

Background and Purpose—
Cardiovascular risk factors significantly increase the risk of developing Alzheimer disease. A possible mechanism may be via ischemic infarction–driving amyloid deposition. We conducted a study to determine the presence of β-amyloid in infarct, peri-infarct, and hemispheric areas after stroke. We hypothesized that an infarct would trigger β-amyloid deposition, with deposition over time.
Methods—
Patients were recruited within 40 days of acute ischemic stroke and imaged with computed tomographic or magnetic resonance imaging and Pittsburgh compound B (11C-PiB) positron emission tomographic scans. Follow-up positron emission tomographic scanning was performed in a subgroup ≤18 months after the stroke event. Standardized uptake value ratios for regions of interest were analyzed after coregistration.
Results—
Forty-seven patients were imaged with 11C-PiB positron emission tomography. There was an increase in 11C-PiB accumulation in the stroke area compared with a reference region in the contralesional hemisphere, which was not statistically significant (median difference in standardized uptake value ratio, 0.07 [95% confidence interval, −0.06 to 0.123]; P=0.452). There was no significant increase in the accumulation of 11C-PiB in the peri-infarct region or in the ipsilesional hemisphere (median difference in standardized uptake value ratio, 0.04 [95% confidence interval, −0.02 to 0.10]; P=0.095). We repeated 11C-PiB positron emission tomography in 21 patients and found a significant reduction in accumulation of 11C-PiB between regions of interest (median difference in standardized uptake value ratio, −0.08 [95% confidence interval, −0.23 to −0.03]; P=0.04).
Conclusions—
There was no significant increase in 11C-PiB accumulation in or around the infarct. There was no increase in ipsilesional hemispheric 11C-PiB accumulation over time. We found no evidence that infarction leads to sustained or increased β-amyloid deposition ≤18 months after stroke.
Keywords: Alzheimer disease, follow-up studies, positron emission tomography, risk factors
There is increased incidence of cognitive dysfunction after stroke. One in 10 develops vascular cognitive impairment after a single stroke and 1 in 3 after recurrent stroke.1 Risk factors that predispose to atherosclerosis are also independently associated with an increased risk of dementia. In a review, we summarized the evidence supporting an increased risk of developing Alzheimer disease (AD) and vascular cognitive impairment in those with cardiovascular risks.2 Although there is evidence of considerable overlap between AD and vascular cognitive impairment based on histopathologic survey,3 the concomitant presence of AD and cerebrovascular pathology is not regarded as proof of a causal relationship between the 2.
It became possible to quantify cerebral β-amyloid (Aβ) in vivo using the radioligand N-methyl-11C[2-(4′-methylaminophenyl)-6-hydroxybenzothiazole] with positron emission tomographic (PET) imaging in 2004. This compound is generally referred to as Pittsburgh compound B (11C-PiB).4 There are now many radioligands available for amyloid PET imaging, labeled with F-18 rather than C-11.5 There have been many studies on the use of 11C-PiB-PET in differentiating AD from other dementias and in predicting conversion of mild cognitive impairment to AD.6,7 The latest AD diagnostic guidelines recommend the use of 11C-PiB-PET as an ancillary diagnostic test.8
Extracellular neuritic plaque, consisting Aβ deposits, is a core pathological feature of AD. What triggers Aβ deposition is unknown, but it is hypothesized that hypoxia and ischemia may play a role. The association between acute ischemia or hypoxia and cerebral Aβ deposition has been largely based on animal studies, whereas human studies have been mostly negative.9,10 Lee et al11 reported an increase in serum Aβ in patients with acute stroke compared with stroke-free controls. Imaging studies by Marchant et al12 showed no correlation between the burden of cerebral vascular lesions and the degree of 11C-PiB accumulation.
The link between vascular risk factors, acute vascular events, and Aβ formation is the subject of much debate,13,14 but it is now generally accepted that vascular risk factors, such as hypertension and diabetes mellitus, independently increase the risk of developing AD.15 It has been suggested that a synergistic relationship may exist between the presence of vascular risk factors/stroke and the development of AD.16 The recent and ongoing research by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) also suggests that a relationship may exist between vascular risk factors and the development of AD.17 However, there is no research to date that has been able to demonstrate a causative link between vascular risk factors and acute vascular lesions in the brain and formation of Aβ.18
We hypothesized that an acute ischemic stroke would trigger the formation and deposition of Aβ in the infarct and peri-infarct areas. In view of the recent literature on vascular risk factors, stroke, and AD,19 we further hypothesized that accumulation of Aβ would increase over time, with higher global Aβ load both in the ipsilesional cerebral hemisphere and throughout the brain. To ascertain the presence of Aβ deposition, we used 11C-PiB-PET. We previously published that 11C-PiB accumulation was acutely increased in the region of the infarct but not in the whole brain generally.20 Our findings were based on a cross-sectional analysis of 11C-PiB-PET in 21 patients with acute stroke. In this study, we describe the assessment of the cross-sectional component in a larger cohort of 47 patients with ischemic stroke and present findings from a longitudinal comparison in patients with repeat 11C-PiB-PET in ≤18 months after stroke. To our knowledge, this is the first study that evaluates both acute and long-term effects of ischemic stroke on amyloid formation.
