all but one participant exhibited increases in these parameters with volume loading.
Blood hemoglobin concentration dropped from a mean of 14.9 ± 0.4 g/dl (range 12.5–17 g/dl) to 12.7 ± 0.4 g/dl (range 10.5–14.2 g/dl) over the course of the intravascular volume expansion (P < 0.0001). Total urine output during and immediately after the 3 hours of this study was minimal (mean < 250 ml).
Discussion
In addition to reinforcing observations of known cardiac responses to fluid resuscitation (modest or no increase in HR and blood pressure, 15–30% increase in SV and CO), the present study yielded several novel observations regarding the mechanisms of that response. The most intriguing of these pertain to the ventricular responses resulting in increased SV. Standard clinical teaching suggests that the increase in SV associated with volume loading is caused by an increase in ventricular preload (i.e. an increase in EDV). Despite consistent and highly significant increases in CO and SV, diastolic ventricular volumes did not respond in parallel (Fig. 1a, Fig. 2a,2f). Group 1 participants in fact demonstrated no mean change in LVEDVI (Table 1, Fig. 1a), whereas group 2 participants exhibited only a modest increase (Table 2, Fig. 2f). The reasons for the absence of a consistent increase in EDV in both ventricles, despite substantial increases in CVP (about 40%) and PWP (about 80%) in the invasively studied group 2 participants, are discussed elsewhere [9]. Novertheless, the decrease in LVESVI in both groups contributed significantly to the increase in SVI (>90% in group 1 and 43% group 2; Fig. 1b, Fig. 2g). Similarly, in group 2 participants, a significant fall in RVESVI contributed substantially (38%) to the increase in SVI (Table 2, Fig. 2b). This latter observation may be entirely novel because it appears that no other study has directly examined volumetric right ventricular response to volume infusion in normal animals or humans. The other novel finding of this study was that volume loading resulted in an increase in indices of ventricular contractility, including EF, stroke work parameters, and the SBP/ESV ratios (Fig. 1c,1d, Fig. 2c,2d,2e,2h,2i,2j). Again, with respect to the normal right ventricle, these data have not previously been described.
The substantial contribution of non-preload-dependent mechanisms to increased SV in both groups of participants examined is intriguing in its inconsistency with the standard teaching of resuscitative physiology. Starling's law of the heart has been interpreted to suggest that administration of large volumes of fluid should result in increased SV through an increase in EDV. However, data from both groups of participants in the present study demonstrate that, despite uniform increases in SV with volume loading, EDV increased only inconsistently (8/12 in group 1 and 13/24 in group 2), and that EDV-related increases in SV are relatively modest (<10% in group 1, and 57% left and 62% right ventricle in group 2). These data suggest that a significant component of the increased SV and CO associated with fluid loading is due to mechanisms that are unrelated to an increase in ventricular preload and the concomitant Starling response. Given the lack of significant alteration in HR, volume-related changes in contractility or afterload are implicated.
The increased ventricular stroke work indices and EF seen in virtually all patients are potentially compatible with a volume-related increase in cardiac contractility (Fig. 1c, Fig. 2c,2d,2h,2i). In addition, the uniform decrease in ESVs in the absence of evidence of increased sympathetic tone (with mean HR unchanged) support this possibility (Fig. 1b, Fig. 2b,2g) [10-13]. Although these parameters cannot differentiate between increased contractility and decreased afterload because of their load dependence, the relatively load-independent peak systolic pressures/ESV (for both the right and left ventricles) appear to support the former (Fig. 1d, Fig. 2e,2j).
