

Abstract
Human noroviruses (HunoVs) are a leading cause of foodborne disease and severe childhood diarrhea, and they cause a majority of the gastroenteritis outbreaks worldwide. However, the development of effective and long-lasting HunoV vaccines and therapeutics has been greatly hindered by their uncultivability. We recently demonstrated that a HunoV replicates in human B cells, and that commensal bacteria serve as a cofactor for this infection. In this protocol, we provide detailed methods for culturing the GII.4-sydney HunoV strain directly in human B cells, and in a coculture system in which the virus must cross a confluent epithelial barrier to access underlying B cells. We also describe methods for bacterial stimulation of HunoV B cell infection and for measuring viral attachment to the surface of B cells. Finally, we highlight variables that contribute to the efficiency of viral replication in this system. Infection assays require 3 d and attachment assays require 3 h. analysis of infection or attachment samples, including rna extraction and rt-qpcr, requires ~6 h.
Introduction
HuNoVs are globally prevalent pathogens. They are the principal cause of gastroenteritis outbreaks in industrialized and developing nations1,2, causing over 20 million symptomatic infections in the United States each year3. HuNoVs are now recognized as the leading cause of severe childhood diarrhea in parts of the world where an effective rotavirus vaccine has been introduced4,5, and they are the most common cause of foodborne disease outbreaks globally6. Despite the clinical importance of these viruses, relatively little is known about their pathogenic mechanisms. One of the most notable obstacles to investigating HuNoVs has historically been their uncultivability. Considering the enteric nature of HuNoVs, intestinal epithelial cells (IECs) lining the gut are a hypothesized cellular target. Yet extensive efforts to cultivate HuNoVs in epithelial cells have been thus far unsuccessful7–10, although NoVs can be internalized by IECs and transcytosed across them11–14. The closely related murine NoVs (MuNoVs) are well established to display tropism for macrophages and dendritic cells in vitro and in vivo, yet they do not seem to replicate in epithelial cells15–17. Although efforts to infect primary blood-derived human macrophages and dendritic cells with a HuNoV were unsuccessful18, several pieces of data led us to recently test whether B cells represent a novel NoV target cell: viral antigen was visualized in the B cell zones of Peyer’s patches in intestinal sections of MuNoV-infected mice17,19; acute MuNoV titers were lower in B cell–deficient animals compared with wild-type mice20; and viral antigen was detected in intestinal B cells in HuNoV-infected chimpanzees21. Indeed, we demonstrated that multiple MuNoV strains and one HuNoV strain infect cultured B cells22,23. Infection of B cells did not result in a detectable loss in cell viability22,23; this finding was unexpected considering that NoVs are nonenveloped and that MuNoV infection of macrophages and dendritic cells is lytic15,22. Importantly, HuNoV replication in B cells is productive, as evidenced by increases in viral genome copies, translation of viral nonstructural and structural proteins and production of infectious progeny virus22,23. This represents the first cultivation system for a HuNoV. Overall evidence thus suggests that NoVs are transcytosed across the intestinal epithelium where they then access their underlying target immune cells, including innate and adaptive cell types24. Here we provide a detailed description of the methods for culturing a HuNoV either in B cells directly or in a coculture system in which the virus must cross a confluent epithelial monolayer to access underlying B cells. A major strength of this assay is its technical simplicity, as it comprises a commonly used cell line maintained in standard medium. A major drawback is the modest level of viral replication achieved in its current state. We also discuss variables influencing the efficiency of HuNoV replication in B cells and the status of reproducibility of this system in other laboratories.
Comparison with other methods
Many groups have unsuccessfully attempted to culture HuNoVs in a variety of cell types, most notably epithelial cells, because this is the first cell type encountered by pathogens that enter the host along mucosal surfaces7–10. One study failed to detect infection of primary human blood-derived macrophages and dendritic cells with a HuNoV18 in spite of the established tropism of MuNoVs for these cell types15. A majority of previous studies focused on one HuNoV genotype (Norwalk virus, a genogroup I, genotype 1 (GI.1) strain) because of the availability of large volumes of Norwalk-positive stool samples, although other genotypes have been tested in more limited analyses7. We instead tested a GII.4 HuNoV strain, specifically GII.4-Sydney22. This stool sample was collected from a 69-year-old male passenger presenting with symptoms of HuNoV infection during a cruise ship outbreak in January of 2013. Another major difference between our methodology and all other studies investigating the cultivability of HuNoVs to the best of our knowledge is that we used unfiltered, unprocessed stool as inoculum, whereas other groups filtered virus-positive stool before applying it onto cultured cells and, in many cases, they further purified virions through sucrose cushions or density gradients. This is a major consideration when comparing methodologies on the basis of our finding that commensal bacteria present in unfiltered inoculum serve as cofactors for infection22,23. Our experiments implicate bacterial surface-expressed histo-blood group antigen (HBGA) as the stimulatory factor, as a HBGA-expressing bacteria could fully rescue the infectivity of filtered inoculum, whereas a non-HBGA-expressing bacteria could not; additionally, synthetic HBGA was also able to rescue infectivity22,23. Although the presence of exogenous HBGA facilitated HuNoV attachment to the surface of B cells and infection22,23, HBGA expression is not sufficient for in vitro permissivity considering that HBGAs expressed on IECs do not render the cells susceptible to viral infection25. Thus, available data indicate that HuNoVs use HBGAs, possibly as attachment factors, and a yet-to-be-identified B cell receptor for viral entry. It is possible that other attempts to culture HuNoVs were unsuccessful because they focused on cell types not expressing the appropriate receptor and/or because they lacked the appropriate commensal bacterial cofactor for infection. It is also feasible that additional cell types including enterocytes will support HuNoV infection when grown under key (yet elusive) conditions.
