

Abstract
Retinoid X receptor-alpha (RXRα), a unique member of the nuclear receptor superfamily, is a well-established drug target, representing one of the most important targets for pharmacologic interventions and therapeutic applications for cancer. However, how RXRα regulates cancer cell growth and how RXRα modulators suppress tumorigenesis are poorly understood. Altered expression and aberrant function of RXRα are implicated in the development of cancer. Previously, several studies had demonstrated the presence of N-terminally truncated RXRα (tRXRα) proteins resulted from limited proteolysis of RXRα in tumor cells. Recently, we discovered that overexpression of tRXRα can promote tumor growth by interacting with tumor necrosis factor-alpha-induced phosphoinositide 3-kinase and NF-κB signal transduction pathways. We also identified nonsteroidal anti-inflammatory drug Sulindac and analogs as effective inhibitors of tRXRα activities via a unique binding mechanism. This review discusses the emerging roles of tRXRα and modulators in the regulation of cancer cell survival and death as well as inflammation and our recent understanding of tRXRα regulation by targeting the alternate binding sites on its surface.
Keywords: tRXRα, RXRα modulators, nongenomic action, inflammation, PI3K
Introduction
Retinoid X receptor-alpha (RXRα) is a unique and important member of the nuclear receptor superfamily (Fig. 1A). As a master regulator, RXRα acts through homodimerization with itself or via serving as obligatory partner for many other nuclear receptors, including retinoic acid receptor (RAR), thyroid hormone receptor (T3R), vitamin D receptor (VDR), Nur77, peroxisome proliferator-activated receptors (PPARs), liver X receptor (LXR), and farnesoid X receptor [1–9]. Many naturally occurring small molecules have been shown to bind to RXRα and modulate its activities [2–4,10–12]. 9-cis-Retinoic acid (9-cis-RA) was the first one that was identified as a natural RXRα ligand (Fig. 1B). Subsequently, several dietary fatty acids were found to bind to RXRα and act as natural RXRα ligands. These include docosahexaenoic acid (DHA), oleic acid, and phytanic acid. However, none of these molecules have been proved to be the bona fide endogenous ligands of RXRα [13,14].
Figure 1.
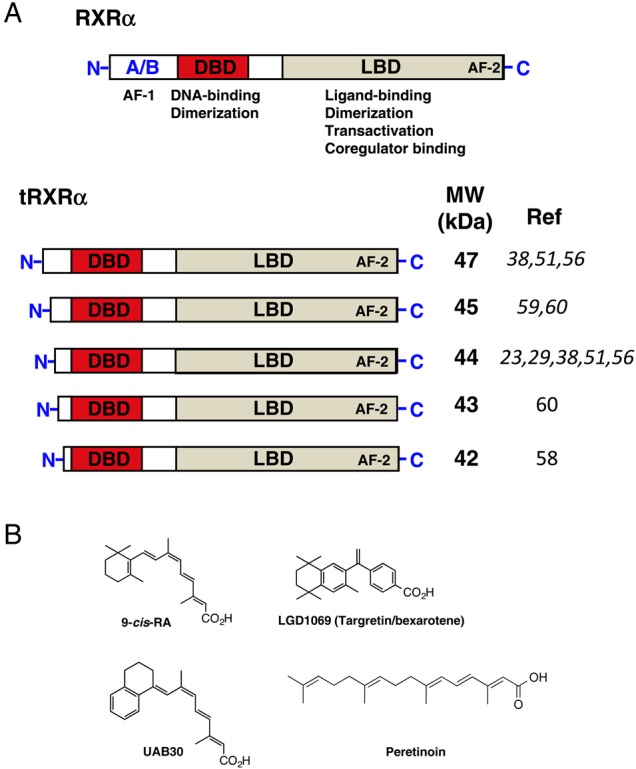
The scheme of RXRα/tRXRα and its ligands (A) Schematic representation of RXRα and tRXRα. DBD, DNA-binding domain; LBD, ligand-binding domain; AF-1, activation function 1; AF-2, activation function 2. (B) Chemical structures of 9-cis-RA, Targretin, UAB30 and peretinoin.
RXRα possesses a common structural organization that is shared by the nuclear receptor family: a disordered N-terminal A/B region containing activation function 1 (AF-1), a DNA-binding domain (DBD) containing two zinc fingers, and a C-terminal ligand-binding domain (LBD) composed of 12 α-helices and a short β-turn (Fig. 1A). The LBD consists of a canonical ligand-binding pocket (LBP), an activation function 2 (AF-2), a co-regulator-binding surface groove, and a dimerization surface (Fig. 1A). A well-described mechanism of RXRα action is that RXRα and its partners act as ligand-dependent transcription factors through binding to specific DNA-response elements of the target genes [1–9]. Ligand binding induces a conformational change that triggers a cascade of events such as co-regulator exchanging or binding, leading to positive or negative gene transcription and subsequent biological activities [1–9].
Genetic analysis demonstrates that RXRα is involved in a plethora of developmental and physiological pathways. Knockout of RXRα resulted in embryonic lethality [15]. Tissue-specific inactivation of RXRα has demonstrated a major role of RXRα in hepatocytes [16], skin [17], prostate [18], or adipose tissue [19]. Strong phenotypes observed in most RXRα mutant mice may be related to alternations in pathways regulated by its heterodimerization partners. The role of RXRα homodimer in vivo was unraveled recently. Ligand-activated RXRα homodimers up-regulate p21 expression through the direct binding of RXRα homodimers to the p21 promoter [20]. Characterization of mice lacking RXRα in myeloid cells reveals an important role of RXRα homodimers in the innate immune response to inflammatory stimuli [21].
Aside from its role in DNA binding and transactivation, RXRα also exerts extranuclear actions through transcription-independent mechanisms [22–26]. RXRα resides in the cytoplasm at different stages during development [27]. In response to differentiation [24], survival [28,29], apoptosis [22], and inflammation [25,26,28,29], RXRα migrates from the nucleus to the cytoplasm. For example, RXRα is translocated from the nucleus to the cytoplasm in response to endotoxin and other inflammatory mediators to inhibit its transactivation function [25,30], while an altered localization of RXRα to the splicing factor compartments occurs in highly malignant human breast cancer cells [31]. These observations revealed the intricacy of RXRα functions and underscored the importance of the RXRα nongenomic signaling.
Post-translational modifications also play a critical role in the regulation of RXRα activities. Phosphorylation of the N-terminal domain of RXRα by mitogen-activated protein kinases occurs in response to several stress agents such as UV radiation, oxidative damage, or ribotoxic agents [32–37], leading to the inhibition of the transcriptional activity of RXRα heterodimers. RXRα could also be phosphorylated at Ser260 in its LBD [33,38]. In human hepatocellular carcinoma (HCC) cells, RXRα is heavily phosphorylated at Ser260, making it resistant to ubiquitination and proteasome-mediated degradation [38,39]. Nuclear export of RXRα in response to inflammatory signaling also involves c-Jun N-terminal kinase (JNK) phosphorylation of Ser260 [26]. RXRα is also a substrate for modification with small ubiquitin like modifier (SUMO) [40]. SUMOylation of RXRα at Lys108 in its AF-1 domain inhibits its transcriptional activity [40]. Interestingly, a recent study showed that inflammatory mediators increase SUMOylation of RXRα in a JNK-dependent manner [41]. Unlike SUMOylation, acetylation of RXRα by p300 promotes its DNA binding, thereby increasing its transcriptional activity [42].
We recently reported that an N-terminally truncated form of RXRα (tRXRα) (Fig. 1A) produced in cancer cells resides in the cytoplasm to promote the growth of tumor cells [29]. Our investigation of tRXRα action in the cytoplasm revealed an extensive interaction between tRXRα and tumor necrosis factor-α (TNFα) signaling. In this review, we will briefly summarize the role of RXRα in cancer and the cancer therapeutic effect of RXRα ligands (rexinoids). We will focus our discussion on the identification of tRXRα in cancer cells and its implication in the regulation of cancer survival and death as well as inflammation through its nongenomic interaction with TNFα-induced phosphoinositide 3-kinase (PI3K)/AKT and NF-κB signal transduction pathways. Finally, we will summarize recent advances in the discovery of tRXRα modulators with new RXRα-binding mechanisms.
RXRα and Cancer
Altered expression and function of RXRα are implicated in the development of a number of cancers and diseases. Although RXRα-knockout fetus dies at embryonic days [15], targeted disruption of RXRα gene leads to preneoplastic lesions in prostate [18], alopecia, epidermal interfollicular hyperplasia, keratinocyte hyperproliferation, and aberrant terminal differentiation in skin [17], the development of cervical malignant lesions [43], alteration of fatty acid oxidation and hepatocyte lifespan in liver [16], and the resistance to diet-induced obesity due to impaired adipocyte differentiation in adipose tissue [19]. Consistently, diminished RXRα expression is associated with the development of certain malignancies, such as thyroid carcinoma [44,45] and liver cancer [46], and levels of RXRα protein are often reduced in cancer cells and tumor tissues [45,47–51]. In addition to reduced levels of RXRα protein, altered RXRα function by phosphorylation is associated with the development of human HCC [38] and colon cancer [52]. Furthermore, several studies have demonstrated that alteration of the subcellular localization of RXRα is linked to the development of cancer [31]. Recent studies showed that RXRα binding to PML/RARα is absolutely required for the development of acute promeylocytic leukemia (APL) in transgenic mice [53–55], demonstrating the oncogenic potential of this protein when it acts inappropriately. Several groups have demonstrated that RXRα is proteolytically cleaved in cancer cells [23,45,51,56–60], and our illustration that tRXRα could enhance TNFα activation of PI3K/AKT and NF-κB pathways revealed that aberration in RXRα signaling by limited proteolysis plays an active role in cancer development [28,29,61–65].
