

Abstract
Sepsis is a complex syndrome that develops when the initial, appropriate host response to an infection becomes amplified, and is then dysregulated. Among other factors, the innate immune system is of central importance to the early containment of infection. Death from infection is strongly heritable in human populations. Hence, genetic variations that disrupt innate immune sensing of infectious organisms could explain the ability of the immune system to respond to infection, the diversity of the clinical presentation of sepsis, the response to current medical treatment, and the genetic predisposition to infection in each individual patient. Such genetic variations may identify patients at high risk for the development of sepsis and organ dysfunction during severe infections. Single base variations, known as single nucleotide polymorphisms (SNPs), are the most commonly used variants. There has been great interest in exploring SNP in those genes involved in the inflammatory cascade resulting from the systemic inflammatory response to micro organisms. The rationale for studying gene SNPs in critical illnesses seeks to identify potential markers of susceptibility, severity, and clinical outcome; seeks to identify potential markers for responders and non-responders in clinical trials, and seeks to identify targets for therapeutic intervention. In this review, we focus on the current state of association studies of those genes governing the powerful bacterial infection-induced inflammation and provide guidelines for future studies describing disease associations with genetic variations based on current recommendations. We envision a time in the near future when genotyping will be include in the standard evaluation of critically ill patients and will help to prioritize a therapeutic option.
Keywords: genetic susceptibility, haplotype, infection, inflammation, polymorphism
Introduction
Sepsis describes a complex clinical syndrome as a result of a systemic inflammatory response to live bacteria and/or bacterial products. This response is expressed as a compendium of a variety of different clinical signs and symptoms such as fever, increased blood leukocyte counts, unexplained thrombocytopenia, mental confusion, transient hypotension, and organ stress and dysfunction. The individual response is determined by many factors, including the virulence of the organism, the size of the inoculum, and the patient's coexisting conditions.
Sepsis develops when the initial, appropriate host response to an infection becomes amplified, and is then dysregulated [1]. The most common sites of infection are the lungs, the abdominal cavity, the urinary tract, and primary infections of the bloodstream. If untreated, septic patients may develop acute respiratory or renal failure, multiorgan dysfunction, shock, and death. The exact cause of death in patients with sepsis remains elusive. No autopsy studies have yet revealed why patients with sepsis die. Sepsis is estimated to affect 18 million people worldwide each year and to kill 1400 people each day. According to a recent epidemiological study [2] sepsis affects about 700,000 people annually in the United States alone, with an overall mortality rate of 30% and above 50% in patients with septic shock and/or multiple system organ failure.
Gram-positive organisms, endotoxin-containing Gram-negative organisms, fungi, malarial parasites, and other microbial pathogens can trigger sepsis. Gram-negative bacteria are responsible for most clinical sepsis, although in the past decade the spectrum of invading microorganisms appears to be shifting to Gram-positive bacteria and fungi. From a financial perspective, sepsis represents a major burden to the health care system in most developed countries since septic patients require admission and aggressive treatment in the intensive care units and are generally hospitalized for longer than 3 weeks.
There have been significant advances in the understanding of the fundamental principles governing bacterial–host interactions. However, the clinical management of sepsis is very complicated because of the nonhomogeneous nature of the patient populations and because of the difficulties in precise clinical classification of septic patients [3]. Resistance to bacterial infection is a heritable trait that seems to be controlled by multiple genes. The innate immune system is of central importance to the early containment of infection. Hence, genetic variations or mutations that disrupt innate immune sensing of infectious organisms could explain the ability of the immune system to respond to infection, the diversity of the clinical presentation of sepsis, the response to current medical treatment, and the genetic predisposition to infection in each individual patient.
The identification, by means of association studies, of such variations in those genes involved in the bacteria-induced cellular response might allow the development of a new classification of sepsis and a more accurate determination of patient outcome. Such genetic variations may ultimately be used to identify patients at high risk for the development of sepsis and organ dysfunction during severe infections. We focus in the present review on the current state of association studies of those genes governing the powerful Gram-negative bacterial infection-induced inflammation that results in a deleterious inflammatory and coagulopathic state, terminating in severe sepsis and septic shock. In addition, we provide guidelines for future studies describing disease associations with genetic variations based on current recommendations.
Cell recognition of bacterial endotoxin
Early recognition of bacterial products is critical for our survival. It is not clear how the host distinguishes between signals from pathogens. Before a microbe has direct contact with a mammalian cell, it is able to induce chemical reactions that reveal its presence. It is the cellular response to the microbes that produces the septic syndrome.
The innate immune system has both a recognition function, which detects bacterial products in tissues, and an effector function, which attracts phagocytic leukocytes to the sites of bacterial entry [4]. Bacterial lipopolysaccharide (LPS), or endotoxin, is one of the major pattern recognition molecules that activate the innate immunity in Gram-negative infections, although whether LPS is a major cause of sepsis in humans has not been confirmed in clinical trials. There is no endotoxin in Gram-positive bacteria, but their cell walls contain peptidoglycan and lipoteichoic acid that account for their biological activity [5].
Soluble LPS binds to a lipopolysaccharide-binding protein (LBP), which is essential for the inflammatory response to LPS [6]. LBP is a type I acute phase response protein that is produced by hepatocytes, epithelial cells, and other cells, and its production is regulated by inflammatory cytokines produced at the onset of acute inflammatory responses [4]. It is now well known that activation of the host cells is dependent on the presence of a LPS–LBP complex and the opsonic receptor CD14 (Fig. 1). In CD14-negative cells (fibroblasts, vascular endothelial cells, dendritic cells), soluble CD14 can accept LPS from LPS–LBP complexes. CD14 does not cause direct cellular activation by LPS.
Figure 1.