Limitations
A limitation of this system is the modest level of viral output achieved, ranging from 0.5 to 3.5 logs in a given experiment (Fig. 1). Another limitation is the nature of the inoculum used as a source of virus, which is specifically unfiltered fecal material. This complicated matrix probably delivers signals of an indeterminate nature to the B cells that could influence their susceptibility to viral infection, possibly contributing to the experimental variability inherent to the system. Indeed, we have observed an inverse correlation between viral input levels and infection efficiency (Fig. 1) that could be explained by the presence of an inhibitor in the unfiltered stool sample used as a source of virus. An alternative possible explanation is that viral genome replication is masked by high input levels because of a threshold effect in viral replication. Finally, in spite of the technical simplicity of this method, successful replication of a HuNoV in B cells in other laboratories has been proven to be difficult. As a result, we are working closely with several laboratories to identify key variables influencing viral infection efficiency. We have most extensively collaborated with the Vinjé research group at the United States Centers for Disease Control and Prevention (CDC). Although we have yet to achieve successful infections at this location in spite of intensive efforts and many experimental repeats, we have excluded numerous variables that could influence infection efficiency including differences in medium components, tissue culture plasticware, RNA extraction methods and RT-qPCR analysis. We have also excluded user-variability as being a contributing factor, as members of research groups from the CDC, the University of Michigan (Wobus research group) and Erasmus Medical Center (EMC; Koopmans research group) have successfully infected human BJAB cells with the GII.4-Sydney HuNoV strain when performing infections at the University of Florida, where this system was developed (Fig. 2a), whereas a member of the Karst laboratory (University of Florida) was unsuccessful in infecting BJAB cells at the CDC (data not shown). The research group at EMC has not achieved successful infection at their institute, potentially because of differences in FBS source or virus stock. However, research groups at the University of Michigan (Fig. 2b) and St. Jude Children’s Research Hospital (Schultz-Cherry research group; data not shown) have successfully infected BJAB cells with a HuNoV at their own institutions, although infection efficiency at St. Jude’s has been variable. We have discovered that the source of FBS in medium can influence viral infection efficiency (Fig. 2c), as is true for a variety of other viruses. Empirical evidence suggests that the state of the B cells at the time of viral inoculation is key to achieving successful infection (described in more detail in the protocol).
Figure 1.
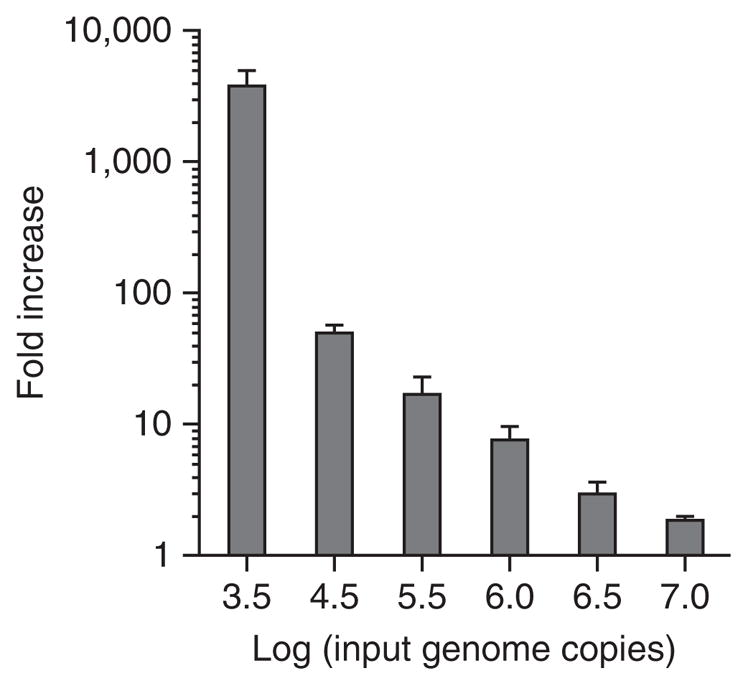
Viral input levels inversely correlate with infection efficiency. BJAB cells were inoculated with increasing numbers of viral genome copies in unfiltered GII.4-Sydney HuNoV-positive stool, as indicated on the x axis. Viral genomes were enumerated at 0 and 3 d.p.i., and they are reported as the log of the fold increase in copies over time. Duplicate samples were tested per condition, and the entire experiment was repeated two times. Values from all replicates per condition were averaged. Error bars denote s.e.m.
Figure 2.

HuNoV infection of B cells is reproducible. (a) Research personnel from the Centers for Disease Control and Prevention (CDC), Erasmus Medical Center (EMC) and the University of Michigan (UM) have successfully infected BJAB cells with GII.4-Sydney HuNoV when performing infections at the University of Florida (UF). Each bar represents the average fold-increase from duplicate or triplicate wells of a single experiment performed at UF. (b) Research personnel at UM have successfully infected BJAB cells with the same stock of GII.4-Sydney HuNoV at UM. Data are reported as the fold increase in genome copies from 0 to 3 d.p.i. at differing inoculation volumes, as indicated on the x axis. Quadruple samples were tested per condition and the entire experiment repeated three times. Values from all replicates per condition were averaged. (c) BJAB cells were cultured in medium containing FBS from three different sources, labeled A, B and C, and then inoculated with GII.4-Sydney HuNoV. Viral genomes were enumerated and the data are reported as fold increase between 0 and 3 d.p.i. Duplicate wells per condition were tested, and the entire experiment was repeated twice. Error bars denote s.e.m. in all panels.
