

Abstract
In-vitro-derived platelets (PLTs) could potentially overcome problems associated with donated PLTs, including contamination and alloimmunization. Although several groups have produced functional PLTs from stem cells in vitro, the challenge of developing this technology to yield transfusable PLT units has yet to be addressed. The asynchronous nature of in vitro PLT generation makes a single harvest point infeasible for collecting PLTs as soon as they are formed. The current standard of performing manual centrifugations to separate PLTs from nucleated cells at multiple points during culture is labor-intensive, imprecise, and difficult to standardize in accordance with current Good Manufacturing Practices (cGMP). In an effort to develop a more effective method, we adapted a commercially-available, spinning-membrane filtration device to separate in-vitro-derived PLTs from nucleated cells and recover immature megakaryocytes (MKs), the precursor cells to PLTs, for continued culture. Processing a mixture of in-vitro-derived MKs and PLTs on the adapted device yielded a pure PLT population and did not induce PLT pre-activation. MKs recovered from the separation process were unaffected with respect to viability and ploidy, and were able to generate PLTs after reseeding in culture. Being able to efficiently harvest in-vitro-derived PLTs brings this technology one step closer to clinical relevance.
Introduction
Several million units of donor platelets (PLTs) are transfused annually in the US and Europe. Prior to transfusion, PLTs must be stored at 20–24°C to avoid irreversible changes in receptor conformation that occur during refrigeration and lead to PLT clearance after infusion (Rumjantseva and Hoffmeister 2010). Although skin bacteria introduced during PLT collection can rapidly proliferate at 20–24°C, the FDA does not require testing PLT products for bacterial contamination prior to transfusion. AABB standards require accredited facilities to implement methods that limit and detect bacterial contamination in PLT products, but contaminated PLTs remain a potentially very dangerous and costly occurrence. It is estimated that 2,000–4,000 contaminated PLTs are transfused each year, making them the leading cause of transfusion-related disease (Lindholm et al. 2011). Recipient alloimmunization against HLA class I antigens, which results in rapid destruction of transfused PLTs, is also a concern. Importantly, the risk of alloimmunization increases in patients receiving long-term PLT therapy, with one study reporting a 55% prevalence in multi-transfused patients (Marwaha and Sharma 2009).
PLTs produced in vitro from megakaryocytes (MKs) derived from hematopoietic stem cells could potentially overcome some of the limitations of donated platelets. Induced pluripotent stem cell (iPSC) lines engineered to be HLA-null could give rise to “universal” PLTs that would lessen the risk of alloimmunization (Zimmermann et al. 2012). In addition, the use of current Good Manufacturing Practices (cGMP) during in vitro PLT production could nearly eliminate the risk of bacterial contamination, as well as the typical 2-day quarantine period assigned to donor PLTs while disease test results are pending. While improving safety, this would also decrease the time between PLT generation and transfusion. Since younger PLTs are thought to be more physiologically active (Guthikonda et al. 2008), this could result in an enhanced post-transfusion benefit for the patient.
We and others have successfully generated MKs and functional PLTs in vitro from a starting population of CD34+ hematopoietic stem and progenitor cells (HSPCs) from mobilized peripheral blood (mPB) (Choi et al. 1995; Panuganti et al. 2013) or umbilical cord blood (Lasky and Sullenbarger 2011; Matsunaga et al. 2006; Pallotta et al. 2011; Robert et al. 2011; Ungerer et al. 2004). However, the heterogeneity of the starting population, despite CD34 enrichment, and the stochastic nature of cell differentiation result in asynchronous MK commitment and maturation. As a result, we find that PLTs are shed into culture over a period of up to 5 days. Functional PLTs have also been generated from MK progenitor cell lines (Nakamura et al. 2014), iPSCs (Nakagawa et al. 2013; Takayama et al. 2010) and embryonic stem cells (ESCs) (Fujimoto et al. 2003; Lu et al. 2011; Takayama et al. 2008), but these cell types also exhibit heterogeneity that would presumably lead to asynchronous PLT generation. Ideally, in-vitro-derived PLTs would be removed from culture soon after generation, and either transfused immediately or placed into optimal storage conditions to preserve PLT function. The harvest would also preferably yield a pure PLT population for transfusion, especially if iPSCs or ESCs are used as the starting cell types, as these cells may persist in differentiation cultures (Fu et al. 2012) and have the ability to form teratomas (Liu et al. 2013). Although the PLT-containing product could be irradiated to destroy any remaining nucleated cells, transfusion of large numbers of apoptotic cells could induce an adverse immune response (Martelli et al. 2000).
In-vitro-derived PLTs are often harvested from 2D culture (i.e., plastic culture dishes) via successive centrifugations to pellet cell populations of decreasing size (Choi et al. 1995; Fujimoto et al. 2003; Lu et al. 2011; Panuganti et al. 2013; Robert et al. 2011; Takayama et al. 2008; Takayama et al. 2010; Ungerer et al. 2004). However, this approach is labor-intensive, subject to error-prone manual manipulation, challenging to implement in a cGMP method, and unlikely to generate a pure PLT population. As an alternative, we adapted a commercially-available, closed-system, spinning-membrane filtration system for rapid, automated separation of in-vitro-derived PLTs from nucleated cells. As part of this development process, we modified the material and pre-wetting procedure for the collection containers used during the process, as well as the membrane rotation speed for improved separation of in-vitro-derived PLTs, which are larger than normal blood PLTs. Importantly, we also showed that MKs recovered from this process retain the ability to form proplatelets (proPLTs) and shed PLTs, suggesting that this separation process could be used to harvest PLTs at multiple time points during in vitro culture.
Materials and Methods
See “Supplemental methods” for information on culture of PMA- and nicotinamide-treated CHRF cells; apheresis PLT collection; analysis of CHRF cell and apheresis PLT recovery; flow cytometeric analysis of MK apoptosis, MK ploidy, apheresis PLTs, and in-vitro-derived MKs and PLTs; measurement of cell size; pre-wetting of PL-1813 and PL-2410 containers and quantification of cell recovery; quantification of RBC recovery; primary HSPC culture for PLT production; harvest and concentration of in-vitro-derived MKs and PLTs; reseeding of concentrated MKs; and immunocytochemistry of in-vitro-derived MKs and PLTs.
Spinning-membrane filtration device and separation process
We used a laptop-controlled, prototype spinning-membrane filtration device (Lovo; Fresenius Kabi, Lake Zurich, IL) that consisted of 3 pumps, 5 clamps, 4 weigh scales, and a spinning-membrane driver mechanism, as described in Fig. 1.
Figure 1. Spinning-membrane filtration device.

