

Summary
Human L3MBTL1, which contains three malignant brain tumor (MBT) repeats, binds monomethylated and dimethylated lysines, but not trimethylated lysines, in several histone sequence contexts. In crystal structures of L3MBTL1 complexes, the monomethyl- and dimethyllysines insert into a narrow and deep cavity of aromatic residue-lined pocket 2, while a proline ring inserts into shallower pocket 1. We have also engineered a single Y to E substitution within the aromatic cage of the BPTF PHD finger, resulting in a reversal of binding preference from trimethyl- to dimethyllysine in an H3K4 sequence context. In both the “cavity insertion” (L3MBTL1) and “surface groove” (PHD finger) modes of methyllysine recognition, a carboxylate group both hydrogen bonds and ion pairs to the methylammonium proton. Our structural and binding studies of these two modules provide insights into the molecular principles governing the decoding of lysine methylation states, thereby highlighting a methylation state-specific layer of histone mark readout impacting on epigenetic regulation.
Introduction
Mono-, di-, and trimethylated forms of particular histone lysine residues are selectively found in different regions of chromatin, thereby implicating specialized and separate biological functions for these marks (Martin and Zhang, 2005). For example, histone H3K4 methylation has been linked to transcriptional regulation, but only H3 trimethylated at lysine 4 (H3K4me3) is tightly coupled to transcription start sites of active genes, while lower lysine methylation states (H3K4me2 and H3K4me1) display different distribution patterns (Ruthenburg et al., 2007). In agreement with discrete signaling functions of specific lysine methylation states, histone methyllysine marks are under the control of site- and state-specific enzyme systems. For instance, the histone methyltransferase (HKMT) SET7/9 protein only generates monomethylation marks at H3K4 (Xiao et al., 2003), while the human MLL1 complex mediates H3K4 trimethylation (Dou et al., 2005). The recent identification of various histone lysine demethylases revealed site- and state-specific lysine demethylation enzymatic activities (Shi and Whetstine, 2007; Klose and Zhang, 2007). Taken together, these data suggest the operation of highly regulated epigenetic pathways, in which writing and erasing of lysine methylation marks occur in a site- and state-specific manner.
The molecular basis for higher lysine methylation state recognition by “reader” or “effector” modules has been well documented in a series of structural studies on chromodomain (Nielsen et al., 2002; Jacobs and Khorasanizadeh, 2002; Min et al., 2003; Fischle et al., 2003), double chromodomain (Flanagan et al., 2005), double tudor (Huang et al., 2006), and PHD finger (Li et al., 2006; Pena et al., 2006; Taverna et al., 2006) modules in complex primarily with trimethyllysine-containing peptides. From these analyses, a general principle has emerged in which an aromatic cage motif containing two to four aromatic residues is responsible for state-specific trimethyllysine binding with the trimethylammonium group stabilized by methyl-π and cation-π (Ma and Dougherty, 1997) interactions. The loss of methyl-π hydrophobic interactions is thought to cause the observed reductions in affinity for lower lysine methylation states in these complexes (Li et al., 2006). An ongoing challenge has been to decipher whether effector modules exist that specifically read lower lysine methylation states in a state-specific manner. To date, few such proteins have been described (Kim et al., 2006; Trojer et al., 2007) and their complexes are just beginning to yield to structural insights into how this discrimination is achieved (Botuyan et al., 2006).
Human lethal (3) malignant brain tumor-like protein 1, L3MBTL1, belongs to a group of factors containing so called malignant brain tumor (MBT) repeats of ∼70 amino acids that are conserved from lower multicellular organisms (i.e., C. elegans) to man. The protein functions as a transcriptional repressor and is requisite for mitotic progression (Boccuni et al., 2003). Overexpression of L3MBTL1 leads to improper chromosome segregation and cytokinesis, thereby inducing multinucleated cell formation (Koga et al., 1999). In a candidate screen for effector proteins, the MBT region of L3MBTL1 was found to bind methylated lysine residues in the context of histone tails (Kim et al., 2006). Recent functional studies have further demonstrated that the three MBT domains of L3MBTL1 can contribute to compaction of nucleosomal arrays within the context of monomethylated and dimethylated states, but not trimethylated states, of H4K20 and H1bK26 (alternately designated H1.4K26) (Trojer et al., 2007).
In agreement with a modification state selective methyllysine binding capacity of MBT domains, Drosophila SFMBT, a four MBT repeat-containing protein, has been reported to bind mono- and dimethylated H3K9 and H4K20 peptides, but discriminates against their trimethylated and unmodified counterparts (Klymenko et al., 2006). In addition, the four tandem MBT repeats of human SFMBT were shown to be both necessary and sufficient for nuclear matrix association and transcriptional repression (Wu et al., 2007).
We have previously reported the crystal structure of the three MBT repeats of L3MBTL1 (Wang et al., 2003), a segment essential for its transcriptional repression function. Each of the three MBT domains of L3MBTL1 contains an aromatic cage motif (Wang et al., 2003), a feature also found in chromo and tudor domains, all of which have been classified as “Royal family” proteins (Maurer-Stroh et al., 2003). In the current study, we conducted quantitative binding and crystallographic studies that firmly establish pocket 2 of L3MBTL1 as a methyllysine-binding module that targets mono- and dimethyllysine marks but discriminates against the trimethylation state. We further find that a proline ring in a Pro-Ser-Ser/Thr sequence context can insert into pocket 1 of L3MBTL1, while the ligand specificity of pocket 3 of L3MBTL1 remains unknown.
The detailed insights into the lower methylation state binding mode of L3MBTL1 led us to engineer specific mutations in the human BPTF PHD finger, a module that preferentially binds Kme3 in the context of H3K4. A single Y to E substitution within the aromatic cage of the recognition pocket of the PHD finger alters the methyllysine recognition specificity such that it prefers to bind Kme2 over Kme3, with improved binding affinity for Kme1 as well. Comparison of the different three-dimensional structures and binding modes highlights two distinct types of methylated lysine recognition by effector domains: a “cavity insertion” recognition mode that is used by L3MBTL1, and a “surface groove” recognition mode that is observed in the engineered PHD finger complex.
Results
L3MBTL1, a Lower State-Specific Methyllysine Mark Reader
To gain insight into L3MBTL1 methyllysine binding specificity, we have measured the binding of the MBT-containing segments of human L3MBTL1 to a series of histone peptides that were either unmodified, or mono-, di-, or trimethylated, at specific lysine residues using fluorescence polarization. The binding data for L3MBTL1197–526 (Figure 1A) to different H3K9 methylation states are plotted in Figure 1B (left panel). The data reveal that L3MBTL1 specifically binds to monomethyl- and dimethyllysine in this histone sequence context, with apparent dissociation constants (KDs) typically at least one order of magnitude weaker for the trimethylated and unmodified counterparts (Figure 1B, left panel).
Figure 1. Structural and Functional Characterization of L3MBTL1 as a Lower Lysine Methylation State-Specific Effector Module.

(A) Domain architecture of the L3MBTL1197–526 used for structural and functional study in this figure.
(B) (Left panel) Fluorescence polarization titration curves for wild-type L3MBTL1197–526 with H31–15 peptides containing either unmodified (blue) or mono- (red), di- (green), and tri- (purple) methyllysine at position 9. (Right panel) Fluorescence polarization titration curves for H31–15K9me1 peptide binding to L3MBTL1197–526 proteins containing Asp to Asn mutants in each of the three binding pockets. The binding curves are black for wild-type L3MBTL1, orange for pocket 1 D248N mutant, red for pocket 2 D355N mutant, and cyan for pocket 3 D459N mutant. The apparent dissociation constants (KD) are listed in each panel. In each case, parameters are reported as the mean (±average deviation from the mean) obtained from three independent titration experiments.
(C) Surface representation of crystal structure of L3MBTL1197–526-H1.523–27K27me2 complex with “PS”-containing C-terminal tail of a symmetry-related L3MBTL1 molecule in pocket 1, K27me2 in pocket 2, and PEG ligand in pocket 3. The red to blue coloring encodes an electrostatic potential distribution ranging from −20 to 20 kT/e on the protein surface.
(D to F) Structural details of Pro insertion of PS peptide into pocket 1 (D), K27me2 insertion into pocket 2 (E), and PEG ligand insertion into one of the entrances to pocket 3 (F).The aromatic residues of each pocket are highlighted in the “dotted” van der Waals radius representation. The hydrogen bonds involving the Asp residue lining the aromatic cage are shown as red dashes, with bond length listed nearby.
