

Abstract
Cardiac structural changes associated with dilated cardiomyopathy (DCM) include cardiomyocyte hypertrophy and myocardial fibrosis. Connective Tissue Growth Factor (CTGF) has been associated with tissue remodeling and is highly expressed in failing hearts. Our aim was to test if inhibition of CTGF would alter the course of cardiac remodeling and preserve cardiac function in the protein kinase Cε (PKCε) mouse model of DCM. Transgenic mice expressing constitutively active PKCε in cardiomyocytes develop cardiac dysfunction that was evident by 3 months of age, and that progressed to cardiac fibrosis, heart failure, and increased mortality. Beginning at 3 months of age, PKCε mice were treated with a neutralizing monoclonal antibody to CTGF (FG-3149) for an additional 3 months. CTGF inhibition significantly improved left ventricular (LV) systolic and diastolic function in PKCε mice, and slowed the progression of LV dilatation. Using gene arrays and quantitative PCR, the expression of many genes associated with tissue remodeling were elevated in PKCε mice, but significantly decreased by CTGF inhibition. However total collagen deposition was not attenuated. The observation of significantly improved LV function by CTGF inhibition in PKCε mice suggests that CTGF inhibition may benefit patients with DCM. Additional studies to explore this potential are warranted.
Keywords: Heart failure, remodeling, gene array, quantitative PCR
1. Introduction
Cardiac remodeling occurs when myocardial injury or hemodynamic overload initiates a series of structural changes throughout the myocardium that ultimately leads to a global deterioration of cardiac function [1]. The most prominent structural changes include compensatory cardiomyocyte hypertrophy and myocardial fibrosis. Like cardiomyocyte hypertrophy, the remodeled ECM may initially represent an adaptive response to cardiac stress, but may ultimately compromise cardiac structure and function. For example, cardiac ECM expansion has been associated with adverse outcomes [2] and may contribute to cardiac morbidity and mortality by affecting mechanical, conductive and vasomotor functions. Factors that regulate ECM remodeling are therefore important therapeutic targets to protect against heart failure (HF) and sudden death.
Connective Tissue Growth Factor (CTGF) is a member of the CCN (Cyr61, CTGF, and Nov) family of multifunctional matricellular proteins [3]. CTGF has been implicated in mediating a variety of cellular processes, including adhesion, proliferation, differentiation, migration, angiogenesis, and apoptosis [4,5], all of which are common features of cardiac remodeling. CTGF is also involved in initiating processes underlying fibrosis of several organs and tissues, including the heart [6]. However, data on its regulation and its functional role in the pathogenesis of cardiac remodeling and fibrosis in the failing heart are limited and controversial [7-15].
Recently, we demonstrated that left ventricular (LV) CTGF mRNA levels were increased 3-to-5-fold in patients undergoing heart transplantation for end-stage dilated cardiomyopathy (DCM) [16]. CTGF up-regulation coincided with increased expression of TGF-β1, type I collagen (Col1a1), type III collagen (Col3a1), matrix metalloproteinase 2 (MMP2), and MMP9 mRNAs. Although CTGF, TGF-β1, Col1a1, and Col3a1 mRNA levels were reduced by LV assist device (LVAD) implantation and hemodynamic unloading, there was only a modest reduction in tissue fibrosis and no difference in protein-bound hydroxyproline concentration between pre-and post-LVAD tissue samples. We speculated that the persistent fibrosis may have been related to slow turnover of ECM proteins, and a concomitant reduction in MMP9 mRNA and protein levels after unloading. Nevertheless, our results, like those of other investigators [17-20] were still consistent with the notion that CTGF plays an important role in regulating tissue fibrosis during maladaptive cardiac remodeling and progression to HF. However, additional studies were needed to determine whether inhibition of CTGF signaling could prevent or slow the progression of LV dysfunction and tissue remodeling.
In the present study, we use a transgenic mouse model of DCM and HF [21] and FG-3149, a chimeric mouse mAb that contains the same CTGF epitope-recognition region as the CTGF-neutralizing human mAb FG-3019 [22], to interrogate the causal role of CTGF in LV remodeling, fibrosis, and HF progression.
2. Methods
2.1. Reagents
A detailed list of reagents used in this study is available in the Online Data Supplement.
2.2. Transgenic mouse studies
All mice were handled in accordance with NIH guidelines and all treatments were approved by the Loyola University Chicago Institutional Animal Care and Use Committee. Mice with cardiomyocyte-specific expression of a constitutively active (A159E) rabbit PKCε transgene driven by the mouse Myh6 gene promoter on an FVB/N background (PKCε mice) were kindly provided by Dr. Peipei Ping, University of California, Los Angeles. Homozygous offspring of low-to-intermediate-expressing transgenic line 344 were generated as previously described [21]. Nontransgenic FVB/N (FVB) mice (Charles River Laboratories, Wilmington, MA) served as controls. To examine the effects of CTGF inhibition, 3-month-old mice underwent baseline echocardiography under general anesthesia (2% isoflurane in O2) and were randomly assigned to one of four groups: FVB mice that received nonimmune mouse immunoglobulin (IgG) (n=10), FVB mice that received FG-3149 (n=12), PKCε mice that received IgG (n=10), and PKCε mice that received FG-3149 (n=12). IgG and FG-3149 antibodies (FibroGen, San Francisco, CA) were administered by intraperitoneal injection twice weekly at 30 mg/kg for three months. After three months of treatment, mice underwent repeat echocardiography and LV catheterization under isoflurane general anesthesia, after which hearts were excised, rapidly frozen in liquid N2, and stored at -80°C for further analysis. Additional, non-antibody-treated mice were also analyzed at 1, 3, 6, 9 and 12 months of age.
2.3 Echocardiography
Transthoracic M-mode and 2D echocardiography were performed on isoflurane-anesthetized mice using an Acuson 15L8 Microson High-Resolution Transducer and an Acuson Sequoia C256 Echocardiography System (Siemens Medical Solutions; Malvern, PA). 2D parasternal long- and short-axis images were analyzed in real-time or saved on 230Mb magneto-optical discs for off-line analysis. Interventricular septal thickness, LV posterior wall (PW) thickness, and LV internal dimension were measured from M-mode images at the end of diastole (d) and systole at maximum sweep speed. LV fractional shortening (LVFS, %) was calculated from digital images as ((LV end-diastolic dimension – LV end-systolic dimension) / LV end-diastolic dimension) × 102. End-systolic, end-diastolic, and stroke volumes (LVESV, LVEDV, and LVSV in μL, respectively) were estimated using the spherical formula. LV mass (mg) was calculated as LV tissue volume (mm3) × 1.04. LV Relative Wall Thickness was calculated as 2 × (LV PW thickness,d / LV internal dimension,d). Left atrial area (mm2) was measured using the tracing function from the parasternal long-axis image. LV Remodeling Index (LVRI, mg/μL) was calculated as the ratio of LV mass (mg) to LV volume (μL). Further details are provided in the Online Data Supplement.