Methods
We included patients who presented with a first-ever acute ischemic stroke and could be scanned with 11C-PiB-PET within 40 days of the event. Patients were recruited from the stroke unit, Austin Hospital, Melbourne, Victoria, Australia. We excluded patients who had a contraindication to PET imaging or a history of brain stem stroke, dementia or other neurodegenerative disorders, delirium, previous head trauma, tumors, previous brain surgery, and previous symptomatic intracerebral hemorrhage. Stroke was classified based on topography.21 The sample included 21 patients, previously published.20
At baseline, we collected demographic details, vascular risk factor profiles, and imaging details; stroke topography and hemorrhagic transformation of infarct, the presence of white-matter lesions (WMLs), and the presence of carotid stenosis using carotid Doppler. This study was approved by the Austin Human Research Ethics Committee, and all participants provided written informed consent before participation.
Imaging
All subjects underwent a baseline brain 11C-PiB-PET and magnetic resonance imaging (MRI) for anatomic coregistration. The MRI sequences included T1- and T2-weighted imaging, diffusion-weighted imaging, apparent diffusion coefficient, and gradient echo images. In selected patients, the MRI was performed with gadolinium contrast administration. Patients were requested to return for a second 11C-PiB-PET ≤18 months after a stroke.
PET Image Acquisition
Each subject received ≈370 MBq of 11C-PiB intravenously for 1 minute. A 30-minute acquisition in 3-dimensional (3D) mode 40 minutes after the injection was performed with a Phillips Allegro PET camera (Philips Medical Systems, Cleveland, OH). A transmission scan was performed for attenuation correction. PET images were reconstructed using a 3D row action maximum likelihood algorithm.
Image Analysis
Images were processed using a semiautomatic region-of-interest (ROI) method. A preset template of narrow cortical ROI was applied to the 11C-PiB scan via placement on the subjects’ coregistered MRI by an operator who was blinded to the subject’s clinical status. Manual adjustments on the MRI were made to ensure minimal overlap with white matter and cerebrospinal fluid. Coregistration of MRI with the PET images was performed with SPM5 (Wellcome Trust). The ROI template was placed on the coregistered MRI and transferred to the coregistered PET images.
Follow-up 11C-PiB images were coregistered with the initial images. The same ROI templates were applied to baseline and follow-up scans. Standardized uptake value values (SUVs) for 11C-PiB were calculated for brain regions examined, and SUV ratios (SUVRs) were generated by dividing regional SUVs by the cerebellar cortex SUV. Neocortical Aβ burden was expressed as the average SUVR of the area-weighted mean of frontal, superior parietal, lateral temporal, lateral occipital, and anterior and posterior cingulate regions.7 ROI measurements were averaged for each hemisphere (hemisphere-specific SUVR).
Stroke regions were identified on brain imaging, and hand-drawn ROIs were used to delineate the infarct and peri-infarct regions. Mirror image ROIs were mapped onto the contralateral hemisphere for comparison. The SUVRs for the infarct and peri-infarct regions, as well as their respective mirror images, were calculated in the same way. The SUVRs were termed infarct-specific and peri-infarct SUVRs. An example of the infarct and peri-infarct ROIs used in the assessment is shown in Figure 1. The same regions were applied for the analysis of the second 11C-PiB-PET scan.
Figure 1.
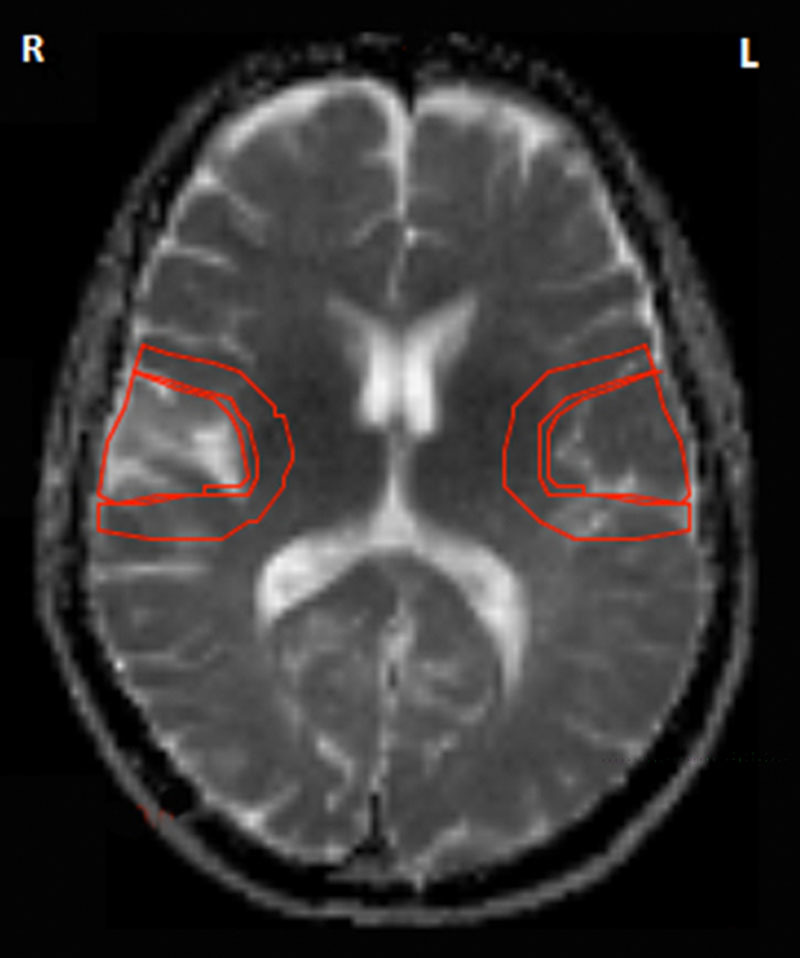
An example of stroke-specific regions of interest showing an area of ischemic infarct (right temporal region) with peri-infarct region and the mirror regions in the contralateral hemisphere. There is no evidence of hemorrhagic transformation. The representative axial image is based on an magnetic resonance apparent diffusion coefficient image at the level of the internal capsule.