Rexinoids and Cancer Therapy
The pleiotropic actions of RXRα under both physiological and pathophysiological conditions suggest RXRα as an important drug target for pharmacologic interventions and therapeutic applications. This is highlighted by the FDA approval of the RXR-based drug Targretin (bexarotene) for treating cutaneous T-cell lymphoma (CTCL) patients who are refractory to at least one prior systemic therapy [66]. Targretin selectively binds to RXRs and does not have significant RAR binding and transactivation [5,67,68]. Side effects have been reported to be associated with the Targretin treatment, including hyperlipidemia and hypothyroidism [66,69]. Hyperlipidemia is thought to be associated with the modulation of RXR heterodimers with PPARs and LXRs [70] and hypothyroidism may be due to the inhibition of the TSH production by Targretin through the thyrotrope-restricted RXRγ isoform [71]. To overcome these side effects, efforts to develop novel RXR-based drug for CTCL treatment are ongoing. Recently, it was reported that certain analogs of Targretin possess improved biological properties [72]. The mechanism of Targretin action has yet to be fully elucidated. It has been shown that the drug can induce apoptosis, differentiation and cellular senescence, inhibit metastasis and angiogenesis, and block cell cycle progression, which was reviewed recently [73–76].
Rexinoids also show significant effect in non-small-cell lung cancer (NSCLC) [77], in which altered RXR signaling has been observed [78]. Preclinical data showed that Targretin can prevent and overcome acquired paclitaxel resistance in NSCLC [79]. Furthermore, Targretin could act synergistically with standard first-line platinium-based chemotherapy [77,78]. In a randomized Phase III trial comparing Targretin in combination with cisplatin/vinorelbine to cisplatin/vinorelbine alone in a total of 623 patients for over survival as the primary efficacy endpoint, a subgroup (32%) of Targretin-treated patients who developed NCI Grade 3/4 hypertriglyceridemia had longer median survival compared with control patients [80]. In another Phase III trial to determine the effects of addition of Targretin to standard first-line carboplatin and paclitaxel therapy, similar results were obtained that increased survival was correlated with the occurrence of Grade 3/4 hypertriglyceridemia in patients treated with Targretin [81].
Rexinoids also induce differentiation in AML patient samples and in various AML cell lines [82,83]. As a differentiation agent, Targretin is being tested for AML treatment [84]. A Phase I dose escalation study in elderly and relapsed AML patients was conducted to investigate whether Targretin could be used in combination with decitabine [84]. It was found that the combination was well tolerated, but produced only modest responses. However, greater AML blast differentiation was observed in patients with clinical response, suggesting a potential of rexinoids in AML therapy. Recent studies suggested that Targretin mediates the RXR/LXR-regulated gene expression that was deregulated in AML cells [85].
Other studies have also revealed an emerging role of RXRα in APL [86]. RXRα has been demonstrated to be a binding partner of PML/RAR. Genome-wide epigenetic studies suggested that PML/RARα/RXR complex acted as a local chromatin modulator [55]. Recruitment of RXRα by the APL fusion protein is crucial for oncogenic transformation and is required for the development of APL in transgenic mice [53–55]. Therefore, rexinoids have great potentials for treating APL.
Malfunction of RXRα due to phosphorylation by the Ras-mitogen-activated protein kinase signaling pathway is profoundly associated with the development of HCC and thus may be a critical target for HCC chemoprevention. Acyclic retinoid (also known as peretinoin), a synthetic retinoid that binds to both RXR and RAR, can prevent phosphorylation of RXRα by inhibiting the activities of Ras–Raf–Erk system through an undefined mechanism [87]. Clinical studies have shown that it is effective in suppressing HCC recurrence and improving patient survival rates following curative therapy [87]. A recent gene expression profiling study using liver biopsy from HCC patients underwent therapy revealed that peretinoin not only enhances the expression of retinoid target genes but also regulates various signal transduction pathways involved in hepatocarcinogenesis [23].
Proteolytic Cleavage of RXRα in Cancer Cells
Numerous studies have shown limited proteolytic cleavage of RXRα protein in tumor cells [23,45,51,56–60]. Matsushima-Nishiwaki et al. [56] showed a 44 kDa tRXRα in liver cells and found that m-calpain could cleave RXRα into the tRXRα lacking N-terminal A/B region in HuH7 HCC cells (Fig. 1A). A recent study showed that tRXRα accumulates more in several HCC cell lines than in normal immortal human hepatocytes [88]. Cathepsin L-type protease could also cleave RXRα at its N-terminal region, producing a 44 kDa tRXRα in rapid growing hepatocytes, which alters thyroid hormone responsiveness [58], and in HCC cells [60]. In osteosarcoma cells, the production of an aberrant 45 kDa tRXRα is implicated in the resistance to the antiproliferative effects of calcitriol and retinoids [59]. Two tRXRα proteins with 47 and 44 kDa were detected in all seven prostate cancer cell lines, which may have altered subcellular localization [51]. Interestingly, a 44 kDa tRXRα was found in mitochondria [23], suggesting that tRXRα may play a nongenomic role. In our study, we found that tRXRα is produced in many different types of cancer cells [29]. Moreover, tRXRα was detected in primary tumors but not in tumor surrounding tissues or distant normal tissues from the same cancer patients [29], demonstrating a close association of tRXRα production with cancer. We also identified calpain II as a protease that could cleave RXRα protein in vitro and in vivo. Activation of calpain II by ionomycin enhances the production of tRXRα in cancer cells, which is regulated in a glycogen synthase kinase 3 beta-dependent manner [63].
Limited proteolytic cleavages of RXRα mainly occur at its N-terminus [23,45,51,56–60]. Comparing with its DBD and LBD, the function and regulation of the N-terminal A/B domains of RXRα have not been well studied. The fact that the N-terminal A/B domains of nuclear receptor family members are highly variable suggests that they may mediate specific functions. Phenotypic analysis of mice expressing RXRα with its N-terminal A/B region deleted indicated that the RXRα AF-1 domain is functionally important for efficiently transducing the retinoid signal during embryonic development [89]. An interesting feature of the N-terminal A/B region is that it contains many consensus phosphorylation sites and is therefore the target of multiple kinases, such as JNK [32,34,36,37,90–92]. Hyperphosphorylation of A/B region (Ser61, Ser75, and Thr87) can induce apoptosis [93], while phosphorylation of RXR at Ser260 has been correlated with the unrestrained growth of certain HCC [38]. However, little is known about the mechanisms through which the N-terminal A/B region and its phosphorylation site participate in the regulation of RXRα activity.
Regulated proteolysis is a key step in a number of different signaling pathways that respond to developmental cues or external stimuli [94–100]. Caspase-mediated cleavage of the BH3-only protein Bid generates a truncated protein (tBid), and the subsequent translocation of tBid to mitochondria is implicated in death receptor signaling [99]. Similarly, caspase-3 mediates retinoic acid-induced degradation of the APL PML/RARα fusion protein [101], and the cleavage product of PML/RARα contributes to ATRA-mediated differentiation in APL [102]. Proteolytic processing of Notch and nuclear translocation of truncated product is a crucial step in the transduction of the Notch signaling [96]. Cleavage of the androgen receptor by calpain produces a truncated receptor protein that may play a role in the development of androgen-independent prostate cancer [103]. Similarly, cleavage of MET, a membrane-bound receptor tyrosine kinase, results in a truncated nMET, which is localized in the nuclei of malignant cells to promote the growth of castration-resistant prostate cancer cells through its activation of both SOX9 and β-catenin [104]. Thus, proteolytic cleavage likely represents an important mechanism that regulates the biological function of RXRα.
Nongenomic Action of tRXRα in Cancer
tRXRα and PI3K/AKT survival signaling
PI3K is a heterodimeric protein composed of a catalytic subunit (p110α/β/γ/δ) and a regulatory subunit (p85α/β) that participate in multiple cellular processes, including cell growth, transformation, differentiation, and survival in a number of cell types and human cancers [105–108]. The p85α/p110α heterodimer is the major form of PI3Ks, in which the p85α regulatory subunit binds to the p110α catalytic subunit to integrate signals from various cellular proteins, providing an integration point for activation of p110α and downstream molecules such as AKT.
We found that tRXRα but not RXRα could act to mediate TNFα activation of PI3K/AKT in a number of cancer cell lines [29] (Fig. 2). Unlike RXRα that resides in the nucleus, tRXRα is cytoplasmic in response to TNFα treatment, and interacts with the p85α regulatory subunit, leading to an enhanced activation of the PI3K/AKT survival pathway and anchorage-independent cell growth in vitro and cancer cell growth in animals (Fig. 2). Abnormal activation of the PI3K/AKT pathway is often observed in cancer cells, contributing to their growth and survival properties and drug resistance [105–108]. Knocking down of tRXRα could reduce basal AKT activation in some cancer cells, demonstrating that tRXRα may play a critical role in the aberrant activation of PI3K/AKT signaling in cancer cells. Interestingly, the interaction of tRXRα with p85α and its activation of PI3K/AKT signaling were induced by inflammatory cytokine TNFα but not by some growth factors such as epidermal growth factor, implying that tRXRα may specifically act in inflammatory environment. These results provided another example that a nuclear receptor can act outside of the nucleus to regulate an important biological process and identified tRXRα as a key molecule involved in the aberrant activation of PI3K/AKT pathway in cancer cells. However, many important questions regarding the nongenomic regulation of the PI3K/AKT pathway by tRXRα and ligands remain to be answered. It is unclear whether the cytoplasmic localization of tRXRα results from its nuclear export or cytoplasmic retention due to its interaction with cytoplasmic proteins such as p85α. As the wild-type RXRα fails to interact with p85α, the N-terminal region deleted from RXRα is expected to play a critical role in regulating RXRα activities, which remains to be determined. As RXRα is often phosphorylated in cancer cells, it will be interesting to examine whether phosphorylation or other modifications of RXRα are involved in the regulation of RXRα cytoplasmic localization and its interaction with p85α. How tRXRα interacts with p85α is still unknown at present. However, it is worth noting that numerous nuclear receptors including estrogen receptor, androgen receptor, glucocorticoid receptor, and RAR have been shown to interact with p85α [109–112], implying the existence of a more general mechanism for their interaction with p85α.
Figure 2.