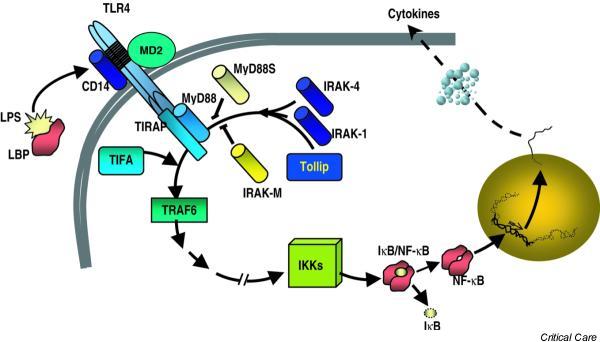
Cell recognition of lipopolysaccharide (LPS). See text for details. LBP, lipopolysaccharide binding protein; TLR4, Toll-like receptor 4; IRAK, IL-1 receptor-associated kinase; Tollip, Toll interacting protein; MD2, myeloid differentiation protein-2; MyD88, myeloid differentiation primary response 88; TIRAP, TIR domain-containing adapter protein (also known as MYD88 adaptor-like protein); TIFA, TRAF-interacting protein with a forkhead-associated domain; TRAF6, TNF receptor-associated factor-6; IKKs, IκB kinases; NF-κB, nuclear factor kappa B; IκB, kappa B inhibitor.
The transfer of LPS–LBP complexes to CD14 results in the activation of a second membrane protein complex, Toll-like receptor (TLR)-4. This complex belongs to an evolutionary conserved family of receptors (TLRs), which seem to be able to combine to form a repertoire capable of distinguishing between closely related ligands [1]. TLR-4 is the LPS receptor while TLR-2 is predominantly responsible for recognizing Gram-positive cell wall structures [1,4,7]. Physical interaction between LPS and TLR-4 is critically important for LPS signal transduction to occur. It has recently been suggested that a cell-surface molecule, MD2, is crucial for the activation of TLR-4, by positioning it correctly on the membrane surface [8].
TLR intracellular signaling is regulated by a group of IL-1 receptor-associated kinases (IRAK) that bind to the TLR intracellular TIR domain, a process that requires the presence of adapter proteins. Five members of TIR adapter proteins have been described to date [9]: myeloid differentiation protein 88 (MyD88), TIRAP/Mal, TRIF/TICAM-1, TRAM, and SARM. Although it seems that MyD88 interacts with all TLRs in their activation, the other adaptors seem to confer certain selectivity to some pathogens. MyD88 short (an alternatively spliced variant of MyD88) and Tollip (Toll interacting protein) have been suggested as suppressors for this activation [10,11]. The binding of IRAK to the downstream adapter tumor necrosis factor (TNF) receptor-associated factor-6, assisted by TIFA, leads to the activation of NF-κB, which involves phosphorylation and degradation of the inhibitors of NF-κB, leading to the translocation of NF-κB heterodimers to the nucleus. The NF-κB system exerts transcriptional regulation on cytokine gene promoters (Fig. 1).
Following the initial host–microbial interactions, there is a widespread activation of the innate immune response involving both humoral and cellular components. LPS interaction with endothelial cells via CD14 and TLRs results in the expression of a variety of adhesion molecules that cause adhesion of neutrophils and monocytes to the vasculature. Under these conditions, leukocytes undergo an additional activation resulting in the production of oxidants, which in turn stimulate endothelial cells to generate inflammatory mediators [12]. In the setting of sepsis, these events result in multiorgan failure, which often leads to death.
Patients with sepsis have features consistent with immunosuppression [13]. The initial immune response is hyperinflammatory with high plasma concentrations of cytokines, but the response rapidly progresses to a hypoinflammatory state with a prolonged depression of the immune system. A wide range of cell types produce the classic proinflammatory cytokines TNF-α, IL-1, and IL-6 and an array of other proinflammatory and anti-inflammatory cytokines. The cytokines act on target cells by binding to specific cytokine receptor ligands, initiating signal transduction and second messenger pathways within the target cell [14]. Although these proinflammatory cytokines are responsible for orchestrating a complex network of secondary cellular responses, the hypothesis that mortality in human bacterial sepsis is caused by an increased production of proinflammatory cytokines seems to be overly simplistic. Death as a result of a cytokine storm, as occurs in animals, does not reflect the clinical picture of human sepsis [15].
Although pharmacologic therapies targeted to specifically block cytokine levels have failed to prevent lethality in experimental and human sepsis [16], anti-inflammatory strategies applied early in patients with a hyperinflammatory immune response may be lifesaving [17].
Why some patients die as a result of an out-of-control septic process whereas other patients recover without problems is still unknown. Some authors have proposed to examine variations in genes that are involved in the signaling cascade during sepsis, in order to establish to which degree variations on those genes controlling the inflammatory and anti-inflammatory responses contribute to the development or fatal outcome of sepsis. Variations in those genes controlling the inflammatory and anti-inflammatory responses could therefore not only be associated with the outcome, but could explain the enormous individual variability during the course of similar infections.
In this vein, during the past decade, some authors have initiated association studies examining variants of those genes involved in innate immunity (TLRs, LBP, CD14, bactericidal/ permeability-increasing protein, angiotensin-converting enzyme, mannose-binding lectin [MBL], heat shock proteins), in acquired immunity (FcγRIIa), in coagulation factors (tissue-type plasminogen activator, plasminogen activator inhibitor-1, factor V Leiden), and cytokines (Table 1). Although most of these studies have not shown positive associations with sepsis, it is important to emphasize that most of them lacked an adequate methodology. Discussion and major limitations of those association studies and present recommendations for further genetic epidemiologic studies will be briefly addressed in the following.
Table 1.