The same trend is observed for L3MBTL1206–519 binding to various histone peptides methylated at distinct lysine residues; their respective KD values are summarized in Table 1. In some cases (H1.4K26 and H4K20), the binding affinities for monomethyl- and dimethyllysine-containing peptides are similar; for others (H3K9, H3K27, and H3K36), the monomethyllysine binds a factor of two tighter than dimethyllysine-containing peptides; and for one (H3K4), the monomethyllysine-containing peptides bind an order of magnitude tighter than dimethyllysine-containing peptides.
Table 1. Binding Constant Summary of L3MBTL1206–519 and Its Gating Loop Mutants.
| Protein | WT | D355N | D355A | N358Q | N358A |
|---|---|---|---|---|---|
| H1.418–32 | H2N-TPVKKKARK26SAGAAK-COOH | ||||
| H1.4unmod | >500 | >500 | >1000 | >1000 | >500 |
| H1.4K26me1 | 12 ± 1 | 67 ± 7 | >500 | 77 ± 10 | 63 ± 4 |
| H1.4K26me2 | 15 ± 2 | 47 ± 8 | >500 | 44 ± 6 | 39 ± 6 |
| H1.4K26me3 | 200 ± 41 | 290 ± 53 | >1000 | >500 | 230 ± 34 |
| H31–15 | H2N-ARTK4QTARK9STGGKAY-COOH | ||||
| H3unmod | >500 | >1000 | >1000 | >1000 | >1000 |
| H3K4me1 | 6 ± 1 | 78 ± 12 | >400 | 30 ± 5 | 66 ± 5 |
| H3K4me2 | 75 ± 12 | 142 ± 13 | >400 | 120 ± 19 | 145 ± 9 |
| H3K4me3 | >500 | 350 ± 23 | >1000 | 200 ±11 | 250 ± 24 |
| H3K9me1 | 10 ± 2 | 120 ± 24 | >500 | 63 ± 7 | 170 ± 11 |
| H3K9me2 | 20 ± 3 | 91 ± 19 | >500 | 72 ± 9 | 200 ±11 |
| H3K9me3 | 400 ± 68 | >500 | >1000 | 250 ±15 | >1000 |
| H3K9me1S10ph | 120 ± 19 | — | — | — | — |
| H3K9me3S10ph | >500 | — | — | — | — |
| H31–15scram | H2N-TAGASRKGKme1QRKTATY-COOH | ||||
| H3Kme1scram | 41 ± 5 | 266 ± 31 | >1000 | 71 ± 5 | 125 ± 13 |
| H319–35 | H2N-QLATKAARK27SAPATGGVY-COOH | ||||
| H3unmod | >1000 | >1000 | — | >1000 | — |
| H3K27me1 | 41 ± 7 | 500 ±121 | — | 110 ± 11 | — |
| H3K27me2 | 75 ± 19 | 500 ± 99 | — | 140 ± 13 | — |
| H3K27me3 | >500 | >1000 | — | >500 | — |
| H328–43 | H2N-SAPATGGVK36KPHRYRPY-COOH | ||||
| H3unmod | >700 | >1000 | >1000 | >1000 | >1000 |
| H3K36me1 | 16 ± 2 | 84 ± 10 | >500 | 83 ± 18 | 180 ± 15 |
| H3K36me2 | 27 ± 5 | 86 ± 18 | >500 | 85 ± 22 | 130 ± 28 |
| H3K36me3 | 400 ±115 | >500 | >500 | >500 | >500 |
| H412–27 | H2N-KGGAKRHRK20VLRDNIQ-COOH | ||||
| H4unmod | 410 ±131 | >500 | >500 | >500 | >500 |
| H4K20me1 | 5 ± 1 | 60 ± 9 | 420 ± 75 | 26 ± 6 | 100 ± 13 |
| H4K20me2 | 6 ± 2 | 43 ± 6 | 205 ±18 | 23 ± 5 | 50 ± 11 |
| H4K20me3 | 190 ± 31 | 290 ± 35 | 500 ± 67 | 160 ± 20 | 190 ± 23 |
| H4K16acK20me1 | 16 ± 1 | — | — | — | — |
| H4K16ac | >500 | — | — | — | — |
| H412–27scram | H2N-LNRQDIAGKme1GKHVKRR-COOH | ||||
| H4Kme1scram | 14 ± 2 | 165 ± 35 | >500 | 52 ± 10 | 150 ± 24 |
Binding constants were obtained from fluorescence polarization-based binding assay and shown at μM scale. Fluorescence anisotropy of the fluorescein-labeled peptide was used to determine the apparent dissociation constant KD (based on an assumption of a 1:1 binding stoichiometry). In each case, parameters are reported as the mean (±average deviation from the mean) obtained from three independent titration experiments. WT, wild type; —, not measured.
Lower Lysine Methylation Marks Target L3MBTL1 Pocket 2
Mutation of a conserved Asp residue to Asn in one of the pockets of Drosophila Scm, a two MBT-repeat-containing protein, resulted in abnormal developmental phenotypes (Bornemann et al., 1998). We therefore generated single Asp to Asn point mutations within the aromatic cage motifs of each of the three pockets of L3MBTL1197–526, and monitored binding of the H3K9me1 peptide by fluorescence polarization (Figure 1B, right panel). Only an Asp to Asn mutation within pocket 2 (D355N) resulted in a significant loss in binding affinity to the H3K9me1 peptide (Figure 1B, right panel). The loss in binding affinity for the D355N mutant of L3MBTL1206–519 was observed for both monomethyl- and dimethyllysine-containing H3K9 peptides, and the same trend persisted with monomethyl-and dimethyllysine-containing H1.4K26, H3K4, H3K27, H3K36, and H4K20 peptides (Table 1).
Pocket Occupancy in the Crystal Structures of L3MBTL1 Bound to H1.5K27me2 and Kme2
To gain insights into the methylation state-specific binding mode of human L3MBTL1, we solved the crystal structures of L3MBTL1197–526 (Figure 1A) bound to H1.523–27 K27me2 peptide (K23 ATKKme2 sequence) and dimethyllysine (Kme2) amino acid. The crystals of the peptide complex diffracted to 1.66 Å (refinement statistics listed in Table 2) and allowed unambiguous identification of the bound ligands in each of the pockets associated with the three MBT modules in the triangular-shaped architecture of the L3MBTL1 protein (Figure 1C and see Figure S1A in the Supplemental Data available with this article online). A proline ring from the C-terminal tail of an adjacent L3MBTL1 protomer in the crystal is inserted into pocket 1 (Figure 1D), the dimethylammonium group of H1.5K27me2 is inserted into pocket 2 (Figure 1E), and a molecule of polyethyleneglycol (PEG) precipitant/cryoprotectant is found threaded into pocket 3 (Figure 1F). Pocket 3 is unusual in that it has two entrances that are partitioned by a bridge formed by the opposing side chains of R461 and Y490 residues (Figure S2); this pocket will not be analyzed further in this study.
