

Abstract
Background
Patients with Duchenne muscular dystrophy (DMD) are at risk of developing cardiomyopathy and cardiac arrhythmias. Studies in a mouse model of DMD revealed that enhanced sarcoplasmic reticulum (SR) Ca2+ leak contributes to the pathogenesis of cardiac dysfunction. In view of recent data suggesting the involvement of altered phosphorylation and oxidation of the cardiac ryanodine receptor (RyR2)/Ca2+ release channel, we hypothesized that inhibition of RyR2 phosphorylation in a mouse model of DMD can prevent SR Ca2+ leak by reducing RyR2 oxidation.
Methods and Results
Confocal Ca2+ imaging and single RyR2 channel recordings revealed that both inhibition of S2808 or S2814 phosphorylation, and inhibition of oxidation could normalize RyR2 activity in mdx mice. Moreover, Western blotting revealed that genetic inhibition of RyR2 phosphorylation at S2808 or S2814 reduced RyR2 oxidation. Production of reactive oxygen species (ROS) in myocytes from mdx mice was reduced by both inhibition of RyR2 phosphorylation or the ROS scavenger 2-mercaptopropionyl glycine (MPG). Finally, it was shown that ROS production in mdx mice is proportional to the activity of RyR2-mediated SR Ca2+ leak, and likely generated by Nox2.
Conclusions
Increased ROS production in the hearts of mdx mice drives the progression of cardiac dysfunction. Inhibition of RyR2 phosphorylation can suppress SR Ca2+ leak in mdx mouse hearts in part by reducing RyR2 oxidation.
Keywords: Duchenne muscular dystrophy, oxidation, phosphorylation, reactive oxygen species, ryanodine receptor type 2
Introduction
Duchenne muscular dystrophy (DMD) is the most common and severe form of muscular dystrophy affecting approximately 1 of every 3,500 male births. DMD is X-linked recessive disorder caused by mutations in the dystrophin gene. Dystrophin and its associated glycoproteins provide a structural link between the myocyte cytoskeleton and extracellular matrix, connecting contractile proteins to the cell membrane [1]. Patients with DMD have a high incidence of cardiovascular morbidity and mortality due to cardiomyopathy and cardiac arrhythmias. The mdx mouse model of DMD also displays a phenotype consisting of age-dependent cardiomyopathy, with older mice showing cardiac dilatation, reduced contractility, fibrosis and arrhythmias [2-4].
Several studies have demonstrated profound changes in excitation-contraction coupling and intracellular Ca2+ handling in hearts of mdx mice [5]. For example, expression levels of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2a) and sarcoplasmic reticulum (SR) luminal proteins such as calsequestrin are reduced, whereas resting [Ca2+ ] are increased [5]. Enhanced activity of the SR Ca2+ release channel known as ryanodine receptor type-2 (RyR2) has been linked to the pathogenesis of cardiac dysfunction in DMD [3, 4, 6]. Changes in the phosphorylation status of Ca2+ handling proteins such as RyR2 are thought to contribute to SR Ca2+ leak, reduced Ca2+ transients, and increased likelihood of diastolic Ca2+ waves [4, 6]. Recent studies suggest a sequential or potentially synergistic contribution of both phosphorylation and oxidation of RyR2 as a contributor of progressive cardiac deterioration in mdx mice [7]. However, at this time the differential roles of RyR2 phosphorylation and oxidation in DMD-related cardiac disease pathogenesis remains unclear.