Markers of candidate genes used for association studies in sepsis
| Gene | Marker | Association | Reference |
| BPI | Lys-Glu (216) | - | [28] |
| BPI | Noncoding PstI | - | [28] |
| BPI | Synonymous TaqI (545) | - | [28] |
| HSP70-2 | Coding PstI (position?) | - | [38] |
| HSP70-HOM | Met-Thr (493) | - | [38] |
| LBP | Cys-Gly (98) | + (male) | [28] |
| LBP | Leu-Pro (436) | - | [28] |
| LBP | Synonymous HpaII (291) | - | [29] |
| MBL | Exon 1 (codons 52, 54, 57) | + | [25] |
| CD14 | -260 (-159) promoter | - | [31,32] |
| TLR-4 | Asp-Gly (299) | + | [34,35] |
| TLR-4 | Thr-Ile (399) | - | [35] |
| TLR-4 | Rare coding mutations | + | [36] |
| TLR-2 | Arg-Gln (753) | + | [39] |
| TLR-2 | Rare coding mutations | - | [36] |
| FCγRIIa | His-Arg (131) | + | [40] |
| Angiotensin-converting enzyme | Noncoding indela | + | [41] |
| Factor V Leiden | Arg-Gln (506) | + | [42] |
| Plasminogen activator inhibitor-1 | Promoter single base indela | + | [43] |
| Tissue-type plasminogen activator | Noncoding indela | - | [44] |
| IL-1 receptor antagonist | Noncoding microsatellite | + | [27] |
| IL-1β | Synonymous TaqI (297) | - | [27] |
| IL-6 | -174 promoter | - | [45] |
| IL-10 | -1082 promoter | + | [40] |
| TNF-α | -308 promoter | + | [23] |
| TNF-β (LTA) | Noncoding NcoI | + | [27,46] |
BPI, bactericidal/permeability-increasing protein; FcgRIIa, immunoglobulin G Fc receptor II; HSP, heat shock protein; LBP, lipopolysaccharide-binding protein; LTA, lymphotoxin-alpha; MBL, mannose-binding lectin; TLR, Toll-like receptor; TNF, tumor necrosis factor. a Insertion/deletion.
Genetic variability, susceptibility to sepsis, and outcome
Genetic differences between people may affect the likelihood of developing diseases. Sørensen and colleagues [18] were the first to report that premature death from infection is strongly heritable in human populations, more so than premature death from any cause, including cardiovascular diseases and cancer. Little is known about the genes that contribute to this heritability and hence are responsible for fatal outcome caused by systemic infectious diseases such as sepsis.
Single base variations, known as single nucleotide polymorphisms (SNPs), are the most commonly used variants. On average, two unrelated people differ at about one base in every 1000 of the 3 × 109 or so bases in their genome. Of the more than 10 million SNPs so far mapped and deposited in public and private databases, only 4% are within genes. By comparing SNPs in patients and healthy controls it should be possible to track down those genetic differences. Once this has been achieved, any person can be genotyped for this limited set of SNPs.
There has been great interest in exploring polymorphisms in TNF, IL-1, and IL-6 genes and their association with serum concentration of those cytokines and severity of critical illness [19,20]. TNF-α is one of the key cytokines of the inflammatory cascade and a central mediator of sepsis. The gene coding for TNF-α is located on chromosome 6 in the region known as the major histocompatibility complex, close to other genes coding for cell cycle. A polymorphism in the promoter region of the TNF-α gene has been associated with an increased synthesis of TNF-α and with a higher mortality rate in patients with malaria, meningococcal disease, and leishmaniosis [21].
Two studies in critically ill patients [22,23] have revealed association between polymorphisms in the TNF genes and sepsis. Stüber and colleagues [22] determined the allele frequency and genotype distribution of a bi-allelic polymorphism in the TNF-β gene and TNF-α plasma concentrations in 40 patients with sepsis. They found that the allele frequency distribution was similar to that found in 105 healthy individuals. However, patients homozygous for the allele TNFB2 showed higher circulating TNF-α levels, a higher organ dysfunction, and a higher mortality rate than heterozygous patients.
In the second study, of 89 patients with septic shock and 87 healthy unrelated blood donors, Mira and colleagues [23] studied the frequency of the polymorphism located at nucleotide position -308 inside the TNF-α promoter region (consisting of a single base replacement, guanosine versus adenosine), which results in two allelic forms. These authors found that several other polymorphisms at positions -419, -244, and -49 were always associated with the TNF2 allele. This variant was found in 39% of septic patients and only in 18% of the controls. In addition, the TNF2 allele was significantly more frequent in the nonsurvivors (54% versus 24%), although plasma TNF-α concentrations were not statistically different between both alleles.
Meningococcal sepsis is characterized by exceptionally high levels of LPS in the blood or cerebrospinal fluid [24]. Hibberd and colleagues [25] have studied the association between the variants of the MBL gene with the susceptibility to develop meningococcal disease. Although 10% of the general population are carriers of the pathogenic strains of Neisseria meningitides in their nasopharinx, it is unknown why they cause fatal illness in few of them. In addition to the acquired immune response, there also exists an innate immune response that activates the alternative complement pathway via the MBL pathway. The amount of MBL in plasma is genetically determined and there are three different alleles coding for structurally different proteins. Hibberd and colleagues studied stored blood samples from hospitalized children with meningococcal disease and controls, and determined the frequency of variants of the MBL gene in the two groups. The prevalence of homozygous variants of MBL was sevenfold higher in children with meningococcal disease than in controls. This study showed that children who are homozygous for the MBL variant allele have a higher chance of suffering from meningococcal disease. The data suggested that genetic variants of MBL could be responsible for one-third of all cases of meningococcal disease.
Koch and colleagues [26] examined the relation of genetic variations of MBL with the susceptibility to common respiratory infections in children. They found that children with genetic variants that result in lower levels of the MBL protein have a significantly greater risk for acute respiratory infections.
The family of genes encoding IL-1 has several members. IL-1A and IL-1B encode the proinflammatory mediators IL-1α and IL-1β, respectively, whereas IL-1RN encodes the anti-inflammatory IL-1 receptor antagonist. Fang and colleagues [27] studied 93 patients with severe sepsis and 261 healthy blood donors to determine whether allele frequencies and genotype distributions of SNPs in IL-1 receptor antagonist gene intron 2 and IL-1β gene exon 5 were associated with the susceptibility to, and outcome of, severe sepsis. They found that the frequency of the allele IL-1 receptor antagonist A2 was increased in patients with severe sepsis compared with healthy individuals, but no association with outcome was observed (Table 1).