2.4. Pressure/volume analysis by LV catheterization
LV catheterization was performed on isoflurane-anesthetized mice using a 1.4F Millar Pressure-Volume Catheter (SPR-839, Millar Instruments, Houston, TX) connected to an MPVS-300 System and interfaced to a PowerLab 8/35 High-Performance Data Acquisition System (ADInstruments, Colorado Springs, CO). A detailed description of these methods are included in the Online Data Supplement.
2.5. Histology
Portions of excised heart were fixed in formalin and 6 μm paraffin sections were stained with Mallory-Trichrome stain. Randomly located photomicrographs were acquired using an Olympus BH2 microscope and a Nikon D200 digital camera. Fibrosis area (%) was quantified by color deconvolution (to separate red from blue staining) using Adobe Photoshop (Adobe Systems, Inc. San Jose, CA) and ImageJ (National Institutes of Health, Bethesda, MD) for quantitative analysis of image areas. Calculations were made on 10× images of each sample.
2.6. Tissue hydroxyproline analysis
Protein-bound hydroxyproline content (μg) was measured in LV homogenates as previously described [16], and normalized to total protein content (mg) determined by bicinchoninic acid protein assay (Pierce Chemical Co, Rockford IL) using bovine serum albumin as standard.
2.7. Immunoblotting
LV lysates were resolved on 10% polyacrylamide gels and blotted to nitrocellulose [16]. Blots were probed at 4°C overnight with mouse mAbs to CTGF, PKCε, or GAPDH; detected with HRP-conjugated goat anti-mouse IgG; and visualized by enhanced chemiluminescence (Pierce Biotechnology, Rockford, IL). Developed films were scanned and band intensities quantified using UN-SCAN-IT Gel Software (Silk Scientific, Orem, UT). Additional methods and Western blotting results are provided in the Online Data Supplement.
2.8. mRNA expression analysis
Total RNA was isolated from frozen LV tissue using TRIzol Reagent (Life Technologies, Carlsbad, CA) and purified on RNeasy Mini Spin Columns (Qiagen, Valencia, CA) with on-column DNase digestion to remove residual genomic DNA. RNA quantity and quality were assessed using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE) and 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA), with all samples having optical density (OD260/280) ratios ≥ 1.9 and RNA integrity (RIN) values ≥ 6.7. LV RNA profiling was performed on Affymetrix MOE 430A 2.0 arrays. GCRMA-processed, median-normalized data were analyzed using Agilent GeneSpring GX software and are available at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68857. Low intensity probesets with mean raw fluorescence values ≤50 in all groups were filtered from further analysis. FG-3149-responsive cardiac injury genes were defined as genes with a >2-fold difference in expression in IgG-treated PKCε mice vs. IgG-treated FVB mice at P<0.05 that were also resolved >1.5-fold at P<0.05 in FG-3149-treated vs. IgG-treated PKCε mice. Expression of select genes was also determined by TaqMan® quantitative RT-PCR. Additional details are provided in the Online Data Supplement.
2.9. Statistical analysis
All results were expressed as mean±SEM. Data for multiple groups were compared by 2-way or 1-way ANOVA followed by the Holm-Sidak test or Bonferroni post-test. Kaplan-Meier survival curves were compared by the Log-Rank test and Cox regression analysis. Data were analyzed using SigmaStat, PRISM and R statistical software packages and P-values <0.05 were considered significant.
3. Results
3.1. Aging PKCε mice have a DCM with interstitial fibrosis accompanied by CTGF up-regulation
As indicated previously [21] and in the Online Data Supplement, LV PKCε expression was substantially increased in transgenic animals, followed an autosomal inheritance pattern, and did not vary with age (Online Figure IA). PKCε mice did, however, show significantly shorter survival than FVB mice (Log-rank and Cox proportional hazard ratios=3.0 and 3.6; P=0.001 and 0.003, respectively), with 91% of FVB mice alive at 12 months vs. 56% of PKCε mice, and with most deaths occurring suddenly and unexpectedly (Online Figure IB). M-mode and 2D echocardiography (Online Figures IC, IIA-H) and pressure/volume analysis by LV catheterization (Online Table I) further revealed that PKCε mice developed a DCM beginning at ∼3mo of age with all four cardiac chambers affected, and which progressed to HF and sudden death.
LV tissue analysis by Mallory-Trichome staining revealed numerous areas of LV interstitial fibrosis in PKCε mice, but not in age-matched FVB mice (Figure 1A). Protein-bound hydroxyproline (a marker of fibrillar collagen accumulation) also progressively increased in the LV of PKCε mice over time (Figure 1B), and at 12 months, 18S rRNA-normalized LV Col1a1 and Col3a1 mRNA expression was 4.7- and 3.2-fold higher in PKCε than FVB mice (P<0.001 each; Figure 1C), with both interstitial collagen transcripts being coordinately expressed across individual animals (r=0.81, P<0.001; Figure 1D). Interstitial fibrosis and fibrillar collagen accumulation were associated with a progressive increase in CTGF mRNA expression, with 6 and 12 month-old PKCε mice exhibiting ∼3- and 6-fold higher LV CTGF mRNA levels than FVB controls, respectively (Figure 1E). The increase in CTGF mRNA was accompanied by an increase in CTGF protein, which was detectable in LV tissue extracts of 12 month-old FVB controls, but was markedly increased in age-matched PKCε mice (Figure 1F).
Figure 1. PKCε mice develop interstitial fibrosis.