Table 2. Data Collection and Refinement Statistics of Peptide/Ligand-Protein Complexes.
| Crystal | L3MBTL1197–526-H1.523–27K27me2 | L3MBTL1-H3.328–34 chimera-Kme2 | L3MBTL1197–526-Kme2 | L3MBTL1206–519-Kme1 | BPTF(Y17E)-H31–9-K4me2 |
|---|---|---|---|---|---|
| Data Collection | |||||
| Beamline | APS-24ID-C | CuKα | APS-24ID-C | CuKα | APS-24ID-C |
| Wavelength (Å) | 1.0858 | 1.54 | 0.9792 | 1.54 | 0.9792 |
| Space group | P21212 | P21212 | P21212 | P21212 | P212121 |
| Unit cell a, b, c (Å) | 85.9, 93.3, 58.6 | 85.2, 94.0, 59.2 | 86.3, 93.1, 59.3 | 85.5, 92.6, 58.7 | 38.3, 64.4, 85.6 |
| Resolution (Å) | 20–1.66 (1.72–1.66)a | 20–1.9 (1.97–1.90) | 20–2.1 (2.17–2.10) | 20–1.9 (1.97–1.90) | 20–1.45 (1.50–1.45) |
| Rsym (%)b | 6.5 (46.0) | 6.8 (41.4) | 8.2 (57.2) | 7.6 (48.5) | 7.0 (49.0) |
| I/σ (I) | 31.5 (5.1) | 26.8 (5.0) | 20.3 (2.7) | 24.7 (3.7) | 39.1 (5.6) |
| Completeness (%) | 100.0 (100.0) | 100.0 (100.0) | 99.4 (99.9) | 99.8 (99.3) | 100 (100) |
| Redundancy | 10.0 (9.8) | 6.4 (6.3) | 3.8 (3.7) | 5.7 (5.5) | 10.3 (10.3) |
| Refinement (F > 0) | |||||
| Number of unique reflections | 54,919 | 37,183 | 26,938 | 35,569 | 38,183 |
| Rwork/Rfree (%)c | 18.7/20.1 | 19.4/21.5 | 19.6/22.6 | 21.0/23.0 | 18.9/19.2 |
| Number of non-H atoms | |||||
| Protein | 2622 | 2623 | 2630 | 2572 | 1410 |
| Water | 564 | 522 | 306 | 459 | 337 |
| Other ligands | 48 | 37 | 45 | 18 | 63 |
| Average B factors (Å2) | |||||
| Protein | 18.0 | 26.5 | 36.6 | 30.2 | 13.8 |
| Water | 29.0 | 40.1 | 38.0 | 43.1 | 28.9 |
| Other ligands | 33.7 | 45.2 | 50.0 | 46.0 | 19.4 |
| Rmsd bonds (Å) | 0.005 | 0.007 | 0.006 | 0.006 | 0.006 |
| Rmsd angle (°) | 1.41 | 1.49 | 1.43 | 1.44 | 1.42 |
Numbers in parentheses refer to the highest resolution shells.
Rsym = ΣhΣi | Ii(h) − < I(h) > | / ΣhΣiIi(h), where Ii(h) is the ith measurement of reflection h, and < I(h) > is the weighted mean of all measurement of h.
R = Σh‖Fobs| − |Fcal‖/Σh|Fobs|, where Fobsand Fcal are the observed and calculated structure factor, respectively. Rworkand Rfree were calculated by using the working and test set reflections, respectively.
Structural Basis for Pro Ring Readout by L3MBTL1 Pocket 1
In the crystal structures of L3MBTL1197–526 bound to Kme2 and H1.523–27K27me2, the C-terminal “REPSSA” peptide segment of a symmetry-related L3MBTL1 molecule (Figure 2A) forms a type II β turn centered about the “Pro-Ser” step and inserts its Pro ring into pocket 1 (Figures 2B and 2C) (Wang et al., 2003). Pocket 1 is lined by aromatic rings of F256, F272, Y275, and H279 and the carboxylate of D248 (Figure 1D). The surface cut-away view highlights the shallowness of pocket 1 and the snug fit between the nonplanar pyrrolidine ring and the walls of the pocket mediated by stacking and hydrophobic interactions (Figure 2B).
Figure 2. Pro-Ser Step-Containing Peptide Binding by L3MBTL1 Pocket 1.
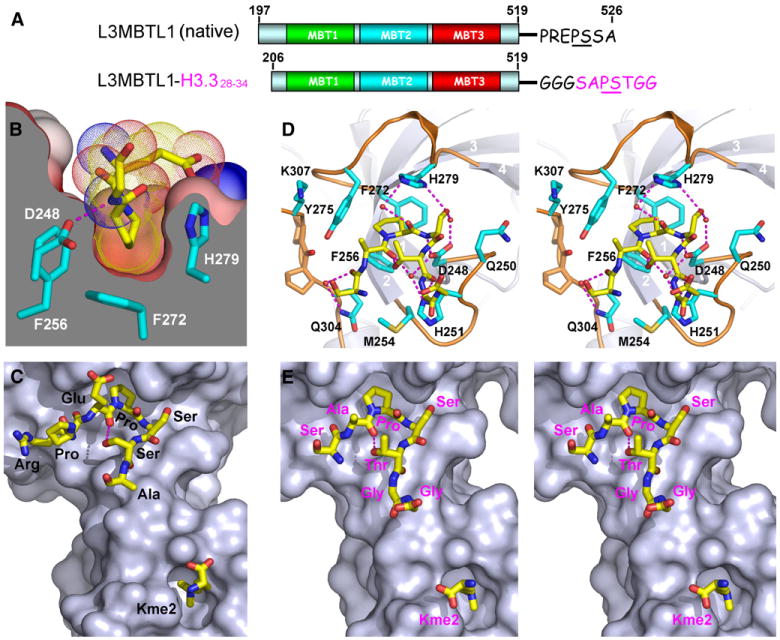
(A) Domain architecture of the L3MBTL1 constructs used for structural and functional study in this figure. For the L3MBTL1-H3.3 construct, a Gly-Gly-Gly linker was used to separate L3MBTL1 and the covalently attached histone 3.328–34 segment.
(B) Insertion of proline into a shallow cavity of symmetry-related L3MBTL1 pocket 1 in the Kme2-L3MBTL1 complex is shown. The Pro ring is sandwiched within the narrow walls at the base of the pocket. The interior parts of the surface are colored in gray, and exterior parts are colored by their electrostatic potential as described for Figure 1C. The Pro ligand is shown in the dotted van der Waals radius representation. The “EPS” peptide segment is shown, with part of the Ser omitted from the drawing for clarity.
(C) Relative positioning of PS step containing C-terminal L3MBTL1 segment in pocket 1 and Kme2 in pocket 2 on the same L3MBTL1 surface in the Kme2-L3MBTL1 complex.
(D) Stereo view of the PS step containing H3.3 SAPSTGG segment with its Pro ring inserted into pocket 1 of the Kme2-L3MBTL1-H3.328–34 complex. Key residues participating in pocket formation are shown in stick representation (cyan) with main-chain atoms omitted for clarity. Water molecules are shown as small red spheres and hydrogen bonds indicated by dashed red lines.
(E) Stereo view of the relative positioning of PS step containing H3.3 SAPSTGG segment in pocket 1 and Kme2 in pocket 2 on the same L3MBTL1 surface in the Kme2- L3MBTL1-H3.328–34 complex. Note that the type II β-turn formed at the “APST” motif in pocket 1 helps to direct the C-terminal GG segment of H3.3 toward the Kme2-bound pocket 2. Water molecules are shown as small red spheres and hydrogen bonds indicated by dashed red lines.
Pockets 1 and 2 of L3MBTL1 are separated by a fixed distance, with pocket 1 binding the Pro ring in a Pro-Ser-Ser sequence context and pocket 2 binding Kme2 (Figure 2C). We therefore searched for natural histone peptides where a Pro in a Pro-Ser-Ser/Thr sequence context could be separated by the appropriate distance from a Lys residue, so that Pro and methyllysine marks could potentially simultaneously bind into pockets 1 and 2, respectively on the same L3MBTL1. Intriguingly, the most likely candidate was histone H3.326–40, containing the sequence AA25RKSAP30STGGV35KKPHR40, where the separation between P30 and K36/37 in an extended conformation could match the distance between L3MBTL1 pockets 1 and 2.
To test whether the natural Pro-Ser-Thr sequence in the context of H3.3 could insert its proline ring into L3MBTL1 pocket 1, we covalently linked L3MBTL1206–519 to the SAP30STGG sequence of H3.3, bridged by a Gly-Gly-Gly linker segment (Figure 2A). We successfully grew crystals and solved the structure of the complex of L3MBTL1-H3.328–34 chimera bound to Kme2 at 1.9 Å resolution (refinement statistics listed in Table 2). The covalently linked SAPSTGG H3.3 peptide segment can be readily traced in the complex (stereo view in Figure 2D and Figure S1C), with the Pro ring inserted into pocket 2 of a symmetry-related molecule in the crystal. In addition, the peptide adopts a type II β turn at the Pro-Ser step and the C-terminal Gly-Gly residues are directed toward pocket 2, which contains bound Kme2 in the complex (stereo view in Figure 2E). The complex is stabilized by direct and water-mediated intermolecular hydrogen bonds, with the recognition specificity notably associated with specific hydrogen bonds between the Pro-Ser-Thr segment of H3.3 and residues D248, H251, and H279 that line the walls of pocket 1 (Figure 2D).