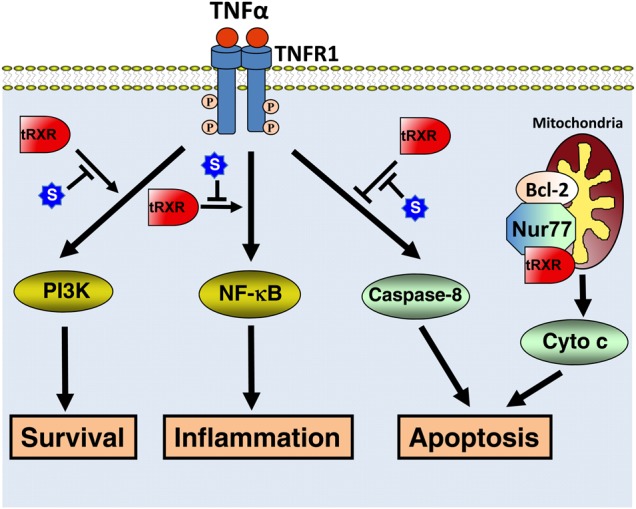
The nongenomic tRXRα actions The cytoplasmic tRXRα acts through its interaction with TNFα-signaling proteins to regulate cell survival, inflammation, and apoptosis. tRXRα effect is negatively regulated by its modulator Sulindac (S). In addition, tRXRα may target mitochondria through heterodimerization with Nur77 to modulate mitochondria-dependent apoptosis.
Because of its role in oncogenesis and drug resistance, the PI3K/AKT pathway has therefore been targeted extensively to develop therapeutics against cancer and related diseases, and to overcome drug resistance. However, current targeting strategies that rely on direct inhibition of PI3K/AKT activities have posed profound adverse effects and are thus far confined to the preclinical and clinical evaluation due to toxicity and lack of selectivity. Thus, the identification of tRXRα-mediated activation of PI3K/AKT signaling pathway in cancer cells may provide new strategies to inhibit the activation of PI3K/AKT in cancer cells by targeting tRXRα. Such tRXRα-based PI3K/AKT inhibitors are likely to be more specific and tumor selective than conventional PI3K/AKT inhibitors.
tRXRα and apoptosis
Apoptosis, programed cell death, is abnormally regulated in cancer cells and the efficacy of chemotherapeutic drugs depends largely on their ability to induce apoptosis [113,114]. Apoptosis often occurs following either triggering of cell surface death receptors (the extrinsic pathway) or perturbation of mitochondria (the intrinsic pathway) [115]. The intrinsic pathway is initiated by the release of apoptogenic factors such as cytochrome c from mitochondria, while the extrinsic pathway often involves the activation of the initiator caspase-8 through stimulation of death receptors of the TNF-receptor superfamily. It is likely that tRXRα is involved in the regulation of both intrinsic and extrinsic apoptotic pathways.
Previous studies showed that RXRs and ligands are implicated in the regulation of mitochondria-dependent apoptosis through modulating the Nur77-Bcl-2 apoptotic pathway due to its heterodimerization with Nur77 [116] (Fig. 2). Nur77, also known as TR3 or NGFI-B, is an immediate-early response gene and an orphan member of the nuclear receptor superfamily [117–119]. It mediates apoptosis of numerous types of cancer cells by a variety of apoptotic stimuli including retinoid-related apoptotic molecules, TPA, calcium ionophore, and many chemotherapeutic agents, perhaps being the most potent pro-apoptotic member in the nuclear receptor superfamily [22,120–130]. We discovered that Nur77 could migrate from the nucleus to the cytoplasm where it targets mitochondria through its interaction with Bcl-2, leading to cytochrome c release and apoptosis [131,132]. The unique property of the Nur77-Bcl-2 apoptotic pathway is the conversion of Bcl-2 from an anti-apoptotic molecule to a pro-apoptotic one upon binding by Nur77 [132,133]. RXRα cotranslocates with Nur77 from the nucleus to the cytoplasm in response to NGF [24], apoptotic stimuli [22,134], and IGFBP-3 [135]. Interestingly, Casas et al. [23] detected a 44 kDa tRXRα in mitochondria. Whether and how cytoplasmic tRXRα modulates the Nur77-Bcl-2 pathway remain to be investigated.
tRXRα has also been shown to modulate the extrinsic apoptotic pathway upon ligand binding [29]. TNFα is a multifunctional cytokine that controls diverse cellular events such as cell survival and death that control the destiny of cancer cells [136–141]. The diverse cellular effects of TNFα are mediated by TNFα receptor-1 (TNFR1). Upon binding to TNFα, TNFR1 forms a complex that consists of TNF-receptor-associated death domain (TRADD) protein, receptor interacting protein (RIP), and Fas-Associated protein with Death Domain (FADD). When TNFR1 signals apoptosis, FADD binds to pro-caspase-8, resulting in its activation and apoptosis. Alternatively, when TNFR1 signals survival, TNF-receptorassociated factor 2 (TRAF2) is recruited to the complex, leading to activation of AP-1 and NF-κB pathways [136–141]. TNFα binding to TNFR1 could also result in activation of the PI3K/AKT survival pathway [142]. Although TNFα is capable of inducing apoptosis of cancer cells through death receptor-dependent mechanism, such an effect is often antagonized by its own survival function through its activation of NF-κB and PI3K/AKT pathways [143–148]. The balance of TNFα-induced survival- and death-signaling is therefore pivotal in determining the fate of TNFα-responding cells [136–142,144,146,149–152]. Since TNFα is produced by malignant or host cells in the tumor microenvironment but not in normal cells, there has been tremendous interest in developing strategies to shift TNFα signaling from survival to death. Although tRXRα is required for TNFα activation of PI3K/AKT to promote the survival of cancer cells, we found that tRXRα binding to nonsteroidal anti-inflammatory drug (NSAID) Sulindac (Fig. 3) or analogs resulted in caspase-8 activation and apoptosis, demonstrating that tRXRα and ligands could regulate the extrinsic apoptotic pathway [29]. The activation of caspase-8-dependent apoptosis by Sulindac and analogs was attributed to their inhibition of tRXRα-dependent activation of the PI3K/AKT survival signaling. This is supported by the observation that Sulindac and analogs inhibit the binding of tRXRα to p85α [29]. Similarly, inhibition of tRXRα-mediated activation of NF-κB survival pathway also leads to apoptosis [65]. These results demonstrate that inhibition of tRXRα-mediated activation of PI3K/AKT and NF-κB pathways could effectively shift TNFα signaling from survival to death. However, currently it cannot be excluded that tRXRα may be directly involved in the regulation of the death-inducing signaling complex comprised of FADD and pro-caspase-8.
Figure 3.

Chemical structures of molecules that act via binding to novel binding regions in RXRα
tRXRα and inflammation
Recent reports suggest a strong link between chronic inflammation and the development of cancer [153,154]. Chronic inflammation due to infection, autoimmune disease, malignant and benign tumors, or other pathologies has become a recognized risk factor for epithelial-derived malignancies [155]. The transcription factor NF-κB is known as a master inflammatory transcriptional regulator, which is highly active in cancer cells. In quiescent cells, NF-κB proteins are sequestered in the cytoplasm by inhibitory molecules, the inhibitor of NF-κB (IκB) proteins [136–141]. NF-κB requires a signaling pathway for activation. Such NF-κB-activating pathways are triggered by a variety of extracellular stimuli and lead to the phosphorylation and subsequent proteasome-mediated degradation of IκB proteins [136–141]. Activated NF-κB migrates into the nucleus to regulate the expression of multiple target genes associated with cancer cell migration, proliferation, anti-apoptosis, angiogenesis, and metastasis [136–141]. In cancer cells, NF-κB is often constitutively activated as a result of undergoing inflammation or the consequence of formation of an inflammatory microenvironment during malignant progression.
The role of RXRα in regulating inflammation was suggested by findings that several anti-inflammatory agents act as RXR ligands. Dietary omega-3 fatty acids, such as DHA, exert their beneficial effects primarily through their anti-inflammatory effects. DHA induces growth inhibition and apoptosis by inhibiting NF-κB activity [156] and suppressing cytokine production in macrophages [157], whereas R-etodolac which is known to bind to RXRα [158] decreases constitutive and RANKL-stimulated NF-κB activation in macrophages and suppresses TNFα-induced IκB-kinase (IKK) phosphorylation and subsequent NF-κB activation in human multiple myeloma cells [159]. LGD1069 down-regulates COX-2 expression in breast cancer cells [160] and inhibits angiogenesis and metastasis in solid tumors [161]. Oral administration of Targretin reduces inflammation in a group of patients with plaque-type psoriasis [162]. Thus, the anti-inflammatory effects of RXRα and its ligands in various cell types underscore its function in the prevention and treatment of cancer and diseases.
Both genomic and nongenomic actions of RXRα could account for the modulation of inflammation in macrophages and cancer cells by RXRα and ligands. For genomic action, interactions between RXRα and proinflammatory transcription factors, particularly NF-κB and AP-1, have been well described in several reviews [163,164]. RXRα may also employ nongenomic action to negatively regulate the NF-κB signaling pathway. The role of nongenomic action of RXRα in the regulation of inflammation is also suggested by mounting evidence that subcellular localization of RXRα is altered in response to inflammation [25,30]. Lipopolysaccharide treatment of animals altered the subcellular location of RXRα [25]. RXRα undergoes rapid nuclear export in response to signals initiated by the proinflammatory cytokine IL-1β in hepatoma cells [26]. Our recent discovery that TNFα could induce cytoplasmic localization of tRXRα underscores the significance of tRXRα cytoplasmic action in the regulation of inflammation (Fig. 2). As discussed above, TNFα is a multiple cytokine that induces not only the extrinsic apoptotic and PI3K/AKT pathways but also the NF-κB inflammatory pathway through its sequential recruitment of various adaptors including TRADD, RIP1, and TRAF2 to the cytoplasmic membrane [136–141]. This is followed by the recruitment and activation of the classical IKK complex [136–141]. Once activated, the IKK complex phosphorylates IκBα, which is subsequently ubiquitinated and degraded via the proteasome pathway. We recently reported that tRXRα could promote TNFα activation of NF-κB pathway through its interaction with TRAF2 and enhance TNFα-induced RIP1 ubiquitination [65]. A nitrostyrene derivative that binds to tRXRα could inhibit tRXRα interaction with TRAF2 and its induction of NF-κB activation. These results demonstrate that tRXRα is directly involved in the activation and regulation of the inflammasome. It remains to be clarified whether other RXRα modulators known to have anti-inflammatory effect also target tRXRα-mediated activation of the NF-κB pathway.