Several cellular and physiologic mechanisms could potentially account for an increase in cardiac contractility in response to high-volume saline infusion. Hypertonic saline exerts significant inotropic effects based on modulation of sarcolemmal calcium fluxes [14]. Because the sodium concentration of normal saline is 5–10 mEq/l, which is higher than normal values for serum, infusion of large volumes of normal saline may afford a similar but attenuated myocardial stimulation. Alternately, Bainbridge described a neurologically mediated reflex of increased HR and contractility in response to rapid volume infusions in large animals [15,16]. More recently, Lew [17,18] described volume-induced increases in myocardial contractility in denervated dogs. Although these reflexes were elicited with liters of fluid administered within minutes in both cases, an attenuated form of the responses could explain our findings.
Although the hemodynamic parameters utilized in the present study are commonly accepted among intensivists as indices of contractility, they are substantially load-dependent. Augmentation of SV due to increases in EDV will necessarily (as a function of the mathematical relationship) be associated with elevations in EF and SWI, even if there is no increase in actual contractility. Only if preload and afterload hold constant can improved contractility be inferred from increases in these parameters. Even the ostensibly load-independent parameter peak SBP/end-systolic volume index, although relatively insensitive to preload alterations, has been shown to be affected by alterations in afterload [19,20]. For these reasons, the preload-independent element of the increase in SV could be due to decreases in afterload that mimic increased contractility when assessed using the hemodynamic indices of this study.
Calvin and colleagues [21] have implicated volume-induced vasodilation after noting significant decreases in end-systolic volume index after fluid resuscitation in a subgroup of critically ill patients. Several contributory mechanisms can be proposed. A decrease in endogenous catecholamines, particularly in stressed participants, could generate a relative loss of vasoconstrictor tone. However, healthy, unstressed adults were studied in our study. Centrally mediated vasomotor relaxation responses mediated by low pressure baroreceptors could mediate peripheral vasodilation in healthy individuals [22]. Alternately, increased release of the vasodilatory peptide atrial natriuretic factor has been described with rapid volume expansion [23,24]. Large volume infusion of saline could also cause alterations in blood electrolytes with increased chloride and decreased bicarbonate (nonanion gap metabolic acidosis), which could also result in vasomotor responses. Finally, a simple mechanical effect related to decreased blood viscosity associated with transient hypervolemic hemodilution could play a role. The participants in this study exhibited a 14.3 ± 0.8% drop in blood hemoglobin during the course of fluid infusion. At least one study has suggested decreased whole blood viscosity as a cause of decreased systolic left ventricular cross-sectional area during volume loading [25].
Unfortunately, the design of the present study does not allow definitive differentiation between altered contractility and afterload as a cause of the preload-independent element of the SV response. Systemic vascular resistance and TPR will be decreased as a mathematical consequence of increased CO with a maintained MAP, whereas EF, stroke work index, and peak SBP/end-systolic volume index are sensitive to one or both of preload and afterload alterations.
Studies of ventricular response to acute increases in volume status in normal humans are extremely limited. Nixon and colleagues [26] used a tilt table to alter preload in healthy volunteers. A head-down tilt increased echocardiographically determined left ventricular EDV without a significant change in ESV; LVEF also increased but mean velocity of circumferential cardiac fiber shortening – a relatively preload-independent index of cardiac contractility – was unchanged. Mangano and coworkers [27] examined the ventricular responses of patients with normal ventricular function following coronary artery bypass grafting to graded infusion of whole blood using radionuclide ventriculography and invasive hemodynamic monitoring. They found that EDV increased (mostly at lower ventricular filling pressures) and EF fell, implying an increase in ESV. In contrast, Van Daele and colleagues [25] demonstrated a sequential increase in EDV with a concurrent decrease in ESV during graded volume loading with a fixed crystalloid/colloid fluid regimen in preoperative patients undergoing orthopedic or oncologic surgery. Calvin and colleagues [21] also demonstrated an increase in EF (associated with a decrease in ESV) as the dominant mechanism of augmented SVI in response to fluid loading with 5% albumin in about almost half of a group of critically ill patients. One of the differentiating elements between the contribution of decreased ESV to SV in the two latter mentioned studies is that both were performed using a standard crystalloid or colloid solution, whereas those with an increase or no change in ESV involved whole blood transfusion or internal blood volume recruitment.
