

Abstract
Autophagy is a major catabolic process in which intracellular membrane structures, protein complexes, and lysosomes are formed as lysoautophagosome to degrade and renew cytoplasmic components. Autophagy is physiologically a strategy and mechanism for cellular homeostasis as well as adaptation to stress, and thus alterations in the autophagy machinery may lead to diverse pathological conditions. The role of autophagy in cancer is complex, and the current literature reflects this as a ‘double-edged sword’. Autophagy shows promise as a novel therapeutic target in various types of breast cancer, inhibiting or increasing treatment efficacy in a context- and cell-type-dependent manner. This review aims to summarize the recent advances in the understanding of the mechanisms by which key modulators of autophagy participate in cancer metastasis, highlight different autophagy-deficient murine models for breast cancer study, and provide further impetus for the modulation of autophagy in anticancer therapy.
Keywords: autophagy, mTOR, breast cancer
Introduction
Metastasis is an advanced stage of cancer progression that generally indicates poorer prognosis and lower survival rate. Multiple cellular events, including the dysfunction of programmed cell death, epithelial-to-mesenchymal transition (EMT) [1–3], the development of tumor-favorable microenvironment [4], and tumor angiogenesis [5] are implicated in cancer metastasis. Cell death occurs when mistakes are made by solitary disseminating cell during these processes [6]. Typically, the initial steps of metastasis proceed rapidly, whereas the final step, colonization, is less so efficient as an estimate of only ∼0.01% of circulating tumor cells eventually form metastatic foci [7]. This inefficiency may be principally attributed to the activation of cell death machinery by various stresses within the circulation or after metastatic cells approach a new environment. Such stresses involve the loss of cell–cell contacts, incapability of extravasation [8], the destruction of solitary tumor cell either by the immune system or hemodynamic forces [9], and the lack of essential growth factors, all of which may trigger programmed cell death, including autophagy [10]. Therefore, tight regulation of cell death is crucial for cancer cells to survive during metastasis.
During the past decade, pioneering studies led to the classification of autophagy as a type of programmed cell death. Autophagic cell death was known to be triggered primarily when the embryonic developmental programs or homeostatic processes in adulthood reach the stage that requires massive cell elimination [11]. Besides this ‘self-disposal’ purpose, however, autophagy is now more accepted as a fundamental cell survival mechanism to combat environmental stressors [12]. Tumor cells evolve multiple strategies to evade programmed cell death via genetic mutations or epigenetic modifications in the key players of programmed cell death, which include components from the autophagic pathway [6]. This review provides an overview of the interplay of autophagy with breast cancer metastasis and summarizes a list of autophagy-deficient murine models and current anticancer therapeutics.
An Introduction to Autophagy
To date, three common types of autophagy (macroautophagy, microautophagy, and chaperone-mediated autophagy) are identified, and they are mediated by autophagy-related genes (ATG) and their associated enzymes [13]. Macroautophagy (hereafter referred to as autophagy) is the best characterized pathway, and it is a highly conserved multistep process that mediates the turnover of intracellular protein aggregates and damaged organelles to maintain energetic homeostasis [12]. To self-digest these proteins and organelles, the cell first undergoes nucleation by de novo formation of a lipid-based double-membrane structure phagophore, which depends on the coordinated activity of a multi-protein complex involving mammalian orthologue of yeast Atg6 (Beclin-1), class III phosphoinositide-3-kinase (Vps34), ultraviolet irradiation resistant-associated gene (UVRAG), B cell CLL/lymphoma 2 (Bcl-2), Bax-interacting factor 1 (Bif-1), and other partners such as activating molecule in beclin-1-regulated autophagy 1 (Ambra1), vacuole membrane protein 1 (VMP1), the viral protein-infected cell protein 34 (ICP34), and viral form of Bcl-2 (Fig. 1). The elongation of phagophore is primarily driven by two ubiquitination-like reactions, which will be discussed in detail in the next section. Additionally, maturation of mammalian autophagosomes involves encapsulation of targeted cargo by the elongating phagophore and subsequent fusion with different endosomal compartments. Delivery of autophagosomal cargo for degradation requires further fusion between autophagosomes with lysosomes [14]. These fusion processes require the assembly of several soluble NSF attachment protein receptor (SNARE)-like proteins to help eventually form a mature lysoautophagosome that executes degradation and recycling [15] (Fig. 2). Basal level of autophagy is generally described as a basic cell survival or cytoprotective response to circumvent stress conditions, such as toxic stimuli, gamma radiation, chemotherapy, and starvation. It is also of particular importance during developmental process, as it is suggested to maintain normal metabolism by providing an alternative cellular source for energy production and biomolecular synthesis. In the canonical starvation-induced pathway, autophagosome formation is induced by Beclin-1, PI3K, and ubiquitin-like conjugation reactions. In contrast to canonical autophagy, non-canonical autophagy does not require the complete set of ATG proteins to form autophagosomes. Typically, Beclin-1-independent autophagy is dependent on the activity of the Unc-51 like kinase 1/2 (Ulk1) complex to induce autophagy and LC3 for phagophore formation [17]. Recently, injury-induced autophagy and mitophagy have been established as non-canonical pathway, as they are PI3K/Beclin-1 independent [18]. Another protective role has been reported that autophagy engages in the defense against pathogens and T cell repertoire shaping during immune response [19].
Figure 1.

Beclin-1 interaction complexes Atg14L and UVRAG/Bif-1 activate the Beclin-1 complexes and induce the formation of autophagosomes in a mutually exclusive manner. UVRAG also potentially functions to promote autophagosome maturation and endocytic trafficking through pathways independent of its interaction with Beclin-1. VMP-1 and PINK1 are two other interacting partners and inducers of autophagy. NIF-1 is a component of the PI3K complex contributing to the interaction of Beclin-1 and Bcl-2 at the ER surface. Bcl-2/Bcl-XL has similar function in Beclin-1 binding and autophagy inhibition. JNK1 and DAPK are autophagy inducers that phosphorylate Bcl-2 and Beclin-1, respectively, to disrupt their interaction with each other. HMGB1 (high-mobility group box 1), a p53 interacting chromatin protein, induces autophagy for cell protection against damage in a similar fashion. TRAF6 and USP13 are reported to play a role in Beclin-1 ubiquitination. Survivin is a Beclin-1-binding anti-apoptotic protein to regulate TRAIL-induced apoptosis. Rubicon binds Beclin-1 to inhibit autophagosome formation and maturation. Dissociation of upstream activators TAB1/2 in TAK1-IKK pathway is thought to be necessary for autophagy induction. GAPR-1 binds Beclin-1 to inhibit autophagy; however, the mechanism is not entirely clear.
Figure 2.

General stages of autophagy Autophagy is characterized by the induction of phagophore by the ULK complex, which is activated by being liberated from the mTORC1 complex due to its inactivity. This dissociation event results in the dephosphorylation of inhibitory sites of Ulk1/2 and autophosphorylation of its activating sites, which leads to concomitant activation of its interacting partners mAtg13 and FIP200 and localization of the ULK complex from the cytosol to the ER. Activation of the ULK complex also mediates the activation and ER assembly of class III PI3K complex that consists of Beclin-1, Vps34, and p150 during the nucleation phase [16]. Complete elongation of the phagophore generates a structure called ‘autophagosome’, and this process involves two ubiquitin-like conjugation pathways. The autophagosome sequesters its cargo and fuses with the lysosome on a Beclin-1-dependent manner to form the lysoautophagosome and where the contents are digested by lysosomal enzymes for degradation or recycle. Details are provided in the text.