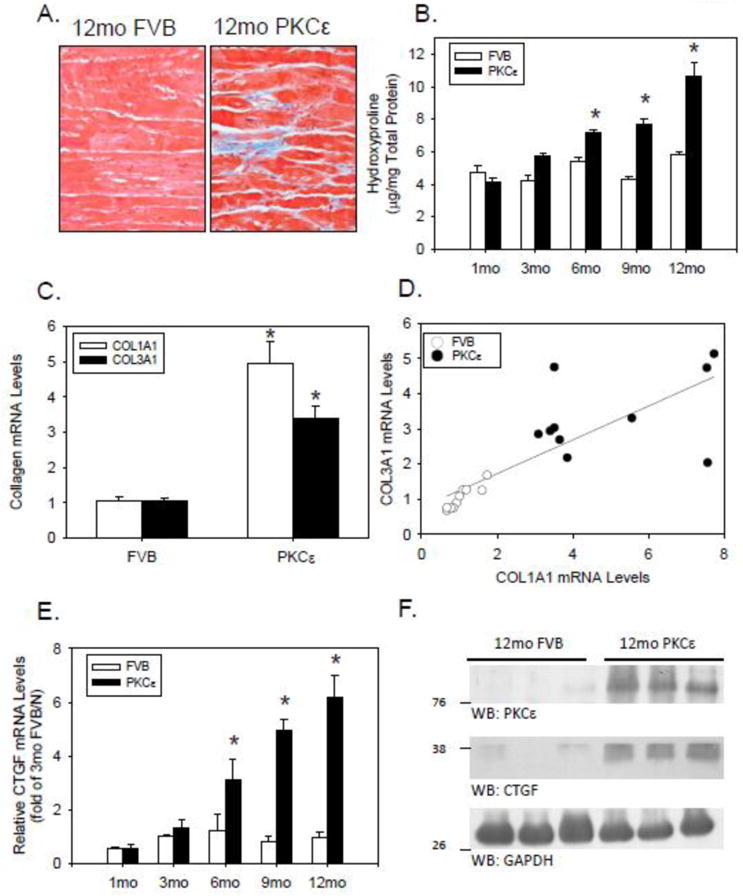
(A) Mallory-Trichome staining of representative LV tissue sections from 12-month-old FVB and PKCε mice. Both images were captured at 400×. (B) Hydroxyproline concentration (μg/mg total protein) in LV hydrolysates from FVB (open bars) and PKCε (closed bars) mice of increasing age. Means±SEM are shown for n=5-9 mice per group. *P<0.05 for PKCε vs. FVB mice at the indicated age. (C) 18S rRNA-normalized mRNA levels for Col1a1 (open bars) and Col3a1 (closed bars) in LV tissue of 12 month old FVB and PKCε mice. Means±SEM are shown for n=10 mice per group. *P<0.05 for PKCε vs FVB mice at the indicated age. (D) 18S rRNA-normalized mRNA levels for Col1a1 and Col3a1 mRNA for each FVB (open circles) and PKCε (closed circles) mice are plotted, and fit via linear regression analysis. (E) 18S rRNA-normalized CTGF mRNA levels in FVB and PKCε mice of increasing age. Mean±SEM are for n=4-10 mice per group. *P<0.05 for PKCε vs FVB mice at a given age. (F) Western blot analysis of LV tissue extracts (100μg/lane) from 12-month-old FVB and PKCε mice.
3.2. CTGF neutralizing antibody maintains LV function and slows the progression of LV remodeling in PKCε mice
To determine if CTGF plays a causal role in the development of LV remodeling and tissue fibrosis, 3 month-old FVB (n=22) and PKCε (n=22) mice underwent baseline echocardiography and were randomly assigned to receive either nonimmune, control IgG (IgG; 30 mg/kg IP) or the CTGF-neutralizing mAb FG-3149 (30 mg/kg IP) [22] twice weekly for a period of 3 months. Initial body weights were similar, and all animals appeared healthy and gained similar amounts of weight during the treatment period (Table 1). FG-3149 had no effect on PKCε expression in either FVB or PKCε mice (Online Figure III). FG-3149 also significantly increased (rather than decreased) the enhanced myofilament phosphorylation of cTnI at Thr143 observed in PKCε mice [23], and had no effect on the modestly increased phosphorylation of MyBP-C at Ser302 (Online Figure IV) [24,25]. Nevertheless, the mAb had significant effects in terms of preserving LV function in PKCε mice. Prior to treatment, no parameter exhibited a significant difference between mice randomized to receive IgG and those randomized to receive FG-3149 in either genotype (Figure 2). Baseline echocardiography did, however, indicate that 3 month-old PKCε mice had a somewhat lower LVFS (Figure 2B) due to an increased LVESV (Figure 2C) and LVEDV (Figure 2D), as well as a modestly increased LV mass (Figure 2A) as compared to non-transgenic FVB controls. After the 3 month treatment period (i.e., at 6 months of age), neither LVFS nor LVESV changed in healthy, control FVB mice receiving either FG-3149 or IgG. However, there was a small but significant reduction in LVEDV (Figure 2D) in FVB mice treated with FG-3149. In diseased PKCε mice, LVFS (Figure 2B) significantly declined in mice receiving the control IgG, due to a progressive increase in LVESV (Figure 2C) and LVEDV (Figure 2D). In contrast, FS did not decline in FG-3149-treated PKCε mice. This preservation of LV structure and function was not accompanied by a further increase in LV mass (Figure 2A). Rather, FG-3149 prevented LV remodeling, as indicated by a significant, 21% decrease in relative wall thickness of IgG-treated PKCε mice, as compared to a nonsignificant 12% increase in FG-3149-treated PKCε mice (Figure 2F). The substantial increase in left atrial area was also completely prevented by FG-3149 treatment (Figure 2G). Similarly, the significant reduction in PKCε LVRI was prevented by FG-3149 (Figure 2H). The response of individual animals before and after each treatment is depicted in the Online Data Supplement (Online Figure V).
Table 1. Systolic and Diastolic Function in 6 month-old FVB and PKCε Mice After 3 months of Treatment with Control IgG or CTGF Neutralizing Monoclonal Antibody.
| Parameter | FVB | PKCε | ||
|---|---|---|---|---|
| Control IgG | FG-3149 | Control IgG | FG-3149 | |
| n= | 10 | 12 | 10 | 12 |
| Initial Body Weight (g) | 25.6±0.9 | 25.7±1.3 | 26.5±1.3 | 27.1±1.5 |
| Terminal Body Weight (g) | 32.2±2.3 | 30.5±1.3 | 30.2±1.5 | 32.5±1.9 |
| Heart Rate (bpm) | 545±13 | 531±12 | 504±17 | 516±9 |
| Maximum Systolic Pressure (mmHg) | 122±10 | 140±13 | 101±13 | 120±7* |
| +dP/dtmax (mmHg × sec-1) | 7280±827 | 10029±663* | 5902±965 | 8008±525* |
| Contractility Index (sec-1) | 123±5 | 119±7 | 95±5 | 108±6 |
| -dP/dtmax (mmHg × sec-1) | -6988±584 | -9864±573* | -4772±915 | -6952±495* |
| Tau (msec-1) | 6.9±0.4 | 6.0±0.3 | 14.5±1.8 | 9.2±0.5* |
| End-diastolic Pressure (mmHg) | 2±1 | 0.3±1 | 7±3 | 2±1* |
| End-diastolic volume (μl) | 49±5 | 48±2 | 56±4 | 58±5 |
3 month-old FVB (n=22) and PKCε (n=22) mice were randomly assigned to receive nonimmune mouse IgG (30mg/kg IP) or FG-3149 (30mg/kg IP), a mouse-specific, neutralizing mAb to CTGF. Animals were treated twice weekly for a period of 3 months, and were then subjected to LV catheterization. n, number of experimental animals in each group; bpm, beats per minute; Contractility Index, defined as dP/dtmax / P at dP/dtmax;
P<0.05 for Control IgG vs. FG-3149 within each genotype by 2-way ANOVA followed by the Holm-Sidak test.