Correction of SUVR
In calculating the neocortical SUVR, it was possible that the SUVR may have been affected by the overlap of a predetermined ROI on a stroke region. This potential bias was corrected by reviewing all the images in the patient group and manually removing ROIs on the 11C-PiB-PET scan images that overlapped with infarct regions. The neocortical SUVR was then calculated using all the original ROIs and then recalculated excluding ROIs that overlapped an infarct region. The mean values for the neocortical SUVR were then compared to assess correlation. The resulting Pearson coefficient was 1.00, suggesting that overlap of infarct ROI had no significant effect on neocortical SUVR.
Hemorrhagic Transformation
In our previous study, we noted that patients with an increased SUVR in the infarct region were likely to have variable degrees of hemorrhagic transformation within the infarct.22 We collected data in all patients to determine the presence and degree of hemorrhagic transformation, so that this could be included in analyses of 11C-PiB retention as a confounding variable. We analyzed the stroke-specific SUVR of the infarct and peri-infarct areas to study 11C-PiB retention over time using the same template for infarct and peri-infarct areas. The degree of hemorrhage was based on the European Cooperative Against Stroke Study (ECASS) classification.23 The ECASS classification defines petechial infarction as HI1 (minimal petechiae) or HI2 (confluent petechiae), whereas a hematoma is classified as PH1 (≤30% of the infarcted area with some mild space-occupying effect) and PH2 (>30% of the infarcted area with significant space-occupying effect or clot remote from infarcted area). In our classification, no differentiation was made between the 2 subclassifications of either petechial hemorrhage or hematoma (details are in given Table 1).
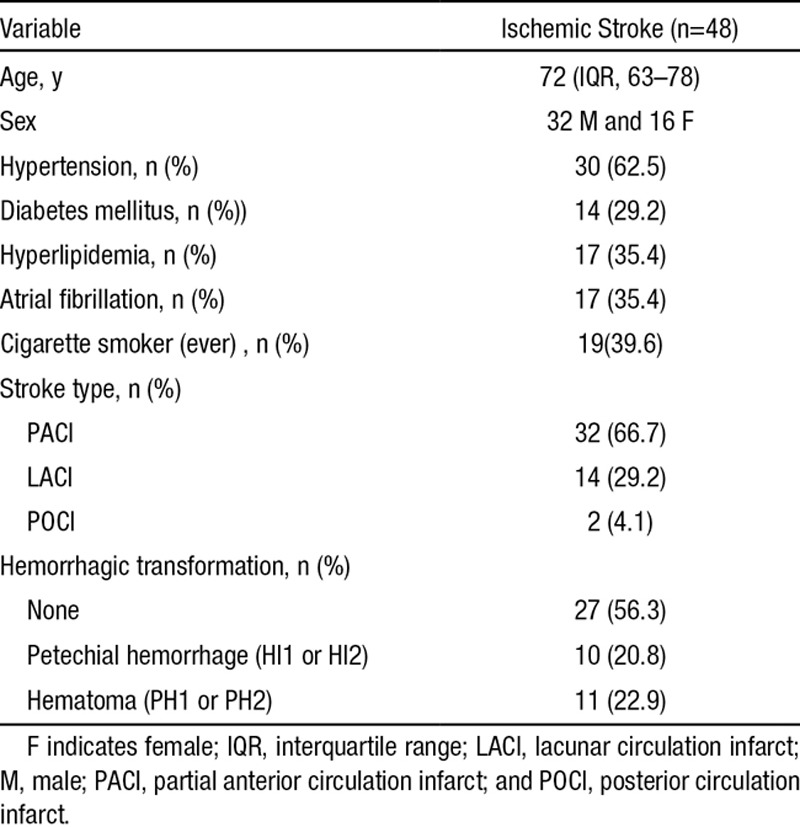
Table 1.
Patient Demographic Details and Risk Factor Profiles: The Stroke Classification Is Based on the Oxfordshire Classification,21 and the Degree of Hemorrhagic Transformation Is Based on the Criteria Used in the European Cooperative Against Stroke Study I Trial23
Outcome Measures
The outcome measure used was the difference in SUVRs. A cutoff of SUVR of 1.40 was used to dichotomize high and low 11C-PiB retention, based on previous studies performed by our group.24 In stroke-specific SUVR, this was calculated in the infarct and peri-infarct areas by subtracting the SUVR for the mirror reference regions from that of the infarct and peri-infarct regions. In hemisphere-specific SUVR, the difference was the SUVR between the contralesional and ipsilesional hemispheres. In the follow-up studies, we compared the difference in stroke-specific SUVR (infract and peri-infarct) and hemisphere-specific SUVR between the first and second PET scans.