Applications
Although the HuNoV cell culture system described here is in the initial stages of development and currently supports only modest levels of viral replication, the availability of permissive cells supporting in vitro HuNoV replication has tremendous potential to transform NoV research. Extrapolating to the long and equally challenging history of developing hepatitis C virus (HCV) cell culture systems, the development of first-generation low- efficiency cell culture systems facilitated the identification of both host and viral factors contributing to enhanced HCV replication, and it was directly linked to the development of successful anti-HCV drugs26. Moreover, even in its current state, the HuNoV B cell culture system has led to the discovery of a cofactor for viral infection (i.e., commensal bacteria expressing appropriate HBGA glycans), and it has been used to demonstrate the neutralizing activity of a polyclonal anti-capsid antibody22. In addition, because we have developed viral attachment (Box 1) and infection (see PROCEDURE) assays, it should be possible to gain insight into the mechanisms by which antibodies neutralize HuNoVs. Other applications of this first-generation cell culture system include viral receptor identification and testing of putative antiviral compounds for in vitro activity.
Box 1. B cell attachment assay ● TIMING 2 h.
On –4 d.p.i., split a BJAB culture 1:4 in a six-well dish and bring it to a total volume of 2 ml with complete RPMI; incubate the plate at 5% CO2 in a 37 °C incubator.
On 0 d.p.i., chill a 48-well plate at 4 °C.
Quick-thaw the virus-positive stool sample in a 37 °C water bath, dilute it to 1:10 in complete RPMI and immediately place it on ice.
-
Add 2 × 105 BJAB cells and 1.5 × 106 viral-genome copies of diluted stool sample into each microcentrifuge tube, bring it up to a total volume of 360 μl using chilled complete RPMI, mix briefly by pipetting and immediately place the tube on ice.
▲ CRITICAL STEP The samples should be kept on ice for the entirety of the experiment.
Add 120 μl of the virus:cell mixture into three wells of the prechilled 48-well plate and incubate it at 4 °C for 1 h.
Transfer the contents of one well into a microcentrifuge tube, add 1 ml of TRIzol and store it at −80 °C for future RNA extraction according to the manufacturer’s instruction, as described in PROCEDURE Step 2A(i) (this sample will allow you to confirm the input number of viral genomes).
Transfer the contents of the other two wells into two microcentrifuge tubes, and centrifuge at 730g for 7.5 min at 4 °C.
Discard the supernatant and resuspend the pellet in 200 μl of chilled 1× PBS; mix by pipetting.
Wash the cells two more times, as described in steps 7 and 8.
After the third wash, add 1 ml of TRIzol to each supernatant and store it at −80 °C for future RNA extraction according to the manufacturer’s instructions, as described in PROCEDURE Step 2A(i) (these samples will contain only genomes associated with virus attached to the B cells).
Experimental design
Maintenance of B cell lines
The BJAB cell line has been used for a majority of our experiments, and we focus on it exclusively in this protocol. However, we have tested a panel of other human B cell lines and determined that the Raji line (kindly provided by G. McFadden, University of Florida) also supports GII.4-Sydney infection, whereas the Namalwa and HuNS1 lines (also provided by G. McFadden) do not support infection to appreciable levels (Fig. 3). We obtained the BJAB cell line from R. Renne (University of Florida), and we have recently verified that BJAB cells from another source (B. Chandran, Rosalind Franklin University of Medical School and Sciences) support comparable levels of infection (data not shown). B cell lines should be maintained in RPMI with L-glutamine supplemented with 10% (vol/vol) FBS and 1× penicillin/streptomycin (pen/strep) (complete RPMI). Seed cells into six-well plates at an approximate density of 5 × 105 cells per ml in a total volume of 2 ml and incubate them at 5% CO2 in a 37 °C incubator, splitting cells 1:4 every 3–4 d. We use cells in infections 4 d after being split; we split the cells a minimum of two times after thawing before use in experiments, and we have never used cultures that are older than 25 passages. We have observed that BJAB cells are sensitive to cell density, with the cells appearing unhealthy when the density falls below 2.5 × 105 cells per ml or above 3 × 106 cells per ml. When BJAB cells are initially passaged, they will form small clumps of ~10–20 cells; after an additional 1–2 d of incubation, clump size should increase to >40 cells (Fig. 4). We always carry our B cells in six-well plates because they appear healthier than when expanded to larger volumes. Moreover, we only perform BJAB infections after large clumps are visible in the cultures.
Figure 3.
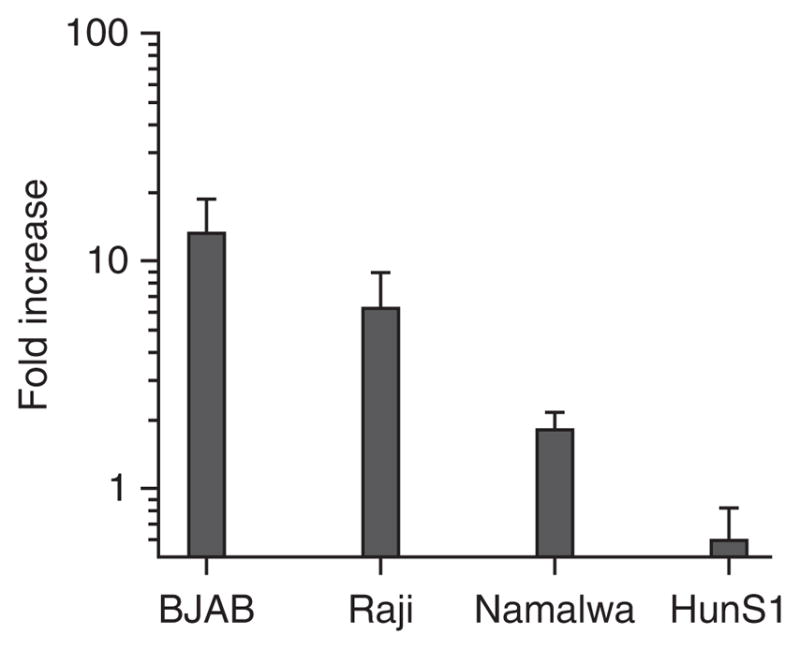
HuNoV replication in B cells is cell line–specific. The Burkitt lymphoma B cell lines BJAB (EBV-negative), Raji (EBV-positive) and Namalwa (EBV-positive) and the myeloma line HuNS1 (EBV-negative) were inoculated with unfiltered GII.4-Sydney HuNoV-positive stool inoculum. Viral genome copy numbers were determined at 0 and 3 d.p.i. for each cell line. Duplicate samples were tested per condition, and the entire experiment was repeated two times. The data are reported as the log of the fold increase in copy numbers over time, and values from all replicates per condition were averaged. Error bars denote s.e.m.