Structural Basis for Kme2 and Kme1 Marks Readout by L3MBTL1 Pocket 2
We also solved the crystal structure of L3MBTL1206–519 bound to monomethyllysine (Kme1) at 1.9 Å (refinement statistics listed in Table 2), which allowed us to compare the structural basis for Kme2 and Kme1 readout by L3MBTL1. The side chain of Kme2 in its complex is perpendicularly inserted into pocket 2 of L3MBTL1 (stereo view in Figure 3A), resulting in complete burial of the dimethylammonium group at the base of the pocket (Figure 3B), where it is positioned within an aromatic-lined cage composed of F379, W382, and Y386 rings. More importantly, the dimethylammonium proton is directly hydrogen bonded and ion paired to the carboxylate of D355 (Figures 1E and 3B). Pocket 2 is both deep and narrow and functions as a size-selective filter (cut-away view in Figure 3B). We refer to such encapsulation of the side chain of Kme2 by pocket 2 of L3MBTL1 (Figure 3B) as a cavity insertion recognition mode.
Figure 3. Structural Details for Specific Readout of Kme1 and Kme2 by L3MBTL1 Pocket 2.
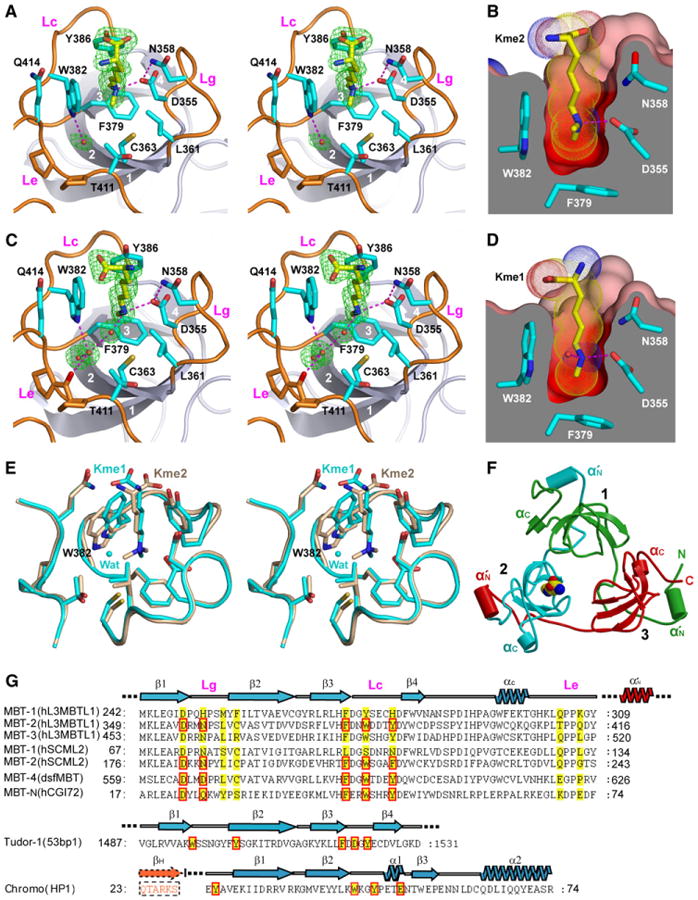
(A and C) Stereo view of Kme2 (A) and Kme1 (C) bound to L3MBTL1 pocket 2. Four strands of the β subunit core are numbered 1,2,3, and 4 and colored in gray. The gating (Lg), caging (Lc), and extra (Le) loops are highlighted in orange. Key residues participating in pocket formation are shown in stick representation (cyan) with main-chain atoms omitted for clarity. Water molecules that mediate hydrogen bonding between L3MBTL1 and Kme2/Kme1 are shown as small red spheres (C). The Fo − Fc omit electron densities (green) are contoured at 4.5 σ level.
(Band D) Protein surface cut-away views of Kme2(B) and Kme1 (D). The color code is as described for Figure 2B. Kme2(B)and Kme1 (D) are inserted into a deep, narrow, and negatively charged cavity in L3MBTL1 pocket 2.
(E) Superpositioned views of Kme2 (beige) and Kme1 (cyan) inserted into pocket 2, based on the least-squares fitting of Cα atoms within the second MBT module (349–416) of L3MBTL1.
(F) Cartoon representation of the three-leaved propeller architecture of L3MBTL1. The MBT repeats 1, 2, and 3 are colored green, cyan, and red, respectively. The bound Kme1 ligand within pocket 2 is represented in CPK mode. Two α helices in each of the three MBT modules are labeled α′N and αC and shown as cylinders.
(G) Sequence comparison among different MBT modules and other related methyllysine reader modules within the Royal family. Key residues participating or predicted to participate in pocket formation are shaded in yellow, and those that are directly involved in methyllysine recognition are boxed in red. Accession numbers are as follows: hL3MBTL1, AAH39820.1; hSCML2, AAH64617; dsfMBT, NP_723786.1; hCGI72, NP_057102; 53BP1, PDB_id: 2IG0; and HP1, PDB_id:1KNA. All listed modules (MBT, tudor, and chromo) generate their reader pockets at one end of a common “SH3-like” β-barrel. βH denotes the histone H3(5–10)K9me peptide.
The monomethyllysine side chain in its complex is also inserted into pocket 2 (stereo view in Figure 3C), with one methylammonium proton forming a direct hydrogen bond to D355, while several water molecules form bridging hydrogen bonds between the other methylammonium proton and a backbone carbonyl. We observe similar dimensions for pocket 2 in cut-away views of the Kme1 (Figure 3D) and Kme2 (Figure 3B) complexes. The superimposed structures of L3MBTL1 pocket 2 containing bound Kme2 (in beige) and bound Kme1 (in cyan) are shown in stereo in Figure 3E.
Mutational Analysis of Gating Residues Lining L3MBTL1 Pocket 2
A ribbon version of the L3MBTL1 crystal structure, with Kme1 in a space-filling view inserted into pocket 2, is shown in Figure 3F. Three loops (Figure 3A) contribute to forming the walls of L3MBTL1 binding pocket 2. Residues 355–361 form a “gating loop” (Lg), which controls the dimensions of the binding pocket; residues 379–386, which contain all three aromatic residues, form a “caging loop” (Lc); and residues 407–423 form an “extra loop” (Le), which is unique among MBT modules within Royal family members (Figures 3A, 3C, and 3G). We have undertaken structural and binding studies on mutants of amino acids D355 and N358, which project from the gating loop and line the walls of L3MBTL1 pocket 2.
The fluorescence polarization-based dissociation constants for binding of monomethyl-, dimethyl-, and trimethyllysine-containing H1.4K26, H3K4, H3K9, H3K27, H3K36, and H4K20 peptides to the D355N, D355A, N358Q and N358A mutants of L3MBTL1206–519 are listed in Table 2. In agreement with an essential role of these amino acids for methyllysine binding, there is a loss in binding affinity for all mutants, with more pronounced losses for the Ala mutants.
To obtain further insight into the role of the gating loop and its role in methyllysine selectivity, we have solved the crystal structures (2.0–2.2 Å resolution) of D355N, D355A, N358Q, and N358A mutants of L3MBTL1206–519 with added Kme2 (refinement statistics listed in Table 3, stereo views in Figures S3A–S3D) and compared these with the structure of wild-type L3MBTL1 containing bound Kme2. Superpositioned comparisons of pocket 2 conformations are shown in stereo for D355N in Figure 4A, for D355A in Figure 4B, for N358Q in Figure 4C, and for N358A in Figure 4D. Although all mutant L3MBTL1 crystals were grown under similar conditions in the presence of Kme2, this ligand was not found inserted into pocket 2 in any of the mutant L3MBTL1 complexes (Figures S3A–S3D). Among the L3MBTL1 mutant structures, D355A (Figure 4B) and N358A (Figure 4D) exhibited significant changes in pocket 2 gating loop conformations, while for D355N and N358Q, the overall conformations of the gating loop remain largely the same, except for increased flexibility as reflected by high B factor distributions and weak electron density (Figures S3A and S3C).