At baseline, we determined the difference in the hemisphere-specific and stroke-specific SUVRs by calculating the difference in values generated from both sides of the brain. In the follow-up scans, the difference in neocortical, hemisphere-specific, and stroke-specific SUVRs was calculated. We also assessed whether the differences in SUVR were influenced by patient factors, such as the presence of chronic cerebral ischemia, using the burden of WMLs based on Fazekas scores25 and the presence of carotid disease as surrogate markers.
Statistical Analyses
Statistical analyses were conducted using Stata version 11 IC software (StataCorp, College Station, TX). Patient characteristics and demographics were described using descriptive statistics. Because of the nature of the distributions, the SUVR values were expressed as median (interquartile range) values and tested using Wilcoxon signed-rank test. P values of <0.05 were considered as indicative of statistical significance.
Results
Patient Details
Forty-eight patients with acute unilateral anterior circulation ischemic infarction were recruited for the study. In analysis of the stroke-specific SUVR, we excluded 1 patient with bilateral infarcts. The demographic data and other details of the sample are shown in Table 1. The mean age of the sample was 70.38 years (interquartile range, 63–78), and 32 (67%) were men. Hypertension was the most prevalent risk factor, affecting 62.5%, and diabetes mellitus the least common (29.2%). The commonest stroke type was partial anterior circulation infarct observed in 32 patients. Twenty patients received intravenous recombinant tissue-type plasminogen activator. Hemorrhagic transformation of the infarct was observed in 21 patients (43.3%) and consisted of those with either petechial hemorrhage or hematoma formation seen in 10 and 11 patients, respectively. Thirteen of the patients with hemorrhagic transformation were treated with recombinant tissue-type plasminogen activator. The median time to scan was 12 days (range, 2–113).
Repeat Scan
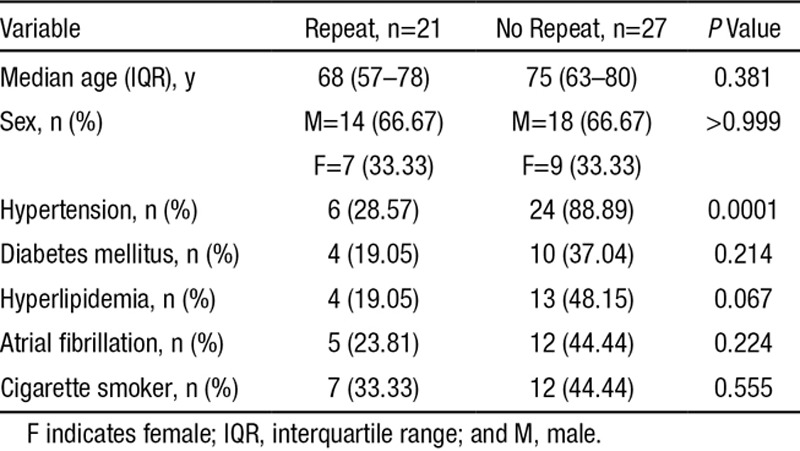
Twenty-one patients returned for a repeat scan. The 27 who did not have repeat scans were deceased, unwell, or unwilling to return for medical or social reasons. There was no significant difference between people who did and did not have a second 11C-PiB-PET scan in the baseline demographics except that there were significantly more patients with hypertension in the group who declined to return (P=0.0001; Table 2). The median time to the second scan was 222 days (range, 8–455). In the repeat group, 8 of the 21 had evidence of hemorrhagic transformation during their initial presentation.
Table 2.
Comparison of the Demographic Features Those of Patients With and Without Repeat 11C-Pittsburgh Compound B Positron Emission Tomography Scanning
Baseline Results of 11C-PiB-PET Scans
Stroke-Specific SUVR
The SUVRs of the infarct and peri-infarct areas were >1.4 in 27 (57.9%) and 29 (61.8%) patients, respectively. In contrast, the corresponding SUVRs in the reference region increased above the cutoff value in 24 (51.1%) and 29 (61.8%) patients, respectively. In 25 (53.2%) patients, the stroke-specific SUVR for the infarct region was higher than that for the reference region (Figure 1). The median difference in the SUVR for the infarct region and the contralateral mirror region was 0.07 (95% confidence interval [CI], −0.06 to 0.13), suggesting the SUVR for the infarct region that was nominally higher but not statistically significant (P=0.452; Table 3).
Table 3.