Characterization of autophagy pathway was initially performed in yeast with identification of almost 30 Atg genes [20] as well as their mammalian homologs, and seminal experiments defining the induction of autophagy in mammalian hepatocytes revealed another eight ATG genes regulating the steps of autophagosome formation and decomposition [21]. These studies established the significance of nutrient levels, energy status, and hormonal regulation as key modulators of autophagy. In addition, environmental stressors such as hypoxia, heat stress, and reactive oxygen species accumulation can also induce autophagy. The endoplasmic reticulum (ER) is a robust and finely regulated organelle in charge of protein folding fidelity and a veritable menagerie of cellular processes. If the ER microenvironment is disrupted, the unfolded protein response will be activated to trigger autophagy through eukaryotic translation initiation factor 2-alpha kinase 3 (EIF2AK3) and activating transcription factor 6 (ATF6) pathways in an effort to ameliorate the accumulation and aggregation of misfolded proteins. Therefore, ER stress is also a potent autophagy inducer. In general, autophagy is implicated in various aspects including longevity, disease prevention, and promotion, as well as mammalian development [2].
Core Machinery Involved in Autophagy
Among the various ways to modulate autophagy, one autophagic signaling pathway of great importance involves mammalian target of rapamycin (mTOR), a serine and threonine kinase. As a part of the energy-sensing mechanism and stress response, the mTOR kinase is an important repressor of autophagy and a controller of cell growth and proliferation. mTORC1 and mTORC2 are two mTOR complexes that are located and regulated differently but are both induced by nutrient starvation, stress, and reduced growth factor signaling. The mTOR pathway inhibits autophagy under normal conditions, but it regulates metabolic stress signals to drive cell growth in the presence of abundant nutrients. Upstream of mTOR is the putative class I PI3K that receives internal or external signals (insulin, growth factors, amino acids, etc.) and functions to facilitate phosphorylation of protein kinase B (Akt) by 3-phosphoinositide-dependent kinase 1 (PDK1). Phosphorylated Akt activates the mTOR complex to inhibit autophagy in nutrient-rich conditions by promoting the activation of downstream Ulk1 complex and disrupting the interaction of Ulk1 and AMP-activated protein kinase (AMPK) complex [22]. Typically, mTORC2 affects cell metabolism and survival by phosphorylating serine/threonine protein kinase Akt/PKB at the serine residue S473, which leads to consequent activation of Akt by PDK1. Under low-energy state, the key energy sensor heterotrimeric enzyme AMPK is turned on to effectively induce autophagy by directly rendering MTORC1 inactive or by phosphorylating Ulk1 [23], which in turn phosphorylates Atg13 and RB1CC1/FIP200 [24] to activate the nucleation stage. The Bcl-2 family proteins suppress autophagy by disrupting the interaction of Beclin-1 with PI3K complex, thus inhibiting early formation of autophagosomes (Fig. 3). The nucleation stage is an orchestrated process relying on the PtdIns3K complex that serves to phosphorylate phosphatidylinositol (PI) to phosphatidylinositol 3-phosphate (PI3P), the latter of which recruits protein complexes and lipids to expand the autophagosome membrane. The PtdIns3K complex anchors two core interacting partners PIK3C3/Vps34 and Beclin-1, which exhibit distinct functions depending on the components of ancillary proteins in the complex. UVRAG/Bif1 and ATG14 are found in the Beclin-1 complex in a mutually exclusive manner. Moreover, the ULK1/2 and Vps34 complexes recruit the two ubiquitin-like protein conjugation systems essentially required to accomplish the expansion of the growing autophagosome membrane (Fig. 4): Atg5–Atg12–Atg16 and LC3-phosphatidylethanolamine (PE) systems [26,27]. Atg12 is initially activated in an ATP-based reaction with Atg7 (E1-like enzyme) and then conjugated to Atg5 by Atg10. Atg16 then becomes covalently bound to the Atg12–Atg5 conjugate, forming an Atg12–Atg5–Atg16L1 trimeric complex on the forming membrane. When elongation completes, components of the complex dissociate from the autophagosome and return to the cytoplasm. Mammalian homolog of yeast protein ATG8 (MAP1LC3) is the modified target of the second conjugation pathway. Atg4B cleaves the C-terminal 22 residues of precursor LC3, producing cytoplasmic LC3-I that is subsequently conjugated with PE by Atg7 and Atg3, an E2-like enzyme. The lipidated form of LC3 (LC3-II) is selectively incorporated into the forming autophagosomal membrane with the help of adaptor p62/SQSTM1 via its C-terminal ubiquitin-binding domain and LC3-interacting region (LIR) [28]. Sequestosome 1 (SQSTM1) is a scaffolding protein constitutively expressed and turned over by autophagy-induced selective degradation. Fusion with the lysosome results in degradation of targeted cargo proteins along with the LC3-II associated on the inner membrane of autophagosome (LC3-II on the outer membrane dissociates) for recycling of macromolecule. Fine-tuning of signals is crucial for fidelity of functional autophagy, as deregulation at any of the above-mentioned steps could result in too much or too little autophagy, which is linked to distinct types of diseases, including cancer.
Figure 3.

Autophagy and some of its regulatory pathways Binding of insulin or growth factors to the insulin receptor triggers the PI3K pathway, converting PIP2 into PIP3 that recruits PDK1 and Akt to the plasma membrane. Activation of Akt by insulin binding or direct mTORC2 stimulation inactivates TSC1/2 (tuberous sclerosis 1/2) [25], resulting in the activation of Rheb and mTORC1. Under starvation or rapamycin application, Ulk1 is phosphorylated by AMP-activated protein kinase and detaches from mTORC1 complex, rendering it inactive and triggers the autophagy pathway. The mTORC1 complex and AMP-activated protein kinase sense the nutrient state of the cell and accordingly maintains homeostasis by controlling levels of amino acids through various pathways, including autophagy. LC3 mediates selective autophagy by recruiting several adapter proteins, including p62, to the autophagosome. The activity of PI3K complex can be manipulated by several pharmacological modulators such as activators like BH3 mimetics and inhibitors such as spautin-1. Other modulators of autophagy are also shown in the figure.
Figure 4.
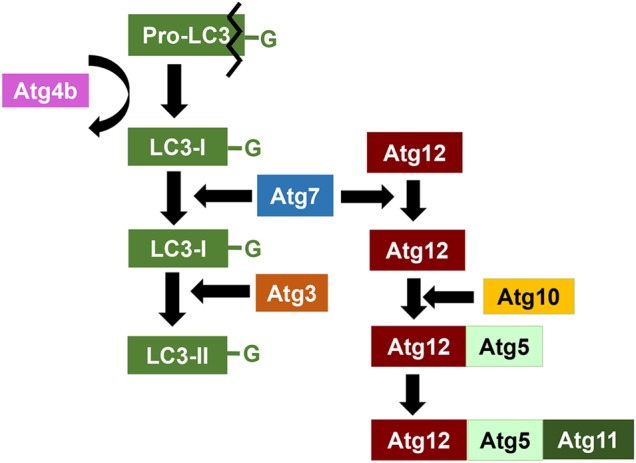
Molecular regulation of the two ubiquitin-like conjugation system The elongation of the autophagosome membrane is dependent on the formation of Atg12–Atg5–Atg11 complex and phosphotidylethanolamine (PE)-conjugated LCIII (Atg8), the major products of the two ubiquitin-like conjugation pathways. Black line indicates the cleavage of pro-LC3. Curved arrow represents the recycling of Atg4b. Details are included in the text.
Autophagy also has its double-sword effect, however, as progressive autophagy under certain environment can be detrimental and substitute for apoptosis in induction of cell death. Thus, it is crucial to differentiate between cytoprotective autophagy and the cellular settings under which autophagy could cause cell death, where autophagy is cytotoxic [29,30].
Autophagy and Breast Cancer
Alteration in autophagy is involved in several types of cancer including breast cancer. In cancer, autophagy is believed to possess both tumor-suppressive and tumor-promoting functions, the paradoxical roles of which may be contributed to distinct context and stages of tumorigenesis. During tumor initiation, autophagy is thought to serve a beneficial cancer-preventing role by, for instance, limiting inflammation, tissue damage, and genome instability, and by preventing oncogene-induced senescence, thereby restricting the invasion and dissemination of cancer cells from the primary site.