The aim of this study was to delineate the potential interactions between RyR2 post-translational modifications, in particular phosphorylation and oxidation, in the mdx mouse model of DMD. Our findings revealed that both inhibition of two major RyR2 phosphorylation sites as well as RyR2 oxidation could normalize SR Ca2+ release activity in cardiomyocytes from mdx mice. Western blotting revealed, surprisingly, that genetic inhibition of RyR2 phosphorylation at S2808 or S2814 reduced RyR2 oxidation, suggesting a potential interaction between these post-translational pathways. The production of reactive oxygen species (ROS) in myocytes from mdx mice was reduced both by inhibition of RyR2 phosphorylation and the ROS scavenger 2-mercaptoproppionylglycin (MPG), suggesting functional synergy between these post-translational modifications. Finally, we demonstrated that ROS production in mdx mice is proportional to the activity of RyR2-mediated SR Ca2+ leak. In conclusion, our study suggests that increased ROS production in the hearts of mdx mice drives the progression of cardiomyopathy, and that inhibition of RyR2 phosphorylation can suppress oxidation-induced SR Ca2+ leak in mdx mouse hearts.
Methods
Animals
The mdx mice were obtained from Jackson laboratories (C57BL/10ScSn-Dmdmdx/J). The mdx:S2808A mice were generated by crossing mdx mice with RyR2-S2808A, in which S2808 on RyR2 is substituted by alanine to inhibit PKA phosphorylation of RyR2 in mdx mice [3]. The mdx:S2814A mice were generated by crossing mdx mice with RyR2-S2814 mice, in which S2814 on RyR2 is genetically inactivated to prevent CaMKII phosphorylation of RyR2 [4]. Only male mice were used for experiments, and all lines were backcrossed for at least 20 generations. All studies were performed according to protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine conforming to the Guide for the Care and Use of Laboratory Animals published by the U.S. National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Mouse Ventricular Myocyte Isolation
Mouse ventricular myocytes isolation was performed as described [8]. Briefly, hearts were removed from anesthetized mice and rinsed in 0 Ca2+ Tyrode solution (137 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 5 mM HEPES, 10 mM glucose, 3 mM NaOH, pH 7.4). The heart was cannulated through the aorta and perfused using a Langendorff system with 0 Ca2+ Tyrode for 5 minutes at 37 °C, followed by 0 Ca2+ Tyrode containing 20 μg/ml Liberase (Roche, Indianapolis, IN) for 10 to 15 minutes at 37 °C. After digestion, the heart was rinsed with 5 ml KB solution (90 mM KCl, 30 mM K2HPO4, 5 mM MgSO4, 5 mM pyruvic acid, 5 mM β-hydroxybutyric acid, 5 mM creatine, 20 mM taurine, 10 mM glucose, 0.5 mM EGTA, 5 mM HEPES, pH 7.2). The digested heart was minced in KB solution and agitated, then filtered through a 210 mm polyethylene mesh. The isolated ventricular myocytes were washed once and stored in KB solution at room temperature before use.
Confocal Calcium Imaging
Confocal imaging was performed as described [8]. Ventricular myocytes were incubated with 2 mM Fluo-4-acetoxymethyl ester (Fluo-4 AM, Invitrogen, Carlsbad, CA) in normal Tyrode (NT) solution containing 1.8 mM Ca2+ for 1 hour at room temperature, followed by 15 min for de-esterification with dye-free NT solution and loaded on a laser scanning confocal microscope (LSM 510, Carl Zeiss, Thornwood, NY). Fluorescence images were recorded in line-scan mode with 1024 pixels per line at 500 Hz. After being paced at 1 Hz (5 ms, 10 V) for 2 minutes, only rod-shaped myocytes showing clear striation and normal contractility were selected for further experiments. Once steady state Ca2+ transient was observed, pacing was stopped and Ca2+ sparks were counted.
ROS Production Assay
For the quantification of ROS production, myocytes were loaded with 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Invitrogen) [9]. Briefly, isolated ventricular myocytes were loaded with 10 mM CM-H2DCFDA in NT solution containing 1.8 mM Ca2+ for 30 min, followed by 15 min for de-esterification with dye-free NT solution. Cells loaded with CM-H2DCFDA were excited at 488 nm using an Argon laser, and emitted fluorescence signals were collected above 500 nm. After a conditioning 1-Hz pacing train, pacing was stopped and ROS production was measured. A time series of 160 cell fluorescence images with 2s interval was acquired for each cell. The laser power was minimized to reduce laser light induced production of ROS.