(A) The device was outfitted with disposable poly(vinyl chloride) (PVC; PL-2286) tubing and a track-etched, polycarbonate filter membrane with either the standard 4-μm pore-size filter or a modified, 5-μm pore-size filter. Standard di-2-ethylhexyl phthalate (DEHP)-plasticized PVC (PL-1813; Fresenius Kabi) or ethyl-vinyl acetate blend, plasticizer-free (PL-2410; Fresenius Kabi) containers were used to collect cells, as indicated. PAS-V was used to pre-wet the device tubing. A syringe outfitted with a 17G cannula (BD, Franklin Lakes, NJ) was used to transfer the input cell suspension to the PreWash container. The PL-2410 PreWash container was modified with an Interlink blood bag spike (Baxter, Deerfield, IL) prior to cell addition. The volume in the PreWash container was increased to 200 mL with PAS-V. Containers for the PreWash, In-Process, MK fraction, and PLT fraction, as well as a container of PAS-V (buffer) were sterile-docked (SCD 312; Terumo BCT, Lakewood, CO) to the device. During the automated separation process, cell solution was drawn from the PreWash container into the device using default flow rate settings and spin speeds set to either 2000 or 3000 rpm. The “No Dilution” option was selected, as cell concentrations were lower than those requiring dilution. Cells that passed through the pores of the spinning membrane were diluted in PAS-V and immediately collected into the PLT fraction container. After all of the PreWash solution had been processed, the device was flushed with PAS-V. Cells that did not pass through the membrane were collected during this flush and directed to the In-Process container. The contents of the In-Process container were drawn back into the device and the separation process was repeated. After all of the In-Process solution had been processed, the device was flushed with PAS-V and any cells present were directed to the MK fraction container. A target final volume of 100 mL was used for the MK fraction. Finally, the device tubing was washed with PAS-V and the rinsate was collected in the PreWash container (lost cells fraction). (B) Schematic of the case housing the polycarbonate filter membrane. (C) Close-up of the filter membrane with track-etched, 4-μm pores that may be perpendicular or deviate up to 45° from the perpendicular.
Separation of PMA- and nicotinamide-treated CHRF cells and apheresis PLTs
The human megakaryoblastic CHRF-288-11 (CHRF) cell line was cultured with phorbol 12-myristate 13-acetate (PMA; Calbiochem, Whitehouse Station, NJ) and nicotinamide (Nic; Sigma Aldrich, St. Louis, MO) to promote MK differentiation and polyploidization, respectively, as described in “Supplemental methods”. On day 5, ~1 × 107 PMA- and Nic-treated CHRF cells were transferred to a PL-1813 (Fresenius Kabi) container and processed using the spinning-membrane filtration device outfitted with PL-1813 collection containers and the standard 4-μm pore-size membrane rotating at 3000 rpm. PLT additive solution-V (PAS-V; Fresenius Kabi) was prepared, as previously described (Radwanski et al. 2012), and used as the resuspension fluid. Apheresis PLTs in 5% plasma/95% PAS-V that had been stored for 1 day (see “Supplemental methods” for collection procedure) were added to the resultant CHRF cell concentrate to a final ratio of 1000 PLTs per CHRF cell (~1 × 1010 PLTs total). The CHRF cell/PLT mixture was then processed using the same settings as for CHRF cells only.
Separation of basal CHRF cells and RBCs
Red blood cells (RBCs) were obtained from an 8-day-old, whole-blood-derived, leukoreduced RBC concentrate stored in additive solution-1 (AS-1; Fenwal, Lake Zurich, IL). Basal CHRF cells in IMDM+10% FBS were transferred to conical tubes and transported for ~1 hour at room temperature from Northwestern University to Fresenius Kabi. There, PL-2410 containers (Fresenius Kabi) to be used during the separation were pre-wet with 1% BSA in PBS. The CHRF cells were transferred to a PL-2410 container, which was then spiked with a sample of RBC concentrate to give a final ratio of 1000 RBCs per CHRF cell. The CHRF cell/RBC mixture was processed on the spinning-membrane filtration device outfitted with either a 4- or 5-μm pore-size membrane rotating at either 2000 or 3000 rpm, using PAS-V as the resuspension fluid.
Separation of in-vitro-derived MKs and PLTs
MKs and PLTs were derived in vitro from CD34+ HSPCs from mPB, harvested, and transported to Fresenius Kabi as described in “Supplemental methods”. On the day of separation, ~1–4 × 107 in-vitro-derived MKs and ~0.1–1 × 107 in-vitro-derived PLTs were transferred to PL-1813 or PL-2410 containers that were unmodified or pre-wetted with 1% BSA in PBS, as described. The cells were separated using the spinning-membrane filtration device outfitted with a 4-μm pore-size membrane rotating at 2000 or 3000 rpm, with PAS-V as the resuspension fluid.
Statistical Analysis
Results are expressed as mean ± standard deviation (SD). Statistical comparisons were done using a Student’s t-test. P values < 0.05 were considered significant.
Results
Separation of apheresis platelets from a differentiated megakaryoblastic cell line
The spinning-membrane filtration device separates cells of different size by pumping a solution of cells into a chamber housing a cylindrical, spinning, porous membrane (Fig. 1). The rotating membrane helps reduce fouling compared to a stationary membrane. Cells that pass through the pores of the membrane are recovered in one container, while larger cells are retained and collected into a separate container.
To evaluate spinning-membrane filtration for separating MKs and PLTs and determine the effect of the separation process on PLT activation and subsequent MK maturation, we first used a mixture of PMA-treated (up to 32N) CHRF megakaryoblastic cells and day-5, donor apheresis PLTs as a model system. We have previously observed that PMA-treated CHRF cells cultured on a surface that inhibits protein adsorption undergo polyploidization, but do not extend proPLT-like extensions until transferred to a surface with adsorbed serum proteins (ACS, unpublished data). Five days prior to separation, CHRF cells were seeded on a neutral, hydrophilic, PolyHEMA-coated surface and treated with PMA and Nic to generate a mix of cells of varying ploidy, similar to what is observed during culture of primary MKs. The CHRF cells (~1 × 107) were transferred to a PL-1813 (DEHP-plasticized PVC), disposable container, which is part of the spinning-membrane filtration device consumables kit. Knowing that blood PLTs are 2–4 μm in diameter and using previous data for removing PLTs from diluted cell suspensions (CW, unpublished data), we chose a membrane with 4-μm pores and a 3000 rpm membrane rotation speed. Because we were unsure how FBS might affect apheresis PLTs, we first used the filtration device to resuspend the CHRF cells in serum-free PAS-V before adding ~1 × 1010 PLTs. When we processed a mixture of CHRF cells and PLTs, we observed ≥ 90% overall recovery of both cell types (Fig. S1A, C), with 97% and 91% separation of recovered CHRF cells and PLTs into the MK and PLT fractions, respectively. The remaining recovered CHRF cells were collected as lost cells (Fig. S1B, D), meaning that they remained in the tubing of the disposable kit after the automated procedure finished. Processed PLTs showed little increase in pre-activation, as measured by surface CD62P expression, but still activated in response to ADP and TRAP-6 agonists (Fig. S2A). CHRF cell viability was unaffected by the separation process (Fig S2B), and CHRF cells with a similar ploidy distribution to the input population were recovered in the MK fraction (Fig. S2C; mean ploidy: PreWash: 6.3 ± 0.3 N, MK fraction: 6.3 ± 0.6 N, n = 3). In addition, recovered CHRF cells reseeded in culture on a surface with adsorbed proteins from serum formed proPLT-like extensions comparable to those generated by cells that were not processed on the device (Fig. S2D).