Table 3. Data Collection and Refinement Statistics of L3MBTL1206–519 Gating Loop Mutants.
| Crystal | D355N | D355A | N358Q | N358A |
|---|---|---|---|---|
| Data Collection | ||||
| Beamline | CuKα | CuKα | CuKα | CuKα |
| Wavelength (Å) | 1.54 | 1.54 | 1.54 | 1.54 |
| Space group | P21212 | P21212 | P21212 | P21212 |
| Unit cell a, b, c (Å) | 86.0, 91.2, 58.8 | 85.5, 92.7, 58.4 | 86.0, 90.7, 58.5 | 85.4, 90.1, 58.4 |
| Resolution (Å) | 20–2.2 (2.28–2.20)a | 20–2.2 (2.28–2.20) | 20–2.0 (2.07–2.00) | 20–2.0 (2.07–2.00) |
| Rsym (%)b | 8.2 (49.6) | 6.6 (56.8) | 5.9 (42.2) | 8.7 (43.2) |
| I/σ (I) | 23.4 (4.8) | 35.3 (4.9) | 32.9 (4.6) | 14.8 (3.7) |
| Completeness (%) | 100.0 (100.0) | 96.0 (98.0) | 100.0 (100.0) | 92.2 (95.7) |
| Redundancy | 6.2 (6.3) | 6.2 (6.2) | 6.7 (6.5) | 4.2 (4.1) |
| Refinement (F > 0) | ||||
| Number of unique reflections | 22,778 | 21,948 | 30,390 | 27,545 |
| Rwork/Rfree (%)c | 22.3/25.1 | 22.5/25.7 | 21.3/24.8 | 20.6/23.1 |
| Number of non-H atoms | ||||
| Protein | 2572 | 2563 | 2573 | 2563 |
| Water | 352 | 330 | 422 | 343 |
| Other ligands | 7 | 37 | 7 | 34 |
| Average B factors (Å2) | ||||
| Protein | 39.4 | 40.7 | 34.4 | 31.9 |
| Water | 51.4 | 50.2 | 48.4 | 44.3 |
| Other ligands | 63.2 | 66.7 | 58.6 | 56.7 |
| Rmsd bonds (Å) | 0.006 | 0.007 | 0.006 | 0.007 |
| Rmsd angle (°) | 1.39 | 1.47 | 1.36 | 1.47 |
Numbers in parentheses refer to the highest resolution shells.
Rsym = ΣhΣi | Ii(h) − < I(h) > | / ΣhΣiIi(h), where Ii(h) is the ith measurement of reflection h, and < I(h) > is the weighted mean of all measurement of h.
R = Σh‖Fobs| − | Fcal‖/Σh|Fobs|, where Fobsand Fcal are the observed and calculated structure factor, respectively. Rwork and Rfree were calculated by using the working and test set reflections, respectively.
Figure 4. Stereo Views of Pocket 2 Comparing Superpositioned Kme2-Bound Wild-Type and Mutant L3MBTL1 Complexes.
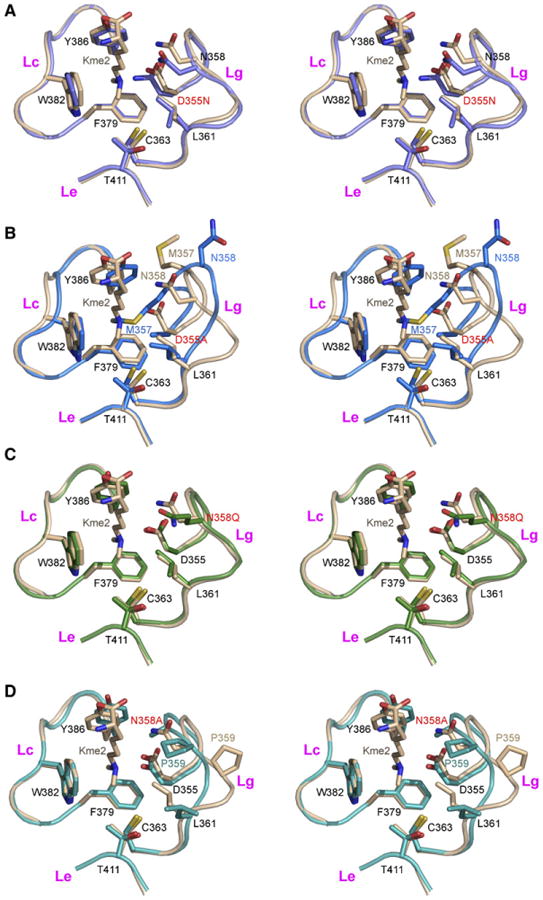
(A–D) The wild-type L3MBTL1 complex with bound Kme2 is shown in beige (A–D). The D355N mutant complex is shown in light blue (A), the D355A mutant is shown in dark blue (B), the N358Q mutant is shown in light green (C), and the N358A mutant is shown in dark green (D). Kme2 was not detected in pocket 2 for complexes with D355N (A), D355A (B), N358Q (C), and N358A (D).
Superpositioning is based on least-squares fitting of Cα atoms within the second MBT module (349–416) of L3MBTL1.
Engineering State-Specific Readout of Kme2 Marks by the BPTF PHD Finger
We have previously monitored the binding of histone H3 peptides containing methylation marks at H3K4 to the PHD finger of BPTF, the largest subunit of nucleosome remodeling factor (NURF) (Wysocka et al., 2006), by surface plasmon resonance and fluorescence polarization experiments. Both methods yielded the same trend in binding affinities, namely H3K4me3 > H3K4me2 > H3K4me1 > H3K4 (Figures 5A, 5B, and 5E) (Li et al., 2006). Three-dimensional structural analysis of the H3K4me3 bound to the BPTF PHD finger established that the trimethylammonium group of H3K4me3 was positioned within a “YYYW” aromatic cage motif that defined a shallow cleft in the protein surface, with the bound trimethylammonium group stabilized by methyl-π and cation-π interactions (Li et al., 2006).
Figure 5. Structural and Functional Analysis of State-Specific Readout of Kme2 Marks by an Engineered Y17E BPTF PHD Finger.
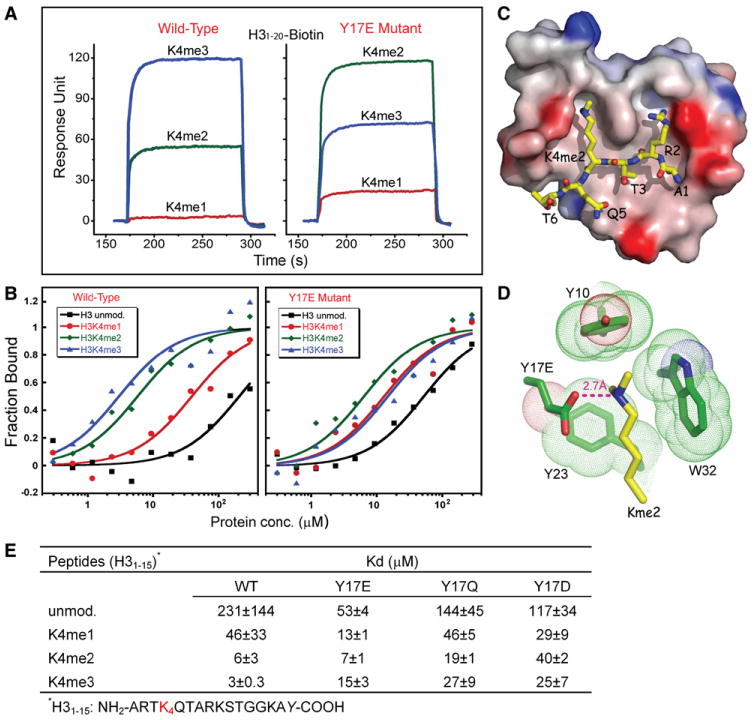
(A) Surface plasmon resonance curves monitoring the methylation state-dependent binding and release of H3K4 peptides by the BPTF PHD finger (left panel, previously reported in Li et al., 2006 and included for comparison purposes) and its Y17E mutant (right panel). Color representations are K4me1 (red), K4me2 (green), and K4me3 (blue).
(B) Fluorescence polarization curves monitoring the methylation state-dependent binding of H3K4 peptides by the BPTF PHD finger and its Y17E mutant (right panel). Color representations are unmodified K4 (black), K4me1 (red), K4me2 (green), and K4me3 (blue).
(C) Electrostatic potential surface representation of BPTF Y17E PHD finger in complex with H31–9K4me2 peptide. Red to blue coloring represents a surface charge distribution from −20 to 20 kT/e. This view emphasizes the surface groove recognition mode of complex formation.