Figure 2. CTGF neutralizing antibody maintains LV function and slows the progression of LV remodeling in PKCε mice.
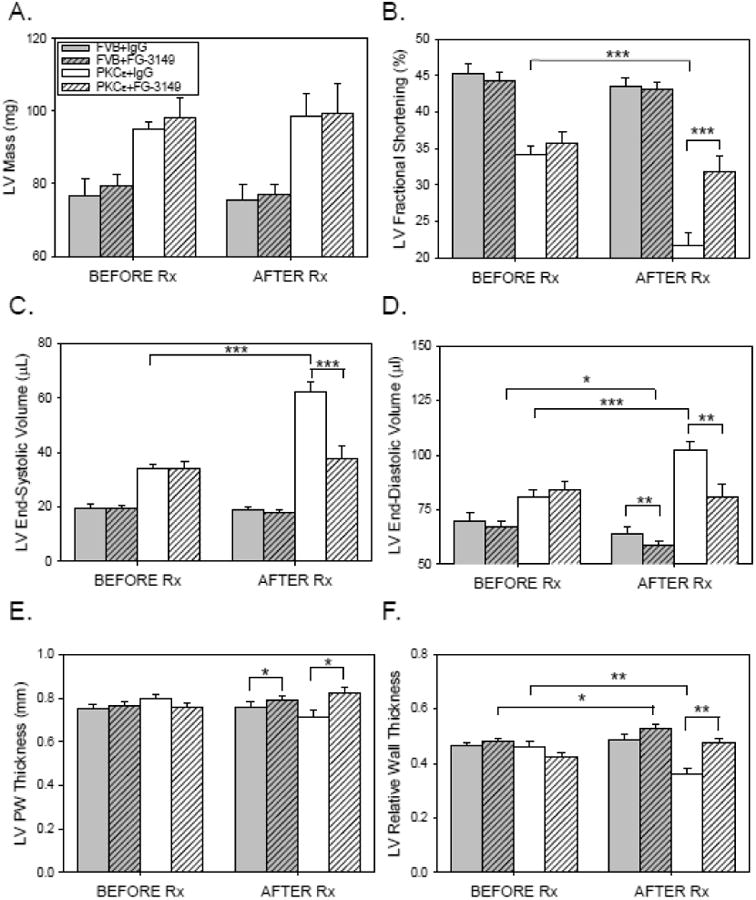
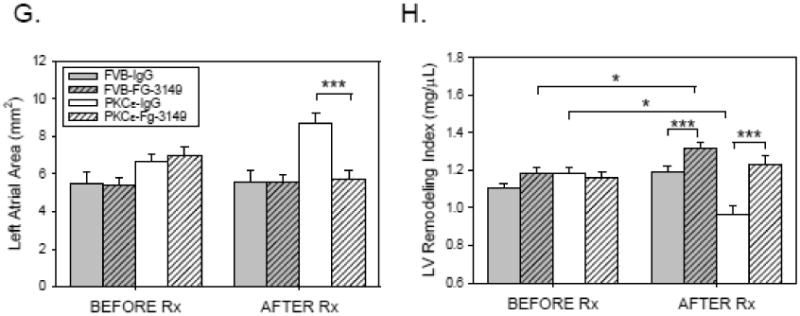
3 month-old FVB (n=22; filled bars) and PKCε (n=22; open bars) mice underwent baseline M-mode and 2-D echocardiography (BEFORE Rx), and were then randomly assigned to receive nonimmune mouse IgG (IgG; 30mg/kg IP; no cross-hatch) or FG-3149 (30mg/kg IP; cross-hatched), a neutralizing mAb to CTGF. Animals were treated twice weekly for a period of 3 months, and were then subjected to repeat M-mode and 2-D echocardiography (AFTER Rx). (A) LV Mass (mg); (B) LV Fractional Shortening (%); (C) LV End-systolic Volume (μL); (D) LV End-diastolic Volume (μL); (E) LV Posterior Wall (PW) Thickness (mm); (F) LV Relative Wall Thickness; (G) Left Atrial Area (mm2); and (H) LV Remodeling Index (mg/μl). Data are means±SEM for n=10-12 mice in each treatment group. Data from the 4 groups BEFORE and AFTER Rx were compared by 2-way ANOVA followed by the Holm-Sidak test. Data for each group BEFORE and AFTER Rx were compared by paired t-test. P<0.05 (*), 0.01 (**), or 0.001 (***).
Pressure/volume analysis by LV catheterization confirmed the beneficial effects of FG-3149 on cardiac function (Table 1). Several indicators of systolic and diastolic function were significantly compromised in 6 month-old PKCε mice compared to IgG-treated FVB controls, and many tended to improve in response to FG-3149 treatment. Some indices (dP/dtmax, -dP/dtmax) also appeared to improve in FG-3149-treated FVB mice. However, other key measures of cardiac function (LVEDP, maximum systolic pressure, and Tau; as well as echo-derived LVFS, LVRI and LVESV; Figure 2) were only significantly affected by FG-3149 in PKCε hearts.
3.3. FG-3149 ameliorates an ECM remodeling gene signature in PKCε hearts
To explore the mechanism by which CTGF inhibition improved cardiac function in PKCε mice, we performed microarray-based gene expression profiling. Cardiac injury genes were identified with substantial and significant expression changes (>2-fold, P<0.05) in IgG-treated PKCε hearts vs. IgG-treated FVB hearts. Cardiac injury genes normalized or partially normalized by FG-3149 were further identified (>1.5-fold at P<0.05 in the opposite direction in FG-3149-treated vs. IgG-treated PKCε hearts; Figure 4A). Approximately two-thirds (67%) of the FG-3149-responsive cardiac injury mRNAs (Online Table II) were elevated in PKCε hearts (122 probesets, 99 named genes) while the remainder were diminished (60 probesets, 47 named genes). The expression changes of selected genes were confirmed by quantitative RT-PCR (Figure 4B and Online Figure VI).