The two subject groups in the present study differed in the relative contribution of a decrease in ESV to augmentation of SV. Group 1 participants studied echocardiographically exhibited almost complete dependence of SV response on decreased LVESVI, whereas in group 2 participants studied with PAC/radionuclide cineangiography increases in ESV accounted for almost half of the SV response. The reasons for this difference may include random population variation, sex differences between the groups, or methodologic differences in the techniques used to measure cardiac responses. We believe that the former is more likely because a third, larger subject group has recently been examined echocardiographically, yielding EDV changes intermediate to the two groups in the present study [28] In addition, examination of the responses of different sexes in group 2 demonstrated similar patterns. In either case, both groups in the present study exhibited similar results with respect to decreases in ESV and increases in contractility indices. An increase in EDV was only inconsistently noted.
Although interesting, the findings of the study should only be extrapolated to critically ill patients with substantial caution. The healthy volunteers in the study were euvolemic and had normal cardiac and vascular function. Critically ill patients, in contrast, may have an abnormal volume status depending on the nature of the underlying disorder and the degree of resuscitation. In addition, alterations in systolic contractility, diastolic lusitropy, and vascular impedance may exist in such patients. Nevertheless, these data suggest that the effect of large volume resuscitation in critically ill patients should be reexamined with a view to developing a better understanding of the cardiovascular mechanics of response. Standard medical teaching suggesting that increased SV following fluid resuscitation results solely from increased cardiac preload (increased EDVI) may overlook significant elements of the cardiovascular response that are independent of preload (diastolic ventricular volume).
Conclusion
The mechanism of the cardiovascular responses noted in the present study cannot be fully delineated based on the available data. However, several conclusions may be drawn. First, infusion of saline at a volume consistent with clinical resuscitation produces a substantial increase in SV and CI in normal individuals. Second, in contrast to standard dogma, the increase in SV associated with such aggressive saline loading is substantially generated by a decrease in ESV with modest increases in EDV. Third, these changes in ventricular volumes and performance occur in parallel in both the right and left ventricles. Finally, these data suggest that resuscitative level volume loading leads to an increase in indices of right and left ventricular contractility, including those that are often considered to be load-independent. However, these responses can potentially be explained by either increased contractility or decreased afterload. Further studies will be required if we are to fully understand the cardiovascular mechanics of volume loading in normal individuals as well as critically ill patients in the intensive care unit.
Competing interests
None declared.
Key messages
• Increased stroke volume associated with aggressive saline infusion in normal subjects is substantially generated through a decrease in end-systolic volumes rather than increases in end-diastolic volume
• This response is consistent between both the right and left ventricles.
• Large volume fluid infusion leads to increases in basic indices of biventricular contractility although these could be explained by changes in inotropy or afterload.
Abbreviations
CI = cardiac index; CO = cardiac output; CVP = central venous pressure; EDV = end diastolic volume; EF = ejection fraction; ESV = end systolic volume; HR = heart rate; LVEDVI = left ventricular end-diastolic volume index; LVEF = left ventricular ejection fraction; LVESVI = left ventricular end-systolic volume index; MAP = mean arterial pressure; PAC = pulmonary artery catheter; PWP = pulmonary artery wedge pressure; RVESVI = right ventricular end-systolic volume index; SBP = systolic blood pressure; SV = stroke volume; TPR = total peripheral resistance; SVI = stroke volume index.