Difference in Stroke-Specific SUVR Between the Regions of Interest (Infarct and Peri-Infarct Regions) Compared With the Reference Region in the Contralesional Hemisphere
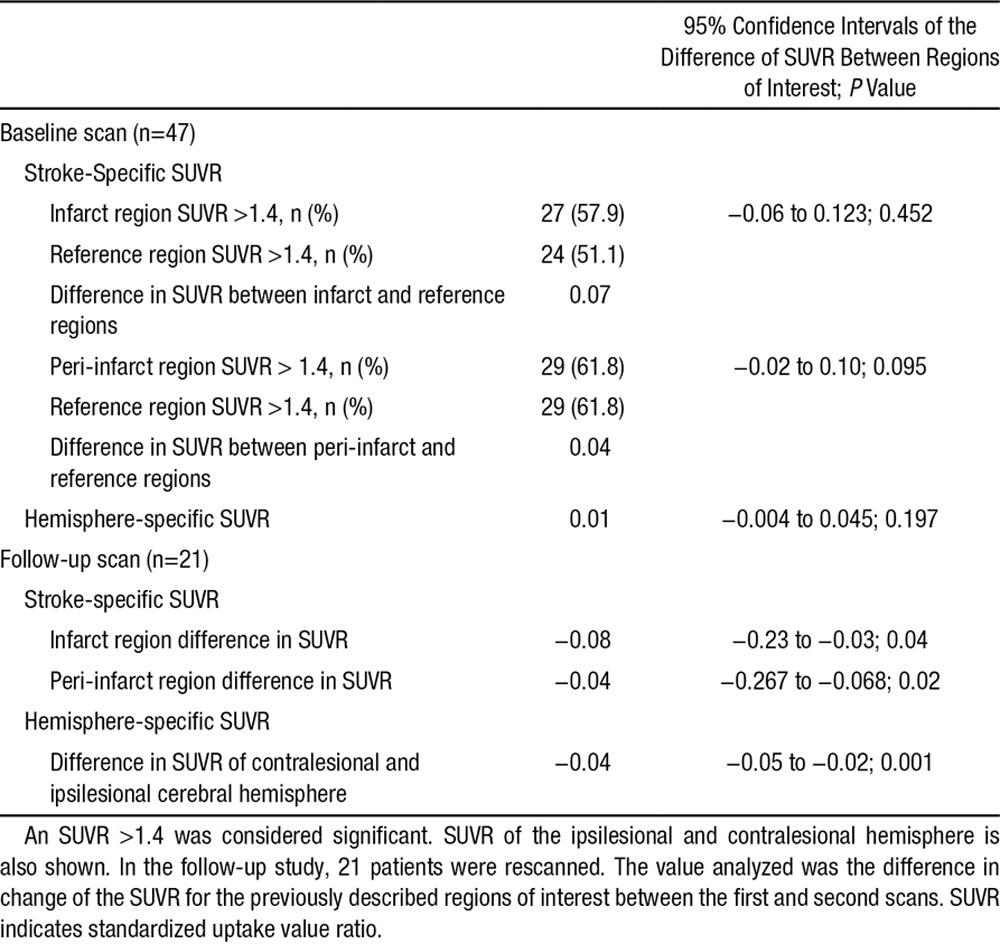
Similarly, in the peri-infarct region, we found that 28 (56.2%) patients had a higher SUVR compared with the contralateral side. The difference in the stroke-specific SUVR for the peri-infarct region was 0.04 (95% CI, −0.02 to 0.10). Similar to the SUVR for the infarct region, the difference in the peri-infarct SUVR was not significant (P=0.095).
Hemisphere-Specific SUVR
The hemisphere-specific SUVR was analyzed based on the difference between the 2 cerebral hemispheres in the same patient. Twenty-nine (61.7%) patients had a higher SUVR for the stroke-affected hemisphere. The median difference of the hemisphere-specific SUVR was 0.01 (95% CI, −0.004 to 0.045), and although marginally higher, the higher SUVR in the stroke-affected hemisphere was not significant (P=0.197).
Hemorrhagic Transformation
Twenty-one of 47 patients had hemorrhagic transformation of the infarct as defined above. Visual assessment of the scans showed that 16 of these 21 patients had higher 11C-PiB focal accumulation in the region of the infarct (Figure 2). The breakdown of patients is shown in Table 1. There was no statistical difference in the number of patients with and without hemorrhagic transformation (P=0.576). There was no significant effect of hemorrhage on the difference of stroke-specific SUVR or the hemisphere-specific SUVR.
Figure 2.
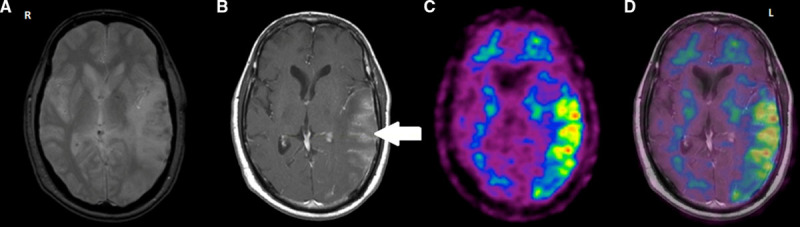
An example of significant 11C-Pittsburgh compound B (PiB) accumulation in an area of infarct with hemorrhagic transformation. There is an area of hypointensity on gradient echo (GRE) seen in the left temporoparietal region (2a) that corresponds to the region of contrast enhancement within the infarct indicated by the white arrow on T1-weighted imaging (2b). The contrast enhancement suggests increased permeability of the blood–brain barrier, and the hypointense areas on GRE indicate microbleeds. The higher accumulation of PiB is represented by the yellow and red areas in the left temporoparietal region (2c) where the ischemic infarct occurred. There is no tracer retention seen in the contralateral mirror region or in other parts of the brain. The infarct-specific standardized uptake value ratios in this case was ≈1.9. The overlap (2d) of the positron emission tomographic and T1-weighted images shows the exact match of the area of increased vascularity and PiB accumulation.
Effect of Carotid Stenosis
Carotid stenosis was assessed using Doppler ultrasound in all 47 patients. The patients were dichotomized based on a neocortical SUVR cutoff of 1.4 and divided into those with nonsignificant or significant stenosis (>70% occlusion of the internal carotid artery). There were 6 patients with an SUVR >1.4, and 2 of these patients had significant carotid disease. The effect of carotid disease on neocortical SUVR was found to be nonsignificant (P=0.573). Analysis of the presence of significant carotid stenosis on stroke-specific SUVR was also nonsignificant (P=0.350).