Bactericidal/permeability-increasing protein and LBP have a high affinity for LPS. Hubacek and colleagues [28] sought to determine whether the genotype frequencies of five SNPs in bactericidal/permeability-increasing protein genes and in LBP genes in 204 patients with sepsis and in 250 healthy control blood donors were associated with the incidence and lethality of sepsis. No differences were found between patients and controls in the polymorphism distributions in either bactericidal/permeability-increasing protein or LBP. However, the presence of LBP genotypes with the less frequent Gly98 allele (SNP at nucleotide 292 of the proximal coding region resulting in an amino acid substitution of cystine to glycine at the 98th residue in the LPB protein) was found to be associated with sepsis in male patients. A recent report by Barber and O'Keefe [29] in 37 patients with sepsis out of a group of 151 trauma patients, however, found that the SNP in the LBP coding region reported to exist at the 292 position and to result in an amino acid substitution actually exists at the adjacent 291 position and does not result in an amino acid substitution. Furthermore, that SNP did not appear to be associated with severe sepsis, with septic shock, or with death.
Recognition of LPS by host cells is mediated by either a membrane-bound form or a soluble form of CD14, a myeloid cell differentiation antigen expressed primarily on monocytes, macrophages, and neutrophils [30]. A common polymorphism within the promoter region of CD14 has not been associated with sepsis development or mortality [31,32]. Heesen and colleagues [32] studied the genotype distribution of the -260 (cytosine versus thymidine) SNP of the CD14 gene and its relation with the development of sepsis in 58 trauma patients. The genotype distribution in trauma patients was similar to that of healthy blood donors, and did not differ in the 14 patients with severe sepsis compared with those with an uncomplicated post-traumatic course.
TLR-4 is the major receptor for LPS in mammals. Structural variations of the gene coding for TLR-4 would be expected to impair host responses to microbial pathogens. Arbour and colleagues [33] identified two common missense SNPs (Asp299Gly and Thr399Ile) affecting the extracellular domain of the TLR-4 receptor associated with hyporesponsiveness to inhaled endotoxin in humans. They investigated whether these specific SNPs were associated with a predisposition to a more severe disease outcome in 91 patients with septic shock compared with 73 healthy blood donor controls [34]. They found that the 299Gly allele was exclusively present in patients with septic shock.
In a cohort of 77 consecutive critically ill patients, Agnese and colleagues [35] studied the association between these two structural TLR-4 SNPs and the outcome. They found that in this population at risk for sepsis, TLR-4 variants were associated with an increased incidence of Gram-negative infections (79% in patients with mutations versus 17% in patients without them).
Smirnova and colleagues [36] studied 197 unrelated children with systemic meningococcal infections and two control groups of 127 and 256 healthy, unrelated children to determine whether variants affecting the TLR-4 structure render humans more susceptible to meningococcal sepsis. They found that no single variant of TLR-4 was significantly over-represented in the meningococcal population, but that an overwhelmingly significant excess of rare heterozygous missense mutations of TLR-4 was observed among individuals with disease (P < 0.00001; odds ratio, 27.0).
TLRs transduce their signals through MyD88 and the serine/threonine kinase IRAK. Although the exact function of each IRAK protein remains controversial, it seems that IRAK-4 is required for the formation and activation of signaling complexes involving TNF receptor-associated factor-6. Picard and colleagues [37] have recently reported an inherited IRAK-4 deficiency in three unrelated children with recurrent pyogenic infections and poor inflammatory response. Three different mutations were detectable in those three children. Their cells did not activate NF-κB and failed to induce downstream cytokines. Other association studies [38-46] are presented in Table 1.
Genetic association studies: limitations and recommendations
The rationale for studying gene polymorphisms in critical illnesses seeks to identify potential markers of susceptibility, severity, and clinical outcome, seeks to identify potential markers for responders and nonresponders in clinical trials, and seeks to identify targets for therapeutic intervention. Genetic association studies have become one of the most common forms of experimental design in the medical literature and remain perhaps some of the hardest to interpret [47]. Association is sought between a specific SNP and the clinical outcome by direct comparison of an individual genotype and the clinical features of the disease. Case–control association studies have been widely used in the search for genetic variants that predispose to sepsis, in which the frequencies of marker alleles in groups of patients and healthy controls are compared, and the difference is subjected to statistical analysis [19]. Judging the results of association studies is problematic. Although association analysis promises to be a useful tool for shedding light on the genetic basis of disease predisposition and outcome, their value is diminished by multiple limitations regarding their use and the interpretation of results.
Detecting SNPs is no longer a difficult technical task; the challenge is to make sense of all the data. In considering how to compare allelic frequencies, there is an understandable tendency to just apply the chi-square test and obtain the 'truth' [47]. The P values do not provide a marker of truth, when in some instances the 'truth' comes from just a single, unconfirmed publication. All probability is conditional; judgment and profound knowledge of the specific topic are critical to accept that the findings of association studies are plausible in the light of what is known. Weak genetic effects combined with underpowered studies lead to significant numbers of falsely negative reports. Special attention has to be paid to the issues of lack of power and small sample size, to disease classification or status, to problems derived from chance, to bias, and to confounding factors.
Since most association studies are small in size (less than a few hundred cases and controls), when a statistical significance is achieved it is almost certain to have overestimated the true effect of the variant being tested. On the contrary, failure to observe an association of the magnitude of effect reported in a previous study should not be taken as a rejection of the association. Since many alleles have weak genetic effects, testing the variant in a large population will be required to determine whether the association achieves statistical significance. An alternative explanation for a positive statistically significant association could be chance. This is particularly the case of multiple testing for different markers without using a correction strategy, which could lead to an overinterpretation of a false positive [48].
Apart from real associations, other factors can produce allele frequency differences between cases and controls. Different sources of bias could be responsible for such artifacts. Most systematic errors (bias) in molecular epidemiological studies occur because of imperfect sampling or classification procedures that cause the dataset to misrepresent the true relationship being studied [49]. Bias in the selection of samples can be resolved using clearly defined inclusion criteria for cases and controls. Among clinicians, the choice of phenotype is critical to the success of association studies. Most case–control association studies employ a dichotomous variable (affected/unaffected). If the cases include a non-homogeneous sample of patients or if the control group includes subjects who were affected in the past or are currently affected but undiagnosed, the power to detect a significant association will be reduced [50]. A biologic plausibility of a candidate gene for involvement in the pathogenesis of sepsis is important, but errors in genotyping can have serious implications for genetic association studies.