References
- Committee on Trauma Research of the American College of Surgeons . Advanced Trauma Life Support Course Manual. 6. Chicago: Committee on Trauma Research, American College of Surgeons; 1997. [Google Scholar]
- Huntsman LL, Stewart DK, Barnes SR, Franklin SB, Colocousis JS, Hessel EA. Noninvasive doppler determination of cardiac output in man. Clinical validation. Circulation. 1983;67:593–601. doi: 10.1161/01.cir.67.3.593. [DOI] [PubMed] [Google Scholar]
- Lewis JF, Kuo LC, Nelson JG, Limacher MC, Quinones MA. Pulsed Doppler echocardiographic determination of stroke volume and cardiac output: clinical validation of two new methods using the apical window. Circulation. 1984;70:425–531. doi: 10.1161/01.cir.70.3.425. [DOI] [PubMed] [Google Scholar]
- Takenaka A, Iwase M, Sobue T, Yokota M. The discrepancy between echocardiography, cineventriculography and thermodilution. Evaluation of left ventricular volume and ejection fraction. Int J Card Imaging. 1995;11:255–262. doi: 10.1007/BF01145194. [DOI] [PubMed] [Google Scholar]
- Gordon EP, Schnittger I, Fitzgerald PJ, Williams P, Popp RL. Reproducibility of left ventricular volumes by two-dimensional echocardiography. J Am Coll Cardiol. 1983;2:506–513. doi: 10.1016/s0735-1097(83)80278-2. [DOI] [PubMed] [Google Scholar]
- Morrison DA, Turgeon J, Ovitt T. Right ventricular ejection fraction measurement: Contrast ventriculography versus gated blood pool and gated first-pass radionuclide methods. Am J Cardiol. 1984;54:651–653. doi: 10.1016/0002-9149(84)90266-2. [DOI] [PubMed] [Google Scholar]
- Port SC. Recent advances in first-pass radionuclide angiography. Cardiol Clin North Am. 1994;12:359–372. [PubMed] [Google Scholar]
- Nivatpumin T, Katz S, Scheuer J. Peak left ventricular systolic pressure/end-systolic volume ratio: a sensitive detector of left ventricular disease. Am J Cardiol. 1979;43:969–674. doi: 10.1016/0002-9149(79)90361-8. [DOI] [PubMed] [Google Scholar]
- Kumar A, Anel R, Bunnell E, Habet K, Zanotti S, Marshall S, Neumann A, Ali A, Kavinsky C, Cheang M, Parrillo JE. PWP and CVP fail to predict ventricular filling volume, cardiac performance or the response to volume infusion in normal subjects. Crit Care Med. 2004;32:691–699. doi: 10.1097/01.CCM.0000114996.68110.C9. [DOI] [PubMed] [Google Scholar]
- Taylor RR, Cingolani HE, McDonald RH. Relationship between left ventricular volume, ejected fraction, and wall stress. Am J Physiol. 1966;211:674–680. doi: 10.1152/ajplegacy.1966.211.3.674. [DOI] [PubMed] [Google Scholar]
- Weber KT, Janicki JS, Reeves RC, Hefner LL. Factors influencing left ventricular shortening in isolated canine heart. Am J Physiol. 1976;230:419–426. doi: 10.1152/ajplegacy.1976.230.2.419. [DOI] [PubMed] [Google Scholar]
- Weber KT, Janicki JS, Hefner LL. Left ventricular force-length relations of isovolumic and ejecting contractions. Am J Physiol. 1976;231:337. doi: 10.1152/ajplegacy.1976.231.2.337. [DOI] [PubMed] [Google Scholar]
- Ilebekk A, Kiil F. Role of preload and inotropy in stroke volume regulation at constant heart rate. Scand J Clin Lab Invest. 1979;39:71–78. doi: 10.3109/00365517909104941. [DOI] [PubMed] [Google Scholar]