During advanced cancer stages, autophagy may play either pro- or anti-metastatic roles depending on the context. It has been suggested that transformation, mediated by certain oncogenes or/and loss of tumor suppressor genes, induces a metabolic switch [31] in cancer cells that predispose them to autophagy under basal and starvation conditions. Autophagy tends to facilitate metastatic process by sustaining spreading cell survival and colonization at a secondary site and by inducing these cells to enter dormancy if they fail to establish stable contact with the extracellular matrix in the new environment. Interestingly, however, in fully transformed cancer cells, it appears that defective autophagy is associated with malignant transformation and carcinogenesis. Additionally, it may also contribute to breast cancer development in a manner independent of genotoxic stress and genomic instability through the induction of ER stress. Based on a study of Kongara et al. [32] in 2010, autophagy plays a role in p62-dependent keratin 8 homeostasis in mammary epithelial cells and low Beclin-1 expression level is linked to phosphor (Ser73) accumulation in human breast carcinoma.
In later stages of cancer development, however, when cancer cells are exposed to metabolic and genotoxic stress during progression, metastasis, and cancer therapy, autophagy shifts to tumor-promoting mechanisms by enabling survival of tumor cells [33]. In essence, unfavorable conditions of hypoxia and acidity within the tumor microenvironment can put cells under metabolic stress [34], while chemotherapeutic drugs and radiotherapies function to damage the genome of tumor cells [35]. In this situation, autophagy is often triggered within the neoplasm, proffering the tumor cell resistance to circumvent the lethal attacks ‘executed’ by chemotherapy and radiotherapy. Typically, excessive and sustained autophagy leads to a type of programmed cell death known as autophagic cell death, represented by early degradation of organelles and a lack of caspase activation or DNA fragmentation and preservation of cytoskeleton elements. Therefore, it has been proposed with this regard that autophagy should be inhibited during cancer treatment to increase the efficacy of therapy [36].
Being the second leading cause of cancer-related death in women in the United States [37], breast cancer is one of the most common malignancies where accumulation of abnormal cells is possibly ascribed to disordered autophagy regulation and imbalanced cell proliferation and apoptosis [38]. Breast carcinoma is well known for its propensity to relapse after a long latency period following initial treatment [39]. Recurrence of the disease is often highly metastatic and exhibits resistance to available treatments. Targeted therapy in breast cancer is often dictated by the expression of certain markers such as hormone or growth factor receptors. Therefore, some types of breast cancer such as triple-negative breast cancer (TNBC) that lack such molecules cannot be treated with hormonal therapies or respective antibodies [40]. In addition, there are also other types of breast cancer that display distinct abnormalities in apoptotic pathways, which confer resistance to many forms of chemotherapy [41]. In regard of the fundamental importance of autophagy in cancer development and progression as well as its influence on treatment response, this review mainly focuses on the pivotal roles of autophagy protein Beclin-1 contributing to the malignant transformation in breast cancer and its impact on various therapeutic options.
Autophagy Protein Beclin-1 and Breast Cancer
Beclin-1 is a protein encoded by the Becn1 gene, which is a mammalian ortholog of the yeast autophagy-related gene 6 (Atg6). Beclin-1 was originally identified as a Bcl-2 interacting protein, and this binding inhibits the association of Beclin-1 with class III PI3K and hence prevents autophagy nucleation phase. Subsequent studies mapped Becn1 gene in close proximity with BRCA1 region on chromosome 17, a locus frequently deleted in breast, ovarian, and prostate cancers [42]. While complete deficiency of Beclin-1 is embryonically lethal, mice harboring mono-allelic deletion of Becn1 exhibited high susceptibility to mammary hyperplasia and suffered increased incidence of spontaneous tumors at various sites. Early studies reported allelic deletion of autophagy regulator Becn1 in breast cancers implicating Becn1 loss, and likely defective autophagy, in tumorigenesis. Consistently, breast cancer tissues showed lower expression profile of Beclin-1 compared with normal breast tissue and ectopic expression of Beclin-1 by mono-allelic Becn1 in MCF-7 breast cancer cells showed decreased proliferation and in vivo tumor formation. These studies identified pro-autophagic protein Beclin-1 as a haplo-sufficient tumor suppressor. Since transformed cells may not tolerate complete loss of Beclin-1, it is indicated that maintaining a single copy and low level of Beclin-1 serves a beneficial way to ensure intact pro-survival autophagy machinery to overcome stress conditions often encountered by cancer cells. Despite the lethality of Becn1−/− during early embryonic stage, mice lacking several other ATGs (e.g. Atg3, Atg5, Atg7, and Atg16) actually survive until birth, ultimately die of metabolic deficiencies during neonatal starvation period [43]. Further observations suggest that the role of autophagy in cancer may be context-dependent and tissue-specific. Remarkably, autophagy promotes tumorigenesis by increasing proliferation of transformed cells rather than facilitating cell survival, and the underlying mechanism remains to be explored. Intriguingly, autophagy increases glucose uptake and facilitates glycolysis in transformed breast cancer cells, as defect in autophagy leads to reduced glycolysis. A speculation involves a supportive role for autophagy in maintaining a pool of healthy mitochondria by degradation of defective mitochondria and thus sustaining the tricarboxylic acid cycle.
Loss of autophagy is associated with a wide array of diseases such as liver failure, inflammatory bowel disease, aging, and cancer. Implication of defective autophagy in human cancer was first reported with a mono-allelic deletion of crucial autophagy regulator BECN1/Beclin-1. Restoration of normal level of Beclin-1 expression in MCF-7 cells suppresses xenograft tumor growth [44], and mono-allelic Becn1+/− mice develop spontaneous lung and liver carcinomas, lymphomas, and even mammary hyperplasia [45]. Complete loss of UVRAG-binding protein Bif-1, a positive regulator of autophagy and Beclin-1-interacting protein, increases spontaneous tumor formation in mice [46].
Recent studies in different tumor cell lines (e.g. breast tumor) have corroborated that tumor resistance to anticancer therapies such as radiation therapy and chemotherapy is often associated with up-regulation of autophagy. Since Becn1 and Atg5 are two essential genes required for autophagy promotion, interfering with Beclin-1 and/or Atg5 expression reduces autophagy and protects against autophagic cell death. However, increasing evidence implicates a paradoxical role of autophagy following anticancer treatment that autophagy induction also mediates antitumor action of therapeutics. Thus, it is critical to understand the pathophysiology of the disease along with functional relevance of autophagy within the tumor to prevent resistance and enhance the effects of anticancer therapies for cancer patients.
Anticancer Therapies Correlated with Autophagy
Since autophagy has been associated with diverse diseases including cancer, it has been a very promising target in breast cancer treatment. Indeed, persistent research on identifying genes, proteins, small molecule compounds, and related molecular mechanisms involved in autophagy regulation and breast cancer eradication has been intensive over the past few years. Despite the uncertainty, substantial progress has been made to prove autophagy-based pharmacotherapy as an attractive new target of great interest to the pharmaceutical industry. Autophagy can be pharmacologically modulated through either stimulation or suppression in numerous pathways. Table 1 shows clinical studies testing the effects of autophagy modulators on breast cancer therapies [47].
Table 1.
Clinical studies testing the effects of autophagy modulators on breast cancer therapiesa
| Autophagy modulation | Phase | Therapeutic regimen | Identifier |
|---|---|---|---|
| Activation | II | Rapamycin+trastuzumab | NCT00411788 |
| I/II | Temsirolimus+neratinib | NCT01111825 | |
| Inhibition | II | HCQ | NCT01292408 |
| I/II | HCQ + ixabepilone | NCT00765765 | |
| I/II | CQ + tamoxifen | NCT01023477 |
aBased on Table 4 of [47].