A relatively easy method for estimating genotyping error is testing some individuals twice and counting the discrepancies [51]. In the most widely used technique for genotyping, restriction fragment length polymorphism, the patterns are produced by the target SNP.
However, restriction fragment length polymorphism is more prone to errors than minisequencing or hybridization (Fig. 2) because in the latter methods the observed genotypes are not influenced by the efficiency of the reaction, and they use more information from sequences adjacent to the SNP. For a proper interpretation of the results it is important to assess the Hardy–Weinberg equilibrium at least within the control group. This test indicates that the genotype frequencies can be determined directly from the allele frequencies. A recent work by Xu and colleagues [52] detected an association between positive results in association studies and deviations from Hardy–Weinberg equilibrium due to genotyping errors.
Figure 2.
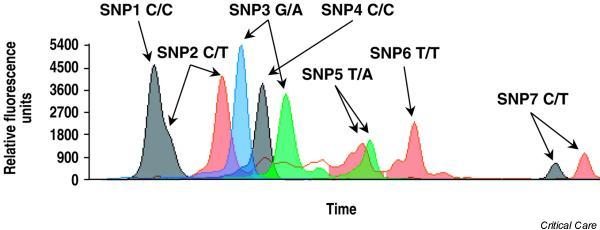
An example of the simultaneous detection of seven known single nucleotide polymorphisms (SNPs) with a minisequencing method. Each peak corresponds to a SNP allele (blue, G; green, A; black, C; red, T).
Linkage disequilibrium is the best known confounding factor affecting case–control studies [53-55], and can be defined as a nonrandom association of alleles at different loci. If linkage disequilibrium is present, the possibility exists that the original marker tested is not the causal allele, and further studies of the region are warranted. In this regard, one SNP or a few SNPs selected on the basis of producing an amino acid change are often typed in a candidate gene. If there are negative results, then the gene is regarded as having no implication with the disease. The opposite would be concluded if the typed SNP results in a positive association. It must be noted, however, that only subsets of the variants of the gene have been explored.
Although multiple polymorphisms have been described within the TNF gene locus, and interpreted to indicate that it is associated with prognosis [22,23], none of those polymorphisms seem to directly alter the TNF-α transcription rate. It is more probable that those associations are not direct, but result from linkage disequilibrium with other genes on chromosome 6 [53,54]. If hundreds of thousands of SNPs were identified across the genome, then it would be possible to perform genome-wide association studies to identify the regions of linkage disequilibrium around disease susceptibility genes [55].
The need to explore nearby variants and surrounding haplotypes, which is the combination of alleles from the different loci, is therefore crucial. SNPs are typically analyzed in isolation, whereas it may be the precise combination of SNPs on a given chromosome (the haplotype) that determines its significance. The most appropriate way to proceed in association studies would be to characterize the linkage disequilibrium distribution in particular regions of interest, and then use these data to extract the maximum information by typing a selected subset of the most informative SNPs, called tagging SNPs [56,57]. The haplotypes constructed from tagging SNPs would produce a modest reduction in power in comparison with direct assays with all common SNPs in the same genomic region. Moreover, it has been suggested that studies based on genotypes or haplotypes from several SNPs may reduce the sample sizes needed to detect association [58]. A drawback of this method is that the tagging SNPs identified in one population may not necessarily perform well in another population [56].
A possible cause of a false-positive association study includes an admixture resulting in population stratification. Ethnicity has been the most common way to match cases and controls based on self-reported ancestry. If cases and controls are drawn from different ethnic groups or subgroups, allele frequencies will tend to differ among the sub-populations for most randomly chosen loci with no causal association with the disease. If one of these subgroups has a higher disease prevalence than the others then stratification occurs, because that subgroup will be over-represented in the cases and will be under-represented in the controls [59]. Stratification can also occur in a single admixed population where the individuals have varying degrees of genetic contributions from two or more population groups.
There are currently several methods for controlling for confounding in a stratified population. One approach for reducing this effect is the use of family-based controls. Since sepsis predominantly affects middle-age adults, it would be difficult to recruit relatives. As an alternative, the population structure can be empirically determined by individually genotyping all potential cases and controls across a set of unlinked marker loci, although an optimum number of these markers have not been established. A family-based approach would be more powerful when the population structure is significant, as in African-Americans, while the approach based on the typing of unlinked markers would be more efficient for populations with low levels of structure, as in Europeans [60].
Association studies are plagued by the impression that they are not consistently reproducible either due to false positives, to false negatives, or to variability in association among different populations [61]. Lohmueller and colleagues [62] proposed three recommendations. First, a single, nominally significant association should be viewed as tentative until it has been independently replicated at least once, and preferably twice. Second, large studies should be encouraged, with collaborative efforts in order to achieve sample sizes of several thousands of case–control pairs. Finally, the authors estimated that one-quarter of previously published associations represent real associations with common diseases.
Using an adequate sample size to test all previously reported associations that have already been replicated at least once would probably identify a significant number of variants that affect the risk of common diseases [63]. Complementing these recommendations, publication bias should be avoided so that both positive and negative results are accessible to the public, as long as they fulfill minimal methodological criteria. Once such a power is reached, findings should be judged on additional functional evidences. Since no published reports to date meet all methodological requirements for supporting a causative relationship with those reported candidate genes (SNP hunting technique, population stratification, linkage disequilibrium, sample size, and lack of power), any conclusions still remain speculative. Results from most genetic case–control association studies need to be confirmed in future studies.