Figure 4. Inhibition of CTGF signaling does not prevent or reduce the interstitial fibrosis seen in 6 month-old PKCε hearts.
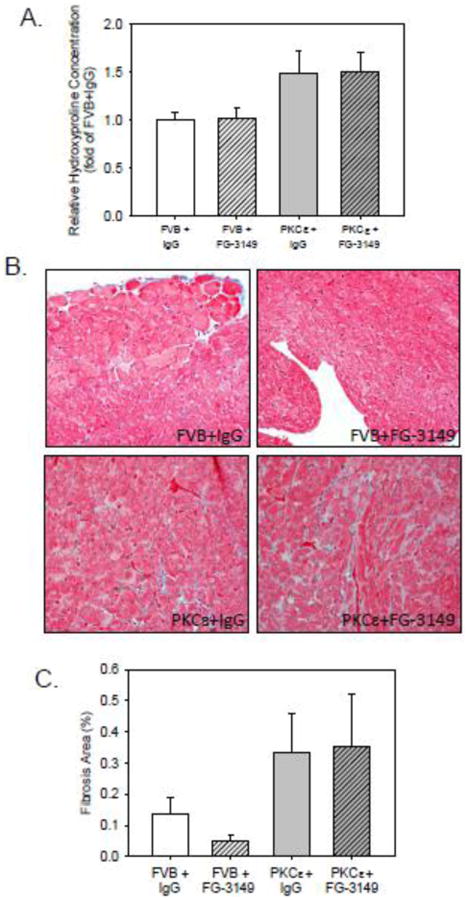
(A) Quantitative analysis of protein-bound hydroxyproline in FVB and PKCε LV tissue homogenates after 3 months of treatment with either nonimmune IgG (IgG) or FG-3149. Data are means±SEM; data were compared by 2-way ANOVA and Holm-Sidak test. Although genotype (i.e., FVB vs. PKCε) was a significant factor (P=0.007) for differences in hydroxyproline content amongst the 4 groups, treatment (i.e., IgG vs. FG-3149) was not a significant factor (P=0.883) in the observed differences in tissue hydroxyproline. (B) Representative Mallory-Trichrome stained LV tissue sections from each of the 4 treatment groups. All images were captured at 400×. (C) Quantitative analysis of fibrosis area (%) obtained from 3 hearts. Data are means±SEM; data were compared by 2-way ANOVA and Holm-Sidak test. Although genotype (i.e., FVB vs. PKCε) was a significant factor for differences in fibrosis area (P=0.05) amongst the 4 groups, treatment (i.e., IgG vs. FG-3149) was not a significant factor (P=0.779) in the observed differences.
To investigate functional relationships among FG-3149-responsive injury genes, gene ontology (GO) analyses were performed. The majority of the GO categories associated with FG-3149-responsive cardiac injury genes elevated by PKCε related to ECM structural proteins and remodeling enzymes, including collagens (Col1a1, Col1a2, Col3a1, Col4a1, Col5a1, Col5a2, Col6a1, Col6a2, Col6a3, Col8a1), collagen processing enzymes (Lox, Loxl1, Loxl2, Loxl3, Pcolce), non-collagen ECM molecules (Bgn, Cspg4, Emilin, Fbln2, Fbn1, Fstl1, Postn, Thbs4, Vcan), proteinase inhibitors (Pi16, Serpine2, Timp1), TGFβ and other fibrosis-associated signaling molecules (Igfbp7, Ltbp2, Pdgfc, Ptn, Sfrp1, Tgfb3), and regulators of mesenchymal cell differentiation (Meox1, Runx2, Sox9, Twist1). “Collagen fibril organization” (GO:30199) was the most significantly over-represented GO term (42-fold enrichment, P<0.001), Thus, the make-up of the FG-3149-responsive gene set indicates that increased ECM remodeling activity is a major feature of cardiac injury and that it is regulated by CTGF.
FG-3149-responsive cardiac injury genes diminished by PKCε were associated with GO ontology terms related to energy metabolism, with “acyl-CoA metabolism” (GO:6637) being most significantly over-represented (61-fold enrichment, P<0.001). The PKCε-diminished metabolism genes that were responsive to FG-3149 included Acaa2, Acot1, Acot2, Acsl1, Adhfe1, Amy1, Ces1d, Dgat2, Fbp2, Gcat, Gstk1, Mccc2, Mettl20, Pah, Pfkfb1, Phkg1 and Tst.
Key FG-3149-responsive measures of cardiac function were exceptionally closely related to gene expression changes observed upon CTGF inhibition, as revealed by permutation analysis of gene-functional outcome correlations (Online Figure VII). Cardiac injury parameters EF, FS and Tau were correlated to FG-3149 treatment-responsive genes at P<0.001 at a frequency of 620- to 720-fold greater than expected by chance. Conversely, there was no evidence of enrichment for parameters like LVSV and HR that were unaffected by PKCε or FG-3149.
3.4. Collagen content was not altered by FG-3149
To assess the effects of CTGF inhibition on cardiac fibrosis, the amount of collagen in the hearts of PKCε mice was determined by hydroxyproline assay (Figure 4). PKCε mice exhibited about a 50% increase in collagen content relative to FVB mice (Figure 4A). FG-3149 treatment did not appear to alter hydroxyproline levels, despite changes in expression of several collagen genes. Similarly, increased cardiac collagen content was observed in PKCε mice by Mallory-Trichrome staining (Figures 4B and 4C), but did not appear affected by FG-3149 treatment.
4. Discussion
4.1 CTGF and cardiac function
Numerous studies have demonstrated that increased CTGF expression accompanies myocardial injury, reduced contractile function, and cardiac fibrosis [26-34]. However, CTGF up-regulation also occurs in response to pressure and volume overload [35, 36], and in the uninjured, mechanically overloaded myocardium following acute myocardial infarction [17]. Once secreted into the cardiac ECM, CTGF does not function as a typical growth factor or cytokine, but rather functions as a matricellular protein that modulates the interaction of cardiomyocytes and cardiac fibroblasts with the ECM in order to modify their phenotype [37]. In so doing, CTGF is thought to interact with other ECM proteins, cytokines, growth factors, and cell-surface receptors, and thereby facilitate or inhibit the signal transduction pathways in which they participate. Because CTGF modulates multiple pathways simultaneously, including those for TGFβs, BMPs, WNTs, VEGFs and integrins, it acts as an integrator of multiple environmental factors that impact cellular response. Consequently, the effects of CTGF are context-dependent and complex.