Follow-Up 11C-PiB-PET Scan
Stroke-Specific SUVR
The median difference in stroke-specific SUVR of the infarct zone was −0.08 (95% CI, −0.23 to −0.03; P=0.04): evidence of a substantial decrease over time. The stroke-specific SUVR of the peri-infarct region also decreased with time with a median difference of −0.04 (95% CI, −0.267 to −0.068; P=0.02). Both of these decreases were statistically significant and were not influenced by the presence of hemorrhage in the initial scan (Table 3).
Hemisphere-Specific SUVR
The median change in the difference of the hemisphere-specific SUVR of the first and second scans was −0.04 (95% CI, −0.05 to −0.02), again evidence of a decrease with time. The decrease was statistically significant (P=0.001), and this decrease was similarly not influenced by the presence of hemorrhage.
Effect of WML
The possible influence of periventricular and subcortical WML on the change in SUVR was analyzed in 18 of the 21 patients with follow-up 11C-PiB-PET. The remaining 3 patients did not have suitable MRI to allow for a proper visual assessment of WML using the Fazekas score. On the basis of analyses, the decrease in hemisphere-specific SUVR was significantly affected by the presence of periventricular WML (P=0.03) but not by the presence of deep WML (P=0.06). The presence of periventricular or deep WML did not have a significant effect on the difference in SUVR of the infarct (P=0.09 and 0.14, respectively) or peri-infarct (P=0.54 and 0.74, respectively) regions.
Discussion
In this study, we explored the possible relationship between an acute ischemic infarct and Aβ deposition using 11C-PiB-PET. We hypothesized that acute ischemia would trigger Aβ formation and deposition in the infarct area, including the ipsilateral cerebral hemisphere. In addition, we theorized that the increased Aβ deposition would continue over time. In our previous article, we presented preliminary results that seemed to support this hypothesis in a group of patients with ischemic stroke and hemorrhagic transformation.22 We found an increased accumulation of 11C-PiB in the infarct core but no increased accumulation of 11C-PiB in the ipsilateral cerebral hemisphere. In addition to the standard SUVR used in the previous study, we included manually drawn ROIs to include specific infarct and peri-infarct areas. The same approach was applied to the follow-up studies.
Our results show that there was no significant increase of PiB accumulation in the infarct and peri-infarct areas compared with their mirror regions in the contralateral hemisphere. This differs from our initial findings.20 There are many possible reasons for this difference. In this study, we assessed a larger number of participants and applied a more accurate determination of SUVR. We also obtained follow-up imaging in a subgroup of patients, allowing direct comparison of findings in individual patients over time. We corrected for potential bias in the event of coincidental overlap of the infarct area and neocortical regions used in determining the overall SUVR in each hemisphere.
We previously reported a significant increase in SUVR in the infarct region, which was not reflected in a general increase in SUVR globally.20 We found that 16 of the 47 patients showed a significant accumulation of 11C-PiB in the stroke region (Figure 2). When the images were closely analyzed, we found that 81% of patients with a higher infarct SUVR had variable amounts of hemorrhagic transformation. The large number of patients with hemorrhagic transformation in our group is likely due to the fact that assessment was mostly based on gradient echo images. These are more sensitive to the presence of blood products. In addition, we made no distinction between the degree or type of hemorrhagic transformation.
The role of vascular pathology in patients with suspected dementia is complex, and there are no clear answers yet on the likely role of vascular risk factors in the development of AD-related pathology. We assessed the effect of vascular disease on 11C-PiB retention and found no association between the presence of significant carotid disease (based on carotid Doppler) and Aβ deposition based on neocortical SUVR. Our findings are in contrast to a previous study that demonstrated a significant association between carotid stenosis and ipsilateral Aβ deposition based on PET imaging.26 We have demonstrated no association in a larger cohort of patients, and we would also argue that concordance between laterality of carotid stenosis and Aβ deposition on PET should not be associated given the collateral cerebral arterial network. However, the primary aim of our study was not to examine the relationship between carotid stenosis and Aβ deposition and as such, the results should be interpreted accordingly.
We also investigated the potential role of chronic small vessel ischemia and Aβ deposition. We found no significant association between the presence of WML and change in stroke-specific SUVR; however, we did find that significant decrease in hemisphere-specific SUVR was more likely in patients with some degree of WML. Our study was not designed to determine an association between WML and neocortical SUVR. There is growing evidence linking WML and Aβ deposition, but the association has been variable.27,28
The finding of a nonsignificant higher 11C-PiB retention in some patients is intriguing. This finding most likely reflects a breakdown of the blood–brain barrier (BBB),23 suggesting that the higher 11C-PiB accumulation is attributable to the leak of the radioactive ligand across the damaged BBB.15,29 It has been demonstrated that the BBB is impaired in the setting of an ischemic infarct29 and that this damage is likely to permit diffusion of 11C-PiB across the BBB.30 The fact that significant decrease in the hemisphere-specific SUVR was more likely in patients with some degree of WML may also indicate the role of inherent weakness of the small vessels because of arteriolosclerosis resulting from the effects of vascular risk factors, such as hypertension and diabetes mellitus.