Autophagy stimulation
Table 2 lists a set of compounds identified as inducers of autophagy (also based on Table 3 of Cheng et al. [47]). Antiestrogen tamoxifen has long been established as a potent inducer of autophagy in various breast cancer cells [59–61]. Screens for chemical modulators of autophagy have revealed a wide array of therapeutic inhibitors of mTORC signaling [48,50,53], including three drugs approved for use in humans (amiodarone, niclosamide, and perhexiline) [62]. mTORC1 inhibitor rapamycin and its analogs (called rapalogs) such as everolimus (RAD001) are shown to enhance the sensitivity of tumors to radiation by induction of autophagy [63,64]. In the everolimus trials, adverse effects (AEs) such as fatigue, stomatitis, anorexia, diarrhea, and metabolic disorders are often observed in patients but usually can be tolerated with dose reduction. Similar AEs are also noticed in the temsirolimus trials, with some milder cases of headache and fever, and severe cases of hyperglycemia, hyperlipemia, and asthenia [65,66]. Natural products such as cyclovirobuxine D (CVB-D), an alkaloid component isolated from the roots of Buxus microphylla var. Sinica, are often used as direct or indirect sources to enhance the efficacy of chemotherapy or ameliorate its side effects [67]. A recent study reported carnosol, a naturally occurring polyphenol, being an inducer of ROS-mediated Beclin-1-independent autophagy and apoptosis in triple negative MDA-MB-231 cell line [68].
Table 2.
Representative compounds known to activate autophagya
| Mode of action | Type of agent | Compound | Chemical structure | Ref. |
|---|---|---|---|---|
| Inhibits mTORC pathway or facilitates autophagosome formation | Inhibitors of mTORC | Amiodarone | 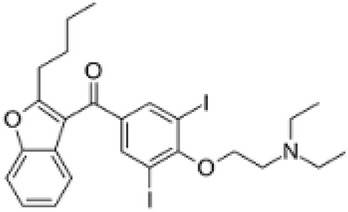 |
[47] |
| Perhexiline | 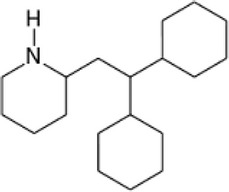 |
[47] | ||
| Mollugin | 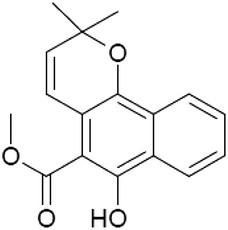 |
[48] | ||
| Inhibitors of tyrosine kinase | Rottlerin |  |
[47] | |
| Thiotanib |  |
[49] | ||
| Inhibitor of Rheb GTPase | Cysmethynil |  |
[50] | |
| Inhibitors of HDAC | Panobinostat |  |
[47] | |
| Vorinostat (SAHA) | 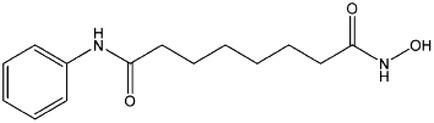 |
[47] | ||
| Givinostat | 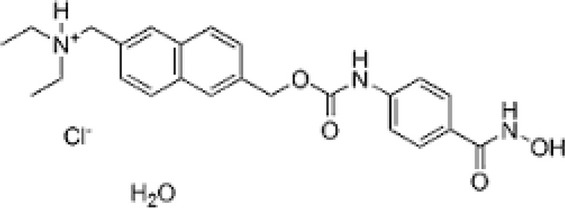 |
[51] | ||
| MRJF4 | 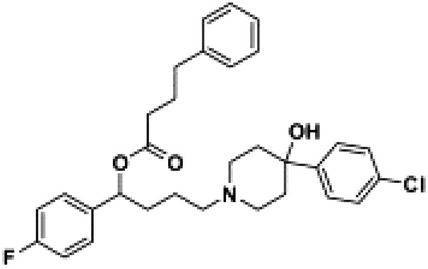 |
[52] | ||
| Activator of MPK | Avicin D | 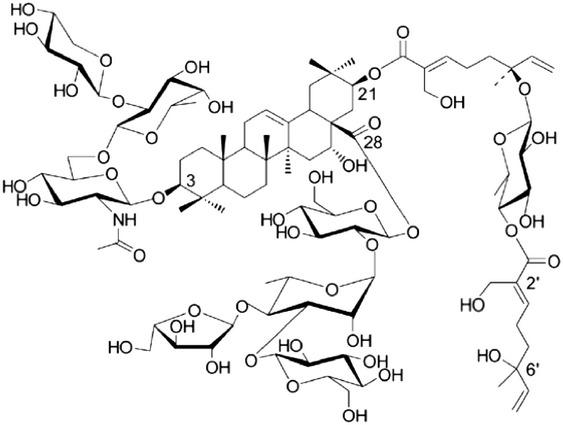 |
[53] | |
| Activates autophagy to eradicate damaged proteins | Inhibitors of proteasome | Bortezomib | 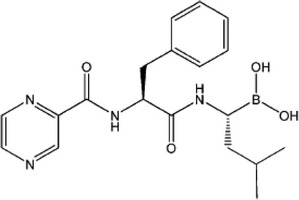 |
[47] |
| Niclosamide | 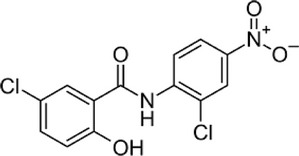 |
[47] | ||
| Disrupts Bcl-2 and Beclin-1 interaction and induces autophagy | BH3 domain mimetics | ABT-737 | 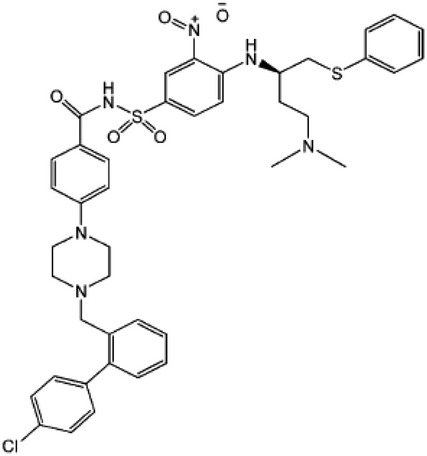 |
[47] |
| HA14-1 | 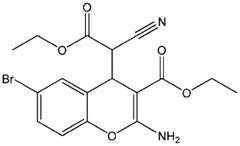 |
[47] | ||
| Inhibitor of Bcl-XL | Z36 |  |
[54] | |
| Increases Beclin-1 and induces autophagy | Antiestrogen | Tamoxifen |  |
[47] |
| Inhibitor of glycolysis | 2-Deoxyglucose |  |
[47] | |
| Promotes caspase-dependent apoptosis or autophagic cell death | Inhibitor of MMP-2 | ARP101 |  |
[55] |
| Stimulator of ER stress | Glucosamine | 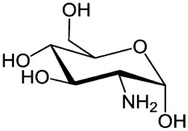 |
[56] | |
| Inhibitor of caspase-9 | FR122047 | 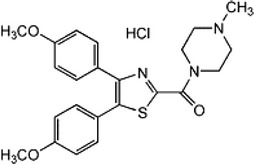 |
[57] | |
| Inhibitor of PI3K | AS605240 | 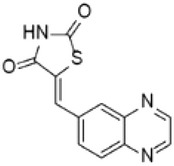 |
[58] |
aModified from Table 3 of [47].
Autophagy can be induced by most of the anticancer drugs due to cellular stress [69]. Some inducers of autophagy, including chemotherapy, augment cell death as a response and eventually cause autophagic cell death [54–57,70]. A recent report on a cell-permeable autophagy-inducing peptide, derived from an evolutionally conserved domain of Beclin-1, confirmed its autophagy-inducing role and anti-viral activity in mice. Other types of drugs with an autophagy-inducing effect also possess potential application in cancer treatment [49,51,58,71]. For instance, histone deacetylase (HDAC) inhibitors (givinostat, vorinostat, panobinostat, etc.) execute anticancer action through induction of autophagic cell death [52]. Niclosamide contributes to the clearance of ubiquitinated proteins through lysosomal pathway caused by proteasome inhibition [72].