Summary
The age of the genome is with us. Current therapies for critically ill patients are selected on the basis of 'standard' patterns and expected responses. However, physicians have long known that every patient has a different response to drugs and is at a different risk for a particular event or bad outcome. We are now discovering that the individual risks and cellular responses can be related to each patient's unique DNA. Genetics seeks to correlate the variation in DNA sequence with phenotypic differences. Genotyping is likely to become increasingly important in clinical medicine. The recognition of genetic predisposition to sepsis might facilitate the search for therapeutic targets in patients with an impaired innate immune system. Establishing a catalogue of all common variants in the human population will facilitate studies to establish relationships between genotype and biological function.
SNPs can work as predictive tools to assist clinical decisions. The challenges that lie beyond include detecting the clinical significance of variations in genetic sequences, identifying different functions of DNA, and other molecular systems in the cell, and unraveling the complexities of gene–gene and gene–environment interactions. In this genetic New World, physicians might be able to use genetic information to dictate immune-based therapies to modulate the response in a given patient.
We envision a time in the near future when genotyping will be included in the standard evaluation of patients and will help to prioritize a therapeutic option. Those who are found to carry a genetic susceptibility will constitute a new class of individuals for medicine: a class that might be designated as 'unpatients'; neither patients in the usual sense of being under treatment, nor nonpatients in the sense of being free of a medically relevant condition [64]. By systematically collecting DNA from every patient in every clinical trial, scientists will analyze it for variations and then, at the end of the trial, perform association studies between the genetic variation, the efficacy, and the adverse effects of therapeutic measures. Careful attention to genotype assignment will be required to maximize the benefits to individual patients in a new era for investigating the genetic bases of human disease and drug response. SNP research is paving a track to personalized medicine.
Competing interests
None declared.
Abbreviations
IL = interleukin; IRAK = IL-1 receptor-associated kinases; LBP = lipopolysaccharide-binding protein; LPS = lipopolysaccharide; MBL = mannose-binding lectin; MyD88 = myeloid differentiation protein 88; NF = nuclear factor; SNP = single nucleotide polymorphism; TLR = Toll-like receptor; TNF = tumor necrosis factor.
Acknowledgments
Acknowledgements
Supported in part by research grants from FUNCIS (37/02) and DGUI (02/209).
References
- Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Martin GS, Mannino DM, Eaton S, Moss M. The epidemiology of sepsis in the United States from 1979 through 2000. N Engl J Med. 2003;348:1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Riedemann NC, Guo RF, Ward PA. The enigma of sepsis. J Clin Invest. 2003;112:460–467. doi: 10.1172/JCI200319523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TR. Recognition of bacterial endotoxin in the lungs. Am J Respir Cell Moll Biol. 2000;23:128–132. doi: 10.1165/ajrcmb.23.2.f189. [DOI] [PubMed] [Google Scholar]
- Morath S, Geyer A, Hartung T. Structure–function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med. 2001;193:393–397. doi: 10.1084/jem.193.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann RR, Leong SR, Flaggs DW, Gray PW, Wright SD, Mathison JC, Tobias PS, Ulevitch RJ. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1431. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- Opal S, Huber CE. Bench-to-bedside review: Toll-like receptors and their role in septic shock. Crit Care. 2002;6:125–136. doi: 10.1186/cc1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K, Nagai Y, Akashi S, Nagafuku M, Ogata M, Kosugi A. Essential role of MD-2 in B-cell responses to lipopolysaccharide and Toll-like receptor 4 distribution. J Endotoxin Res. 2002;8:449–452. doi: 10.1179/096805102125001055. [DOI] [PubMed] [Google Scholar]
- O'Neill LA, Fitzgerald KA, Bowie AG. The Toll-IL-1 receptor adaptor family grows to five members. Trends Immunol. 2003;24:286–290. doi: 10.1016/S1471-4906(03)00115-7. [DOI] [PubMed] [Google Scholar]
- Burns K, Janssens S, Brissoni B, Olivos N, Beyaert R, Tschopp J. Inhibition of interleukin 1 receptor/toll-like receptor signaling through the alternatively spliced, short form of MyD88 is due to its failure to recruit IRAK-4. J Exp Med. 2003;197:263–268. doi: 10.1084/jem.20021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by tollip. J Biol Chem. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- Riedemann NC, Ward PA. Oxidized lipid protects against sepsis. Nat Med. 2002;8:1084–1085. doi: 10.1038/nm1002-1084. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Dinarello CA, Oppenheim JJ, Hertzog P. Recent advances in cytokines, cytokine receptors and signal transduction. Cytokine Growth Factor Rev. 1998;9:175–181. doi: 10.1016/S1359-6101(98)00006-9. [DOI] [PubMed] [Google Scholar]
- Pruitt JH, Welborn MB, Edwards PD, Harward TR, Seeger JW, Martin TD, Smith C, Kenney JA, Wesdorp RI, Meijer S, Cuesta MA, Abouhanze A, Copeland EM, 3rd, Giri J, Sims JE, Moldawer LL, Oldenburg HS. Increased soluble interleukin-1 type II receptor concentrations in postoperative patients and in patients with sepsis syndrome. Blood. 1996;87:3282–3288. [PubMed] [Google Scholar]