Figure 4.

BJAB morphology. BJAB cells at 4 d after being split 1:4 should have a smooth, round morphology and clumps consisting of >40 cells. Cells were imaged using a Leica DFC350 FX microscope and Leica Application Suite LAS AF AF6000 software.
Maintenance of HT-29 cultures and FITC-dextran assay to test for confluency
HT-29 cells (ATCC HTB-38) should be maintained in DMEM medium containing glucose, glutamine and sodium pyruvate supplemented with 1× Pen/Strep and 10% (vol/vol) FBS (complete DMEM). To set up cocultures using Transwells, trypsinize a confluent flask of HT-29 cells, seed 5 × 104 cells per Transwell insert and bring the final volume of the apical supernatant to 1 ml; the basal chamber should contain 750 μl of medium. Incubate the cells at 5% CO2 in a 37 °C incubator. Change the medium in apical and basal chambers every 3 d. Confluent monolayers should form within 2–3 weeks. The confluence of the monolayer should be confirmed before use in experiments using a FITC-dextran assay: prepare a fresh 4.0 μg/ml solution of FITC-dextran in 1× PBS, aspirate the medium from the apical chamber of each hanging well and add 250 μl of the FITC-dextran solution. Incubate the plate at room temperature (25 °C) and remove 50 μl of medium from the basal chamber at 0, 15 and 45 min. Determine fluorescence of the samples using a fluorometer fitted with 484/20 (excitation) and 528/20 (emission) filter sets and compare it with a standard curve that comprises a serial dilution series of FITC-dextran included on the same plate. HT-29 monolayers are considered confluent when absorbance readings do not increase more than threefold over the 45-min period. If readings increase over this threshold, extend incubation of the Transwell up to 10 d, by checking wells periodically for confluence. If confluence is not reached by 1 month from the date of seeding, discard the Transwell. Aspirate the FITC-dextran solution from the apical chamber and replace it with 1 ml of complete DMEM.
Controls
For all infection and attachment experiments, mock-inoculated cells (i.e., prepared identically to HuNoV-infected cells but inoculated with media only) should be tested in parallel to HuNoV-infected cells to serve as a negative control. For all experiments supplementing filtered stool with a stimulatory factor such as appropriate bacteria, filtered stool in the absence of the stimulatory factor should be included as a negative control and unfiltered HuNoV-positive stool should be included as a positive control.
Materials
REAGENTS
Virus-positive stool sample ! CAUTION HuNoVs are biosafety level 2 (BSL2) pathogens, so standard BSL2 practices should be adhered to strictly whenever you are working with live virus. Acquisition of virus-positive stool samples from infected subjects should only be performed in accordance with a protocol approved by an appropriate institutional review board.
TRIzol (Invitrogen, cat. no. 15596018) ! CAUTION Phenol is toxic and flammable. Use it in a designated hood only.
Chloroform (Acros Organics, cat. no. 423550010) ! CAUTION Chloroform is toxic and flammable. Use it in a designated hood only.
Glycogen (Ambion, cat. no. AM9510)
Isopropanol (Fisher Scientific, cat. no. A451-1) ! CAUTION Isopropanol is toxic and flammable.
Ethanol (Decon Labs, cat. no. 2716) ! CAUTION Ethanol is toxic and flammable.
Nuclease-free water (Fisher Scientific, cat. no. BP2484-100)
Superscript III first-strand synthesis SuperMix (Invitrogen, cat. no. 11752-050); SYBR Green qPCR master mix (Fisher Scientific, cat. no. FERK0243)
Forward genogroup II (GII)–specific primer (NKP2F: ATG TTY AGR TGG ATG AGA TTC TC)25 (Integrated DNA Technologies)
Reverse GII–specific primer (NKP2R: 5′-TCG ACG CCA TCT TCA TTC AC-3′)25 (Integrated DNA Technologies)
AgPath-ID One-step RT-PCR kit (Ambion, cat. no. 4387391)
NdeI restriction enzyme (New England Biolabs, cat. no. R0111S)
MEGAscript T7 transcription kit (Life Technologies, cat. no. AM1334)
Proteinase K (Fisher BioReagents, cat. no. BP1700100)
3 M sodium acetate, pH 5.5 (Ambion, cat. no. AM9740)
10% (wt/vol) SDS solution (Ambion, cat. no. AM9822)
GII-specific probe (RING2-TP: 5′-FAM-TGG GAG GGC GAT CGC AAT CT-TAMRA-3′)25 (Life Technologies, special order)
Trypsin (HyClone, cat. no. SH30042.01)
Balanced salt solution DPBS (HyClone, cat. no. SH30028.02)
Dextran fluorescein (FITC-dextran), 3,000 MW (Invitrogen, cat. no. D3305)
Enterobacter cloacae (ATCC PTA-3882)
Difco nutrient agar (BD Biosciences, cat. no. 213000)
Difco nutrient broth (BD Biosciences, cat. no. 234000)
Growth medium and supplements
RPMI with L-glutamine (Corning Cellgro, cat. no. 10-040-CM)
Penicillin/streptomycin (pen/strep; Corning, cat. no. 30-002-Cl)
FBS, heat-inactivated at 56 °C for 30 min (Atlanta Biologicals, cat. no. S11550)
DMEM with glucose, L-glutamine and sodium pyruvate (Corning Cellgro, cat. no. 10-013-CM)
Cells
BJAB cell line (provided by R. Renne, University of Florida) ! CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
HT-29 cell line (ATCC HTB-38)
EQUIPMENT
Heat block
Water bath
Class II biological safety cabinet appropriate for work with BSL2 pathogens
P1000 barrier pipette tips
P200 barrier pipette tips
P20 barrier pipette tips
Shaking 37 °C incubator
iCycler iQ (Bio-Rad)
qPCR plates (Bio-Rad, cat. no. 2239441)
Optical PCR film (Bio-Rad, cat. no. 2239444)
Tissue culture plates (Costar, cat. no. 3506, 3512, 3548)
T-25 culture flasks (Corning, cat. no. 430168)
1.0-μm hanging cell culture inserts (Millipore, cat. no. PIRP15R48)
3-ml syringes (Fisher, cat. no. 14-823-435)
1.5-ml microcentrifuge tubes
0.22-μm filters (Fisher, cat. no. 09-719C)
Refrigerated microcentrifuge
Fluorometer with 520-nm and 492-nm wavelength, such as Synergy 2 fluorometer (Biotek)
REAGENT SETUP
Complete RPMI
Complete RPMI is RPMI with L-glutamine supplemented with 10% (vol/vol) FBS and 1× pen/strep.