Pre-wetting containers and decreasing membrane rotation speed improve separation of in-vitro-derived PLTs
After characterizing the separation process using our model system, we separated MKs and PLTs derived from mPB CD34+ HSPCs. To obtain in vitro-derived PLTs, CD34+ HSPCs were cultured using a protocol that we have previously shown promotes MK differentiation (Panuganti et al. 2013). On day 7 of culture, MKs were isolated using immunomagnetic separation, then reseeded for PLT production. Day-12 cultures of MKs (~1–4 × 107) and PLTs (~0.1–1 × 107) were transferred to PL-1813 containers and processed on the filtration device with the same settings used for the model system (4-μm pore-size membrane rotating at 3000 rpm). Although we observed nearly exclusive separation of recovered, in-vitro-derived MKs into the MK fraction (Fig. S3B), similar to what we had observed with our model system, overall recoveries of MKs and PLTs were substantially lower (~50% and 30% for MKs and PLTs, respectively; Fig. S3A, C). Furthermore, only ~70% of all recovered in-vitro-derived PLTs were found in the PLT fraction (Fig. S3D), in contrast to > 90% with apheresis PLTs (Fig. S1D). Promisingly, though, we saw little increase in culture-derived PLT activation with processing (data not shown), which mirrored our results with apheresis PLTs.
Because the HSPCs were cultured in serum-free medium, whereas the CHRF cells were cultured with FBS, we wondered whether non-specific cell adsorption to the hydrophobic surface of the PL-1813 containers (Gabriel et al. 2012) might be responsible for the low overall cell recovery. To explore this further, we pre-wetted PL-1813 containers, as well as PL-2410 containers, with PBS alone or 1% BSA in PBS, then filled them with a solution of washed, basal CHRF cells in PBS. PL-1813 plastic is not gas-permeable and is used for plasma and RBC storage, while PL-2410 is very gas-permeable and is used for PLT storage (Prowse et al. 2014). CHRF cell recovery after 1 hour was less than 60% for both types of containers in the absence of pre-wetting (Fig. S4), which mirrored what we saw in our first separation experiment with in-vitro-derived MKs and PLTs (Fig. S3). Pre-wetting the containers with 1% BSA in PBS resulted in ~100% cell recovery, while pre-wetting with PBS alone gave no improvement. Based on these results, and the fact that gas-permeable PL-2410 containers would be preferable for PLT storage after separation, we chose to use PL-2410 containers pre-wetted with 1% BSA in PBS for all subsequent experiments.
To better understand the poorer separation of in-vitro-derived PLTs, compared to apheresis PLTs, into the PLT fraction, we determined the average diameter of each type of PLT. Day-5 apheresis PLTs, which have increased in size during storage (Schwertz et al. 2010), were 3.3±0.8 μm in diameter, while in-vitro-derived PLTs were significantly larger at 5.6±2.3 μm (Fig. S5A, B). Interestingly, in-vitro-derived PLTs displayed a unimodal size distribution (Fig. S5C), suggesting that culture-derived PLTs are uniformly larger than blood PLTs. Although the enucleate nature of PLTs allows for increased flexibility, it is likely that some in-vitro-derived PLTs were impeded from traversing the 4-μm pores of the membrane due to their larger size. To overcome this problem in future separations, we proposed to increase the membrane pore size and/or decrease the membrane rotation speed, which would allow larger cells to move closer to the membrane and increase their probability of passing through. We compared the effects of these changes using a mixture of basal CHRF cells (12–13-μm diameter) and RBCs. Basal CHRF cells are exclusively 2N and 4N, and are therefore smaller than mature, polyploid in-vitro-derived MKs and closer in size to non-MKs that could arise during culture of CD34+ HSPCs. RBCs, with an average diameter of 7.3±1.3 μm, are 30% larger than in-vitro-derived PLTs (Fig. S5). Therefore, we reasoned that using basal CHRF cells and RBCs would present a “worst-case scenario” with respect to size, and that any improvements we observed in RBC recovery into the PLT fraction, in the absence of CHRF cell contamination, would translate to future separations of smaller, in-vitro-derived PLTs and larger, in-vitro-derived MKs. Note that enucleated RBCs are not present in our primary MK cultures (ACS, WMM; unpublished data) and therefore would not potentially contaminate the PLT fraction. We chose to evaluate 5-μm pores and 2000 rpm because the nucleus of a 2N CHRF cell is ~5.5 μm (ACS, unpublished data) and data from the separation of dilute blood cell suspensions showed little increase in white blood cell (10–12-μm-diameter) contamination of the PLT fraction at 2000 rpm (CW, unpublished data). Upon separating basal CHRF cells and RBCs, we saw that decreasing the membrane rotation speed to 2000 rpm and increasing the pore size to 5 μm resulted in the highest RBC recovery in the PLT fraction (92%), but also allowed 20% of basal CHRF cells to pass through the pores and into the PLT fraction (Fig. S6). Since generating a PLT population free of nucleated cells was one of our key objectives, we felt that this level of nucleated cell contamination of the PLT fraction was too high. However, reducing the membrane rotation speed to 2000 rpm, while keeping the 4-μm pore size, gave 80% RBC recovery in the PLT fraction without considerable nucleated cell contamination; only 7% of basal CHRF cells were recovered in the PLT fraction. Using 1500 rpm and 4-μm pores, PLT recovery in the PLT fraction was increased to 89%, however recovery of nucleated cells was also increased to 15%. As with 2000 rpm and 5-μm pores, we decided that this nucleated cell contamination was too high and decided to proceed with 2000 rpm and 4-μm pores. Importantly, enucleate RBCs have been shown to easily pass through 4-μm-diameter micropipettes (Jay et al. 1972), which is in line with our results with 4-μm-diameter pores.