Autophagy inhibition
Numerous functional studies have reported that autophagy inhibition can be combined with extant therapies in breast cancer to improve clinical outcome. Table 3 (also based on Table 2 of Cheng et al. [47]) lists a set of compounds identified as inhibitors of autophagy. The inhibitors that act on later stage of autophagy include chloroquine (CQ) and its derivative hydroxychloroquine (HCQ), which can increase lysosomal pH and compromises the digestive activity of hydrolases, ultimately leading to inhibition of fusion of autophagosome with lysosome and degradation of auto-lysosome. However, HCQ appears to be preferred over CQ owing to its more tolerable side effects [74]. It was shown recently that the combination of rapamycin and resveratrol effectively blocks autophagy and induces apoptosis in both estrogen receptor positive and negative breast cancer cells [75]. Given the prevalence of resistance in ER+ breast cancers, autophagy inhibition might serve as an advantageous combination strategy for these subsets of breast cancer patients. Similarly, in a small cohort of breast cancer patients with HER2/c-neu amplification, the concomitant loss of Beclin-1 is significantly associated with better clinical response to trastuzimab, indicative of the cell death-promoting aspect of autophagy in response to the targeted treatment [76]. This evidence supports the hypothesis that defective autophagy functions as a modifier, potentially a fundamental driver, of genomic damage during tumor progression. Doxorubicin predominantly induces autophagy at low doses and apoptosis at high doses. The combination of Bcl-2 siRNA treatment with a low dose of doxorubicin was reported to enhance autophagic cell death and tumor inhibition [77]. Notably, in vitro finding on MDA-MB-231 cell lines by co-treatment of autophagy inhibitor such as CQ and chemotherapeutic drugs with docetaxel-loaded dendritic copolymer nanoparticles has provided evidence for the development of nanomedicine as a valuable method to enhance cancer cell killing [78]. Microtubule stabilizing agents (taxanes) are common anticancer drugs partially for their profound effects on autophagy. Microtubules support the whole autophagosome formation and trafficking process and regulate the two major complexes involved in autophagy initiation: mTORC1 and class III PI3K complex. The cytotoxic effect of taxanes (docetaxel and paclitaxel) has been demonstrated to induce autophagic cell death possibly by blocking autophagosome transport and maturation. Other microtubule-targeting drugs like vinblastine disrupt microtubules and thus decrease autophagic flux, which is important since autophagy is always induced in response to stress posed by chemotherapy [73].
Table 3.
Representative compounds known to suppress autophagya
| Mode of action | Type of agent | Compound | Chemical structure | Ref. |
|---|---|---|---|---|
| Blocks the formation of autophagosomes | Inhibitor of Class III PI3K | 3-Methyladenine (3-MA) | 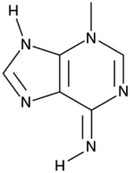 |
[47] |
| Promotes Vps34 complex degradation by promoting Beclin-1 ubiquitination | Inhibitor of ubiquitin-specific peptidase | Spautin-1 | 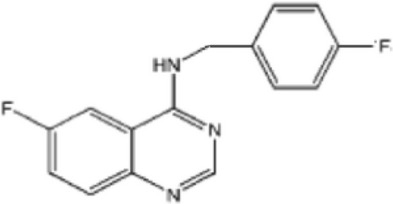 |
[47] |
| Blocks the fusion of autophagosome with lysosome or affects lysosomal proteolysis | Inhibitors of microtubule formation | Nocodazole | 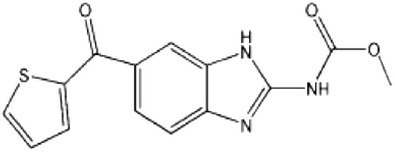 |
[47] |
| Bafilomycin A1 | 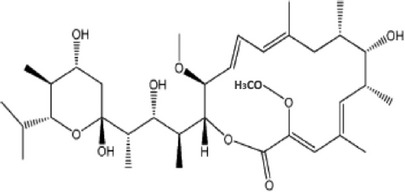 |
[47] | ||
| Paclitaxel (Taxol) | 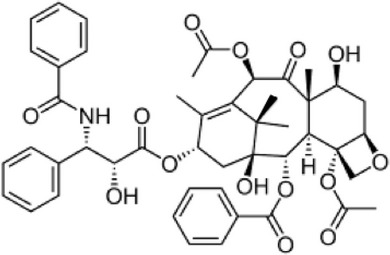 |
[73] | ||
| Docetaxel (Taxotere) |  |
[73] | ||
| Inhibitors of lysosome acidification | CQ |  |
[47] | |
| HCQ |  |
[47] | ||
| Quinacrine (QN) |  |
[70] | ||
| 33b |  |
[70] | ||
| 34b |  |
[70] | ||
| ARN5187 |  |
[71] |
aModified from Table 2 of [47].
bThe names of compounds ‘33’ and ‘34’ were unidentified in literature as of April 2015.
Autophagy-deficient Models for Breast Cancer Study
The majority of proteins that participate in the modulation of autophagy are either tumor suppressors or oncogenes. Therefore, it is not surprising that mechanisms involved in this process largely overlap with signaling pathways implicated in the control of cancer. Before the advent of transgenic techniques, early studies on cancer were largely modeled by tissue culture of cell lines established from human and animal tumors, as well as by inoculation of such cells lines under the skin of immune-deficient mice. However, these models failed to fully recapitulate the subtleties observed in human tumors [79]. It was the eminent developmental biologists Rudolf Jaenisch and Beatrice Mintz who sought to introduce SV40 DNA tumor virus into mice via viral infection of early embryos that shed light on subsequent generation of tumor-prone oncomice-transgenic mice carrying dominant oncogenes [80]. In addition to such transplant tumor models, an ever-expanding set of research tools have emerged to mimic human cancer development and progression.
Perhaps not surprisingly, our knowledge of genetic contribution to human cancer has been further enhanced with the incorporation of tissue-specific gene deletion techniques. Two distinct types of murine models have been employed to characterize autophagy based on the purpose of research. The first type takes advantage of a reporter model system to detect and quantify the level of autophagy in vivo, while the second type modifies the mouse genome to perform global or tissue-specific gene deletions, thereby creating pathological disease conditions [81]. Accordingly, the effect of autophagy on tumorigenesis could be evaluated using different models that recapitulate the deficiency for specific autophagy factors. This section will discuss several types of autophagy knockout murine models based on the implication of the gene in various stages of autophagy (see Table 4, also refer to Table 2 of [15]), plus an additional MMTV/c-neu knockout model that is heavily involved in breast cancer metastasis.
Table 4.
Autophagy-deficient murine models for breast cancer studiesa
| Gene | Genotype | Phenotype | Ref. |
|---|---|---|---|
| RB1CC1 or FIP200 | Tek-Cre;rb1cc1f/f | Perinatal lethal from severe erythroblastic anemia. | [15] |
| rb1cc1f/+;p62+/− | Viable; mammary tumor growth severely impaired in FIP200-null compared with FIP200 and p62 double-knockout mice. | [15] | |
| rb1cc1−/− | Embryonic lethal at E14.5–15.5. | [15] | |
| rb1cc1f/f;MMTV-Cre;MMTV-PyMT | Develop palpable mammary tumor with T50 of 85d; >60% decrease in epithelial surface covered by hyperplastic nodules and 2.5-fold reduction in the average mass of the mammary glands compared with that for rb1cc1f/f;MMTV-PyMT mice. | [82] | |
| rb1cc1f/+;MMTV-Cre;MMTV-PyMT | Develop palpable mammary tumor with T50 of 62d. | [82] | |
| rb1cc1f/f;MMTV-PyMT | Develop palpable mammary tumor with T50 of 56d. | [82] | |
| Beclin-1 | Becn1+/−;MMTV-Wnt1 | Display significantly shorter mammary tumor-free survival (4 vs. 7.2 months) and overall survival compared with Becn1+/+;MMTV-Wnt1. | [83] |
| Tsc1 | tsc1f/f;MMTV-PyMT | Viable | [25] |
aModified from Table 2 of [15].
Ulk1/Ulk2
Ulk1 (mammalian homolog of yeast Atg1) is a member of the ULK kinase complex, which consists of Ulk1, Atg13, RB1CC1, and C12orf44/Atg101. During nutrient stress and the induction phase of autophagy, Ulk1 and Atg13 are liberated from MTORC1 phosphorylation to induce autophagy pathway. The ulk1 conventional knockout mice are phenotypically normal and viable, indicating that starvation-induced autophagy is not impaired in the mutants. Like ulk1 knockouts, ulk2 knockout mice are also viable and show no overt developmental defects due to the likelihood of functional redundancy in mammalian system. However, ulk1 and ulk2 double knockouts die shortly after birth, suggesting the critical role of Ulk1 in survival [15].