Complete DMEM
Complete DMEM is DMEM medium containing glucose, glutamine and sodium pyruvate supplemented with 10% (vol/vol) FBS and 1× pen/strep.
PROCEDURE
Infection of BJAB cells
-
1|
This step can be performed using option A (direct infection of B cells using unfiltered stool as virus inoculum); option B (direct infection of B cells using filtered virus-positive stool preincubated with stimulatory commensal bacteria); or option C (coculture infection with confluent epithelial cells cultured between the virus inoculum and the B cells). We also include a protocol for enumerating virus attachment to BJAB cells (Box 1).
(A) Direct infection of BJAB cells ● TIMING 7 d
On −4 d post infection (d.p.i.), split a BJAB culture 1:4 in a six-well dish and bring it to a total volume of 2 ml; incubate the plate at 5% CO2 in a 37 °C incubator for 4 d.
-
On 0 d.p.i., quick-thaw the virus-positive stool sample in a 37 °C water bath, dilute it 1:10 in complete RPMI and immediately place it on ice.
▲ CRITICAL STEP Virus-positive stool samples should be divided into single-use aliquots (aliquot volume will need to be determined empirically for each stool sample) and frozen at −80 °C to avoid freezing/thawing the virus.
-
Transfer 1.3 × 105 BJAB cells (determined by counting cells using a hemocytometer) into a microcentrifuge tube.
▲ CRITICAL STEP The cell density and state of the cells at the time of inoculation influences their susceptibility to HuNoV infection; confirm that the BJAB culture is replicating at an appropriate doubling time (33 h) and that large clumps have formed (Fig. 4).
-
Add 104–106 viral genome copies of the stool sample to the B cells and mix briefly by pipetting. Viral genome concentration in a stool sample should be determined before use in infection assays by using the RNA extraction and RT-qPCR protocols outlined in Step 2.
▲ CRITICAL STEP The amount of input virus affects the efficiency of infection (see Fig. 1, explained further in the INTRODUCTION).
Bring the total volume up to 100 μl using complete RPMI. Incubate the mixture for 2 h at 5% CO2 in a 37 °C incubator.
Centrifuge the samples at 730g at 4 °C for 7.5 min, and then discard the supernatant.
Resuspend the pellet in 100 μl of complete RPMI and pipette it up and down using a P200 pipette approximately six times.
Add half of each sample into a well of a 48-well plate (one for 0 d.p.i. collection and the other for 3 d.p.i. collection) and bring each well to a final volume of 1 ml with complete RPMI.
For enumerating input viral genome copies, mix the contents of one well thoroughly and transfer 500-μl aliquots into two microcentrifuge tubes. Add 1 ml of TRIzol to one aliquot for RNA extraction, and reserve the second aliquot for future use if needed.
-
Store both aliquots at −80 °C for future use.
▪ PAUSE POINT The TRIzol mixture can be stored at −80 °C indefinitely (we have stored samples up to 1 month), and it can be extracted simultaneously with samples from later time points (Step 2).
Incubate the 48-well infection plate at 5% CO2 in a 37 °C incubator.
On 3 d.p.i., collect the remaining well, as described in Step 1A(ix–x) for 0 d.p.i. samples.
(B) Bacterial stimulation of viral infection ● TIMING 7 d
On −4 d.p.i., split a BJAB culture 1:4 in a six-well dish and bring it to a total volume of 2 ml with complete RPMI; incubate the plate at 5% CO2 in a 37 °C incubator.
On −2 d.p.i., streak a glycerol stock of E. cloacae onto a nutrient agar plate.
-
On −1 d.p.i., inoculate a loopful of E. cloacae from the overnight plate into 4 ml of nutrient broth and incubate for 18–20 h at 37 °C, with shaking at 220 r.p.m.
▲ CRITICAL STEP Growth conditions (i.e., temperature and medium) can greatly affect the expression of H-type HBGA by E. cloacae. Expression should be confirmed before use in stimulation assays22.
On 0 d.p.i., dilute the overnight culture of E. cloacae to a concentration of 1 × 106 c.f.u. per ml in 10 ml of nutrient broth. Overnight cultures are generally 1 × 109 c.f.u. per ml, and this can be verified by plating a portion of the overnight culture on nutrient agar, by incubating overnight at 37 °C and by counting colonies.