To validate improved PLT separation with 4-μm pores and 2000 rpm membrane rotation speed, we derived MKs and PLTs from CD34+ HSPCs, then split the day-12 population into two containers for processing at either 2000 or 3000 rpm. Size-based flow cytometry plots of the PLT fractions from each run indicate that larger PLTs were able to traverse the membrane at 2000 rpm, compared to 3000 rpm (Fig. 2A, see arrows), which resulted in 80% of all recovered PLTs being collected in the PLT fraction at 2000 rpm, versus only 34% at 3000 rpm (Fig. 2B). Importantly, at either rotation speed, < 1% of the input nucleated cells were recovered in the PLT fraction (Fig. 2C). The fact that a lower percentage of PLTs was recovered in the PLT fraction for 3000 rpm in this experiment compared to our first experiment with in-vitro-derived MKs and PLTs may be attributed to donor-dependent differences in PLT size.
Figure 2. 2000 rpm membrane rotation speed increases in-vitro-derived PLT recovery in the PLT fraction.

MKs and PLTs derived from CD34+ HSPCs were harvested from culture on day 12 and divided into two PL-2410 containers. The containers were processed on the spinning-membrane filtration device outfitted with a 4-μm pore-size membrane rotating at either 2000 or 3000 rpm. Side scatter (SSC) versus forward scatter (FSC) flow cytometry plots show that larger PLTs are retained in the PLT fraction for 2000 vs. 3000 rpm (A, arrows). The percentages of recovered PLTs (B) and MKs (C) collected into each fraction are shown for a single experiment.
Separation of in-vitro-derived MKs and PLTs
We used a 4-μm pore-size membrane and 2000 rpm rotation speed to perform replicate day-12 separations of MKs and PLTs derived from CD34+ HSPCs from two additional donors. From the 3 experiments, we observed ~100% recovery of MKs and PLTs (Fig. 3A). Separation of recovered MKs into the MK fraction was similarly high at 92% (Fig. 3B), with the remainder recovered as lost cells. However, PLT separation was lower, as on average only 62% of PLTs were recovered into the PLT fraction, with 34% being collected into the MK fraction (Fig 3C). PLT recovery into the PLT fraction was also highly donor-dependent, with a range of 50–80%.
Figure 3. Spinning-membrane filtration of in-vitro-derived, day-12 MKs and PLTs with a 4-μm pore-size membrane rotating at 2000 rpm.

Recovery of MKs and PLTs (A) and separation of recovered MKs (B) and PLTs (C) into the various fractions are shown as the mean ± SD of 3 separate experiments. The symbols representing each donor match those shown in Figs. 4 and 9.
Separated PLTs show minimal increase in pre-activation and are responsive to agonist treatment
Upon PLT activation at a wound site, granules containing CD62P are translocated to the PLT surface, and the β3 subunit of the surface fibrinogen (Fg) receptor is converted to its high-affinity conformation to allow binding of soluble Fg. Although PAC-1 antibody surface binding can be used to assess β3 activity, soluble Fg binding is a more physiologically relevant test. In addition, at least two cancer cell lines have been shown to bind PAC-1 antibody, but not soluble Fg (Boudignon-Proudhon et al. 1996; Tohyama et al. 1998), suggesting that soluble Fg binding is a more stringent test for β3 activity. We observed that the percentage of CD62P+ PLTs (Fig. 4A, B) and PLT binding of soluble Fg (Fig. 4A, C) in the PLT fraction was minimally higher than that observed in the PreWash population, indicating that the separation process did not result in pre-activation. There was a somewhat greater increase in % CD62P+ PLTs in the MK fraction, compared to the PreWash population (Fig. 4B). We also analyzed CD62P expression for day-6 apheresis PLTs for comparison (Fig. 4B). In-vitro-derived PLTs from two donors had more resting CD62P+ PLTs than day-6 apheresis PLTs, which were 20–30% CD62P+, a similar level to that observed previously (Radwanski and Min 2013). These results indicate that further steps must be taken to limit pre-activation of in-vitro-derived PLTs in culture, prior to harvesting. Importantly, addition of the agonist TRAP-6 similarly increased CD62P expression of PLTs in the PreWash and PLT fraction to ~100%, similar to day-6 apheresis PLTs. Coupled with the fact that soluble Fg binding increased with TRAP-6 exposure for the PreWash and PLT fractions (Fig. 4C), these results suggest that the recovered PLTs retained their potential for activation. Morphological analysis of PLTs in the PreWash and PLT fractions showed the characteristic ring of β-tubulin, minimal spreading on BSA, formation of actin-containing filopodia on Fg, and stress fiber formation in the presence of thrombin, with little difference between the two populations other than smaller PLT size in the PLT fraction (Fig. 5). Although these assays suggest similar functionality of in vitro-derived PLTs before and after spinning-membrane filtration, more rigorous testing, including aggregometry with collagen, epinephrine, ristocetin, and ADP, would be required to fully assess the functionality of in vitro-derived PLTs.
Figure 4. Activation state of in-vitro-derived, day-12 PLTs before and after separation.

Day-12 PreWash PLTs and those recovered in the MK and PLT fractions after separation were identified by size (side scatter [SSC] vs forward scatter [FSC]), then CD41+CD42b+ PLTs were analyzed for surface CD62P expression and binding of soluble Fg when at rest or after incubation with TRAP-6 agonist. For comparison, day-6 apheresis PLTs were also analyzed for CD62P expression. (A) Representative staining panel for in-vitro-derived PLTs. (B) CD62P results from 3 separate experiments. (C) Fg binding results shown as the mean ± SD of 2 separate experiments. MFI = mean fluorescence intensity. N.S. = not significant; P > 0.05. The symbols representing each donor match those shown in Figs. 3 and 9.
Figure 5. Morphology of in-vitro-derived, day-12 PLTs before and after separation.