Ligands that Bind to a Novel Site to Regulate the Nongenomic Activities of tRXRα
We identified that NASID Sulindac (also called CLINORIL®) currently used for treating pain and inflammation can inhibit the binding interaction between tRXRα and p85α, resulting in caspase-8-dependent apoptosis [29]. Furthermore, we synthesized several Sulindac analogs and identified several Sulindac derivatives including K-80003, K-8008, and K-8012 (Fig. 3), which can also bind to tRXRα to inhibit the TNFα-induced interaction of tRXRα with p85α and activate the TNFα-dependent apoptotic pathway [29]. K-80003, K-8008, and K-8012 are much more effective than Sulindac in inhibiting the growth of various cancer cells, including A549 lung cancer, PC3 prostate cancer, and ZR-75-1 and MB231 breast cancer cells. Both K-80003 and K-8008 inhibit HepG2 tumor growth in animals and do not show apparent toxic effects. Several natural products including CF31 [28] and nitrostyrene derivatives [65] could also activate this death pathway by directly binding to tRXRα.
Our identification of NSAID Sulindac and its analogs as tRXRα binders and modulators prompted us to study their binding mode. The crystal structure of RXRα-LBD in complex with K-8008 or K-8012 [62] demonstrates the existence of different binding site. The complex structures exist as noncrystallographic homotetramer similar to the reported apo-homotetramer [165,166], in which bottoms of two homodimers interface form a tetramer. In a tetramer, two K-8008 molecules were found to bind to one homotetramer in a hydrophobic region that is near the entry and the edge of the cognate LBP [62]. The K-8008 binding region is close to the dimer–dimer interface that does not overlap with the binding region of 9-cis-RA (Fig. 4A). With respect to the monomeric and the dimeric RXRα-LBD, the K-8008 binding region is located on the surface of the RXRα molecules. RXRα has been shown to form transcriptionally silent homotetramers in solution, which rapidly dissociate into active homodimers upon binding of agonists or antagonists [165–167]. Therefore, it is intriguing that binding of K-8008, an RXRα antagonist, to a new region does not induce tetramer dissociation, a similar phenomenon observed in the binding of danthron [168]. The structural basis of K-8008 binding suggests that RXRα tetramerization represents a key mechanism for the regulation of RXR nongenomic actions.
Figure 4.

Novel binding regions in RXRα (A) K-8008 binds to a novel binding region: the K-8008 binding region is away from the 9-cis-RA binding area and located on the surface of monomeric RXRα. It shows the superposition of the monomer of RXRα-LBD/K-8008 complex structure (brown) and the apo protein structure (purple, from PDB entry 1G1U). K-8008 is shown as sticks (carbon in magenta and nitrogen in blue). The classic ligand-binding site is indicated by a VDW ball model of 9-cis-RA (in cyan/red) taken from a superimposed 1FBY of PDB. (B) The proposed binding region for compound 23. The compound 23-binding region overlaps with the coactivator-binding region. Here, compound 23 was docked to the structure 3FUG (in pink) of PDB and the docked conformation (in VDW balls) was displayed with the coactivator peptide (in green) in the structure of 3FUG.
Ligands Targeting the Co-regulator-binding Site of tRXRα
Drug discovery and development efforts targeting RXRα have been focused on identifying and optimizing rexinoids that bind to RXRα canonical LBP. However, as mentioned above, there are key limitations of using rexinoids that include rising of plasma triglyceride levels, suppression of the thyroid hormone axis, and induction of hepatomegaly. Therefore, targeting alternate sites on RXRα for regulating its activities could become a new strategy for RXRα-based drug discovery. Compounds that bind to alternate sites have been identified for other nuclear receptors [169–171], including estrogen receptor, androgen receptor, VDR, and T3R. Among the reported alternate sites on nuclear receptors, the co-regulator binding site is the most studied one. Recently, by employing a docking-based virtual screening approach, we identified some small molecules that bind to the co-regulator-binding surface of RXRα, a region where the binding sites of corepressor and the coactivator overlap (Fig. 4B). One of the identified binder, compound 23 (Fig. 3), could regulate the biological functions of tRXRα. Compound 23 could inhibit the TNFα-induced interaction between tRXRα and p85α, inhibit AKT activation in vitro and in animals, and induce apoptosis [61]. Compound 23 represents the first example of an RXRα modulator that acts via the co-regulator-binding site rather than the classical LBP. Furthermore, compound 23 does not bind to the LBP. Thus, targeting the alternate binding sites on the surface of RXRα for therapeutic intervention may become a new paradigm for nuclear receptor-based drug discovery.
Conclusion and Perspective
RXRα is a unique and important drug target as evidenced by the success of rexinoid Targretin in treating CTCL and the enormous favorable results from testing rexinoids in various cancer models. However, the mechanisms by which RXRα modulate carcinogenesis are complex and remain to be fully defined, which has hampered the exploitation of the therapeutic potential of RXRα. The identification of tRXRα in cancer cells and the illustration of its roles in the control of apoptosis, survival, and inflammation offer new strategies to develop improved therapeutics against cancer by targeting tRXRα.
RXRα-based drug development is also hampered by the side effects associated with targeting its cognate LBP that is highly conserved among many nuclear receptors. Therefore, one of the current challenges in developing RXRα-based drugs is to identify selective RXRα modulators that possess the desired pharmacological activities without unwanted side effects. Many new modulators are being developed. In addition, screening for natural and synthetic RXRα ligand is ongoing. Thus, the findings that RXRα is cleaved in tumor cells and that Sulindac-derived small molecules and others act at the alternate binding sites on the surface of tRXRα will provide new rational for drug design and screening approach. Such an approach may help to identify small molecules specific to tumor- or disease-selective RXRα (i.e. tRXRα or RXRα with abnormal modifications) and may also circumvent side effects associated with binding to the cognate RXRα LBP. However, many questions remain unanswered regarding tRXRα production and function, and the underlying mechanisms of tRXRα actions need to be determined. Binding of Sulindac analogs to the tetrameric form of RXRα LBD is interesting. However, little is known about the biological function of the RXRα tetramer with respect to the regulation of the nongenomic RXRα action. Characterizing the surface-binding sites in tRXRα and developing selective inhibitors targeting the surface-binding sites may support a transition from the traditional paradigm of drugs targeting the LBP to a novel rational approach targeting functionally important surface sites, which may lead to more effective and specific therapeutics.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (Nos. 91129302, 91429306, and U1405229), the Xiamen Science and Technology Project (No. 3502Z20123015), the Xiamen Oceanic Administration (No. 14PYY051SF04), the U.S. Army Medical Research and Material Command (No. W81XWH-11-1-0677), the National Institutes of Health (No. CA179379), and the California Breast Cancer Research Program (No. 20IB-0138).