- Fisher CJ, Jr, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, Abraham E, Schein RM, Benjamin E. Treatment of septic shock with the tumor necrosis factor receptor:Fc fusion protein. The Soluble TNF Receptor Sepsis Study Group. N Engl J Med. 1996;334:1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ, Jr, Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Sørensen TIA, Nielsen GG, Andersen PK, Teasdale TW. Genetic and environmental influences on premature death in adult adoptees. N Engl J Med. 1988;318:727–732. doi: 10.1056/NEJM198803243181202. [DOI] [PubMed] [Google Scholar]
- Bidwell J, Keen L, Gallagher G, Kimberly R, Huizinga T, McDermott MF, Oksenberg J, McNicholl J, Pociot F, Hardt C, D'Alfonso S. Cytokine gene polymorphism in human disease: on-line databases. Genes Immun. 1999;1:3–19. doi: 10.1038/sj.gene.6363645. [DOI] [PubMed] [Google Scholar]
- Villar J, Flores C, Méndez-Álvarez S. Genetic susceptibility toacute lung injury. Crit Care Med. 2003;31(Suppl):S272–S275. doi: 10.1097/01.CCM.0000057903.11528.6D. [DOI] [PubMed] [Google Scholar]
- Hill AV. Genetics and genomics of infectious disease susceptibility. Br Med Bull. 1999;24:381–384. doi: 10.1258/0007142991902457. [DOI] [PubMed] [Google Scholar]
- Stüber F, Petersen M, Bokelmann F, Schade U. A genomic polymorphism within the tumor necrosis factor locus influence plasma tumor necrosis factor-α concentrations and outcome of patients with severe sepsis. Crit Care Med. 1996;24:381–384. doi: 10.1097/00003246-199603000-00004. [DOI] [PubMed] [Google Scholar]
- Mira JP, Cariou A, Grall F, Delclaux C, Losser MR, Heshmati F, Cheval C, Monchi M, Teboul JL, Riche F, Leleu G, Arbibe L, Mignon A, Delpech M, Dhainaut JF. Association of TNF2, a TNF-α promoter polymorphism, with septic shock susceptibility and mortality. A multicenter study. JAMA. 1999;282:561–568. doi: 10.1001/jama.282.6.561. [DOI] [PubMed] [Google Scholar]
- Vermont CL, de Groot R, Hazelzet JA. Bench-to-bedside review: genetic influences on meningococcal disease. Crit Care. 2002;6:60–65. doi: 10.1186/cc1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibberd ML, Sumiya M, Summerfield JA, Booy R, Levin M. Association of variants of the gene for mannose-binding lectin with susceptibility to meningococcal disease. Meningococcal Research Group. Lancet. 1999;353:1049–1053. doi: 10.1016/S0140-6736(98)08350-0. [DOI] [PubMed] [Google Scholar]
- Koch A, Melbye M, Sorensen P, Homoe P, Madsen HO, Molbak K, Hansen CH, Andersen LH, Hahn GW, Garred P. Acute respiratory tract infections and mannose-binding lectin insufficiency during early childhood. JAMA. 2001;285:1316–1321. doi: 10.1001/jama.285.10.1316. [DOI] [PubMed] [Google Scholar]
- Fang XM, Schroder S, Hoeft A, Stuber F. Comparison of two polymorphisms of the interleukin-1 gene family: interleukin-1 receptor antagonist polymorphism contributes to susceptibility to severe sepsis. Crit Care Med. 1999;27:1330–1334. doi: 10.1097/00003246-199907000-00024. [DOI] [PubMed] [Google Scholar]
- Hubacek JA, Stuber F, Frohlich D, Book M, Wetegrove S, Ritter M, Rothe G, Schmitz G. Gene variants of the bactericidal/ permeability increasing protein and lipopolysaccharide binding protein in sepsis patients: gender-specific predisposition to sepsis. Crit Care Med. 2001;29:557–561. doi: 10.1097/00003246-200103000-00015. [DOI] [PubMed] [Google Scholar]
- Barber RC, O'Keefe GE. Characterization of a single nucleotide polymorphism in the lipopolysaccharide binding protein and its association with sepsis. Am J Respir Crit Care Med. 2003;167:1316–1320. doi: 10.1164/rccm.200209-1064OC. [DOI] [PubMed] [Google Scholar]
- Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- Hubacek JA, Stuber F, Frohlich D, Book M, Wetegrove S, Rothe G, Schmitz G. The common functional C(-159)T polymorphism within the promoter region of the lipopolysaccharide receptor CD14 is not associated with sepsis development or mortality. Genes Immun. 2000;1:405–407. doi: 10.1038/sj.gene.6363691. [DOI] [PubMed] [Google Scholar]
- Heesen M, Bloemeke B, Schade U, Obertacke U, Majetschak M. The -260 C→T promoter polymorphism of the lipopolysaccharide receptor CD14 and severe sepsis in trauma patients. Intensive Care Med. 2002;28:1161–1163. doi: 10.1007/s00134-002-1389-0. [DOI] [PubMed] [Google Scholar]
- Arbour NC, Lorenz E, Schutte BC, Zabner J, Kline JN, Jones M, Frees K, Watt JL, Schwartz DA. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat Genet. 2000;25:187–192. doi: 10.1038/76048. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Frees KL, Schwartz DA. Relevance of mutations in the TLR4 receptor in patients with Gram-negative septic shock. Arch Intern Med. 2002;162:1028–1032. doi: 10.1001/archinte.162.9.1028. [DOI] [PubMed] [Google Scholar]
- Agnese DM, Calvano JE, Hahm SJ, Coyle SM, Corbett SA, Calvano SE, Lowry SF. Human toll-like receptor 4 mutations but not CD14 polymorphisms are associated with an increased risk of Gram-negative infections. J Infect Dis. 2002;186:1522–1525. doi: 10.1086/344893. [DOI] [PubMed] [Google Scholar]
- Smirnova I, Mann N, Dols A, Derkx HH, Hibberd ML, Levin M, Beutler B. Assay of locus-specific genetic load implicates rare Toll-like receptor 4 mutations in meningococcal susceptibility. Proc Natl Acad Sci. 2003;100:6075–6080. doi: 10.1073/pnas.1031605100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al-Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Ozinsky A, Casanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076–2079. doi: 10.1126/science.1081902. [DOI] [PubMed] [Google Scholar]
- Schroeder S, Reck M, Hoeft A, Stüber F. Analysis of two human leukocyte antigen-linked polymorphic heat shock protein 70 genes in patients with severe sepsis. Crit Care Med. 1999;27:1265–1270. doi: 10.1097/00003246-199907000-00006. [DOI] [PubMed] [Google Scholar]