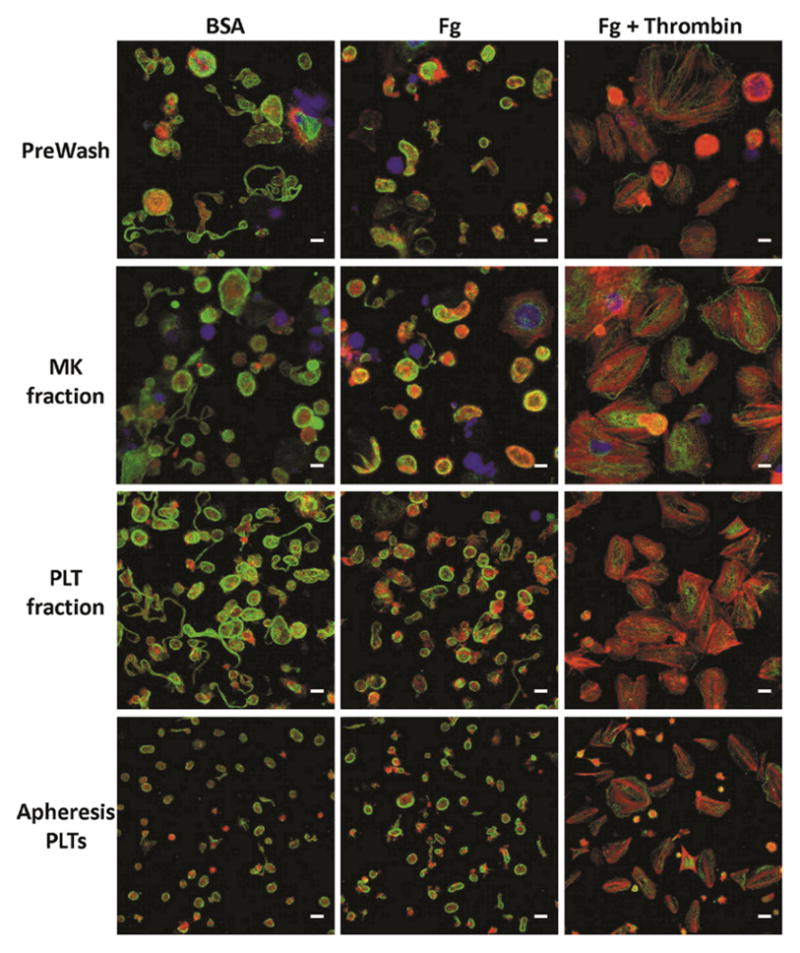
Day-12 PreWash PLTs and those recovered in the MK and PLT fractions after separation, as well as day-6 stored apheresis PLTs were seeded on BSA, Fg, or Fg in the presence of thrombin for 1 hour at 37°C. The PLTs were then stained for β-tubulin (green), F-actin (red), and DNA (blue). Scale bar = 5 μm. Images are from a single representative experiment.
Separated MKs are similar to PreWash MKs with respect to viability and ploidy
To determine whether the separation process damaged MKs, we assayed for viability and apoptosis, and found that the PreWash and MK fractions were very similar in their distribution of viable, apoptotic, dead, and disintegrated cells (Fig. 6A). It remains controversial whether aspects of apoptosis are required for proPLT formation (De Botton et al. 2002; Josefsson et al. 2011; White et al. 2012), but senescent MKs (i.e., those that have shed PLTs) undergo classical apoptosis (De Botton et al. 2002). Therefore, it is not surprising that we observed low (~40%, Fig. 6A) viability of in-vitro-derived MKs in cultures where PLT shedding had begun. We also examined whether the separation process favored recovery of MKs of a particular ploidy class. However, the ploidy distribution of recovered MKs was very similar to that of the PreWash MKs (Fig. 6B; mean ploidy: PreWash: 4.7 ± 0.9N, MK fraction: 4.7 ± 1.1 N, n = 3).
Figure 6. Viability and ploidy of in-vitro-derived, day-12 MKs before and after separation.

Day-12 PreWash MKs and those recovered in the MK fraction after separation were analyzed for viability and apoptosis by DAPI and Annexin V staining (A), as well as ploidy using proidium iodide to stain DNA (B). Viability results are shown as mean ± SD for 3 separate experiments. N.S. = not significant; P > 0.05. Ploidy histograms are from a single representative experiment.
Separated MKs form proPLTs and release functional PLTs after reseeding into culture
Because the separation process would ideally be performed at multiple time points during culture for maximum recovery of newly formed PLTs, we reseeded MKs from the day-12 recovered MK fraction into culture for PLT generation. In an effort to exclude large prePLTs that were recovered in the day-12 MK fraction and make it easier to identify new PLT production that had occurred post reseeding, only large, presumably nucleated, cells that pelleted during a slow centrifugation (100g for 10 minutes) were reseeded. Importantly, proPLT formation was not observed immediately after reseeding, suggesting that any proPLTs observed on subsequent days were newly formed (Fig. S7). Fluorescence microscopy images of proPLTs formed from reseeded day-12 MK fraction cells showed similar morphology compared to proPLTs formed from unprocessed cells, suggesting that the separation process did not affect MK terminal maturation potential (Fig. 7).
Figure 7. ProPLT formation by in-vitro-derived, day-12 MKs reseeded in culture before or after separation.

Day-12 PreWash MKs and those recovered in the MK fraction after separation were resuspended in fresh media and cytokines and reseeded on glass slides. On day 14, the cells were stained for β-tubulin (green), F-actin (red), and DNA (blue). Scale bar = 25 μm. This figure shows two images from a single representative experiment.
On day 14, the day-12-reseeded MKs and their PLT progeny were processed using a 4-μm pore-size membrane rotating at 2000 rpm. In one experiment, we observed low overall recoveries of both MKs and PLTs, but a subsequent experiment showed nearly 100% recovery of both cell types (Fig. 8A, C). Importantly, in both experiments, >93% of all recovered MKs were collected in the MK fraction (Fig. 8B), with less than 3% in the PLT fraction. Similar to the recovery of day-12 PLTs, ~25–30% and 65–70% of recovered PLTs were collected in the MK and PLT fractions, respectively (Fig. 8D). With respect to PLT activation, the day-14 separation process resulted in minimal pre-activation, as evidenced by little change in % CD62P+ PLTs and the amount of Fg binding (Fig. 9). However, all day-14 PLTs appeared to have lessened potential for activation (Fig. 9B, C), as TRAP-6 treatment resulted in only 60–90% CD62P+ PLTs and a smaller increase in Fg binding, compared to the same dose given to day-12 PLTs (Fig. 4B,C). Morphologically, the recovered day-14 PLTs were very similar to the PreWash PLTs (Fig. 10).
Figure 8. Spinning-membrane filtration of in-vitro-derived, day-14 MKs and PLTs with a 4-μm pore-size membrane rotating at 2000 rpm.

Cells recovered in the MK fraction after day-12 separation were centrifuged at 100g for 10 minutes to pellet large cells and exclude any PLTs. The cell pellet was resuspended in fresh media and cytokines, then reseeded into culture. On day 14, MKs and PLTs were harvested and processed on the spinning-membrane filtration device outfitted with a 4-μm pore-size membrane rotating at 2000 rpm. Recovery and separation of recovered MKs (A, B) and PLTs (C, D) are shown for 2 separate experiments.
Figure 9. Activation state of in-vitro-derived, day-14 PLTs before and after separation.

Day-14 PreWash PLTs and those recovered in the MK and PLT fractions after separation were identified by size (side scatter [SSC] vs forward scatter [FSC]), then CD41+CD42b+ PLTs were analyzed for surface CD62P expression and binding of soluble Fg when at rest or after incubation with TRAP-6 agonist. (A) Representative staining panel for in-vitro-derived PLTs. (B) CD62P results from 2 separate experiments. (C) Fg binding results shown as the mean ± SD of 2 separate experiments. MFI = mean fluorescence intensity. N.S. = not significant; P > 0.05. The symbols representing each donor match those shown in Figs. 3 and 4.