In this study, CTGF was pharmacologically inhibited in transgenic DCM mice by administering an antibody to CTGF from the ages of 3 to 6 months. This resulted in significant improvement in both systolic and diastolic function at 6 months, as indicated by increased LVFS, +dP/dtmax, and maximal systolic pressure; and decreased –dP/dtmax, Tau, and LV end-diastolic pressure. It is likely that such improvements in cardiac function would translate to improvements in survival. However, a longer experiment will be necessary to demonstrate such a benefit.
Nevertheless, the beneficial effects of CTGF inhibition appeared unrelated to changes in PKCε expression, or PKCε-induced myofilament phosphorylation. In contrast, a previous study by Scruggs et al. [38] showed that a double transgenic mouse expressing both PKCε and cTnI harboring mutations in putative PKC phosphorylation sites resulted in improved functional endpoints. Thus, CTGF inhibition likely alters more downstream aspects of the PKCε-induced disease state. Similarly, Szabo and coworkers [13] showed that mice subjected to 8 weeks of transverse aortic coarctation (TAC) and treated with the same monoclonal antibody had better preserved LV systolic function and reduced LV dilatation as compared to mice treated with nonimmune IgG. In both their study [13] and ours, the preservation of LV function occurred without a discernable reduction in tissue fibrosis. While bulk collagen was unaffected in these models, gene expression studies in our model predicted some role for collagen synthesis. Thus, qualitative aspects of collagen synthesis may still be important, such that improved function is linked to improved quality of ECM remodeling rather than ECM quantity, in a manner not revealed by the quantitative collagen assays and fibrosis area measurements performed here. Alternatively, CTGF may affect cardiac function through its modulation of multiple signaling pathways. For example, constant infusion of a mixture of recombinant human latent TGF-β1 and domain 4 of human CTGF for 14 days reduced the LVEF of normal rats, which was prevented by concomitant treatment with olmesartan, an angiotensin II receptor antagonist [39].
4.2. CTGF inhibition vs. CTGF over-expression studies
Our results indicate that elevated CTGF contributes to the pathological remodeling of DCM, and that inhibiting CTGF improves contractile function even in the absence of a significant resolution in tissue fibrosis. These results are interesting as studies in which CTGF was overexpressed in cardiomyocytes of transgenic rodents suggested that elevated CTGF would be protective of cardiac function [7,11,12,17,18]. Cardiomyocyte CTGF over-expression studies also indicated that elevated CTGF alone was not sufficient to induce cardiac fibrosis [7], which is consistent with the concept that the pro-fibrotic effects of CTGF require additional co-stimulatory signals arising from the ECM microenvironment such as those produced during tissue injury or inflammation [40]. Our mRNA profiling results suggest that CTGF is necessary for the expression of multiple genes involved in tissue remodeling. Additional co-stimulatory molecules such as TGF-β family members were also generated in response to PKCε overexpression, but were not sufficient to fully maintain the pro-fibrotic gene signature in PKCε mice treated with the CTGF neutralizing mAb. The gene signatures reported here suggest altered fibroblast function or activity, but changes in relative abundance of fibroblasts are also likely to be relevant.
4.3. CTGF inhibition vs. CTGF gene deletion
In contrast to the beneficial effects of CTGF inhibition in TAC [13] and PKCε-induced DCM (this report), targeted deletion of most CTGF in adult cardiomyocytes and nonmuscle cells did not appear to prevent the functional changes or fibrosis induced by TAC [14, 15], or overexpression of a constitutively active form of TGF-β [14]. However, in some experiments, the reported data exhibited trends consistent with a beneficial effect of loss of CTGF function on fibrillar collagen gene expression [15], but were not significant at the small group sizes examined [14]. Thus, although further investigation is necessary to understand the differences observed from the various animal models used, the cumulative data suggest that inhibition of CTGF in patients with cardiovascular disease is unlikely to be harmful and instead, could provide a significant benefit to patients with DCM.
4.4. CTGF and the pro-fibrotic gene signature
To further understand the relevance of the gene signatures observed in the mouse model used here, we compared them to gene expression changes in 74 human cardiomyopathy vs. 14 non-failing heart samples of the Harvard Cardiogenomics dataset (NCBI GEO Series GSE1145; Online Table III). Forty-four percent of the genes elevated by constitutively active PKCε and responsive to CTGF inhibition were also significantly elevated in human cardiomyopathy; a level of overlap that is far greater than would be expected by chance (P<0.001). Moreover, the overlapping genes were common to multiple forms of human cardiomyopathy (dilated, ischemic, hypertrophic, post-partum and viral), and were highly enriched in ECM remodeling genes. FG-3149-response genes that were similarly altered in both PKCε hearts and human hearts appear likely to be most pertinent to human disease. Moreover, the substantial over-representation of FG-3149-responsive PKCε-elevated genes among human cardiomyopathy genes suggests that the PKCε model recapitulates key aspects of the human disease, and suggests injury genes resolved by FG-3149 in the current model may also respond to FG-3019 in treatment of human heart disease.
4.5. Conclusions
The PKCε model of DCM used in this study appears to recapitulate many aspects of human DCM. The observation of progressively increasing expression of CTGF led us to test how inhibition of CTGF would affect disease progression. The improvements in cardiac structure and function observed upon inhibition of CTGF are very encouraging, and suggest that CTGF contributes to DCM progression. Therefore, additional studies to explore this approach as a treatment for DCM are warranted.
Supplementary Material
Figure 3. Inhibition of CTGF signaling reduces the PKCε-induced pro-fibrotic gene signature.
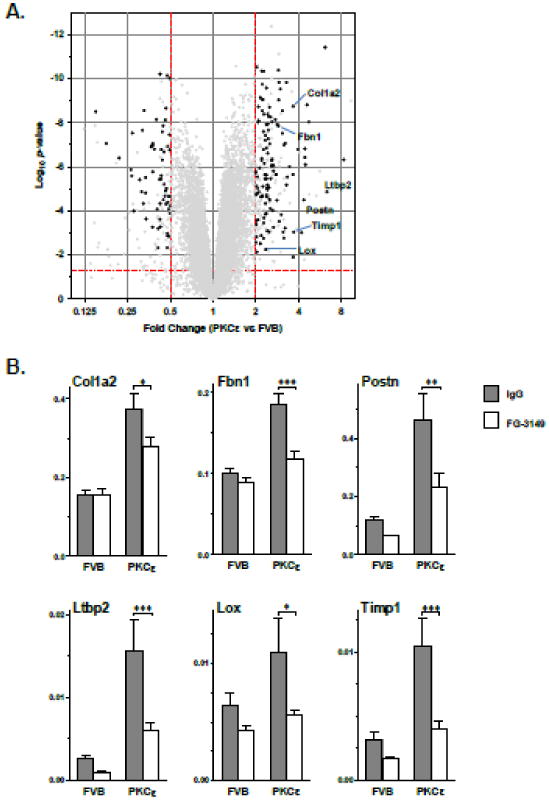
(A) Volcano plot of cardiac gene expression changes in IgG-treated PKCε mice vs. IgG-treated FVB mice for all genes with mean raw fluorescence values >50 in at least one group. Black circles indicate the PKCε-altered genes (Fc>2, P<0.05) that were partially reversed by FG-3149 vs. IgG in PKCε hearts (P<0.05). (B) Validation of select ECM and ECM remodeling genes by qPCR. Grey and white bars indicate IgG and FG-3149 treatments, respectively. Actb-normalized expression (mean ± SEM) was altered by genotype (P<0.0001) and treatment (P<0.05) for all genes (2-way ANOVA). Bonferroni post-test P<0.05 (*), 0.01 (**), or 0.001 (***). Additional qPCR results are provided in Online Figure V.