References
- 1.Evans RM, Mangelsdorf DJ. Nuclear receptors, RXR, and the Big Bang. Cell 2014, 157: 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Szanto A, Narkar V, Shen Q, Uray IP, Davies PJ, Nagy L. Retinoid X receptors: X-ploring their (patho)physiological functions. Cell Death Differ 2004, 11(Suppl 2): S126–S143. [DOI] [PubMed] [Google Scholar]
- 3.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nat Rev Drug Discov 2007, 6: 793–810. [DOI] [PubMed] [Google Scholar]
- 4.Dawson MI, Xia Z. The retinoid X receptors and their ligands. Biochim Biophys Acta 2012, 1821: 21–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dawson MI, Zhang XK. Discovery and design of retinoic acid receptor and retinoid X receptor class- and subtype-selective synthetic analogs of all-trans-retinoic acid and 9-cis-retinoic acid. Curr Med Chem 2002, 9: 623–637. [DOI] [PubMed] [Google Scholar]
- 6.Desvergne B. RXR: from partnership to leadership in metabolic regulations. Vitam Horm 2007, 75: 1–32. [DOI] [PubMed] [Google Scholar]
- 7.Roszer T, Menendez-Gutierrez MP, Cedenilla M, Ricote M. Retinoid X receptors in macrophage biology. Trends Endocrinol Metab 2013, 24: 460–468. [DOI] [PubMed] [Google Scholar]
- 8.Ahuja HS, Szanto A, Nagy L, Davies PJ. The retinoid X receptor and its ligands: versatile regulators of metabolic function, cell differentiation and cell death. J Biol Regul Homeost Agents 2003, 17: 29–45. [PubMed] [Google Scholar]
- 9.Tanaka T, De Luca LM. Therapeutic potential of “rexinoids” in cancer prevention and treatment. Cancer Res 2009, 69: 4945–4947. [DOI] [PubMed] [Google Scholar]
- 10.Kagechika H. Novel synthetic retinoids and separation of the pleiotropic retinoidal activities. Curr Med Chem 2002, 9: 591–608. [DOI] [PubMed] [Google Scholar]
- 11.Nagpal S, Chandraratna RA. Recent developments in receptor-selective retinoids. Curr Pharm Des 2000, 6: 919–931. [DOI] [PubMed] [Google Scholar]
- 12.Perez E, Bourguet W, Gronemeyer H, de Lera AR. Modulation of RXR function through ligand design. Biochim Biophys Acta 2012, 1821: 57–69. [DOI] [PubMed] [Google Scholar]
- 13.Calleja C, Messaddeq N, Chapellier B, Yang H, Krezel W, Li M, Metzger D et al. Genetic and pharmacological evidence that a retinoic acid cannot be the RXR-activating ligand in mouse epidermis keratinocytes. Genes Dev 2006, 20: 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolf G. Is 9-cis-retinoic acid the endogenous ligand for the retinoic acid-X receptor? Nutr Rev 2006, 64: 532–538. [DOI] [PubMed] [Google Scholar]
- 15.Mark M, Ghyselinck NB, Chambon P. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal 2009, 7: e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan YJ, An D, Cai Y, Repa JJ, Hung-Po Chen T, Flores M, Postic C et al. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol Cell Biol 2000, 20: 4436–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Indra AK, Warot X, Brocard J, Messaddeq N, Kato S, Metzger D et al. Skin abnormalities generated by temporally controlled RXRalpha mutations in mouse epidermis. Nature 2000, 407: 633–636. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, Powell WC, Khodavirdi AC, Wu J, Makita T, Cardiff RD, Cohen MB et al. Prostatic intraepithelial neoplasia in mice with conditional disruption of the retinoid X receptor alpha allele in the prostate epithelium. Cancer Res 2002, 62: 4812–4819. [PubMed] [Google Scholar]
- 19.Imai T, Jiang M, Chambon P, Metzger D. Impaired adipogenesis and lipolysis in the mouse upon selective ablation of the retinoid X receptor alpha mediated by a tamoxifen-inducible chimeric Cre recombinase (Cre-ERT2) in adipocytes. Proc Natl Acad Sci USA 2001, 98: 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka T, Suh KS, Lo AM, De Luca LM. p21WAF1/CIP1 is a common transcriptional target of retinoid receptors: pleiotropic regulatory mechanism through retinoic acid receptor (RAR)/retinoid X receptor (RXR) heterodimer and RXR/RXR homodimer. J Biol Chem 2007, 282: 29987–29997. [DOI] [PubMed] [Google Scholar]
- 21.Nunez V, Alameda D, Rico D, Mota R, Gonzalo P, Cedenilla M, Fischer T et al. Retinoid X receptor alpha controls innate inflammatory responses through the up-regulation of chemokine expression. Proc Natl Acad Sci USA 2010, 107: 10626–10631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao X, Liu W, Lin F, Li H, Kolluri SK, Lin B, Han Y-H et al. Retinoid X receptor regulates Nur77/TR3-dependent apoptosis by modulating its nuclear export and mitochondrial targeting. Mol Cell Biol 2004, 24: 9705–9725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casas F, Daury L, Grandemange S, Busson M, Seyer P, Hatier R, Carazo A et al. Endocrine regulation of mitochondrial activity: involvement of truncated RXRalpha and c-Erb Aalpha1 proteins. FASEB J 2003, 17: 426–436. [DOI] [PubMed] [Google Scholar]
- 24.Katagiri Y, Takeda K, Yu ZX, Ferrans VJ, Ozato K, Guroff G. Modulation of retinoid signalling through NGF-induced nuclear export of NGFI-B. Nat Cell Biol 2000, 2: 435–440. [DOI] [PubMed] [Google Scholar]
- 25.Ghose R, Zimmerman TL, Thevananther S, Karpen SJ. Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: a novel mechanism for reduced hepatic gene expression in inflammation. Nucl Recept 2004, 2: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmerman TL, Thevananther S, Ghose R, Burns AR, Karpen SJ. Nuclear export of retinoid X receptor alpha in response to interleukin-1beta-mediated cell signaling: roles for JNK and SER260. J Biol Chem 2006, 281: 15434–15440. [DOI] [PubMed] [Google Scholar]
- 27.Fukunaka K, Saito T, Wataba K, Ashihara K, Ito E, Kudo R. Changes in expression and subcellular localization of nuclear retinoic acid receptors in human endometrial epithelium during the menstrual cycle. Mol Hum Reprod 2001, 7: 437–446. [DOI] [PubMed] [Google Scholar]
- 28.Wang GH, Jiang FQ, Duan YH, Zeng ZP, Chen F, Dai Y, Chen JB et al. Targeting truncated retinoid X receptor-alpha by CF31 induces TNF-alpha-dependent apoptosis. Cancer Res 2013, 73: 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou H, Liu W, Su Y, Wei Z, Liu J, Kolluri SK, Wu H et al. NSAID sulindac and its analog bind RXRalpha and inhibit RXRalpha-dependent AKT signaling. Cancer Cell 2010, 17: 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mey J, Schrage K, Wessels I, Vollpracht-Crijns I. Effects of inflammatory cytokines IL-1beta, IL-6, and TNFalpha on the intracellular localization of retinoid receptors in Schwann cells. Glia 2007, 55: 152–164. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Dancheck BL, Trifiletti LC, Birnkrant RE, Taylor BJ, Garfield SH, Thorgeirsson U et al. Altered localization of retinoid X receptor alpha coincides with loss of retinoid responsiveness in human breast cancer MDA-MB-231 cells. Mol Cell Biol 2004, 24: 3972–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C. Hyperphosphorylation of the retinoid X receptor alpha by activated c-Jun NH2-terminal kinases. J Biol Chem 1999, 274: 18932–18941. [DOI] [PubMed] [Google Scholar]
- 33.Bruck N, Bastien J, Bour G, Tarrade A, Plassat JL, Bauer A, Adam-Stitah S et al. Phosphorylation of the retinoid x receptor at the omega loop, modulates the expression of retinoic-acid-target genes with a promoter context specificity. Cell Signal 2005, 17: 1229–1239. [DOI] [PubMed] [Google Scholar]
- 34.Lee HY, Suh YA, Robinson MJ, Clifford JL, Hong WK, Woodgett JR, Cobb MH et al. Stress pathway activation induces phosphorylation of retinoid X receptor. J Biol Chem 2000, 275: 32193–32199. [DOI] [PubMed] [Google Scholar]
- 35.Mann KK, Padovani AM, Guo Q, Colosimo AL, Lee HY, Kurie JM, Miller WH Jr. Arsenic trioxide inhibits nuclear receptor function via SEK1/JNK-mediated RXRalpha phosphorylation. J Clin Invest 2005, 115: 2924–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solomon C, White JH, Kremer R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J Clin Invest 1999, 103: 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshimura K, Muto Y, Shimizu M, Matsushima-Nishiwaki R, Okuno M, Takano Y, Tsurumi H et al. Phosphorylated retinoid X receptor alpha loses its heterodimeric activity with retinoic acid receptor beta. Cancer Sci 2007, 98: 1868–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsushima-Nishiwaki R, Okuno M, Adachi S, Sano T, Akita K, Moriwaki H, Friedman SL et al. Phosphorylation of retinoid X receptor alpha at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res 2001, 61: 7675–7682. [PubMed] [Google Scholar]
- 39.Adachi S, Okuno M, Matsushima-Nishiwaki R, Takano Y, Kojima S, Friedman SL, Moriwaki H et al. Phosphorylation of retinoid X receptor suppresses its ubiquitination in human hepatocellular carcinoma. Hepatology 2002, 35: 332–340. [DOI] [PubMed] [Google Scholar]
- 40.Choi SJ, Chung SS, Rho EJ, Lee HW, Lee MH, Choi HS, Seol JH et al. Negative modulation of RXRalpha transcriptional activity by small ubiquitin-related modifier (SUMO) modification and its reversal by SUMO-specific protease SUSP1. J Biol Chem 2006, 281: 30669–30677. [DOI] [PubMed] [Google Scholar]
- 41.Schneider Aguirre R, Karpen SJ. Inflammatory mediators increase SUMOylation of retinoid X receptor alpha in a c-Jun N-terminal kinase-dependent manner in human hepatocellular carcinoma cells. Mol Pharmacol 2013, 84: 218–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao WX, Tian M, Zhao BX, Li GD, Liu B, Zhan YY, Chen HZ et al. Orphan receptor TR3 attenuates the p300-induced acetylation of retinoid X receptor-alpha. Mol Endocrinol 2007, 21: 2877–2889. [DOI] [PubMed] [Google Scholar]