Figure 10. Morphology of in-vitro-derived, day-14 PLTs before and after separation.
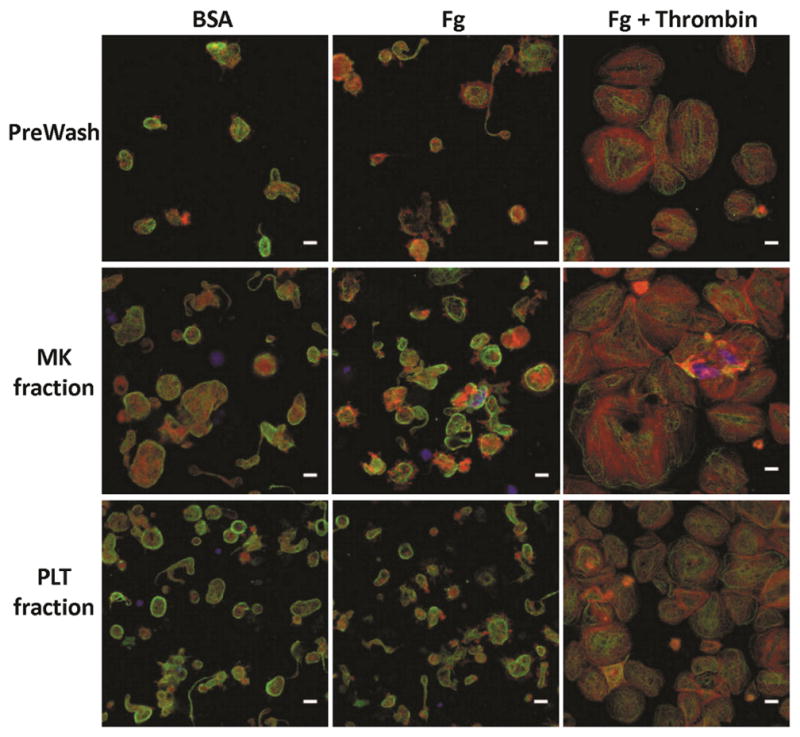
Day-14 PreWash PLTs and those recovered in the MK and PLT fractions after separation were seeded on BSA, Fg, or Fg in the presence of thrombin for 1 hour at 37°C. The PLTs were then stained for β-tubulin (green), F-actin (red), and DNA (blue). Scale bar = 5 μm. Images are from a single representative experiment.
Discussion
Although several groups have successfully produced PLTs in vitro from HSPCs, the challenge to develop a scalable, automated harvest method for the generation of transfusable PLT products still exists. Part of the difficulty in designing such a method lies in how to address the asynchronous nature of PLT production from in-vitro-derived MKs. In order to avoid loss of PLT functionality at 37°C (Bertino et al. 2003; Holme and Heaton 1995), PLTs would ideally be harvested from culture immediately after generation. In 2D culture, PLT harvest can be achieved via successive centrifugations, however this approach is time-consuming and error-prone, and would be difficult to align with cGMP. A few studies have been published on 3D in vitro PLT production (Nakagawa et al. 2013; Pallotta et al. 2011; Sullenbarger et al. 2009), wherein MKs are cultured in the vicinity of a porous membrane, through which they extend proPLTs and release PLTs, which can then be collected into a storage container. Although this approach allows for continuous PLT harvest, the technology is still at an early stage and scale-up has yet to be addressed.
To provide an alternative method for harvesting PLTs from 2D culture, we adapted a commercially-available, spinning-membrane filtration device to separate in-vitro-derived MKs and PLTs. This approach is both automated and efficient and is capable of separating the 1 × 1011 PLTs required for a single unit (70–93% recovery of PLTs from mononuclear cell products that typically contain ≥ 4 × 1011 PLTs; CW, unpublished data). We demonstrated that the process results in a PLT population free of contaminating nucleated cells, and does not pre-activate PLTs nor reduce their activation potential. MKs recovered from the process formed proPLTs and shed PLTs after reseeding in culture. However, despite decreasing the membrane rotation speed to increase the probability that larger PLTs would pass through the membrane, approximately one third of all recovered PLTs were collected into the MK fraction. Thon et al. termed these large PLTs “preplatelets” (prePLTs) and showed that isolated murine prePLTs can mature into PLTs after injection into the circulation (Thon et al. 2010). Their study suggests that flowing culture-derived prePLTs through a circulating flow loop prior to spinning-membrane filtration could promote the conversion of prePLTs to smaller PLTs, and subsequently increase the efficiency of PLT recovery into the PLT fraction. Finally, for the purpose of performing replicate experiments in this study, we chose to first separate in-vitro-derived PLTs and MKs on day 12 of culture, then again on day 14, after reseeding the recovered MKs. However, knowing that the kinetics of MK commitment and maturation vary between donors, the timing of PLT separation would ideally be determined on a case-by-case basis. Earlier recovery of PLTs would minimize exposure at 37°C and would likely decrease the extent of pre-activation observed for PreWash PLTs (Fig. 4B).
Importantly, in our experiments, the liquid initially present in the PreWash bag (i.e., the media in which the in-vitro-derived MKs and PLTs were grown and buffer from the harvesting process), passed through the pores of the spinning membrane and into the PLT fraction. MKs remaining on the outside of the membrane were resuspended in PAS-V solution. In practice, MKs would be resuspended in media for continued culture, while PLTs would be resuspended in PAS-V for transfusion or storage. In order to achieve this, a two-phase separation could be performed wherein media would be used as the wash fluid for the first separation with a 4-μm pore-size membrane rotating at 2000 rpm. The PLT fraction from this separation would be re-processed using a < 2-μm pore-size membrane and a slow membrane rotation speed to allow liquid, but not PLTs, to pass through the membrane, and PLTs would be collected in PAS-V.
In summary, we have demonstrated that spinning-membrane filtration is a viable method for harvesting in-vitro-derived PLTs from culture and recovering immature, in-vitro-derived MKs for further culture and PLT production. With the anticipated adaptation of MK and PLT production to bag culture, the mixed starting population could be easily sterile-docked to the device, thereby ensuring a sterile, closed system. Similarly, MKs collected in the MK fraction bag could be easily transferred to an incubator for continued culture. However, it is important to note that the efficiency of in vitro PLT production must be substantially improved for this technology to be considered clinically relevant. While MKs in vivo can give rise to thousands of PLTs (Trowbridge and et al. 1984), many in vitro studies have reported less than 1 PLT generated per culture-derived MK (Cortin et al. 2005; Proulx et al. 2003; Proulx et al. 2004; Takahashi et al. 2008). These low PLT yields can be attributed to the low percentage of MKs that form proPLTs in culture. Further optimization to increase the efficiency of PLT separation and achieve maximum PLT recovery using spinning-membrane filtration – together with the development of culture conditions that promote proPLT formation – would bring culture-derived PLTs closer to clinical relevance.