FIP200
FIP200 (FAK family-interacting protein of 200 kDa) encodes an evolutionarily conserved protein characterized by a large coiled-coil region containing a leucine zipper motif. FIP200, as a component of ULK1–ATG13–FIP200–ATG101 complex, is shown to be essential for autophagosome formation. FIP200 conditional knockout in the MMTV-PyMT mouse model of human breast cancer showed reduced tumor initiation and progression by both impairing tumor cell proliferation and inducing increased immune surveillance [82].
Ultraviolet irradiation resistant-associated gene
Mono-allelic deletion of ultraviolet irradiation resistant-associated gene (UVRAG) has been reported to be present in numerous human malignancies. Interaction between UVRAG and Beclin-1 via their coiled-coil domain is suggested to promote Vps34 complex binding to and activation by Beclin-1. However, no UVRAG knockout or conditional knockout mouse is currently available [15].
Bif-1
Unlike Becn1 null mice, which are embryonic lethal, Bif-1 knockout mice develop normally but are prone to spontaneous tumors. In contrast to 14.3% of wild-type mice, around 89.7% of Bif-1−/− mice developed spontaneous tumors at 12 months of age [84].
Pik3c3
The main role of PIK3C3 in autophagy is to phosphorylate PI and generate PI3P for phagophore membrane elongation. A global knockout of Pik3c3 in early developmental stage by crossing conditional Pik3c3 allele with Meox1-Cre transgenic strain manifested a high rate of lethality at E7.5, similar to Becn1 null mutants. Like several other autophagy-deficient models, the Pik3c3 heterozygous knockout strain exhibits no obvious phenotype [15].
Bcl-2
In mammals, the Bcl-2 family is categorized into anti-apoptotic members (e.g. Bcl-2 and Bcl-XL) and pro-apoptotic members (e.g. Bax and Bak) with four BH domains, and typically pro-apoptotic Bcl-2 homology 3 (BH3)-only members. The anti-cell death function of Bcl-2 was solely attributed to the inhibition of apoptosis [85,86], until the identification of Beclin-1 as a Bcl-2-binding protein connecting Bcl-2 to a distinct type of cell death—autophagy. Inhibition of autophagy is accomplished by the BH3 domain of Beclin-1 interacting with the BH3-binding groove in Bcl-2/Bcl-XL [87,88]. Bcl-2 mutants with no Beclin-1 binding activity fail to inhibit autophagy under nutrient starvation in Beclin-1-expressing human breast cancer cells. Remarkably, a viral form of Bcl-2, encoded by tumorigenic murine r-herpesvirus 68, binds to Beclin-1 at a much higher level than cellular Bcl-2. Such binding affinity renders the viral Bcl-2 resistant to displacement from Beclin-1 [89].
Atg5 and Atg7
Atg5−/− mice are documented as autophagy deficient, and Atg5−/−;GFP-LC3 mice show no indication of autophagosome formation [15,90]. Mouse models with systemic mosaic deletion of Atg5 or liver-specific Atg7−/− develop liver adenomas. Autophagy-defective hepatocytes in these models exhibit oxidative and genotoxic stress as well as aberrant accumulation of p62/SQSTM1 (p62), damaged mitochondria, ER chaperones, and protein disulfide isomerases due to failure of protein quality control. The accumulation of p62 upon metabolic stress leads to increased ROS production and consequent deregulation of the NF-κB pathway, creating a positive feedback loop for cellular stress. Tumor growth is partially suppressed in Atg7−/− liver devoid of p62, indicating a supporting role of p62 accumulation in liver tumor progression. Spontaneous tumors arisen in these models are benign, autophagy deficient, and uniformly restricted to only one tissue type (fail to exhibit distant metastasis) [33]. Higher-level understanding of autophagy in tumor metastasis has yet to be elucidated, but we know autophagy is required for advanced tumor progression [91].
Beclin-1
Beclin-1 is a core protein constituent of the PtdIns3K complex required for nucleation phase of autophagy, and it contains an N-terminal BH3 domain to be bound and inhibited by Bcl-2/Bcl-2L1. Beclin-1 is expressed at a high level in normal breast epithelial cells but is markedly decreased in breast cancer cell lines such as MCF-7. Perhaps attributed to its cross-talking role in both apoptosis and autophagy, Beclin-1 knockout displays a more severe phenotype than other ATG. Becn1−/− mice die in utero around E7.5d due to developmental failure to close the pro-amniotic canal [92]. Heterozygous deletion of Becn1 promotes spontaneous tumor progression compared with control littermates and that aberrant expression of Beclin-1 in many kinds of tumor tissues correlates with poor prognosis [83,93]. Many breast carcinoma cells lines with deletions of one or more Becn1 alleles and human breast tumors exhibit reduced Beclin-1 levels compared with normal adjacent tissue. Becn1+/− mice, like other pro-autophagy heterozygous mutants of UVRAG or Bif-1, do not show increased frequency of mammary tumors, but are rather susceptible to lymphomas, hepatocellular carcinoma, and lung tumors after long latency through an increase in genetic instability [94]. Additional induced mutant models for Becn1 demonstrated a development-specific role of autophagy in maintaining a pool of undifferentiated lymphocyte progenitor cell [95]. On the other hand, overexpression of Beclin-1 leads to a decrease in MCF-7 cellular proliferation, and breast cancer formation occurs in nude mice. Furthermore, Negri et al. [76] reported that Becn1 heterozygosity suppressed Palb2-associated mammary tumorigenesis by p53-dependent mechanism in Palb2f/f;Wap-Cre;Becn1+/− mice.
To study mammary tumorigenesis in autophagy-defective models, an alternative approach would involve the use of MMTV/c-neu transgenic mice. In contrast to the stochastic occurrence of solitary mammary tumors in transgenic mice carrying the MMTV/c-myc or the MMTV/v-Ha-ras oncogenes, the MMTV/c-neu transgene-bearing mice develop adenocarcinomas that extensively infiltrate the entire mammary epithelium in each gland. Histological analysis of these tumors and surrounding tissues revealed a complete absence of morphologically normal mammary epithelium, suggesting that the expression of the mutant c-neu transgene can sufficiently transform the mammary epithelium [96]. Several cancer cell lines and primary tumor profiling provided evidence for autophagy to be permissive for tumor growth and enhanced in primary tumors, indicating the necessity of autophagy for the quick growth and high metabolic needs of some tumor cells described as ‘addicted to autophagy’. What leads to autophagy upon oncogenic transformation is not precisely known. A proposed genetic model is to generate Becn1−/−;MMTV/c-neu transgenic mice, by crossing Cre-Becn1f/− line with MMTV/c-neu line, in order to assess the characteristics of tumor growth and their susceptibility to apoptosis.
Conclusions and Perspectives
One of the unresolved problems in cancer therapy is the increased tumor resistance to extant treatments, which is a direct consequence of apoptotic defect. Mediating an alternative from of cell death, canonical or non-canonical autophagy, via multiple pathways might be an ultimate solution to maximize cancer cell death.
In this review, we summarized how autophagy affects breast cancer metastasis based on the current literature. However, we are still at the very preliminary phase of understanding the intertwining relationship of autophagy and cancer. As we dig further, it gets clear that autophagy is deeply integrated into diverse biological pathways, involving metabolism, stress response, and programmed cell death. Nowadays, the consensus view is that the role of autophagy in cancer progression is dual-sided with both anti- and pro-metastatic properties at various stages and aspects of cancer. While promoting autophagy to prevent chronic inflammation and persistent tissue damage might benefit cancer prevention, blocking autophagy-mediated survival of tumor cells is probably more promising in the treatment of cancer. Existing antineoplastic regimens including radiation, chemotherapy, HDAC inhibitors (vorinostat, etc.), cytokines (TNFα, IFNγ, etc.), imatinib, rapamycin, arsenic trioxide, and antiestrogen hormonal therapies (tamoxifen, etc.) have been demonstrated to induce autophagy as a pro-survival mechanism for human cancer cell lines. Therefore, the therapeutic efficacy of these agents could be largely improved if autophagy is inhibited. However, we should exercise a reasonable degree of caution in the clinical evaluation of tumor genotype and drug action before turning autophagy inhibitors into all-purpose solution for breast cancer. With this being said, the next decades will inevitably witness the enormous challenge of designing clinical trials to combine targeted drugs for more effective treatment.