- 43.Ocadiz-Delgado R, Castaneda-Saucedo E, Indra AK, Hernandez-Pando R, Gariglio P. Impaired cervical homeostasis upon selective ablation of RXRalpha in epithelial cells. Genesis 2008, 46: 19–28. [DOI] [PubMed] [Google Scholar]
- 44.Mao GE, Reuter VE, Cordon-Cardo C, Dalbagni G, Scher HI, DeKernion JB, Zhang ZF et al. Decreased retinoid X receptor-alpha protein expression in basal cells occurs in the early stage of human prostate cancer development. Cancer Epidemiol Biomarkers Prev 2004, 13: 383–390. [PubMed] [Google Scholar]
- 45.Takiyama Y, Miyokawa N, Sugawara A, Kato S, Ito K, Sato K, Oikawa K et al. Decreased expression of retinoid X receptor isoforms in human thyroid carcinomas. J Clin Endocrinol Metab 2004, 89: 5851–5861. [DOI] [PubMed] [Google Scholar]
- 46.Ando N, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Tsurumi H, Tanaka T, Moriwaki H. Expression of retinoid X receptor alpha is decreased in 3′-methyl-4-dimethylaminoazobenzene-induced hepatocellular carcinoma in rats. Oncol Rep 2007, 18: 879–884. [PubMed] [Google Scholar]
- 47.Ariga N, Moriya T, Suzuki T, Kimura M, Ohuchi N, Sasano H. Retinoic acid receptor and retinoid X receptor in ductal carcinoma in situ and intraductal proliferative lesions of the human breast. Jpn J Cancer Res 2000, 91: 1169–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang SY, Shen SR, Shyu RY, Yu JC, Harn HJ, Yeh MY, Lee MM et al. Expression of nuclear retinoid receptors in normal, premalignant and malignant gastric tissues determined by in situ hybridization. Br J Cancer 1999, 80: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lotan Y, Xu XC, Shalev M, Lotan R, Williams R, Wheeler TM, Thompson TC et al. Differential expression of nuclear retinoid receptors in normal and malignant prostates. J Clin Oncol 2000, 18: 116–121. [DOI] [PubMed] [Google Scholar]
- 50.Picard E, Seguin C, Monhoven N, Rochette-Egly C, Siat J, Borrelly J, Martinet Y et al. Expression of retinoid receptor genes and proteins in non-small-cell lung cancer. J Natl Cancer Inst 1999, 91: 1059–1066. [DOI] [PubMed] [Google Scholar]
- 51.Zhong C, Yang S, Huang J, Cohen MB, Roy-Burman P. Aberration in the expression of the retinoid receptor, RXRalpha, in prostate cancer. Cancer Biol Ther 2003, 2: 179–184. [DOI] [PubMed] [Google Scholar]
- 52.Yamazaki K, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Kanemura N, Araki H, Tsurumi H et al. Synergistic effects of RXR{alpha} and PPAR{gamma} ligands to inhibit growth in human colon cancer cells—phosphorylated RXR{alpha} is a critical target for colon cancer management 1. Gut 2007, 56: 1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeisig BB, Kwok C, Zelent A, Shankaranarayanan P, Gronemeyer H, Dong S, So CW. Recruitment of RXR by homotetrameric RARalpha fusion proteins is essential for transformation. Cancer Cell 2007, 12: 36–51. [DOI] [PubMed] [Google Scholar]
- 54.Zhu J, Nasr R, Peres L, Riaucoux-Lormiere F, Honore N, Berthier C, Kamashev D et al. RXR is an essential component of the oncogenic PML/RARA complex in vivo. Cancer Cell 2007, 12: 23–35. [DOI] [PubMed] [Google Scholar]
- 55.Martens JH, Brinkman AB, Simmer F, Francoijs KJ, Nebbioso A, Ferrara F, Altucci L et al. PML-RARalpha/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 2010, 17: 173–185. [DOI] [PubMed] [Google Scholar]
- 56.Matsushima-Nishiwaki R, Shidoji Y, Nishiwaki S, Moriwaki H, Muto Y. Limited degradation of retinoid X receptor by calpain. Biochem Biophys Res Commun 1996, 225: 946–951. [DOI] [PubMed] [Google Scholar]
- 57.Matsushima-Nishiwaki R, Shidoji Y, Nishiwaki S, Yamada T, Moriwaki H, Muto Y. Aberrant metabolism of retinoid X receptor proteins in human hepatocellular carcinoma. Mol Cell Endocrinol 1996, 121: 179–190. [DOI] [PubMed] [Google Scholar]
- 58.Nagaya T, Murata Y, Yamaguchi S, Nomura Y, Ohmori S, Fujieda M, Katunuma N et al. Intracellular proteolytic cleavage of 9-cis-retinoic acid receptor alpha by cathepsin L-type protease is a potential mechanism for modulating thyroid hormone action. J Biol Chem 1998, 273: 33166–33173. [DOI] [PubMed] [Google Scholar]
- 59.Prufer K, Schroder C, Hegyi K, Barsony J. Degradation of RXRs influences sensitivity of rat osteosarcoma cells to the antiproliferative effects of calcitriol. Mol Endocrinol 2002, 16: 961–976. [DOI] [PubMed] [Google Scholar]
- 60.Nomura Y, Nagaya T, Yamaguchi S, Katunuma N, Seo H. Cleavage of RXRalpha by a lysosomal enzyme, cathepsin L-type protease. Biochem Biophys Res Commun 1999, 254: 388–394. [DOI] [PubMed] [Google Scholar]
- 61.Chen F, Liu J, Huang M, Hu M, Su Y, Zhang XK. Identification of a new RXRalpha antagonist targeting the coregulator-binding site. ACS Med Chem Lett 2014, 5: 736–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen L, Wang ZG, Aleshin AE, Chen F, Chen J, Jiang F, Alitongbieke G et al. Sulindac-derived RXRalpha modulators inhibit cancer cell growth by binding to a novel site. Chem Biol 2014, 21: 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gao W, Liu J, Hu M, Huang M, Cai S, Zeng Z, Lin B et al. Regulation of proteolytic cleavage of retinoid X receptor-alpha by GSK-3beta. Carcinogenesis 2013, 34: 1208–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang ZG, Chen L, Chen J, Zheng JF, Gao W, Zeng Z, Zhou H et al. Synthesis and SAR study of modulators inhibiting tRXRalpha-dependent AKT activation. Eur J Med Chem 2013, 62: 632–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zeng Z, Sun Z, Huang M, Zhang W, Liu J, Chen L, Chen F et al. Nitrostyrene derivatives act as RXRalpha ligands to inhibit TNFalpha activation of NF-kappaB. Cancer Res 2015, 75: 2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schadt CR. Topical and oral bexarotene. Dermatol Ther 2013, 26: 400–403. [DOI] [PubMed] [Google Scholar]
- 67.Jong L, Lehmann JM, Hobbs PD, Harlev E, Huffman JC, Pfahl M, Dawson MI. Conformational effects on retinoid receptor selectivity. 1. Effect of 9-double bond geometry on retinoid X receptor activity. J Med Chem 1993, 36: 2605–2613. [DOI] [PubMed] [Google Scholar]
- 68.Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Berger E, Suto CM et al. Synthesis and structure-activity relationships of novel retinoid X receptor-selective retinoids. J Med Chem 1994, 37: 2930–2941. [DOI] [PubMed] [Google Scholar]
- 69.Smith BD, Wilson LD. Management of mycosis fungoides: Part 2. Treatment. Oncology (Williston Park, NY) 2003, 17: 1419–1428; discussion 1430, 1433. [PubMed] [Google Scholar]
- 70.Moore DD, Kato S, Xie W, Mangelsdorf DJ, Schmidt DR, Xiao R, Kliewer SA. International Union of Pharmacology. LXII. The NR1H and NR1I receptors: constitutive androstane receptor, pregnene X receptor, farnesoid X receptor alpha, farnesoid X receptor beta, liver X receptor alpha, liver X receptor beta, and vitamin D receptor. Pharmacol Rev 2006, 58: 742–759. [DOI] [PubMed] [Google Scholar]
- 71.Sherman SI. Etiology, diagnosis, and treatment recommendations for central hypothyroidism associated with bexarotene therapy for cutaneous T-cell lymphoma. Clin Lymphoma 2003, 3: 249–252. [DOI] [PubMed] [Google Scholar]
- 72.Perez Santin E, Germain P, Quillard F, Khanwalkar H, Rodriguez-Barrios F, Gronemeyer H, de Lera AR et al. Modulating retinoid X receptor with a series of (E)-3-[4-hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-y l)phenyl]acrylic acids and their 4-alkoxy isomers. J Med Chem 2009, 52: 3150–3158. [DOI] [PubMed] [Google Scholar]
- 73.Qu L, Tang X. Bexarotene: a promising anticancer agent. Cancer Chemo Pharmacol 2010, 65: 201–205. [DOI] [PubMed] [Google Scholar]
- 74.Sokolowska-Wojdylo M, Lugowska-Umer H, Maciejewska-Radomska A. Oral retinoids and rexinoids in cutaneous T-cell lymphomas. Postepy Dermatol Alergol 2013, 30: 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen Y, Dokmanovic M, Stein WD, Ardecky RJ, Roninson IB. Agonist and antagonist of retinoic acid receptors cause similar changes in gene expression and induce senescence-like growth arrest in MCF-7 breast carcinoma cells. Cancer Res 2006, 66: 8749–8761. [DOI] [PubMed] [Google Scholar]
- 76.Shilkaitis A, Bratescu L, Green A, Yamada T, Christov K. Bexarotene induces cellular senescence in MMTV-Neu mouse model of mammary carcinogenesis. Cancer Prev Res 2013, 6: 299–308. [DOI] [PubMed] [Google Scholar]
- 77.Hermann TW, Yen WC, Tooker P, Fan B, Roegner K, Negro-Vilar A, Lamph WW et al. The retinoid X receptor agonist bexarotene (Targretin) synergistically enhances the growth inhibitory activity of cytotoxic drugs in non-small cell lung cancer cells. Lung Cancer (Amsterdam, Netherlands) 2005, 50: 9–18. [DOI] [PubMed] [Google Scholar]
- 78.Brabender J, Danenberg KD, Metzger R, Schneider PM, Lord RV, Groshen S, Tsao-Wei DD et al. The role of retinoid X receptor messenger RNA expression in curatively resected non-small cell lung cancer. Clin Cancer Res 2002, 8: 438–443. [PubMed] [Google Scholar]
- 79.Yen WC, Corpuz MR, Prudente RY, Cooke TA, Bissonnette RP, Negro-Vilar A, Lamph WW. A selective retinoid X receptor agonist bexarotene (Targretin) prevents and overcomes acquired paclitaxel (Taxol) resistance in human non-small cell lung cancer. Clin Cancer Res 2004, 10: 8656–8664. [DOI] [PubMed] [Google Scholar]
- 80.Ramlau R, Zatloukal P, Jassem J, Schwarzenberger P, Orlov SV, Gottfried M, Pereira JR et al. Randomized phase III trial comparing bexarotene (L1069–49)/cisplatin/vinorelbine with cisplatin/vinorelbine in chemotherapy-naive patients with advanced or metastatic non-small-cell lung cancer: SPIRIT I. J Clin Oncol 2008, 26: 1886–1892. [DOI] [PubMed] [Google Scholar]