Supplementary Material
Acknowledgments
Imaging work was performed at the NU Biological Imaging Facility. Confocal microscopy was performed on a Leica TCS SP5 laser scanning confocal microscope system purchased with funds from the NU Office for Research. Flow cytometery analysis was performed at the NU RHLCCC Flow Cytometry Facility. This work was supported by NSF Grant CBET-1265029 (WMM). Alaina Schlinker was supported in part by NIH/NCI training grant T32CA09560 and a John N. Nicholson fellowship.
References
- Bertino AM, Qi XQ, Li J, Xia Y, Kuter DJ. Apoptotic markers are increased in platelets stored at 37 degrees C. Transfusion. 2003;43(7):857–66. doi: 10.1046/j.1537-2995.2003.t01-4-00431.x. [DOI] [PubMed] [Google Scholar]
- Boudignon-Proudhon C, Patel PM, Parise LV. Phorbol ester enhances integrin alpha IIb beta 3-dependent adhesion of human erythroleukemic cells to activation-dependent monoclonal antibodies. Blood. 1996;87(3):968–76. [PubMed] [Google Scholar]
- Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets generated in vitro from proplatelet-displaying human megakaryocytes are functional. Blood. 1995;85(2):402–13. [PubMed] [Google Scholar]
- Cortin V, Garnier A, Pineault N, Lemieux R, Boyer L, Proulx C. Efficient in vitro megakaryocyte maturation using cytokine cocktails optimized by statistical experimental design. Exp Hematol. 2005;33(10):1182–91. doi: 10.1016/j.exphem.2005.06.020. [DOI] [PubMed] [Google Scholar]
- De Botton S, Sabri S, Daugas E, Zermati Y, Guidotti JE, Hermine O, Kroemer G, Vainchenker W, Debili N. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100(4):1310–7. doi: 10.1182/blood-2002-03-0686. [DOI] [PubMed] [Google Scholar]
- Fu W, Wang SJ, Zhou GD, Liu W, Cao Y, Zhang WJ. Residual undifferentiated cells during differentiation of induced pluripotent stem cells in vitro and in vivo. Stem Cells Dev. 2012;21(4):521–9. doi: 10.1089/scd.2011.0131. [DOI] [PubMed] [Google Scholar]
- Fujimoto T-T, Kohata S, Suzuki H, Miyazaki H, Fujimura K. Production of functional platelets by differentiated embryonic stem (ES) cells in vitro. Blood. 2003;102(12):4044–4051. doi: 10.1182/blood-2003-06-1773. [DOI] [PubMed] [Google Scholar]
- Gabriel M, Strand D, Vahl CF. Cell adhesive and antifouling polyvinyl chloride surfaces via wet chemical modification. Artif Organs. 2012;36(9):839–44. doi: 10.1111/j.1525-1594.2012.01462.x. [DOI] [PubMed] [Google Scholar]
- Guthikonda S, Alviar CL, Vaduganathan M, Arikan M, Tellez A, DeLao T, Granada JF, Dong JF, Kleiman NS, Lev EI. Role of reticulated platelets and platelet size heterogeneity on platelet activity after dual antiplatelet therapy with aspirin and clopidogrel in patients with stable coronary artery disease. J Am Coll Cardiol. 2008;52(9):743–9. doi: 10.1016/j.jacc.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Holme S, Heaton A. In vitro platelet ageing at 22 degrees C is reduced compared to in vivo ageing at 37 degrees C. Br J Haematol. 1995;91(1):212–8. doi: 10.1111/j.1365-2141.1995.tb05272.x. [DOI] [PubMed] [Google Scholar]
- Jay AW, Rowlands S, Skibo L. The resistance to blood flow in the capillaries. Can J Physiol Pharmacol. 1972;50(10):1007–13. doi: 10.1139/y72-145. [DOI] [PubMed] [Google Scholar]
- Josefsson EC, James C, Henley KJ, Debrincat MA, Rogers KL, Dowling MR, White MJ, Kruse EA, Lane RM, Ellis S, et al. Megakaryocytes possess a functional intrinsic apoptosis pathway that must be restrained to survive and produce platelets. J Exp Med. 2011;208(10):2017–31. doi: 10.1084/jem.20110750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasky LC, Sullenbarger B. Manipulation of oxygenation and flow-induced shear stress can increase the in vitro yield of platelets from cord blood. Tissue Eng Part C Methods. 2011;17(11):1081–8. doi: 10.1089/ten.tec.2011.0108. [DOI] [PubMed] [Google Scholar]
- Lindholm PF, Annen K, Ramsey G. Approaches to minimize infection risk in blood banking and transfusion practice. Infect Disord Drug Targets. 2011;11(1):45–56. doi: 10.2174/187152611794407746. [DOI] [PubMed] [Google Scholar]
- Liu Z, Tang Y, Lu S, Zhou J, Du Z, Duan C, Li Z, Wang C. The tumourigenicity of iPS cells and their differentiated derivates. J Cell Mol Med. 2013;17(6):782–91. doi: 10.1111/jcmm.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S-J, Li F, Yin H, Feng Q, Kimbrel EA, Hahm E, Thon JN, Wang W, Italiano JE, Cho J, et al. Platelets generated from human embryonic stem cells are functional in vitro and in the microcirculation of living mice. Cell Res. 2011;21(3):530–545. doi: 10.1038/cr.2011.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli AM, Tazzari PL, Bortul R, Riccio M, Tabellini G, Santi S, Frabetti F, Musiani D, Bareggi R, Conte R. Nuclear matrix protein is released from apoptotic white cells during cold (1–6 degrees C) storage of concentrated red cell units and might induce antibody response in multiply transfused patients. Transfusion. 2000;40(2):169–77. doi: 10.1046/j.1537-2995.2000.40020169.x. [DOI] [PubMed] [Google Scholar]
- Marwaha N, Sharma RR. Consensus and controversies in platelet transfusion. Transfus Apher Sci. 2009;41(2):127–33. doi: 10.1016/j.transci.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Matsunaga T, Tanaka I, Kobune M, Kawano Y, Tanaka M, Kuribayashi K, Iyama S, Sato T, Sato Y, Takimoto R, et al. Ex Vivo Large-Scale Generation of Human Platelets from Cord Blood CD34+ Cells. STEM CELLS. 2006;24(12):2877–2887. doi: 10.1634/stemcells.2006-0309. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Nakamura S, Nakajima M, Endo H, Dohda T, Takayama N, Nakauchi H, Arai F, Fukuda T, Eto K. Two differential flows in a bioreactor promoted platelet generation from human pluripotent stem cell-derived megakaryocytes. Exp Hematol. 2013;41(8):742–8. doi: 10.1016/j.exphem.2013.04.007. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K, Fujita K, Koike T, Harimoto K, Dohda T, et al. Expandable megakaryocyte cell lines enable clinically applicable generation of platelets from human induced pluripotent stem cells. Cell Stem Cell. 2014;14(4):535–48. doi: 10.1016/j.stem.2014.01.011. [DOI] [PubMed] [Google Scholar]