Funding
This work was supported by the grants from the National Institutes of Health (Nos. RO1s CA125454 and CA188118), DOD Breakthrough Award (BC140733P1), and the Mary Kay Ash Foundation (to B.P.Z.).
Acknowledgements
We apologize to the many authors whose studies are important but could not be cited due to space limitation.
References
- 1.Wang Y, Zhou BP. Epithelial-mesenchymal transition—a hallmark of breast cancer metastasis. Cancer Hallm 2013, 1: 38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi J, Cao J, Zhou BP. Twist-BRD4 complex: potential drug target for basal-like breast cancer. Curr Pharm Des 2015, 21: 1256–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin Y, Dong C, Zhou BP. Epigenetic regulation of EMT: the Snail story. Curr Pharm Des 2014, 20: 1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin 2008, 40: 643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle 2009, 8: 3267–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su Z, Yang Z, Xu Y, Chen Y, Yu Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 2015, 14: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidler IJ. Metastasis: quantitative analysis of the distribution and fate of tumor emboli labeled with 125I-5-iodo-2′-deoxyuridine. J Natl Cancer Inst 1970, 45: 773–782. [PubMed] [Google Scholar]
- 8.Weiss L. Metastatic inefficiency. Adv Cancer Res 1990, 54: 159–211. [DOI] [PubMed] [Google Scholar]
- 9.Weiss L. The biomechanics of cancer cell traffic, arrest, and intravascular destruction. In: Orr FW, Buchanan MR, Weiss L eds. Microcirculation in Cancer Metastasis. Boca Raton: CRC Press, Inc., 1991, 131–144. [Google Scholar]
- 10.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 1998, 153: 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 2004, 6: 463–477. [DOI] [PubMed] [Google Scholar]
- 12.Roy S, Debnath J. Autophagy and tumorigenesis. Semin Immunopathol 2010, 32: 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JS, Kim YJ, Kim CL, Lee GM. Differential induction of autophagy in caspase-3/7 down-regulating and Bcl-2 overexpressing recombinant CHO cells subjected to sodium butyrate treatment. J Biotechnol 2012, 161: 34–41. [DOI] [PubMed] [Google Scholar]
- 14.Qi Y, Zhang M, Li H, Frank JA, Dai L, Liu H, Zhang Z et al. Autophagy inhibition by sustained overproduction of IL6 contributes to arsenic carcinogenesis. Cancer Res 2014, 74: 3740–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB 3rd. Autophagy: regulation and role in development. Autophagy 2013, 9: 951–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 2009, 186: 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hasima N, Ozpolat B. Regulation of autophagy by polyphenolic compounds as a potential therapeutic strategy for cancer. Cell Death Dis 2014, 5: e1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scarlatti F, Maffei R, Beau I, Ghidoni R, Codogno P. Non-canonical autophagy: an exception or an underestimated form of autophagy? Autophagy 2008, 4: 1083–1085. [DOI] [PubMed] [Google Scholar]
- 19.Orvedahl A, Levine B. Eating the enemy within:autophagy in infectious diseases. Cell Death Differ 2008, 130: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett 1994, 349: 275–280. [DOI] [PubMed] [Google Scholar]
- 21.Lippai M, Low P. The role of the selective adaptor p62 and ubiquitin-like proteins in autophagy. Biomed Res Int 2014, 2014: 832704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci USA 2011, 108: 4788–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011, 13: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008, 181: 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Wei H, Liu F, Guan JL. Hyperactivation of mammalian target of rapamycin complex 1 (mTORC1) promotes breast cancer progression through enhancing glucose starvation-induced autophagy and Akt signaling. J Biol Chem 2014, 289: 1164–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 2012, 31: 4304–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007, 282: 37298–37302. [DOI] [PubMed] [Google Scholar]
- 28.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007, 282: 24131–24145. [DOI] [PubMed] [Google Scholar]
- 29.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012, 19: 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7: 457–465. [DOI] [PubMed] [Google Scholar]
- 31.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, Lin Y et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23: 316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kongara S, Kravchuk O, Teplova I, Lozy F, Schulte J, Moore D, Barnard N et al. Autophagy regulates keratin 8 homeostasis in mammary epithelial cells and in breast tumors. Mol Cancer Res 2010, 8: 873–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debnath J. The multifaceted roles of autophagy in tumorsimplications for breast cancer. J Mammary Gland Biol Neoplasia 2011, 16: 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey KM, Wojtkowiak JW, Hashim AI, Gillies RJ. Targeting the metabolic microenvironment of tumors. Adv Pharmacol 2012, 65: 63–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chabner BA, Roberts TG Jr. Timeline: chemotherapy and the war on cancer. Nat Rev Cancer 2005, 5: 65–72. [DOI] [PubMed] [Google Scholar]
- 36.Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011, 8: 528–539. [DOI] [PubMed] [Google Scholar]
- 37.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011, 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 38.Yao Q, Chen J, Lv Y, Wang T, Zhang J, Fan J, Wang L. The significance of expression of autophagy-related gene Becn1, Bcl-2, and Bax in breast cancer tissues. Tumour Biol 2011, 32: 1163–1171. [DOI] [PubMed] [Google Scholar]
- 39.Aguirre-Ghiso JA. Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007, 7: 834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 2010, 7: 683–692. [DOI] [PubMed] [Google Scholar]
- 41.Jain K, Paranandi KS, Sridharan S, Basu A. Autophagy in breast cancer and its implications for therapy. Am J Cancer Res 2013, 3: 251–265. [PMC free article] [PubMed] [Google Scholar]
- 42.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999, 59: 59–65. [DOI] [PubMed] [Google Scholar]
- 43.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005, 169: 425–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402: 672–676. [DOI] [PubMed] [Google Scholar]
- 45.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003, 112: 1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010, 31: 961–967. [DOI] [PubMed] [Google Scholar]
- 47.Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 2013, 65: 1162–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Wang HD, Zhu JH, Xu JG, Ding K. Mollugin induces tumor cell apoptosis and autophagy via the PI3K/AKT/mTOR/p70S6K and ERK signaling pathways. Biochem Biophys Res Commun 2014, 450: 247–254. [DOI] [PubMed] [Google Scholar]
- 49.Fan J, Dong X, Zhang W, Zeng X, Li Y, Sun Y, Wang S et al. Tyrosine kinase inhibitor Thiotanib targets Bcr-Abl and induces apoptosis and autophagy in human chronic myeloid leukemia cells. Appl Microbiol Biotechnol 2014, 98: 9763–9775. [DOI] [PubMed] [Google Scholar]
- 50.Wang M, Tan W, Zhou J, Leow J, Go M, Lee HS, Casey PJ. A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J Biol Chem 2008, 283: 18678–18684. [DOI] [PubMed] [Google Scholar]
- 51.Campbell GR, Bruckman RS, Chu YL, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV Type 1. J Biol Chem 2014, 290: 5028–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marrazzo A, Fiorito J, Zappalà L, Prezzavento O, Ronsisvalle S, Pasquinucci L, Scoto GM et al. Antiproliferative activity of phenylbutyrate ester of haloperidol metabolite II [(±)-MRJF4] in prostate cancer cells. Eur J Med Chem 2010, 46: 433–438. [DOI] [PubMed] [Google Scholar]
- 53.Xu ZX, Liang J, Haridas V, Gaikwad A, Connolly FP, Mills GB, Gutterman JU. A plant triterpenoid, avicin D, induces autophagy by activation of AMP-activated protein kinase. Cell Death Differ 2007, 14: 1948–1957. [DOI] [PubMed] [Google Scholar]
- 54.Lin J, Zheng Z, Li Y, Yu W, Zhong W, Tian S, Zhao F et al. A novel Bcl-XL inhibitor Z36 that induces autophagic cell death in Hela cells. Autophagy 2009, 5: 314–320. [DOI] [PubMed] [Google Scholar]