- 81.Blumenschein GR Jr., Khuri FR, von Pawel J, Gatzemeier U, Miller WH Jr., Jotte RM, Le Treut J et al. Phase III trial comparing carboplatin, paclitaxel, and bexarotene with carboplatin and paclitaxel in chemotherapy-naive patients with advanced or metastatic non-small-cell lung cancer: SPIRIT II. J Clin Oncol 2008, 26: 1879–1885. [DOI] [PubMed] [Google Scholar]
- 82.Altucci L, Rossin A, Hirsch O, Nebbioso A, Vitoux D, Wilhelm E, Guidez F et al. Rexinoid-triggered differentiation and tumor-selective apoptosis of acute myeloid leukemia by protein kinase A-mediated desubordination of retinoid X receptor. Cancer Res 2005, 65: 8754–8765. [DOI] [PubMed] [Google Scholar]
- 83.Kizaki M, Dawson MI, Heyman R, Elster E, Morosetti R, Pakkala S, Chen DL et al. Effects of novel retinoid X receptor-selective ligands on myeloid leukemia differentiation and proliferation in vitro. Blood 1996, 87: 1977–1984. [PubMed] [Google Scholar]
- 84.Welch JS, Niu H, Uy GL, Westervelt P, Abboud CN, Vij R, Stockerl-Goldstein KE et al. A phase I dose escalation study of oral bexarotene in combination with intravenous decitabine in patients with AML. Am J Hematol 2014, 89: E103–E108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanchez PV, Glantz ST, Scotland S, Kasner MT, Carroll M. Induced differentiation of acute myeloid leukemia cells by activation of retinoid X and liver X receptors. Leukemia 2014, 28: 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thomas M, Sukhai MA, Kamel-Reid S. An emerging role for retinoid X receptor alpha in malignant hematopoiesis. Leuk Res 2012, 36: 1075–1081. [DOI] [PubMed] [Google Scholar]
- 87.Shimizu M, Shirakami Y, Imai K, Takai K, Moriwaki H. Acyclic retinoid in chemoprevention of hepatocellular carcinoma: targeting phosphorylated retinoid X receptor-alpha for prevention of liver carcinogenesis. J Carcinog 2012, 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Srivastava J, Robertson CL, Rajasekaran D, Gredler R, Siddiq A, Emdad L, Mukhopadhyay ND et al. AEG-1 regulates retinoid X receptor and inhibits retinoid signaling. Cancer Res 2014, 74: 4364–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mascrez B, Mark M, Krezel W, Dupe V, LeMeur M, Ghyselinck NB, Chambon P. Differential contributions of AF-1 and AF-2 activities to the developmental functions of RXR alpha. Development 2001, 128: 2049–2062. [DOI] [PubMed] [Google Scholar]
- 90.Macoritto M, Nguyen-Yamamoto L, Huang DC, Samuel S, Yang XF, Wang TT, White JH et al. Phosphorylation of the human retinoid X receptor alpha at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J Biol Chem 2008, 283: 4943–4956. [DOI] [PubMed] [Google Scholar]
- 91.Bastien J, Adam-Stitah S, Plassat JL, Chambon P, Rochette-Egly C. The phosphorylation site located in the A region of retinoic X receptor alpha is required for the antiproliferative effect of retinoic acid (RA) and the activation of RA target genes in F9 cells. J Biol Chem 2002, 277: 28683–28689. [DOI] [PubMed] [Google Scholar]
- 92.Rochette-Egly C. Nuclear receptors: integration of multiple signalling pathways through phosphorylation. Cell Signal 2003, 15: 355–366. [DOI] [PubMed] [Google Scholar]
- 93.Tarrade A, Bastien J, Bruck N, Bauer A, Gianni M, Rochette-Egly C. Retinoic acid and arsenic trioxide cooperate for apoptosis through phosphorylated RXR alpha. Oncogene 2005, 24: 2277–2288. [DOI] [PubMed] [Google Scholar]
- 94.Chapman HA. Cathepsins as transcriptional activators? Dev Cell 2004, 6: 610–611. [DOI] [PubMed] [Google Scholar]
- 95.Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278: 1966–1968. [DOI] [PubMed] [Google Scholar]
- 96.Fortini ME. Notch and presenilin: a proteolytic mechanism emerges. Curr Opin Cell Biol 2001, 13: 627–634. [DOI] [PubMed] [Google Scholar]
- 97.Golde TE, Eckman CB. Physiologic and pathologic events mediated by intramembranous and juxtamembranous proteolysis. Sci STKE 2003, 2003: RE4. [DOI] [PubMed] [Google Scholar]
- 98.Hendry L, John S. Regulation of STAT signalling by proteolytic processing. Eur J Biochem 2004, 271: 4613–4620. [DOI] [PubMed] [Google Scholar]
- 99.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94: 491–501. [DOI] [PubMed] [Google Scholar]
- 100.Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature 1999, 398: 525–529. [DOI] [PubMed] [Google Scholar]
- 101.Nervi C, Ferrara FF, Fanelli M, Rippo MR, Tomassini B, Ferrucci PF, Ruthardt M et al. Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein. Blood 1998, 92: 2244–2251. [PubMed] [Google Scholar]
- 102.Jing Y, Xia L, Lu M, Waxman S. The cleavage product deltaPML-RARalpha contributes to all-trans retinoic acid-mediated differentiation in acute promyelocytic leukemia cells. Oncogene 2003, 22: 4083–4091. [DOI] [PubMed] [Google Scholar]
- 103.Libertini SJ, Tepper CG, Rodriguez V, Asmuth DM, Kung HJ, Mudryj M. Evidence for calpain-mediated androgen receptor cleavage as a mechanism for androgen independence. Cancer Res 2007, 67: 9001–9005. [DOI] [PubMed] [Google Scholar]
- 104.Xie Y, Lu W, Liu S, Yang Q, Carver BS, Li E, Wang Y et al. Crosstalk between nuclear MET and SOX9/beta-catenin correlates with castration-resistant prostate cancer. Mol Endocrinol 2014, 28: 1629–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cantley LC. The phosphoinositide 3-kinase pathway. Science 2002, 296: 1655–1657. [DOI] [PubMed] [Google Scholar]
- 106.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta 2008, 1784: 159–185. [DOI] [PubMed] [Google Scholar]
- 107.Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27: 5486–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007, 129: 1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leis H, Page A, Ramirez A, Bravo A, Segrelles C, Paramio J, Barettino D et al. Glucocorticoid receptor counteracts tumorigenic activity of Akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Mol Endocrinol 2004, 18: 303–311. [DOI] [PubMed] [Google Scholar]
- 110.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407: 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun M, Yang L, Feldman RI, Sun XM, Bhalla KN, Jove R, Nicosia SV et al. Activation of phosphatidylinositol 3-kinase/Akt pathway by androgen through interaction of p85alpha, androgen receptor, and Src. J Biol Chem 2003, 278: 42992–43000. [DOI] [PubMed] [Google Scholar]
- 112.Yan TD, Wu H, Zhang HP, Lu N, Ye P, Yu FH, Zhou H et al. Oncogenic potential of retinoic acid receptor-gamma in hepatocellular carcinoma. Cancer Res 2010, 70: 2285–2295. [DOI] [PubMed] [Google Scholar]
- 113.Reed JC. Cytochrome c: can't live with it–can't live without it [comment]. Cell 1997, 91: 559–562. [DOI] [PubMed] [Google Scholar]
- 114.Green DR, Reed JC. Mitochondria and apoptosis. Science 1998, 281: 1309–1312. [DOI] [PubMed] [Google Scholar]
- 115.Reed JC. Apoptosis-based therapies. Nat Rev Drug Discov 2002, 1: 111–121. [DOI] [PubMed] [Google Scholar]
- 116.Moll UM, Marchenko N, Zhang XK. p53 and Nur77/TR3 - transcription factors that directly target mitochondria for cell death induction. Oncogene 2006, 25: 4725–4743. [DOI] [PubMed] [Google Scholar]
- 117.Chang C, Kokontis J. Identification of a new member of the steroid receptor super-family by cloning and sequence analysis. Biochem Biophys Res Commun 1988, 155: 971–977. [DOI] [PubMed] [Google Scholar]
- 118.Hazel TG, Nathans D, Lau LF. A gene inducible by serum growth factors encodes a member of the steroid and thyroid hormone receptor superfamily. Proc Natl Acad Sci USA 1988, 85: 8444–8448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Milbrandt J. Nerve growth factor induces a gene homologous to the glucocorticoid receptor gene. Neuron 1988, 1: 183–188. [DOI] [PubMed] [Google Scholar]
- 120.Dawson MI, Hobbs PD, Peterson VJ, Leid M, Lange CW, Feng KC, Chen G et al. Apoptosis induction in cancer cells by a novel analogue of 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphthalenecarboxylic acid lacking retinoid receptor transcriptional activation activity. Cancer Res 2001, 61: 4723–4730. [PubMed] [Google Scholar]
- 121.Holmes WF, Soprano DR, Soprano KJ. Elucidation of molecular events mediating induction of apoptosis by synthetic retinoids using a CD437-resistant ovarian carcinoma cell line. J Biol Chem 2002, 277: 45408–45419. [DOI] [PubMed] [Google Scholar]
- 122.Holmes WF, Soprano DR, Soprano KJ. Comparison of the mechanism of induction of apoptosis in ovarian carcinoma cells by the conformationally restricted synthetic retinoids CD437 and 4-HPR. J Cell Biochem 2003, 89: 262–278. [DOI] [PubMed] [Google Scholar]
- 123.Jeong JH, Park JS, Moon B, Kim MC, Kim JK, Lee S, Suh H et al. Orphan nuclear receptor Nur77 translocates to mitochondria in the early phase of apoptosis induced by synthetic chenodeoxycholic acid derivatives in human stomach cancer cell line SNU-1. Ann NY Acad Sci 2003, 1010: 171–177. [DOI] [PubMed] [Google Scholar]
- 124.Kolluri SK, Bruey-Sedano N, Cao X, Lin B, Lin F, Han Y-H, Dawson MI et al. Mitogenic effect of orphan receptor TR3 and its regulation by MEKK1 in lung cancer cells. Mol Cell Biol 2003, 23: 8651–8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu S. Induction of apoptosis by TPA and VP-16 is through translocation of TR3. World J Gastroenterol 2002, 8: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wilson AJ, Arango D, Mariadason JM, Heerdt BG, Augenlicht LH. TR3/Nur77 in colon cancer cell apoptosis. Cancer Res 2003, 63: 5401–5407. [PubMed] [Google Scholar]