(D) Position of Kme2 side chain of H31–9K4me2 peptide inserted into an engineered Glu-containing aromatic-lined cage of the Y17E mutant BPTF PHD finger. The aromatic residues are shown in the dotted van der Waals radius representation. The proper positioning and orientation of Y17E side chain results in a direct hydrogen bond (≈2.7 Å) between the dimethylammonium group of K4me2 and the carboxylate of E17.
(E) Summary of fluorescence polarization-based binding constants. Fluorescence anisotropy of the fluorescein-labeled peptide was used to determine the apparent dissociation constant KD (based on an assumption of a 1:1 binding stoichiometry). In each case, parameters are reported as the mean (±average deviation from the mean) obtained from two independent titration experiments.
We pursued systematic mutational studies of residues lining the aromatic cage within the BPTF PHD finger, substituting them with acidic residues. Binding of mutant proteins to mono-, di-, and trimethylated H3K4me peptides was monitored by surface plasmon resonance. These efforts lead to the identification of a modified “YEYW” aromatic cage motif following Y17E substitution, which shows a reversal from tri- to dimethylation state preference and enhanced monomethyllysine binding affinity as monitored by surface plasmon resonance (Figure 5A, right panel) and fluorescence polarization (Figure 5B, right panel) studies. The binding affinities as monitored by fluorescence polarization for the Y17E PHD finger mutant decreased in the order H3K4me2 > H3K4me3 ≈ H3K4me1 > H3K4 (Figures 5B and 5E).
Structural Basis for Kme2 Recognition by BPTF PHD Finger Y17E Mutant
To obtain insights into the binding mode of BPTF Y17E mutant, we solved the 1.45 Å resolution crystal structure of H31–9K4me2 bound to the Y17E mutant PHD finger-linker-bromodomain (2583–2751) construct (refinement statistics listed in Table 2, stereo view in Figure S4). The alignment of the peptide on the complex, in which the side chains of R2 and K4me2 align in adjacent surface channels (Figure 5C), is essentially the same as for the native PHD finger complex (Li et al., 2006). The key recognition difference is localized within the cage region of the binding pocket where we observe a direct hydrogen bond and ion pair between the dimethylammonium group of K4me2 and the carboxylate of Y17E (Figure 5D). We designate the recognition of the side chain of Kme2 by a shallow declivity in the surface of the engineered BPTF PHD finger (Figure 5C)a surface groove recognition mode. We have also generated Y17Q and Y17D mutations of the BPTF PHD finger to investigate the effect of size and charge at position 17 on methylation state-specific H3K4me binding propensity. As shown from the fluorescence polarization binding constants listed in Figure 5E, and plotted in Figure S5, both Y17Q and Y17D mutants exhibited a reduced affinity for H3K4me3 and H3K4me2 marks.
Discussion
Our combined structural and binding studies reinforce a prevailing theme in chromatin-mediated gene regulation—specific effector proteins mediate functional readout by precise molecular recognition of distinct histone modifications (Ruthenburg et al., 2007). The present report expands on this emerging paradigm and provides insights into lower lysine methylation state-specific recognition of histone marks.
Methyllysine Specificity of Histone Tail Readout by L3MBTL1 Pocket 2
Our extensive fluorescence polarization binding studies establish that L3MBTL1 recognizes methyllysine marks in a range of different histone tail sequence contexts (H1.4K26, H3K4, H3K9, H3K27, and H4K20), exhibiting a preference for mono- and dimethylated lysine marks, with greater than an order of magnitude stronger affinity relative to trimethylation marks (Table 1). Further, the binding analysis of mutant 3MBT molecules established that the lower methylation state specificity of L3MBTL1 was exclusively mediated by binding pocket 2 (Figure 1B, right panel). It should be noted that one common feature of the synthetic histone tail peptides used is that their methyllysine marks are positioned within the context of sequences enriched in positively charged residues, which most likely contribute to the binding enthalpy via electrostatic complementarity of the negatively charged surface of pocket 2 (Figure 1C). In support of this view, the binding affinity of L3MBTL1 decreases modestly on simultaneous incorporation of a neutral Kac mark (H4K16ac in the vicinity of H4K20me) and more dramatically on simultaneous incorporation of a negatively charged adjacent Sph mark (H3S10ph next to H3K9me) (Table 1). In agreement with the observed promiscuous binding affinities for methylated peptides containing positively charged residues (Table 1), pocket 2 is the most electrostatically negatively charged (red color) of the three pockets of L3MBTL1 (Figure 1C).
H1.5K27me2-L3MBTL1 Complex
The H1.523–27K27me2 peptide used in the present structural study has the sequence KATKKme2, rich in basic amino acids. In the crystals of its complex with L3MBTL1, we could only trace the TKKme2 segment, with K27me2 inserting into pocket 2 (Figures 1C and 1E). There were no intermolecular hydrogen bonding contacts involving either the backbone of the TKKme2 segment or the side chains of the TK segment with L3MBTL1, reinforcing the contribution of electrostatic complementarity to complex formation.
Comparison of Kme1 and Kme2 Recognition by L3MBTL1 Pocket 2
Our determination of high-resolution crystal structures of Kme2 (Figures 3A and 3B) and Kme1 (Figures 3C and 3D) inserted into pocket 2 of L3MBTL1 establish that mono- and dimethylated side chains adopt similar alignments within pocket 2 and that the narrowness and depth of pocket 2 remain the same in both complexes. Preference for lower lysine methylation states (Kme1 and Kme2) relative to their trimethyllysine (Kme3) counterpart is attributed to the positioning of the mono- and dimethylammonium groups within an aromatic cage, such that the methylammonium proton forms a direct hydrogen bond, facilitated by electrostatic interactions, to the carboxylate of acidic residue D355 lining pocket 2. The other methylammonium proton of bound Kme1 is hydrogen bonded to a water molecule, which in turn is hydrogen bonded to a second water molecule that is held in place by hydrogen bonds to main-chain and side-chain residues (Figure 3C and Figure S6B).
The observed lateral displacement of caging loop residue W382 between the Kme1 and Kme2 complexes (superpositioned in Figure 3E) is accompanied by a reorganization of the hydrogen bonding network within pocket 2 (Figures 3A and 3C and Figures S6A and S6B). The greater accessibility of the W382 side chain in the Kme1 complex may account for the relative preference of L3MBTL1 for H3K4me1 (Table 2), provided that L3MBTL1 binds the H3K4me peptide by sandwiching the tryptophan ring between R2 and K4me side chains as consistently observed in published structures of related complexes (Flanagan et al., 2005; Li et al., 2006; Pena et al., 2006; Huang et al., 2006).
Dimensions and Hydrogen Bonding Network of L3MBTL1 Pocket 2 and Implications for Other Royal Family Members
The dimensions of pocket 2 are defined by the orientations of side chains that project from the gating, caging, and extra loops. Within the gating loop, one carboxylate oxygen of D355 forms strong hydrogen bonds to the side-chain amide of N358 and the methylammonium proton of bound Kme1 (Figure 3C) and Kme2 (Figure 3A), while the other carboxylate oxygen forms a direct hydrogen bond to the backbone amide proton of M357 (stereo views in Figure S6). This hydrogen bonding network significantly stabilizes the optimal gating loop conformation for gauging the lower methylation state-specific readout by L3MBTL1 pocket 2.
The limited circumference of the binding pocket 2 entrance in the L3MBTL1-Kme1/2 complexes (Figures 3B and 3D, this study), similar to that reported previously for the 53BP1-Kme2 complex (Botuyan et al., 2006), restricts access of the larger trimethylammonium group. We suggest that this putative steric exclusion of highly branched structures along the entire length of the pocket (Figures 3B and 3D), together with the observed hydrogen bond and ion pair (Figures 3A and 3C), explains the strong preference of L3MBTL1 pocket 2 for Kme1/2 over Kme3.
A mild preference has been reported for recognition of dimethylated over monomethylated lysines in an H4K20 sequence context by the tandem tudor-containing 53BP1 protein (Botuyan et al., 2006). Four aromatic residues and an acidic residue line the methylated lysine-binding pocket of 53BP1 protein (Figure S7B). This presumably leaves less room available for bound water molecules required for further stabilization of Kme1 ligand, as revealed in the structure of the L3MBTL1206–519-Kme1 complex (Figure 3C), thereby perhaps accounting for the observed methylation state preference. In contrast, HP1 chromodomain has a wider pocket composed of three aromatic and one acidic residue, and the histone peptide binds via a surface groove binding mode, which makes HP1 chromodomain a higher lysine methylation state-specific reader (Figure 3G and Figure S7C).