- Lorenz E, Mira JP, Cornish KL, Arbour NC, Schwartz DA. A novel polymorphism in the Toll-Like Receptor 2 and its potential association with Staphylococcal infection. Infect Immun. 2000;68:6398–6401. doi: 10.1128/IAI.68.11.6398-6401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Pol WL, Huizinaga TWJ, Vidarsson G, van der Linden MW, Jansen MD, Keijsers V, Leppers-van de Straat FGJ, Westerdaal NAC, van de Winkel JGJ, Westendorp RGJ. Relevance of Fcγ receptor and interleukin-10 polymorphisms for meningococcal disease. J Infect Dis. 2001;184:1548–1555. doi: 10.1086/324662. [DOI] [PubMed] [Google Scholar]
- Harding D, Baines PB, Brull D, Vassiliou V, Ellis I, Hart A, Thomson APJ, Humphries SE, Montgomery HE. Severity of meningococcal disease in children and the angiotensin-converting enzyme insertion/deletion polymorphism. Am J Respir Crit Care Med. 2002;165:1103–1106. doi: 10.1164/ajrccm.165.8.2108089. [DOI] [PubMed] [Google Scholar]
- Kerlin BA, Yan SB, Isermann BH, Brandt JT, Sood R, Basson BR, Joyce DE, Weiler H, Dhainaut JF. Survival advantage associated with heterozygous factor V Leiden mutation in patients with severe sepsis and in mouse endotoxemia. Blood. 2003;102:3085–3092. doi: 10.1182/blood-2003-06-1789. [DOI] [PubMed] [Google Scholar]
- Westendorp RGJ, Hottenga JJ, Slagboom PE. Variation in plasminogen-activator-inhibitor-1 gene and risk of meningococcal septic shock. Lancet. 1999;354:561–563. doi: 10.1016/S0140-6736(98)09376-3. [DOI] [PubMed] [Google Scholar]
- Kondaveeti S, Hibberd ML, Levin M. The insertion/deletion polymorphism in the t-PA gene does not significantly affect outcome of meningococcal disease. Thromb Haemost. 1999;82:161–162. [PubMed] [Google Scholar]
- Schluter B, Raufhake C, Erren M, Schotte H, Kipp F, Rust S, Van Aken H, Assmann G, Berendes E. Effect of the interleukin-6 promoter polymorphism (-174 G/C) on the incidence and outcome of sepsis. Crit Care Med. 2002;30:32–37. doi: 10.1097/00003246-200201000-00005. [DOI] [PubMed] [Google Scholar]
- Majetschak M, Flohé S, Obertacke U, Schröder J, Staubach K, Nast-Kolb D. Relation of a TNF gene polymorphism to severe sepsis in trauma patients. Ann Surg. 1999;230:207–214. doi: 10.1097/00000658-199908000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees J. P for Probability and p for p53. J Invest Dermatol. 2003;121:xii–xiii. doi: 10.1046/j.1523-1747.2003.12381.x. [DOI] [PubMed] [Google Scholar]
- Boehringer S, Epplen JT, Krawczak M. Genetic association studies of bronchial asthma – a need for Bonferroni correction? [Letter] Hum Genet. 2000;107:197. doi: 10.1007/s004390000353. [DOI] [PubMed] [Google Scholar]
- Vineis P, McMichael AJ. Bias and confounding in molecular epidemiological studies: special considerations. Carcinogenesis. 1998;19:2063–2067. doi: 10.1093/carcin/19.12.2063. [DOI] [PubMed] [Google Scholar]
- Silverman EK, Palmer LJ. Case–control association studies for the genetics of complex respiratory diseases. Am J Respir Cell Mol Biol. 2000;22:645–648. doi: 10.1165/ajrcmb.22.6.f191. [DOI] [PubMed] [Google Scholar]
- Rice KM, Holmans P. Allowing for genotyping error in analysis of unmatched case–control studies. Ann Hum Genet. 2003;67:165–174. doi: 10.1046/j.1469-1809.2003.00020.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Turner A, Little J, Bleecker ER, Meyers DA. Positive results in association studies are associated with departure from Hardy–Weinberg equilibrium: hint for genotyping error? Hum Genet. 2002;111:573–574. doi: 10.1007/s00439-002-0819-y. [DOI] [PubMed] [Google Scholar]
- van Deventer SJH. Cytokine and cytokine receptor polymorphisms in infectious disease. Intensive Care Med. 2000;26:S98–S102. doi: 10.1007/s001340051125. [DOI] [PubMed] [Google Scholar]
- Knight J, Keating BJ, Rockett KA, Kwiatkowski DP. In vivo characterization of regulatory polymorphisms by allele-specific quantification of RNA polymerase loading. Nat Genet. 2003;33:469–475. doi: 10.1038/ng1124. [DOI] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA. Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet. 2003;33:518–521. doi: 10.1038/ng1128. [DOI] [PubMed] [Google Scholar]
- Weale ME, Depondt C, Macdonald SJ, Smith A, Lai PS, Shorvon SD, Wood NW, Goldstein DB. Selection and evaluation of Tagging SNPs in the Neuronal-Sodium-Channel gene SCN1A: implications for linkage-disequilibrium gene mapping. Am J Hum Genet. 2003;73:551–565. doi: 10.1086/378098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, Schork NJ. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimers disease. Genome Res. 2001;11:143–151. doi: 10.1101/gr.148401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Lohmueller K, Byrne E, Hirschhorn K. A comprehensive review of genetic association studies. Genet Med. 2002;4:45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- Bacanu S-A, Devlin B, Roeder K. The power of genomic control. Am J Hum Genet. 2000;66:1933–1944. doi: 10.1086/302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Bell JI. Association study designs for complex diseases. Nat Rev Genet. 2001;2:91–99. doi: 10.1038/35052543. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Pearce CL, Pike CL, Lander ES, Hirschhorn JN. Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003;33:177–182. doi: 10.1038/ng1071. [DOI] [PubMed] [Google Scholar]
- García-Closas M, Lubin JH. Power and sample size calculations in case–control studies of gene–environment interactions: comments on different approaches. Am J Epidemiol. 1999;149:689–692. doi: 10.1093/oxfordjournals.aje.a009876. [DOI] [PubMed] [Google Scholar]
- Jonsen AR, Durfy SJ, Burke W, Motulsky AG. The advent of the 'unpatients'. Nat Med. 1996;2:622–624. doi: 10.1038/nm0696-622. [DOI] [PubMed] [Google Scholar]