Highlights.
Cardiac structural changes associated with dilated cardiomyopathy (DCM) include cardiomyocyte hypertrophy and myocardial fibrosis. Connective Tissue Growth Factor (CTGF) has been associated with tissue remodeling and is highly expressed in patients with end-stage DCM.
Our aim was to test if inhibition of CTGF with FG-3149, a neutralizing mouse monoclonal antibody to CTGF, would alter the course of cardiac remodeling and preserve cardiac function in the protein kinase Cε (PKCε) mouse model of DCM.
CTGF inhibition significantly improved left ventricular (LV) systolic and diastolic function in PKCε mice, and slowed the progression of LV dilatation.
Using gene arrays and quantitative PCR, the expression of many genes associated with tissue remodeling were elevated in PKCε mice, but significantly decreased by CTGF inhibition. However total collagen deposition and LV fibrosis were not attenuated.
The observation of significantly improved LV function by CTGF inhibition in PKCε mice suggests that CTGF inhibition may benefit patients with DCM. Additional studies to explore this potential are warranted.
Acknowledgments
This work was supported by the National Institutes of Health grant numbers P01 HL62426, 1F32 HL096143, and R01 DA-030996. The authors thank Abhishek Sethi for his help with the echocardiographic measurements.
Footnotes
Disclosures: M.D.S., T.W.S, and K.E.L. are employees and shareholders of Fibrogen, Inc.
Appendix A. Supplementary data: Supplementary data to this article can be found online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cohn JN, Anand IS. Overview: cardiac remodeling and its relationship to the development of heart failure. In: Greenberg B, editor. Cardiac Remodeling: Mechanisms and Treatment. New York: Taylor & Francis; 2006. pp. 1–8. [Google Scholar]
- 2.Kuruvilla S, Adenaw N, Katwal AB, Lipinski MJ, Kramer CM, Salerno M. Late gadolinium enhancement on cardiac magnetic resonance predicts adverse cardiovascular outcomes in nonischemic cardiomyopathy: a systematic review and meta-analysis. Circ Cardiovasc Imaging. 2014;7:250–258. doi: 10.1161/CIRCIMAGING.113.001144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bork P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993;327:125–130. doi: 10.1016/0014-5793(93)80155-n. [DOI] [PubMed] [Google Scholar]
- 4.Hishikawa K, Oemar BS, Tanner FC, Nakaki T, Luscher TF, Fujii T. Connective tissue growth factor induces apoptosis in human breast cancer cell line MCF-7. J Biol Chem. 1999;274:37461–37466. doi: 10.1074/jbc.274.52.37461. [DOI] [PubMed] [Google Scholar]
- 5.Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126:137–145. doi: 10.1093/oxfordjournals.jbchem.a022414. [DOI] [PubMed] [Google Scholar]
- 6.Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Panek AN, Posch MG, Alenina N, Ghadge SK, Erdmann B, Popova E, et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One. 2009;4:e6743. doi: 10.1371/journal.pone.0006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon PO, Lee MA, Cha H, Jeong MH, Kim J, Jang SP, et al. The opposing effects of CCN2 and CCN5 on the development of cardiac hypertrophy and fibrosis. J Mol Cell Cardiol. 2010;49:294–303. doi: 10.1016/j.yjmcc.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed MS, Gravning J, Martinov VN, von Lueder TG, Edvardsen T, Czibik G, et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H1291–302. doi: 10.1152/ajpheart.00604.2010. [DOI] [PubMed] [Google Scholar]
- 10.Tsoutsman T, Wang X, Garchow K, Riser B, Twigg S, Semsarian C. CCN2 plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure. J Mol Cell Cardiol. 2013;62:164–178. doi: 10.1016/j.yjmcc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 11.Gravning J, Ahmed MS, Qvigstad E, Krobert K, Edvardsen T, Moe IT, et al. Connective tissue growth factor/CCN2 attenuates β-adrenergic receptor responsiveness and cardiotoxicity by induction of G protein-coupled receptor kinase-5 in cardiomyocytes. Mol Pharmacol. 2013;84:372–383. doi: 10.1124/mol.113.087312. [DOI] [PubMed] [Google Scholar]
- 12.Gravning J, Ahmed MS, von Lueder TG, Edvardsen T, Attramadal H. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int J Cardiol. 2013;168:2049–2056. doi: 10.1016/j.ijcard.2013.01.165. [DOI] [PubMed] [Google Scholar]
- 13.Szabo Z, Magga J, Alakoski T, Ulvila J, Piuhola J, Vainio L, et al. Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload-induced heart failure. Hypertension Hypertension. 2014;63:1235–1240. doi: 10.1161/HYPERTENSIONAHA.114.03279. [DOI] [PubMed] [Google Scholar]
- 14.Accornero F, van Berlo JH, Correll RN, Elrod JW, Sargent MA, York A, et al. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor β signaling and cardiac remodeling. Mol Cell Biol. 2015;35:2154–2164. doi: 10.1128/MCB.00199-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fontes MS, Kessler EL, van Stuijvenberg L, Brans MA, Falke LL, Kok B, et al. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J Mol Cell Cardiol. 2015;88:82–90. doi: 10.1016/j.yjmcc.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Koshman YE, Patel N, Chu M, Iyengar R, Kim T, Ersahin C, et al. Regulation of connective tissue growth factor gene expression and fibrosis in human heart failure. J Card Fail. 2013;19:283–294. doi: 10.1016/j.cardfail.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed MS, Oie E, Vinge LE, Yndestad A, Oystein Andersen G, Andersson Y, et al. connective tissue growth factor - a novel mediator of angiotensin II-stimulated cardiac fibroblast activation in heart failure in rats. J Mol Cell Cardiol. 2004;36:393–404. doi: 10.1016/j.yjmcc.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Ahmed MS, von Lueder TG, Oie E, Kjekshus H, Attramadal H. Induction of myocardial connective tissue growth factor in pacing-induced heart failure in pigs. Acta Physiol Scand. 2005;184:27–36. doi: 10.1111/j.1365-201X.2005.01416.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Q, Yi QJ, Qian YR, Li R, Deng B, Wang Q. Expression of connective tissue growth factor in cardiomyocyte of young rats with heart failure and benazepril intervention. Zhonghua Er Ke Za Zhi. 2006;44:733–737. [PubMed] [Google Scholar]