Incubate the diluted E. cloacae at 70 °C for 40 min in a water bath, by mixing the culture at 10 min intervals to heat-inactivate the bacteria.
Centrifuge the heat-inactivated cultures at 4,500g at room temperature for 20 min.
Aspirate the supernatant and discard and resuspend the pellet in 100 μl of complete RPMI.
Quick-thaw a virus-positive stool sample in a 37 °C water bath, dilute it to 1:10 in complete RPMI and immediately place it on ice.
-
Filter the diluted stool through a 0.22-μm membrane using a 3-ml syringe.
▲ CRITICAL STEP Volume is reduced by filtration, so filter 1.5–2 times the necessary final volume of stool needed for the experiment.
Aliquot 20 μl of heat-inactivated bacteria and 104–106 viral genome copies of filtered stool sample (genome copy concentration of the virus-positive stool sample used as a source of virus should be determined before use in infection assays using the RNA extraction and RT-qPCR protocols outlined in Step 2) into each microcentrifuge tube, and incubate the tubes at 5% CO2 in a 37 °C incubator for 1 h.
Add 1.3 × 105 BJABs to the bacteria-virus mixture in the microcentrifuge tube from Step 1B(x), and bring the total volume up to 100 μl using complete RPMI; incubate for 2 h at 5% CO2 in a 37 °C incubator.
Perform Step 1A(vi–xii) for direct infection of BJAB cells.
(c) Coculture infection ● TIMING 8 d
On −5 d.p.i., confirm confluency of the HT-29 epithelium cultured in Transwells using the FITC-dextran assay described in Experimental design (INTRODUCTION).
On −4 d.p.i., split a BJAB culture 1:4 in a six-well dish and bring it to a total volume of 2 ml; incubate the plate at 5% CO2 in a 37 °C incubator.
On 0 d.p.i., quick-thaw a virus-positive stool sample in a 37 °C water bath, dilute it 1:10 in complete RPMI and immediately place it on ice.
Transfer 2.5 × 105 BJAB cells per well in a 12-well plate, and bring each well up to a final volume of 1 ml using complete RPMI.
Aspirate the medium from the apical chamber of each hanging well of HT-29 cells, and then transfer the hanging wells to the plate seeded with BJAB cells (the B cells are now in the basal compartment underneath the confluent epithelium); add 100 μl of complete DMEM to the apical side of each hanging well.
Add 5 × 105 viral genome copies of diluted stool sample to the apical chamber.
-
Add an additional 50 μl of DMEM to the apical side and gently mix the medium and virus.
! CAUTION Do not disrupt the HT-29 cells; tilt the plate so that the volume can be gently mixed by pipetting on the edge of the well.
Incubate the plate at 5% CO2 in a 37 °C incubator for 2 h.
Gently remove the supernatant from the apical chamber and replace it with 1 ml of complete DMEM.
Collect the basal contents from one or two wells into TRIzol to determine the viral input associated with the BJAB cells (i.e., 0 d.p.i. titers), as described for direct infections (Step 1A(ix–x)).
Incubate the remaining wells at 5% CO2 in a 37 °C incubator and collect basal contents at 3 d.p.i., as described in Step 1A(ix–x).
Store all aliquots at −80 °C for up to 1 month for future genome copy determination, as described in Step 1A(x).
Enumeration of viral genome replication
-
2|
Viral genomes can be quantified using option A (one-step RT-PCR) or option B (two-step RT-PCR) protocols; these two methods are comparable in sensitivity and linear range of detection.
(A) One-step RT-PCR ● TIMING 5 h
Extract RNA from samples stored in TRIzol (from Steps 1A(ix–xii), 1B(xii) and 1C(x–xii)) according to the manufacturer’s protocol.
-
Using the AgPath-ID One-step RT-PCR kit, set up a master mix using per reaction quantities as follows:
2× RT-PCR buffer 12.5 μl GII.4 NKP2F primer (10 μM) 0.5 μl GII.4 NKP2R primer (10 μM) 0.5 μl GII.4 RING2-TP (10 μM stock) 0.5 μl Enzyme mix 1.0 μl RNA 150 ng Nuclease-free water Up to 25 μl total volume Run reactions on a Bio-Rad iCycler using the following cycling program: 10 min at 42 °C (RT step); 10 min at 95 °C; 45 cycles of 15 s at 95 °C, and 60 s at 60 °C (collect fluorescence data here).
Include a standard curve for viral genome copy number on each plate, prepared as described in Box 2.
Box 2. Preparation of the standard curve template for one-step RT-PCR reactions ● TIMING 13 h.
Linearize 8 μg of a pGII.4 control plasmid containing the amplicon sequence27 by digesting with Ndel for 3 h at 37 °C; inactivate the restriction enzyme by incubating it for 20 min at 65 °C. Remove a 10-μl aliquot of DNA and verify complete digestion by performing electrophoresis on a 1% (wt/vol) agarose gel: Prepare a 1% (wt/vol) agarose gel containing ethidium bromide. Load ~200 ng of digested plasmid with appropriate proportion of loading dye. As a control, include ~200 ng of undigested plasmid. Run the gel at ~120 V for ~45 min using 1× TAE buffer.
Treat the digested plasmid with 200 μg/ml of proteinase K and 0.5% (wt/vol) SDS for 30 min at 50 °C to remove nucleases.
Perform a phenol/chloroform extraction of the digested DNA, ethanol-precipitate and resuspend it in 15 μl of nuclease-free water using standard procedures.
Perform in vitro transcription using the MEGAscript T7 transcription kit as per the manufacturer’s instructions.