However, we do not discount the possibility that the accumulation of 11C-PiB may be due to acute Aβ formation in the regions of the infarct. Garcia-Alloza et al31 demonstrated the formation of Aβ in the infarct region of induced stroke in a transgenic amyloid precursor protein mouse model. The accumulation of Aβ was shown using real-time microscopy. This was a transient phenomenon and likely a reflection of the role of amyloid precursor protein in the acute inflammatory phase. It should be noted that 11C-PiB does not significantly bind to nonsoluble Aβ but rather to aggregated fibrils in neuritic plaque.5 So, although this transient Aβ formation is likely to reflect an amyloid precursor protein–driven response to injury in the first 40 days after stroke, binding of 11C-PiB to soluble species is not, further suggesting that 11C-PiB accumulation in the stroke area does not reflect specific binding but leakage of a radiolabeled substance across the damaged BBB.
We observed no significant 11C-PiB accumulation in the infarct, peri-infarct, or stroke-affected hemispheric areas compared with the normal side in the follow-up phase of the study. The initial increase in 11C-PiB accumulation in the area of infarct was no longer apparent. In fact, we found a significant decrease in the stroke-specific and hemisphere-specific SUVRs. This may have been the result of involutional changes after stroke resulting in decreased binding of 11C-PiB.
Our study saw a high attrition rate because of the inability to attend follow-up and death. This might have affected our findings, but we did not find a significant difference in the baseline demographic or imaging characteristics of the patients who returned for a second scan and those who did not aside from incident hypertension. We also acknowledge that we were able to do repeat scans in only 21 patients and ≤18 months after the initial stroke. A similar study in a larger number of patients with periodic repeat scans over a longer period of time may yield results different from ours. Unlike the study design stage, when the concept of power could be useful, once the study is completed, a much more relevant measure is the precision of the effect observed suggested by the 95% CI, which we have indicated for all the significant observed effects.
Summary
Overall, we conclude that an acute ischemic stroke does not lead to an increase in Aβ deposition in the 18 months after stroke, either in the stroke region or across the brain generally. There was an initial increase in 11C-PiB accumulation observed in the infarct region. On the basis of our findings, the possibility that the increased accumulation was because of transient Aβ accumulation is unlikely. Furthermore, the 11C-PiB accumulation was not sustained over time and most likely represents increased permeability of the BBB caused by ischemia. Our findings are in keeping with the results published by Marchant et al12 and Mok et al32 who also showed no significant increase in 11C-PiB accumulation in patients with chronic cerebral vascular lesions.
Acknowledgments
Statistical analysis was conducted by L. Churilov, Florey Institute of Neuroscience and Mental Health, Melbourne, Australia, and University of Melbourne, Australia.
Sources of Funding
This study was funded, in part, by National Health and Medical Research Council, Australia and support was provided by the Victorian State Government, Australia, via the Operational Infrastructure Scheme to the Florey Institute.
Disclosures
None.
Footnotes
Drs Donnan and Brodtmann contributed equally.
References
- 1.Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol. 2009;8:1006–1018. doi: 10.1016/S1474-4422(09)70236-4. doi: 10.1016/S1474-4422(09)70236-4. [DOI] [PubMed] [Google Scholar]
- 2.Sahathevan R, Brodtmann A, Donnan GA. Dementia, stroke, and vascular risk factors; a review. Int J Stroke. 2012;7:61–73. doi: 10.1111/j.1747-4949.2011.00731.x. doi: 10.1111/j.1747-4949.2011.00731.x. [DOI] [PubMed] [Google Scholar]
- 3.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. J Neurol Sci. 2007;257:80–87. doi: 10.1016/j.jns.2007.01.045. doi: 10.1016/j.jns.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 4.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 5.Rowe CC, Villemagne VL. Brain amyloid imaging. J Nucl Med. 2011;52:1733–1740. doi: 10.2967/jnumed.110.076315. doi: 10.2967/jnumed.110.076315. [DOI] [PubMed] [Google Scholar]
- 6.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 7.Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popa-Wagner A, Schröder E, Walker LC, Kessler C. beta-Amyloid precursor protein and ss-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion: effect of age. Stroke. 1998;29:2196–2202. doi: 10.1161/01.str.29.10.2196. [DOI] [PubMed] [Google Scholar]
- 10.Aho L, Jolkkonen J, Alafuzoff I. Beta-amyloid aggregation in human brains with cerebrovascular lesions. Stroke. 2006;37:2940–2945. doi: 10.1161/01.STR.0000248777.44128.93. doi: 10.1161/01.STR.0000248777.44128.93. [DOI] [PubMed] [Google Scholar]
- 11.Lee PH, Bang OY, Hwang EM, Lee JS, Joo US, Mook-Jung I, et al. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J Neural Transm. 2005;112:1371–1379. doi: 10.1007/s00702-004-0274-0. doi: 10.1007/s00702-004-0274-0. [DOI] [PubMed] [Google Scholar]