- 127.Wu Q, Liu S, Ye XF, Huang ZW, Su WJ. Dual roles of Nur77 in selective regulation of apoptosis and cell cycle by TPA and ATRA in gastric cancer cells. Carcinogenesis 2002, 23: 1583–1592. [DOI] [PubMed] [Google Scholar]
- 128.Maddika S, Booy EP, Johar D, Gibson SB, Ghavami S, Los M. Cancer-specific toxicity of apoptin is independent of death receptors but involves the loss of mitochondrial membrane potential and the release of mitochondrial cell-death mediators by a Nur77-dependent pathway. J Cell Sci 2005, 118(Pt 19): 4485–4493. [DOI] [PubMed] [Google Scholar]
- 129.Lee KW, Ma L, Yan X, Liu B, Zhang XK, Cohen P. Rapid apoptosis induction by IGFBP-3 involves an insulin-like growth factor-independent nucleomitochondrial translocation of RXRalpha/Nur77. J Biol Chem 2005, 280: 16942–16948. [DOI] [PubMed] [Google Scholar]
- 130.Liu S, Wu Q, Ye XF, Cai JH, Huang ZW, Su WJ. Induction of apoptosis by TPA and VP-16 is through translocation of TR3. World J Gastroenterol 2002, 8: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, Lin B et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3 [see comments] [comment]. Science 2000, 289: 1159–1164. [DOI] [PubMed] [Google Scholar]
- 132.Lin B, Kolluri S, Lin F, Liu W, Han YH, Cao X, Dawson MI et al. Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 2004, 116: 527–540. [DOI] [PubMed] [Google Scholar]
- 133.Kolluri SK, Zhu X, Zhou X, Lin B, Chen Y, Sun K, Tian X et al. A short Nur77-derived peptide converts Bcl-2 from a protector to a killer. Cancer Cell 2008, 14: 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lin XF, Zhao BX, Chen HZ, Ye XF, Yang CY, Zhou HY, Zhang MQ et al. RXRalpha acts as a carrier for TR3 nuclear export in a 9-cis retinoic acid-dependent manner in gastric cancer cells. J Cell Sci 2004, 117(Pt 23): 5609–5621. [DOI] [PubMed] [Google Scholar]
- 135.Lee KW, Ma L, Yan X, Liu B, Zhang XK, Cohen P. Rapid apoptosis induction by IGFBP-3 involves an insulin-like growth factor-independent nucleomitochondrial translocation of RXRalpha/Nur77. J Biol Chem 2005, 280: 16942–16948. [DOI] [PubMed] [Google Scholar]
- 136.Balkwill F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev 2006, 25: 409–416. [DOI] [PubMed] [Google Scholar]
- 137.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009, 9: 361–371. [DOI] [PubMed] [Google Scholar]
- 138.Mocellin S, Nitti D. TNF and cancer: the two sides of the coin. Front Biosci 2008, 13: 2774–2783. [DOI] [PubMed] [Google Scholar]
- 139.van Horssen R, Ten Hagen TL, Eggermont AM. TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11: 397–408. [DOI] [PubMed] [Google Scholar]
- 140.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin 2008, 29: 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bertazza L, Mocellin S. Tumor necrosis factor (TNF) biology and cell death. Front Biosci 2008, 13: 2736–2743. [DOI] [PubMed] [Google Scholar]
- 142.Pincheira R, Castro AF, Ozes ON, Idumalla PS, Donner DB. Type 1 TNF receptor forms a complex with and uses Jak2 and c-Src to selectively engage signaling pathways that regulate transcription factor activity. J Immunol 2008, 181: 1288–1298. [DOI] [PubMed] [Google Scholar]
- 143.Rivas MA, Carnevale RP, Proietti CJ, Rosemblit C, Beguelin W, Salatino M, Charreau EH et al. TNF alpha acting on TNFR1 promotes breast cancer growth via p42/P44 MAPK, JNK, Akt and NF-kappa B-dependent pathways. Exp Cell Res 2008, 314: 509–529. [DOI] [PubMed] [Google Scholar]
- 144.Rivas MA, Tkach M, Beguelin W, Proietti CJ, Rosemblit C, Charreau EH, Elizalde PV et al. Transactivation of ErbB-2 induced by tumor necrosis factor alpha promotes NF-kappaB activation and breast cancer cell proliferation. Breast Cancer Res Treat 2010, 122: 111–124. [DOI] [PubMed] [Google Scholar]
- 145.Rubio MF, Werbajh S, Cafferata EG, Quaglino A, Colo GP, Nojek IM, Kordon EC et al. TNF-alpha enhances estrogen-induced cell proliferation of estrogen-dependent breast tumor cells through a complex containing nuclear factor-kappa B. Oncogene 2006, 25: 1367–1377. [DOI] [PubMed] [Google Scholar]
- 146.Zhou BP, Hu MC, Miller SA, Yu Z, Xia W, Lin SY, Hung MC. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J Biol Chem 2000, 275: 8027–8031. [DOI] [PubMed] [Google Scholar]
- 147.Garcia-Tunon I, Ricote M, Ruiz A, Fraile B, Paniagua R, Royuela M. Role of tumor necrosis factor-alpha and its receptors in human benign breast lesions and tumors (in situ and infiltrative). Cancer Sci 2006, 97: 1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Varela LM, Stangle-Castor NC, Shoemaker SF, Shea-Eaton WK, Ip MM. TNFalpha induces NFkappaB/p50 in association with the growth and morphogenesis of normal and transformed rat mammary epithelial cells. J Cell Physiol 2001, 188: 120–131. [DOI] [PubMed] [Google Scholar]
- 149.Lee CW, Lin CC, Lin WN, Liang KC, Luo SF, Wu CB, Wang SW et al. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3 K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007, 292: L799–L812. [DOI] [PubMed] [Google Scholar]
- 150.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401: 82–85. [DOI] [PubMed] [Google Scholar]
- 151.Yamaoka T, Yan F, Cao H, Hobbs SS, Dise RS, Tong W, Polk DB. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci USA 2008, 105: 11772–11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yoon K, Jung EJ, Lee SY. TRAF6-mediated regulation of the PI3 kinase (PI3K)-Akt-GSK3beta cascade is required for TNF-induced cell survival. Biochem Biophys Res Commun 2008, 371: 118–121. [DOI] [PubMed] [Google Scholar]
- 153.Shishodia S, Aggarwal BB. Nuclear factor-kappaB: a friend or a foe in cancer? Biochem Pharmacol 2004, 68: 1071–1080. [DOI] [PubMed] [Google Scholar]
- 154.Tan TT, Coussens LM. Humoral immunity, inflammation and cancer. Curr Opin Immunol 2007, 19: 209–216. [DOI] [PubMed] [Google Scholar]
- 155.Brower V. Feeding the flame: new research adds to role of inflammation in cancer development. J Natl Cancer Inst 2005, 97: 251–253. [DOI] [PubMed] [Google Scholar]
- 156.Narayanan NK, Narayanan BA, Reddy BS. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-kappaB, and steroid hormone receptors. Int J Oncol 2005, 26: 785–792. [PubMed] [Google Scholar]
- 157.Weldon SM, Mullen AC, Loscher CE, Hurley LA, Roche HM. Docosahexaenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J Nutr Biochem 2007, 18: 250–258. [DOI] [PubMed] [Google Scholar]
- 158.Kolluri SK, Corr M, James SY, Bernasconi M, Lu D, Liu W, Cottam HB et al. The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor-selective apoptosis. Proc Natl Acad Sci USA 2005, 102: 2525–2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Feng R, Anderson G, Xiao G, Elliott G, Leoni L, Mapara MY, Roodman GD et al. SDX-308, a nonsteroidal anti-inflammatory agent, inhibits NF-kappaB activity, resulting in strong inhibition of osteoclast formation/activity and multiple myeloma cell growth. Blood 2007, 109: 2130–2138. [DOI] [PubMed] [Google Scholar]
- 160.Kong G, Kim HT, Wu K, DeNardo D, Hilsenbeck SG, Xu XC, Lamph WW et al. The retinoid X receptor-selective retinoid, LGD1069, down-regulates cyclooxygenase-2 expression in human breast cells through transcription factor crosstalk: implications for molecular-based chemoprevention. Cancer Res 2005, 65: 3462–3469. [DOI] [PubMed] [Google Scholar]
- 161.Yen WC, Prudente RY, Corpuz MR, Negro-Vilar A, Lamph WW. A selective retinoid X receptor agonist bexarotene (LGD1069, targretin) inhibits angiogenesis and metastasis in solid tumours. Br J Cancer 2006, 94: 654–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Smit JV, de Jong EM, van Hooijdonk CA, Otero ME, Boezeman JB, van de Kerkhof PC. Systemic treatment of psoriatic patients with bexarotene decreases epidermal proliferation and parameters for inflammation, and improves differentiation in lesional skin. J Am Acad Dermatol 2004, 51: 257–264. [DOI] [PubMed] [Google Scholar]
- 163.Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab 2006, 17: 321–327. [DOI] [PubMed] [Google Scholar]
- 164.Li MD, Yang X. A retrospective on nuclear receptor regulation of inflammation: lessons from GR and PPARs. PPAR Res 2011, 2011: 742785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gampe RT Jr., Montana VG, Lambert MH, Wisely GB, Milburn MV, Xu HE. Structural basis for autorepression of retinoid X receptor by tetramer formation and the AF-2 helix. Genes Dev 2000, 14: 2229–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang H, Chen L, Chen J, Jiang H, Shen X. Structural basis for retinoic X xreceptor repression on the tetramer. J Biol Chem 2011, 286: 24593–24598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kersten S, Reczek PR, Noy N. The tetramerization region of the retinoid X receptor is important for transcriptional activation by the receptor. J Biol Chem 1997, 272: 29759–29768. [DOI] [PubMed] [Google Scholar]
- 168.Zhang H, Zhou R, Li L, Chen J, Chen L, Li C, Ding H et al. Danthron functions as a retinoic X receptor antagonist by stabilizing tetramers of the receptor. J Biol Chem 2011, 286: 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Buzon V, Carbo LR, Estruch SB, Fletterick RJ, Estebanez-Perpina E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol Cell Endocrinol 2012, 348: 394–402. [DOI] [PubMed] [Google Scholar]
- 170.Moore TW, Mayne CG, Katzenellenbogen JA. Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs). Mol Endocrinol 2010, 24: 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Caboni L, Lloyd DG. Beyond the ligand-binding pocket: targeting alternate sites in nuclear receptors. Med Res Rev 2013, 33: 1081–1118. [DOI] [PubMed] [Google Scholar]