Confirm efficient DNase treatment by performing a PCR reaction with an aliquot of RNA in the absence of a reverse transcription step: perform PCR using the GII.4 NKP2F and GII.4 NKP2R primers, ~20 ng of RNA template and a DNA-dependent polymerase according to the manufacturer’s instructions.
Prepare a dilution series of the RNA transcript: determine the copy number of the RNA sample using a ssRNA copy number calculator (such as the one found at http://endmemo.com/bio/dnacopynum.php). Adjust the RNA concentration with nuclease-free water to yield 1010 copies per μl. Include the following dilutions in the one-step RT-PCR reactions in order to generate a standard curve: 109, 107, 105, 103, 102, 101 and 0 (water only) per qPCR reaction.
(B) Two-step RT-PCR ● TIMING 8 h
Extract RNA from samples stored in TRIzol according to the manufacturer’s protocol.
Generate first-strand cDNAs using the Superscript III first-strand synthesis SuperMix according to the manufacturer’s instructions.
-
Set up a qPCR master mix using per reaction quantities as follows:
SYBR Green master mix 12.5 μl GII.4 NKP2F primer (10 μM) 0.5 μl GII.4 NKP2R primer (10 μM) 0.5 μl cDNA 2 μl Nuclease-free water 9.5 μl Run the samples and plasmid dilution series on a Bio-Rad iCycler using the following cycling program: 10 min at 95 °C; 41 cycles of 15 s at 95 °C, 41 cycles of 30 s at 58 °C (collect melt curve data here); and 30 s at 72 °C.
● TIMING
Step 1A(i), cell preparation: 5–15 min
Step 1A(ii–iv), infection preparation: 20–30 min
Step 1A(v), stool:BJAB incubation: 2 h
Step 1A(vi–x), aliquotting of stool:BJAB mixture and 0 d.p.i. collection: 50 min
Step 1A(xi), incubation: 3 d
Step 1A(xii), day 3 collection: 45 min
Step 1B(i), cell preparation: 5–15 min
Step 1B(ii), culturing of E. cloacae: 5 min
Step 1B(iii), inoculation of E. cloacae: 5 min
Step 1B(iv–vii), 1.25 h
Step 1B(viii–x), stool preparation and incubation with bacteria: 70 min
Step 1B(xi), bacteria:stool:BJAB mixture incubation: 2 h
Step 1B(xii), aliquotting of stool:bacteria:BJAB mixture and collection: 50 min
Step 1C(i), FITC-dextran assay: 1.5 h
Step 1C(ii), cell preparation: 5–15 min
Step 1C(iii), stool preparation: 5 min
Step 1C(iv–v), infection preparation: 15 min
Step 1C(vi–viii), inoculation and incubation: 2.25 h
Step 1C(ix–x), 0 d.p.i. collection: 50 min
Step 1C(xi), infection: 3 d
Step 2A(i), RNA extraction: 2.5 h
Step 2A(ii), RT-qPCR setup: 1 h
Box 1, 2 h
Box 2, 13 h
Step 2A(iii), RT-qPCR reaction:1.5 h
Step 2A(iv), standard curve preparation: 8 h
Step 2B(i), RNA extraction: 2.5 h
Step 2B(ii), cDNA synthesis: 2 h
Step 2B(iii), qPCR setup: 45 min
Step 2B(iv), qPCR reaction: 2.75 h
ANTICIPATED RESULTS
We routinely analyze B cell infection data as the fold increase in genome copy number from 0 d.p.i. (this sample is collected after a 2-h incubation of virus with B cells and removal of unbound virus) to 3 d.p.i. Successful infection of B cells results in a fivefold or greater increase in genome copy number when comparing 0 d.p.i. with 3 d.p.i. There is considerable variability in the efficiency of infection within and between experiments, ranging from 0.5- to 3.5-log fold increases in genome copy number (Fig. 1). Thus, we recommend averaging results from at least triplicate reactions per condition in a given experiment and a minimum of two experimental replicates before applying statistical tests. On the basis of our published findings22,23, filtration of the virus-positive stool sample through a 0.22-μm membrane should prevent viral genome replication, and preincubation of filtered stool inoculum with appropriate bacteria should fully restore infectivity. We have also demonstrated that B cells cultured in the basal chamber underlying a confluent epithelial monolayer will support HuNoV infection when virus inoculum is applied to the apical supernatant22,23; B cell infection in this context is also filtration sensitive, and it can be rescued by preincubation of filtered stool inoculum with heat-killed E. cloacae (unpublished results, Karst laboratory). We routinely analyze B cell attachment data as the percentage of viral genomes associated with B cells after washing compared with viral genomes in the unwashed replicate sample. This percentage is typically ~10–50% for unfiltered virus-positive stool inoculum and 1% for filtered inoculum.
Acknowledgments
This work was funded by the US National Institutes of Health (NIH) R01 1R01AI116892 and the University of Florida Opportunity Fund 00093472 for S.M.K., and by NIH R01 AI080611 and R21 AI103961 for C.E.W. We would like to thank G. McFadden (University of Florida), R. Renne (University of Florida) and B. Chandran (Rosalind Franklin University of Medical School and Sciences) for generously providing cell lines used in these studies. The findings and conclusions in this article are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention (CDC). Names of specific vendors, manufacturers or products are included for public health and informational purposes; inclusion does not imply endorsement of the vendors, manufacturers or products by the CDC or the US Department of Health and Human Services.