- 55.Jo YK, Park SJ, Shin JH, Kim Y, Hwang JJ, Cho DH, Kim JC. ARP101, a selective MMP-2 inhibitor, induces autophagy-associated cell death in cancer cells. Biochem Biophys Res Commun 2011, 404: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 56.Hwang MS, Baek WK. Glucosamine induces autophagic cell death through the stimulation of ER stress in human glioma cancer cells. Biochem Biophys Res Commun 2010, 399: 111–116. [DOI] [PubMed] [Google Scholar]
- 57.Jeong HS, Choi HY, Lee ER, Kim JH, Jeon K, Lee HJ, Cho SG. Involvement of caspase-9 in autophagy-mediated cell survival pathway. Biochim Biophys Acta 2011, 1813: 80–90. [DOI] [PubMed] [Google Scholar]
- 58.Petrilli AM, Fuse MA, Donnan MS, Bott M, Sparrow NA, Tondera D, Huffziger J et al. A chemical biology approach identified PI3K as a potential therapeutic target for neurofibromatosis type 2. Ame J Transl Res 2014, 6: 471–U274. [PMC free article] [PubMed] [Google Scholar]
- 59.Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, Gorski SM. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat 2008, 112: 389–403. [DOI] [PubMed] [Google Scholar]
- 60.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy 2009, 5: 400–403. [DOI] [PubMed] [Google Scholar]
- 61.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, Browning D. A role for macroautophagy in protection against 4-hydroxytamoxifeninduced cell death and the development of antiestrogen resistance. Mol Cancer Ther 2008, 7: 2977–2987. [DOI] [PubMed] [Google Scholar]
- 62.Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One 2009, 4: e7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carew JS, Kelly KR, Nawrocki ST. Autophagy as a target for cancer therapy: new developments. Cancer Manag Res 2012, 4: 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benbrook DM, Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp Oncol 2012, 34: 286–297. [PubMed] [Google Scholar]
- 65.Wolff AC, Lazar AA, Bondarenko I, Garin AM, Brincat S, Chow L, Sun Y et al. Randomized phase III placebo-controlled trial of letrozole plus oral temsirolimus as first-line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol 2013, 31: 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P, Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res 2014, 16: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lu J, Sun D, Gao S, Gao Y, Ye J, Liu P. Cyclovirobuxine D induces autophagy-associated cell death via the Akt/mTOR pathway in MCF-7 human breast cancer cells. J Pharmacol Sci 2014, 125: 74–82. [DOI] [PubMed] [Google Scholar]
- 68.Al Dhaheri Y, Attoub S, Ramadan G, Arafat K, Bajbouj K, Karuvantevida N, AbuQamar S et al. Iratni, Carnosol induces ROS-mediated beclin1-independent autophagy and apoptosis in triple negative breast cancer. PLoS One 2014, 9: e109630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.You J, He Z, Chen L, Deng G, Liu W, Qin L, Qiu F et al. CH05–10, a novel indinavir analog, is a broad-spectrum antitumor agent that induces cell cyclearrest, apoptosis, endoplasmic reticulum stress andautophagy. Cancer Sci 2010, 101: 2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang T, Goodall ML, Gonzales P, Sepulveda M, Martin KR, Gately S, MacKeigan JP. Synthesis of improved lysomotropic autophagy inhibitors. J Med Chem 2015, 58: 3025–3035. [DOI] [PubMed] [Google Scholar]
- 71.Torrente E, Parodi C, Ercolani L, De Mei C, Ferrari A, Scarpelli R, Grimaldi B. Synthesis and in vitro anticancer activity of the first class of dual inhibitors of REV-ERBb and autophagy. J Med Chem 2015, 58: 5900–5915. [DOI] [PubMed] [Google Scholar]
- 72.Gies E. Niclosamide prevents the formation of large ubiquitin-containing aggregates caused by proteasome inhibition. PLoS One 2010, 5: e14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maycotte P, Thorburn A. Targeting autophagy in breast cancer. World J Clin Oncol 2014, 5: 224–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruiz-Irastorza G, Ramos-Casals M, Brito-Zeron P, Khamashta MA. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: a systematic review. Ann Rheum Dis 2010, 69: 20–28. [DOI] [PubMed] [Google Scholar]
- 75.Alayev A, Berger SM, Kramer MY, Schwartz NS, Holz MK. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J Cell Biochem 2015, 116: 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Negri T, Tarantino E, Orsenigo M, Reid JF, Gariboldi M, Zambetti M, Pierotti MA et al. Chromosome band 17q21 in breast cancer: significant association between beclin-1 loss and HER2/NEU amplification. Genes Chromosomes Cancer 2010, 49: 901–909. [DOI] [PubMed] [Google Scholar]
- 77.Akar U, Chaves-Reyez A, Barria M, Tari A, Sanguino A, Kondo Y, Kondo S et al. Ozpolat, Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy 2008, 4: 669–679. [DOI] [PubMed] [Google Scholar]
- 78.Zhang X, Yang Y, Liang X, Zeng X, Liu Z, Tao W, Xiao X et al. Enhancing therapeutic effects of docetaxel-loaded dendritic copolymer nanoparticles by co-treatment with autophagy inhibitor on breast cancer. Theranostics 2014, 4: 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanahan D, Wagner EF, Palmiter RD. The origins of oncomice: a history of the first transgenic mice genetically engineered to develop cancer. Genes Dev 2007, 21: 2258–2270. [DOI] [PubMed] [Google Scholar]
- 80.Jaenisch R, Mintz B. Simian virus 40 DNA sequences in DNA of healthy adult mice derived from preimplantation blastocysts injected with viral DNA. Proc Natl Acad Sci USA 1974, 71: 1250–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hale A, Ledbetter D, Gawriluk T, Rucker EB III. Altering autophagy: mouse models of human disease. In: Bailly Y. ed. Autophagy-A Double-Edged Sword-Cell Survival or Death? InTeOpP, 2013, 121–138. [Google Scholar]
- 82.Wei HJ, Wei S, Gan BY, Peng X, Zou WP, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011, 25: 1510–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, Zhong H et al. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy 2014, 10: 2036–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007, 9: 1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hong Y, Zhou Y, Wang Y, Xiao S, Liao DJ, Zhao Q. PPARgamma mediates the effects of WIN55,212–2, an synthetic cannabinoid, on the proliferation and apoptosis of the BEL-7402 hepatocarcinoma cells. Mol Biol Rep 2013, 40: 6287–6293. [DOI] [PubMed] [Google Scholar]
- 86.Wang Y, Zhou YT, Zhao Q. The effect of the activation of cannabinoid receptor on the proliferation and apoptosis of hepatoma HepG2 cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2010, 26: 344–347. [PubMed] [Google Scholar]
- 87.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J 2007, 26: 2527–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Feng W, Huang S, Wu H, Zhang M. Molecular basis of Bcl-xL's target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol 2007, 372: 223–235. [DOI] [PubMed] [Google Scholar]
- 89.Ku B, Woo JS, Liang C, Lee KH, Hong HS, E X, Kim KS et al. Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog 2008, 4: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Terada M, Nobori K, Munehisa Y, Kakizaki M, Ohba T, Takahashi Y, Koyama T et al. Double transgenic mice crossed GFP-LC3 transgenic mice with alphaMyHCmCherry-LC3 transgenic mice are a new and useful tool to examine the role of autophagy in the heart. Circ J 2010, 74: 203–206. [DOI] [PubMed] [Google Scholar]
- 91.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 2011, 193: 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin-1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003, 100: 15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang P, Mizushima N. Autophagy and human diseases. Cell Res 2014, 24: 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zarzynska JM. The importance of autophagy regulation in breast cancer development and treatment. Biomed Res Int 2014, 2014: 710345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Arsov I, Adebayo A, Kucerova-Levisohn M, Haye J, MacNeil M, Papavasiliou FN, Yue Z et al. A role for autophagic protein beclin 1 early in lymphocyte development. J Immunol 2011, 186: 2201–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54: 105–115. [DOI] [PubMed] [Google Scholar]