- 20.Koitabashi N, Arai M, Kogure S, Niwano K, Watanabe A, Aoki Y, et al. Increased connective tissue growth factor relative to brain natriuretic peptide as a determinant of myocardial fibrosis. Hypertension. 2007;49:1120–1127. doi: 10.1161/HYPERTENSIONAHA.106.077537. [DOI] [PubMed] [Google Scholar]
- 21.Goldspink PH, Montgomery DE, Walker LA, Urboniene D, McKinney RD, Geenen DL, et al. Protein kinase Cε overexpression alters myofilament properties and composition during the progression of heart failure. Circ Res. 2004;95:424–432. doi: 10.1161/01.RES.0000138299.85648.92. [DOI] [PubMed] [Google Scholar]
- 22.Alapati D, Rong M, Chen S, Hehre D, Rodriguez MM, Lipson KE, et al. Connective tissue growth factor antibody therapy attenuates hyperoxia-induced lung injury in neonatal rats. Am J Respir Cell Mol Biol. 2011;45:1169–1177. doi: 10.1165/rcmb.2011-0023OC. [DOI] [PubMed] [Google Scholar]
- 23.Koshman YE, Chu M, Kim T, Kalmanson O, Farjah M, Kumar M, et al. Cardiomyocyte- specific expression of CRNK, the C-terminal domain of PYK2, maintains ventricular function and slows ventricular remodeling in a mouse model of dilated cardiomyopathy. J Mol Cell Cardiol. 2014;72:281–291. doi: 10.1016/j.yjmcc.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. J Mol Cell Cardiol. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Solaro RJ, Henze M, Kobayashi T. Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res. 2013;112:355–366. doi: 10.1161/CIRCRESAHA.112.268672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohnishi H, Oka T, Kusachi S, Nakanishi T, Takeda K, Nakahama M, et al. Increased expression of connective tissue growth factor in the infarct zone of experimentally induced myocardial infarction in rats. J Mol Cell Cardiol. 1998;30:2411–2422. doi: 10.1006/jmcc.1998.0799. [DOI] [PubMed] [Google Scholar]
- 27.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by GF-β in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 28.Way KJ, Isshiki K, Suzuma K, Yokota T, Zvagelsky D, Schoen FJ, et al. Expression of connective tissue growth factor is increased in injured myocardium associated with protein kinase C β2 activation and diabetes. Diabetes. 2002;51:2709–2718. doi: 10.2337/diabetes.51.9.2709. [DOI] [PubMed] [Google Scholar]
- 29.Chuva de Sousa Lopes SM, Feijen A, Korving J, Korchynskyi O, Larsson J, Karlsson S, et al. Connective tissue growth factor expression and Smad signaling during mouse heart development and myocardial infarction. Dev Dyn. 2004;231:542–550. doi: 10.1002/dvdy.20162. [DOI] [PubMed] [Google Scholar]
- 30.Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, et al. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:245–1256. doi: 10.1369/jhc.4A6560.2005. [DOI] [PubMed] [Google Scholar]
- 31.Csencsits K, Wood SC, Lu G, Faust SM, Brigstock D, Eichwald EJ, et al. Transforming growth factor β-induced connective tissue growth factor and chronic allograft rejection. Am J Transplant. 2006;6:959–966. doi: 10.1111/j.1600-6143.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 32.Lang C, Sauter M, Szalay G, Racchi G, Grassi G, Rainaldi G, et al. Connective tissue Growth factor: a crucial cytokine-mediating cardiac fibrosis in ongoing enterovirus myocarditis. J Mol Med. 2008;86:49–60. doi: 10.1007/s00109-007-0249-3. [DOI] [PubMed] [Google Scholar]
- 33.Yuan YC, Xia ZK, Mu JJ, Zhang QC, Yin BL. Increased connective tissue growth factor Expression in a rat model of chronic heart allograft rejection. J Formos Med Assoc. 2009;108:240–246. doi: 10.1016/S0929-6646(09)60058-9. [DOI] [PubMed] [Google Scholar]
- 34.Au CG, Butler TL, Sherwood MC, Egan JR, North KN, Winlaw DS. Increased connective tissue growth factor associated with cardiac fibrosis in the mdx mouse model of dystrophic cardiomyopathy. Int J Exp Pathol. 2011;92:57–65. doi: 10.1111/j.1365-2613.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blaauw E, Lorenzen-Schmidt I, Babiker FA, Munts C, Prinzen FW, Snoeckx LH, et al. stretch-induced upregulation of connective tissue growth factor in rabbit cardiomyocytes. J cardiovasc Transl Res. 2013;6:861–869. doi: 10.1007/s12265-013-9489-5. [DOI] [PubMed] [Google Scholar]
- 36.Chemaly ER, Kang S, Zhang S, McCollum L, Chen J, Benard L, et al. Differential patterns of replacement and reactive fibrosis in pressure and volume overload are related to the propensity for ischaemia and involve resistin. J Physiol. 2013;591:5337–5355. doi: 10.1113/jphysiol.2013.258731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipson KE, Wong C, Teng Y, Spong S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012;5:S24. doi: 10.1186/1755-1536-5-S1-S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scruggs SB, Walker LA, Lyu T, Geenen DL, Solaro RJ, Buttrick PM, et al. Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCε phosphorylation. J Mol Cell Cardiol. 2006;40:465–473. doi: 10.1016/j.yjmcc.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto M, Hirohata S, Ogawa H, Ohtsuki T, Shinohata R, Miyoshi T, et al. Connective tissue growth factor induction in a pressure-overloaded heart ameliorated by the angiotensin II type 1 receptor blocker olmesartan. Hypertens Res. 2010;33:1305–1311. doi: 10.1038/hr.2010.189. [DOI] [PubMed] [Google Scholar]
- 40.Brigstock DR. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: lessons from transgenic animals. J Cell Commun Signal. 2010;4:1–4. doi: 10.1007/s12079-009-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.