- 12.Marchant NL, Reed BR, DeCarli CS, Madison CM, Weiner MW, Chui HC, et al. Cerebrovascular disease, β-amyloid, and cognition in aging. Neurobiol Aging. 2012;33:1006.e25–1006.e36. doi: 10.1016/j.neurobiolaging.2011.10.001. doi: 10.1016/j.neurobiolaging.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de la Torre JC. The vascular hypothesis of Alzheimer’s disease: bench to bedside and beyond. Neurodegener Dis. 2010;7:116–121. doi: 10.1159/000285520. doi: 10.1159/000285520. [DOI] [PubMed] [Google Scholar]
- 14.de la Torre JC. Preface: Physiopathology of vascular risk factors in Alzheimer’s Disease. J Alzheimers Dis. 2012;32:517–518. doi: 10.3233/JAD-2012-120830. doi: 10.3233/JAD-2012-120830. [DOI] [PubMed] [Google Scholar]
- 15.Tolppanen AM, Solomon A, Soininen H, Kivipelto M. Midlife vascular risk factors and Alzheimer’s disease: evidence from epidemiological studies. J Alzhceimers Dis. 2012;32:531–540. doi: 10.3233/JAD-2012-120802. doi: 10.3233/JAD-2012-120802. [DOI] [PubMed] [Google Scholar]
- 16.Hachinski V. Stroke and Alzheimer disease: fellow travelers or partners in crime? Arch Neurol. 2011;68:797–798. doi: 10.1001/archneurol.2011.118. doi: 10.1001/archneurol.2011.118. [DOI] [PubMed] [Google Scholar]
- 17.Nettiksimmons J, Beckett L, Schwarz C, Carmichael O, Fletcher E, Decarli C. Subgroup of ADNI normal controls characterized by atrophy and cognitive decline associated with vascular damage. Psychol Aging. 2013;28:191–201. doi: 10.1037/a0031063. doi: 10.1037/a0031063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Wong A, Law AC, Mok VC. Cerebrovascular disease, amyloid plaques, and dementia. Stroke. 2015;46:1402–1407. doi: 10.1161/STROKEAHA.114.006571. doi: 10.1161/STROKEAHA.114.006571. [DOI] [PubMed] [Google Scholar]
- 19.Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–2263. doi: 10.1016/S0140-6736(15)60461-5. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 20.Ly JV, Rowe CC, Villemagne VL, Zavala JA, Ma H, Sahathevan R, et al. Subacute ischemic stroke is associated with focal 11C PiB positron emission tomography retention but not with global neocortical Aβ deposition. Stroke. 2012;43:1341–1346. doi: 10.1161/STROKEAHA.111.636266. doi: 10.1161/STROKEAHA.111.636266. [DOI] [PubMed] [Google Scholar]
- 21.Bamford J, Sandercock P, Dennis M, Burn J, Warlow C. A prospective study of acute cerebrovascular disease in the community: the Oxfordshire Community Stroke Project–1981-86. 2. Incidence, case fatality rates and overall outcome at one year of cerebral infarction, primary intracerebral and subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 1990;53:16–22. doi: 10.1136/jnnp.53.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ly JV, Rowe CC, Villemagne VL, Zavala JA, Ma H, O’Keefe G, et al. Cerebral β-amyloid detected by Pittsburgh compound B positron emission topography predisposes to recombinant tissue plasminogen activator-related hemorrhage. Ann Neurol. 2010;68:959–962. doi: 10.1002/ana.22072. doi: 10.1002/ana.22072. [DOI] [PubMed] [Google Scholar]
- 23.Fiorelli M, Bastianello S, von Kummer R, del Zoppo GJ, Larrue V, Lesaffre E, et al. Hemorrhagic transformation within 36 hours of a cerebral infarct: relationships with early clinical deterioration and 3-month outcome in the European Cooperative Acute Stroke Study I (ECASS I) cohort. Stroke. 1999;30:2280–2284. doi: 10.1161/01.str.30.11.2280. [DOI] [PubMed] [Google Scholar]
- 24.Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149:351–356. doi: 10.2214/ajr.149.2.351. doi: 10.2214/ajr.149.2.351. [DOI] [PubMed] [Google Scholar]
- 26.Huang KL, Lin KJ, Ho MY, Chang YJ, Chang CH, Wey SP, et al. Amyloid deposition after cerebral hypoperfusion: evidenced on [(18)F]AV-45 positron emission tomography. J Neurol Sci. 2012;319:124–129. doi: 10.1016/j.jns.2012.04.014. doi: 10.1016/j.jns.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 27.Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY, et al. Alzheimer’s Disease Neuroimaging Initiative. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. 2013;70:455–461. doi: 10.1001/jamaneurol.2013.1321. doi: 10.1001/jamaneurol.2013.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JH, Seo SW, Kim C, Kim SH, Kim GH, Kim ST, et al. Effects of cerebrovascular disease and amyloid beta burden on cognition in subjects with subcortical vascular cognitive impairment. Neurobiol Aging. 2014;35:254–260. doi: 10.1016/j.neurobiolaging.2013.06.026. doi: 10.1016/j.neurobiolaging.2013.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 30.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol Sci. 2009;30:431–440. doi: 10.1016/j.tips.2009.05.005. doi: 10.1016/j.tips.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, et al. Cerebrovascular lesions induce transient β-amyloid deposition. Brain. 2011;134(Pt 12):3697–3707. doi: 10.1093/brain/awr300. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mok V, Leung EY, Chu W, Chen S, Wong A, Xiong Y, et al. Pittsburgh compound B binding in poststroke dementia. J Neurol Sci. 2010;290:135–137. doi: 10.1016/j.jns.2009.12.014. doi: 10.1016/j.jns.2009.12.014. [DOI] [PubMed] [Google Scholar]