- Pallotta I, Lovett M, Kaplan DL, Balduini A. Three-dimensional system for the in vitro study of megakaryocytes and functional platelet production using silk-based vascular tubes. Tissue Eng Part C Methods. 2011;17(12):1223–32. doi: 10.1089/ten.tec.2011.0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panuganti S, Schlinker AC, Lindholm PF, Papoutsakis ET, Miller WM. Three-Stage Ex Vivo Expansion of High-Ploidy Megakaryocytic Cells: Toward Large-Scale Platelet Production. Tissue Eng Part A. 2013 doi: 10.1089/ten.tea.2011.0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx C, Boyer L, Hurnanen DR, Lemieux R. Preferential ex vivo expansion of megakaryocytes from human cord blood CD34+-enriched cells in the presence of thrombopoietin and limiting amounts of stem cell factor and Flt-3 ligand. J Hematother Stem Cell Res. 2003;12(2):179–88. doi: 10.1089/152581603321628322. [DOI] [PubMed] [Google Scholar]
- Proulx C, Dupuis N, St-Amour I, Boyer L, Lemieux R. Increased megakaryopoiesis in cultures of CD34-enriched cord blood cells maintained at 39°C. Biotechnology and Bioengineering. 2004;88(6):675–680. doi: 10.1002/bit.20288. [DOI] [PubMed] [Google Scholar]
- Prowse CV, de Korte D, Hess JR, van der Meer PF. Commercially available blood storage containers. Vox Sang. 2014;106(1):1–13. doi: 10.1111/vox.12084. [DOI] [PubMed] [Google Scholar]
- Radwanski K, Min K. The role of bicarbonate in platelet additive solution for apheresis platelet concentrates stored with low residual plasma. Transfusion. 2013;53(3):591–9. doi: 10.1111/j.1537-2995.2012.03745.x. [DOI] [PubMed] [Google Scholar]
- Radwanski K, Wagner SJ, Skripchenko A, Min K. In vitro variables of apheresis platelets are stably maintained for 7 days with 5% residual plasma in a glucose and bicarbonate salt solution, PAS-5. Transfusion. 2012;52(1):188–94. doi: 10.1111/j.1537-2995.2011.03252.x. [DOI] [PubMed] [Google Scholar]
- Robert A, Boyer L, Pineault N. Glycoprotein Ibalpha receptor instability is associated with loss of quality in platelets produced in culture. Stem Cells Dev. 2011;20(3):379–90. doi: 10.1089/scd.2010.0041. [DOI] [PubMed] [Google Scholar]
- Rumjantseva V, Hoffmeister KM. Novel and unexpected clearance mechanisms for cold platelets. Transfus Apher Sci. 2010;42(1):63–70. doi: 10.1016/j.transci.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertz H, Koster S, Kahr WH, Michetti N, Kraemer BF, Weitz DA, Blaylock RC, Kraiss LW, Greinacher A, Zimmerman GA, et al. Anucleate platelets generate progeny. Blood. 2010;115(18):3801–9. doi: 10.1182/blood-2009-08-239558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullenbarger B, Bahng JH, Gruner R, Kotov N, Lasky LC. Prolonged continuous in vitro human platelet production using three-dimensional scaffolds. Experimental Hematology. 2009;37(1):101–110. doi: 10.1016/j.exphem.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Monzen S, Yoshino H, Abe Y, Eguchi-Kasai K, Kashiwakura I. Effects of a 2-step culture with cytokine combinations on megakaryocytopoiesis and thrombopoiesis from carbon-ion beam-irradiated human hematopoietic stem/progenitor cells. J Radiat Res (Tokyo) 2008;49(4):417–24. doi: 10.1269/jrr.07132. [DOI] [PubMed] [Google Scholar]
- Takayama N, Nishikii H, Usui J, Tsukui H, Sawaguchi A, Hiroyama T, Eto K, Nakauchi H. Generation of functional platelets from human embryonic stem cells in vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood. 2008;111(11):5298–5306. doi: 10.1182/blood-2007-10-117622. [DOI] [PubMed] [Google Scholar]
- Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, Yamaguchi T, Otsu M, Nishimura K, Nakanishi M, et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med. 2010;207(13):2817–30. doi: 10.1084/jem.20100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, Ehrlicher A, Larson MK, Hoffmeister K, Hartwig JH, Italiano JE., Jr Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol. 2010;191(4):861–74. doi: 10.1083/jcb.201006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama Y, Tohyama K, Tsubokawa M, Asahi M, Yoshida Y, Yamamura H. Outside-In signaling of soluble and solid-phase fibrinogen through integrin alphaIIbbeta3 is different and cooperative with each other in a megakaryoblastic leukemia cell line, CMK. Blood. 1998;92(4):1277–86. [PubMed] [Google Scholar]
- Trowbridge EA, et al. The origin of platelet count and volume. Clinical Physics and Physiological Measurement. 1984;5(3):145. doi: 10.1088/0143-0815/5/3/007. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Peluso M, Gillitzer A, Massberg S, Heinzmann U, Schulz C, Munch G, Gawaz M. Generation of functional culture-derived platelets from CD34+ progenitor cells to study transgenes in the platelet environment. Circ Res. 2004;95(5):e36–44. doi: 10.1161/01.RES.0000141700.96085.2e. [DOI] [PubMed] [Google Scholar]
- White MJ, Schoenwaelder SM, Josefsson EC, Jarman KE, Henley KJ, James C, Debrincat MA, Jackson SP, Huang DC, Kile BT. Caspase-9 mediates the apoptotic death of megakaryocytes and platelets, but is dispensable for their generation and function. Blood. 2012;119(18):4283–90. doi: 10.1182/blood-2011-11-394858. [DOI] [PubMed] [Google Scholar]
- Zimmermann A, Preynat-Seauve O, Tiercy JM, Krause KH, Villard J. Haplotype-based banking of human pluripotent stem cells for transplantation: potential and limitations. Stem Cells Dev. 2012;21(13):2364–73. doi: 10.1089/scd.2012.0088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.