Footnotes
AUTHOR CONTRIBUTIONS M.K.J., K.R.G., S.A.T. and S.M.K. developed and optimized direct and co-culture infections of BJAB cells, attachment assays, RNA extraction and qPCR parameters. C.L.G. and S.M.W. contributed to the development of the coculture infections. M.K.J. performed direct and co-culture infections. J.V. and V.C. provided GII.4-Sydney–positive stool samples and performed extensive numbers of infections in their CDC laboratory, investigating assay variables, and they also performed infections at the University of Florida. A.O.K. and C.E.W. performed infections at the University of Florida and the University of Michigan. P.F. and S.S.-C. performed direct infections at St. Jude Children’s Research Hospital using stool samples provided by the CDC and collected at St. Jude’s. M.d.G. and M.K. performed infections at the University of Florida and EMC. M.K.J., K.R.G. and S.M.K. jointly wrote the manuscript and prepared the figures. All authors reviewed and edited the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Patel MM. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg Infect Dis. 2008;14:1224–1231. doi: 10.3201/eid1408.071114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed SM, et al. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect Dis. 2014;14:725–730. doi: 10.1016/S1473-3099(14)70767-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall AJ, et al. Norovirus disease in the United States. Emerg Infect Dis. 2013;19:1198–1205. doi: 10.3201/eid1908.130465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koo HL, et al. Noroviruses: the most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vaccination. J Pediatr Infect Dis Soc. 2013;2:57–60. doi: 10.1093/jpids/pis070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Payne DC, et al. Norovirus and medically attended gastroenteritis in US children. N Engl J Med. 2013;368:1121–1130. doi: 10.1056/NEJMsa1206589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koo HL, Ajami N, Atmar RL, DuPont HL. Noroviruses: the principal cause of foodborne disease worldwide. Discov Med. 2010;10:61–70. [PMC free article] [PubMed] [Google Scholar]
- 7.Duizer E, et al. Laboratory efforts to cultivate noroviruses. J Gen Virol. 2004;85:79–87. doi: 10.1099/vir.0.19478-0. [DOI] [PubMed] [Google Scholar]
- 8.Herbst-Kralovetz MM, et al. Lack of norovirus replication and histo-blood group antigen expression in 3-dimensional intestinal epithelial cells. Emerg Infect Dis. 2013;19:431–438. doi: 10.3201/eid1903.121029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papafragkou E, Hewitt J, Park GW, Greening G, Vinje J. Challenges of culturing human norovirus in a 3-D organoid cell culture model. PLoS ONE. 2013;8:e63485. doi: 10.1371/journal.pone.0063485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takanashi S, et al. Failure of propagation of human norovirus in intestinal epithelial cells with microvilli grown in three-dimensional cultures. Arch Virol. 2013;159:257–266. doi: 10.1007/s00705-013-1806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marionneau S, et al. Norwalk virus binds to histo-blood group antigens present on gastroduodenal epithelial cells of secretor individuals. Gastroenterology. 2002;122:1967–1977. doi: 10.1053/gast.2002.33661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Hernandez MB, et al. Murine norovirus transcytosis across an in vitro polarized murine intestinal epithelial monolayer is mediated by M-like cells. J Virol. 87:12685–12693. doi: 10.1128/JVI.02378-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamura M, Natori K, Kobayashi M, Miyamura T, Takeda N. Interaction of recombinant Norwalk virus particles with the 105-kilodalton cellular binding protein, a candidate receptor molecule for virus attachment. J Virol. 2000;74:11589–11597. doi: 10.1128/jvi.74.24.11589-11597.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White LJ, et al. Attachment and entry of recombinant Norwalk virus capsids to cultured human and animal cell lines. J Virol. 1996;70:6589–6597. doi: 10.1128/jvi.70.10.6589-6597.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wobus CE, et al. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol. 2004;2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward JM, et al. Pathology of immunodeficient mice with naturally occurring murine norovirus infection. Toxicol Pathol. 2006;34:708–715. doi: 10.1080/01926230600918876. [DOI] [PubMed] [Google Scholar]
- 17.Mumphrey SM, et al. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J Virol. 2007;81:3251–3263. doi: 10.1128/JVI.02096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lay MK, et al. Norwalk virus does not replicate in human macrophages or dendritic cells derived from the peripheral blood of susceptible humans. Virology. 2010;406:1–11. doi: 10.1016/j.virol.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basic M, et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10–deficient mice. Inflamm Bowel Dis. 2014;20:431–443. doi: 10.1097/01.MIB.0000441346.86827.ed. [DOI] [PubMed] [Google Scholar]
- 20.Zhu S, et al. Identification of immune and viral correlates of norovirus protective immunity through comparative study of intra-cluster norovirus strains. PLoS Pathog. 2013;9:e1003592. doi: 10.1371/journal.ppat.1003592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bok K, et al. Chimpanzees as an animal model for human norovirus infection and vaccine development. Proc Natl Acad Sci USA. 2011;108:325–330. doi: 10.1073/pnas.1014577107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones MK, et al. Enteric bacteria promote human and murine norovirus infection of B cells. Science. 2014;346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karst SM. Identification of a novel cellular target and a co-factor for norovirus infection: B cells and commensal bacteria. Gut Microbes. 2015;6:266–71. doi: 10.1080/19490976.2015.1052211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karst SM, Wobus CE. A working model of how noroviruses infect the intestine. PLoS Pathog. 2015;11:e1004626. doi: 10.1371/journal.ppat.1004626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guix S, et al. Norwalk virus RNA is infectious in mammalian cells. J Virol. 2007;81:12238–12248. doi: 10.1128/JVI.01489-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohmann V, Bartenschlager R. On the history of hepatitis C virus cell culture systems. J Med Chem. 2013;57:1627–1642. doi: 10.1021/jm401401n. [DOI] [PubMed] [Google Scholar]
- 27.Park Y, Cho YH, Ko G. A duplex real-time RT-PCR assay for the simultaneous genogroup-specific detection of noroviruses in both clinical and environmental specimens. Virus Genes. 2011;43:192–200. doi: 10.1007/s11262-011-0626-4. [DOI] [PubMed] [Google Scholar]