Sequence (Figure 3G) and structural (stereo view, Figure S7A) alignment reveals striking similarities in the binding pocket architecture of pocket 2 of Kme1-bound L3MBTL1 (this study) and pocket 2 of ligand-free SCML2, a two MBT repeat-containing protein (Sathyamurthy et al., 2003). Sequence alignments (Figure 3G) also suggest that pocket 4 of four-MBT repeat-containing dsfMBT protein (Klymenko et al., 2006), and the MBT domain of hCGI72, a protein composed of flanking MBT and Tudor domains (Kim et al., 2006), could also adopt a similar “caging” strategy for lower lysine methylation state readout.
Role of Acidic D355 and Neutral N358 Residues Lining L3MBTL1 Pocket 2
D355 is a key residue lining L3MBTL1 pocket 2, given that it ion pairs and hydrogen bonds to the methylammonium group of bound Kme2 (Figure 3A) and Kme1 (Figure 3C). We note that the D355N mutation results in a 5- to 10fold loss in binding affinity for Kme1 marks and in a 2- to 8-fold loss in binding affinity for Kme2 marks for all peptides listed in Table 1. Relative to the native structure, there are no changes to the backbone conformation of the gating and caging loops, nor any significant movement of side chains lining the pocket (Figure 4A), indicating that the observed loss in binding affinity (up to 10-fold) results from the critical role of the negative charge of D355 in Kme1 and Kme2 recognition.
Binding is too weak to detect (KD ≥ 500 μM) between the D355A mutant of L3MBTL1 and all Kme1- and Kme2-containing peptides listed in Table 1. This is readily explainable because there is a large conformational change in the gating loop, with concomitant insertion of the side chain of M357 into binding pocket 2 of L3MBTL1 (Figure 4B).
Residue N358 is positioned at the entrance to pocket 2 and its side-chain hydrogen bonded to the side chain of D355 (Figures 3A and 3C). We note that the N358Q mutation results in a 3- to 6-fold loss in binding affinity for Kme1 marks and in a 2- to 4-fold loss in binding affinity for Kme2 marks, depending on histone sequence context (Table 1). Although the gating and caging loops adopt the same fold, as does the alignment of side chains of amino acids lining pocket 2 (Figure 4C), Gln has a longer side chain than Asn and could partially interfere with Kme2/Kme1 insertion.
The N355A mutation results in a 10- to 20-fold loss in binding affinity for Kme1 marks and in a 2- to 10-fold loss in binding affinity for Kme2 marks, depending on histone sequence context (Table 2). We note that there is a conformational change in the gating loop for the N355A mutation, most likely reflecting the breakage of the hydrogen bond to D355 in the mutant, and this could perhaps cause the observed reduction in binding affinity (Figure 4D).
We do not detect any electron density representing Kme2 bound to pocket 2 in the crystal structures of the Kme2-L3MBTL1 complexes containing D355N (Figure 4A), D355A (Figure 4B), N358Q (Figure 4C), and N358A (Figure 4D) mutants. Mutation of either D355 or N358 appears to perturb the hydrogen bonding network stabilizing the integrity of pocket 2, and, in the case of D355A and N358A mutants, the gating loops undergo a conformational change that occludes the entrance to the binding pocket (Figures 4B and 4D). Thus, it appears that integrity and accessibility to the pocket, together with proper alignment for intermolecular hydrogen bonding to the methylammonium proton, are key components that must be satisfied for insertion of Kme2 into pocket 2.
Altering Methyllysine Recognition Specificity within a Surface Groove Recognition Pocket
The specificity for H3K4me3 recognition by the BPTF PHD finger is diminished in the the Y17E mutant compared to the wild-type protein as measured by both surface plasmon resonance (Figure 5A) and fluorescence polarization (KD ≈ 3.0 μM for wild-type and KD ≈ 15 μM for Y17E mutant) (Figures 5B and 5E). By contrast, there is an increase in binding affinity for H3K4me2 by the Y17E mutant compared to wild-type as measured by surface plasmon resonance, though fluorescence polarization (KD ≈ 6 μM for wild-type and KD ≈ 7 μM for Y17E mutant) (Figures 5B and 5E) yielded similar values. There was also an increase in binding affinity for H3K4me1 in the Y17E mutant as measured by both surface plasmon resonance (Figure 5A) and fluorescence polarization (KD ≈ 46 μM for wild-type and KD ≈ 13 μM for Y17E mutant) (Figures 5B and 5E).
The direct hydrogen bond and ion pair between the dimethylammonium group of K4me2 and the carboxylate of E17 in the crystal structure of the Y17E BPTF PHD finger complex (Figure 5D) readily accounts for the stabilization of the K4me2 interaction. This newly formed hydrogen bond and ion pair probably could also account for the increase in K4me1 binding affinity in the Y17E mutant relative to wild-type (Figure 5E). The decreased affinity for H3K4me3 in the Y17E mutant (Figure 5E) could reflect steric repulsion between the charged glutamate side chain and the hydrophobic methyl group in the absence of extra hydrogen bonding capability.
We note that both Y17Q and Y17D mutations displayed overall decreased affinities for the H3K4-methylated peptides compared to their Y17E mutant counterpart (Figure 5E), which most likely indicates that both the negative charge and the proper position of the carboxylate side chain lining the aromatic cage are required for optimal lower methylation state H3K4me readout.
We anticipate that the availability of a properly positioned carboxylate residue lining the walls of the binding pocket could play a role in fine-tuning the biological function of methyllysine-binding PHD finger proteins. Bioinformatic analysis has revealed a subset of wild-type PHD fingers, such as ECM5 and SPP1 from budding yeast (Shi et al., 2007), which contain one acidic residue lining the aromatic cage (Figure S8) and hence are most likely capable of lower lysine methylation state readout.
Cavity Insertion versus Surface Groove Recognition Modes
The extent of discriminative recognition between different methylated lysine states could also be modulated by the nature of the binding mode between the effector module and the methyllysine mark. Two different binding modes have been identified based on the known structures of complexes: the cavity insertion recognition mode that is evident in the L3MBTL1 (this study) and the 53BP1 tandem tudor domain (Botuyan et al., 2006) complexes, and the surface groove recognition mode that exists in the HP1 and Polycomb chromodomain (Nielsen et al., 2002; Jacobs and Khorasanizadeh, 2002; Min et al., 2003; Fischle et al., 2003), CHD1 double chromodomain (Flanagan et al., 2005), PHD fingers (Li et al., 2006; Pena et al., 2006; Taverna et al., 2006), and JMJD2A double tudor domain (Huang et al., 2006) complexes. In the cavity insertion recognition mode, the methylammonium group of methylated lysine is inserted into and buried within a cavity of the protein, thereby endowing the pocket with potential size-sensitive selection filter capabilities (Figures 3B and 3D and Figures S7A and S7B). By contrast, the binding pockets reported for the surface groove recognition mode are both wider and more accessible, such that the methylated lysine side chain lies along a protein surface groove, leaving one side of the methylammonium group open and accessible to solvent (Figure 5C and Figure S7C). Consequently, the latter recognition mode is less stringent in its discrimination preferences, as our substantial body of mutagenesis-mediated perturbations to structure and binding capacities attests.
Potential for Combinatorial Readout by Three MBT Modules of L3MBTL1
The L3MBTL1 protein containing three MBT repeats, each with its own pocket, is especially interesting, as each pocket seems to have evolved a different readout: pocket 1 binds a Pro-Ser step (a dipeptide sequence identified in certain histone peptides and also prevalent in phosphorylation-dependent signaling substrates), pocket 2 binds Kme2- and Kme1-containing peptides, and pocket 3 may play a role in binding an as yet unidentified ligand or histone mark. Thus, a given L3MBTL1 molecule with its three reader modules positioned on the same face of the protein can potentially participate in combinatorial readout of two or more marks.