- Mouren S, Delayance S, Mion G, Souktani R, Fellahi JL, Arthaud M, Baron JF, Viars P. Mechanisms of increased myocardial contractility with hypertonic saline solutions in isolated blood-perfused rabbit hearts. Anesth Analg. 1995;81:777–782. doi: 10.1097/00000539-199510000-00021. [DOI] [PubMed] [Google Scholar]
- Hakumaki MO. Seventy years of the Bainbridge reflex. Acta Physiol Scand. 1987;130:177–185. doi: 10.1111/j.1748-1716.1987.tb08126.x. [DOI] [PubMed] [Google Scholar]
- Boettcher DH, Zimpfer M, Vatner SF. Phylogenesis of the Bain-bridge reflex. Am J Physiol. 1982;242:R244–R246. doi: 10.1152/ajpregu.1982.242.3.R244. [DOI] [PubMed] [Google Scholar]
- Lew WY. Mechanisms of volume-induced increase in left ventricular contractility. Am J Physiol. 1993;265:H1778–H1786. doi: 10.1152/ajpheart.1993.265.5.H1778. [DOI] [PubMed] [Google Scholar]
- Lew WY. Time-dependent increase in left ventricular contractility following acute volume loading in the dog. Circ Res. 1988;63:635–647. doi: 10.1161/01.res.63.3.635. [DOI] [PubMed] [Google Scholar]
- Robotham JL, Takata M, Berman M, Harasawa Y. Ejection fraction revisited. Anesthesiology. 1991;74:172–183. doi: 10.1097/00000542-199101000-00026. [DOI] [PubMed] [Google Scholar]
- Carabello BA, Spann JF. The uses and limitations of end-systolic indices of left ventricular function. Circulation. 1984;69:1058–1064. doi: 10.1161/01.cir.69.5.1058. [DOI] [PubMed] [Google Scholar]
- Calvin JE, Driedger AA, Sibbald WJ. The hemodynamic effect of rapid fluid infusion in critically ill patients. Surgery. 1981;90:61–76. [PubMed] [Google Scholar]
- Oberg B, Thoren P. Studies on left ventricular receptors; signaling in non-medullated vagal afferents. Acta Physiol Scand. 1972;85:145–163. doi: 10.1111/j.1748-1716.1972.tb05246.x. [DOI] [PubMed] [Google Scholar]
- Ohki S, Ishikawa S, Ohtaki A, Takahashi T, Koyano T, Otani Y, Murakami J, Mohara J, Isa Y, Kunimoto F, Morishita Y. Hemodynamic effects of alpha-human atrial natriuretic polypeptide on patients undergoing open-heart surgery. J Cardiovasc Surg. 1999;40:781–785. [PubMed] [Google Scholar]
- Legault L, van Nguyen P, Holliwell DL, Leenen FH. Hemodynamic and plasma atrial natriuretic factor responses to cardiac volume loading in young versus older normotensive humans. Can J Physiol Pharmacol. 1992;70:1549–1554. doi: 10.1139/y92-222. [DOI] [PubMed] [Google Scholar]
- van Daele ME, Trouwborst A, van Woerkens LC, Tenbrinck R, Fraser AG, Roelandt JR. Transesophageal echocardiography monitoring of preoperative acute hypervolemic hemodilution. Anesthesiology. 1994;81:602–609. doi: 10.1097/00000542-199409000-00012. [DOI] [PubMed] [Google Scholar]
- Nixon JV, Murray RG, Leonard PD, Mitchell JH, Blomqvist CG. Effect of large variations in preload on left ventricular performance characteristics in normal subjects. Circulation. 1982;65:698–703. doi: 10.1161/01.cir.65.4.698. [DOI] [PubMed] [Google Scholar]
- Mangano DT, Van Dyke DC, Ellis RJ. The effect of increasing preload on ventricular output and ejection in man: limitations of the Frank–Starling mechanism. Circulation. 1980;62:535–541. doi: 10.1161/01.cir.62.3.535. [DOI] [PubMed] [Google Scholar]
- Kumar A, Anel R, Bunnell E, Habet K, Neumann A, Wolff D, Rosenson R, Cheang M, Parrillo JE. Effect of large volume infusion on left ventricular volumes, performance and contractility parameters in normal volunteers. Intensive Care Med. 2004. [DOI] [PubMed]