Based on the structure of the L3MBTL1-H3.328–34 chi-mera-Kme2 complex (Figure 2E), we speculate that histone H3.3 (AA25RKSAP30STGGV35KKPHR40) peptide could interact with L3MBTL1 in a combinatorial readout, where P30 inserts into pocket1and methylated K36/37 inserts into pocket 2. This hypothesis is potentially interesting given that histone H3.3 gets modified during mitosis, including phosphorylation ofS28 and mono-or dimethylation at K36 (Garcia et al., 2005).
Summary
It is intriguing to note that aromatic-lined cage motifs that read out methyllysine marks have convergently evolved in different effector modules, such as chromo, tudor, and MBT members of Royal family proteins (Maurer-Stroh et al., 2003) and PHD finger domains (Li et al., 2006; Pena et al., 2006). Our data lend support to an extension of this general theme–namely, that cage motifs can be fine-tuned by size-selective filters to allow discrimination against higher lysine methylation states.
Our structural results establish that optimal lower methylation state readout requires a properly positioned carboxylate side chain within an aromatic-lined cage, with the carboxylate group both hydrogen bonded and ion paired to the methylammonium proton of bound mono-and dimethyllysines. Further, gating and caging loops control pocket accessibility such that the bulky side chain of trimethyllysine is sterically excluded along the entire length of pocket 2 of L3MBTL1.
Experimental Procedures
Details of protein expression and purification, peptide synthesis and modification, detailed crystallization conditions, and surface plasmon resonance binding assays are outlined in the Supplemental Data.
Crystallization, Structure Determination, and Refinement
Crystals were successfully grown for L3MBTL1197–526 bound to H1.523–27K27me2 peptide. Crystallization was attempted on complexes of L3MBTL1197-526 construct with mono-, di-, and trimethylated lysine by vapor diffusion at 20°C. Diffraction quality crystals only grew for the Kme2 complex, with detailed conditions outlined in the Supplemental Experimental Procedures.
Crystallization of the L3MBTL1206-519-Kme1 complex, L3MBTL1206-519 D355N/A and N358Q/A mutants, and L3MBTL1-H3.328–34 chimera-Kme2 complex were screened by Mosquito crystallization robot (TTP Labtech, Royston, England) using the sitting-drop method. Using the CP-CUSTOM (I-VI) screening kit (AXYGEN Biosciences), many hits were identified with the use of maleate-containing buffer. Structural determination later revealed that crystal packing is significantly facilitated by an unconventional covalent modification of two surface cysteine residues by maleate, which are potentially catalyzed by an adjacent arginine residue side chain upon crystal packing (Figure S9). Detailed conditions for crystallization are outlined in the Supplemental Experimental Procedures.
Crystallization of BPTF Y17E PHD finger-linker-bromodomain in complex with H31–9K4me2 peptide was also performed by vapor diffusion at 20°C. Detailed crystallization conditions are outlined in the Supplemental Experimental Procedures.
Details of data collection at synchrotron and in-house sites, data processing, model building, and refinement are outlined in the Supplemental Experimental Procedures.
Data processing and structure refinement statistics are listed in Tables 2 and 3.
Fluorescence Polarization Binding Assays
Fluorescence polarization assays were performed essentially as described previously (Fischle et al., 2003). The assays on L3MBTL1 were performed under conditions of 20 mM imidazole (pH 8.0), 25 mM NaCl and 2 mM DTT (pH 8.0), or 25 mM Tris-HCl (pH 8.0) and 2 mM DTT. The wild-type and mutant BPTF PHD fingers were assayed using buffer containing 20 mM Tris-HCl (pH 7.5) and 50 mM KCl. In both cases, 50 μl reactions were incubated with 100 nM C-terminally fluorescein-labeled peptide for at least 1 hr at room temperature. Fluorescence polarization was then measured in a Hidex Chameleon plate reader at room temperature.
Supplementary Material
Acknowledgments
D.J.P. is supported by funds from the Abby Rockefeller Mauze Trust and the Dewitt Wallace and Maloris Foundations, and C.D.A. is supported by an NIH MERIT award and funds from Rockefeller University. We thank Drs. Nagesh Kalakonda, Stephen D. Nimer, Alex Ruthenburg, Sean Taverna, Valentina Tereshko, and Joanna Wysocka for their helpful advice and comments. We thank the Peptide Core Facilities at Sloan-Kettering (S.S. Yi at Microchemistry and Proteomics) and Rockefeller University for the synthesis and purification of K4-methylated H3 peptides. We would like to thank the staff at beamline NE CAT 24ID-C of the Advanced Photon Source at the Argonne National Laboratory, supported by the US Department of Energy, for assistance with data collection.
Footnotes
Supplemental Data: Supplemental Data include nine figures and Supplemental Experimental Procedures and can be found with this article online at http://www.molecule.org/cgi/content/full/28/4/677/DC1/.
Accession Numbers: Coordinates for the various complexes have been deposited into the Protein Data Bank under the following accession codes: L3MBTL1197–526-H1.523–27K27me2, 2RHI; L3MBTL1-H3.328–34 chimera-Kme2, 2RHU; L3MBTL1197–526-Kme2, 2RHX; L3MBTL1206–519-Kme1, 2RHY; L3MBTL1206–519(D355N), 2RHZ; L3MBTL1206–519(D355A), 2RI2; L3MBTL1206–519(N358Q), 2RI3; L3MBTL1206–519(N358A), 2RI5; BPTF(Y17E)-H31–9K4me2, 2RI7.
References
- Boccuni P, MacGrogan D, Scandura JM, Nimer SD. The human L(3)MBT polycomb group protein is a transcriptional repressor and interacts physically and functionally with TEL (ETV6) J Biol Chem. 2003;278:15412–15420. doi: 10.1074/jbc.M300592200. [DOI] [PubMed] [Google Scholar]
- Bornemann D, Miller E, Simon J. Expression and properties of wild-type and mutant forms of the Drosophila sex comb on midleg (SCM) repressor protein. Genetics. 1998;150:675–686. doi: 10.1093/genetics/150.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 2003;17:1870–1881. doi: 10.1101/gad.1110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Garcia BA, Barber CM, Hake SB, Ptak C, Turner FB, Busby SA, Shabanowitz J, Moran RG, Allis CD, Hunt DF. Modifications of human histone H3 variants during mitosis. Biochemistry. 2005;44:13202–13213. doi: 10.1021/bi050906n. [DOI] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- Kim J, Daniel J, Espejo A, Lake A, Krishna M, Xia L, Zhang Y, Bedford MT. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8:307–318. doi: 10.1038/nrm2143. [DOI] [PubMed] [Google Scholar]
- Klymenko T, Papp B, Fischle W, Kocher T, Schelder M, Fritsch C, Wild B, Wilm M, Muller J. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyllysine-binding activities. Genes Dev. 2006;20:1110–1122. doi: 10.1101/gad.377406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga H, Matsui S, Hirota T, Takebayashi S, Okumura K, Saya H. A human homolog of Drosophila lethal(3)malignant brain tumor (l(3)mbt) protein associates with condensed mitotic chromosomes. Oncogene. 1999;18:3799–3809. doi: 10.1038/sj.onc.1202732. [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JC, Dougherty DA. The cation-π interaction. Chem Rev. 1997;97:1303–1324. doi: 10.1021/cr9603744. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci. 2003;28:69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 2003;17:1823–1828. doi: 10.1101/gad.269603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Sathyamurthy A, Allen MD, Murzin AG, Bycroft M. Crystal structure of the malignant brain tumor (MBT) repeats in sex comb of midleg-like 2 (SCML2) J Biol Chem. 2003;278:46968–46973. doi: 10.1074/jbc.M306469200. [DOI] [PubMed] [Google Scholar]
- Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- Shi X, Kachirskaia I, Walter KL, Kuo JH, Lake A, Davrazou F, Chan SM, Martin DG, Fingerman IM, Briggs SD, et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J Biol Chem. 2007;282:2450–2455. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P, Li G, Sims RJ, III, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- Wang WK, Tereshko V, Boccuni P, MacGrogan D, Nimer SD, Patel DJ. Malignant brain tumor repeats: a three-leaved propeller architecture with ligand/peptide binding pockets. Structure. 2003;11:775–789. doi: 10.1016/s0969-2126(03)00127-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Trievel RC, Rice JC. Human SFMBT is a transcriptional repressor protein that selectively binds N-terminal tail of histone H3. FEBS Lett. 2007;581:3289–3296. doi: 10.1016/j.febslet.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 2003;421:652–656. doi: 10.1038/nature01378. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.