Single Channel Recordings
Single channel recordings of native RyR2 from mdx mice and WT mice were acquired under voltage-clamp conditions at 0mV as described [8]. Native cardiac SR preparations were added to be incorporated into lipid bilayer membranes. Once the RyR2 channel as inserted into the membrane, data were collected using a digidata 1322A (Axon instruments, Sunnyvale, CA) and a Warner Bilayer Clamp Amplifier BC-535 (Warner Instruments, Hamden, CT, USA). Ryanodine (5 μM) was applied to the cis chamber to confirm RyR2 identity at the end of each experiment. Data were analyzed using pCLAMP 9.2 software (Axon instruments, Sunnyvale, CA).
Western Blot Analyses
Heart lysates were prepared from flash-frozen mouse hearts as described [8]. Phosphorylation of RyR2 were detected by using custom-made phospho-epitope specific antibodies against RyR2-S2814 (1:1,000) and RyR2-S2808 (1:1,000) [4]. Western blotting was performed using anti-gp91-phox (NADPH oxidase-2 or Nox2, 1:1,000; Abcam, Cambridge, MA), p47-phox (regulatory subunit of Nox2, 1:1,000; Santa Cruz, Dallas, TX), NADPH oxidase 4 (Nox4, 1:1,000; Abcam, Cambridge, MA). Oxidation level of RyR2 was measured by oxyblot (OxyBlot™ Protein Oxidation Detection Kit, 1:150; Millipore, Bedford, MA). Briefly, samples were denatured by SDS and the carbonyl groups in the protein side chains were derivatized to DNP-hydrazone by reaction with DNPH (2,4-dinitrophenylhydrazine). The DNP-derivatized protein samples were separated by polyacrylamide gel electrophoresis followed by Western blotting, and then recognized by an antibody specific to the DNP moiety of the proteins.
Immunoprecipitation
Immunoprecipitation was performed as previously described [8]. Briefly, heart lysates were used for immunoprecipitation by using a commercial RyR2 antibody, the pellets were subjected to Western blotting. The membranes were probed with RyR2 antibody (1:5,000; Thermo Scientific, Rockford, IL), gp91-phox, p47-phox and Nox 4 antibodies. The intensities of both RyR2 and NADPH oxidase bands were measured with Image J software (version 1.44).
Statistical Analysis
Results are expressed as mean ± SEM. Continuous variables were evaluated with an unpaired Student t test or ANOVA test with post hoc Bonferroni correction. P<0.05 was considered statistically significant. All the analyses were done using SPSS.
Results
Inhibition of RyR2 phosphorylation and oxidation both inhibit Ca2+ sparks in mdx mice
To test the effect of inhibition of RyR2 phosphorylation and oxidation on SR Ca2+ leak in mdx mice, Ca2+ imaging studies were performed on ventricular myocytes isolated from 3-month-old wildtype (WT) and mdx mice. Isolated ventricular myocytes were subjected to a 1-Hz pacing to obtain steady-state Ca2+ cycling, after which pacing was stopped and the incidence of Ca2+ sparks was documented. Representative line-scan images (Fig. 1A) and bar graphs containing summary data (Fig. 1B) show an increased frequency of Ca2+ sparks in myocytes from mdx mice (2.8±0.5 sparks/100μm/s) compared to WT mice (1.1±0.3 sparks/100μm/s; P<0.05), consistent with enhanced SR Ca2+ leak in mdx mice. In contrast, genetic inhibition of the primary PKA phosphorylation site on RyR2 (S2808) or primary CaMKII site (S2814) greatly reduced the frequency of Ca2+ sparks, suggesting that inhibition of RyR2 phosphorylation reduces diastolic Ca2+ leak in mdx mice (Fig. 1B). Moreover, the ROS scavenger 2-mercaptopropionyl glycine (MPG) also reduced the Ca2+ spark frequency in mdx mice (1.55±0.35 sparks/100μm/s, P<0.05 vs mdx) (Fig. 1B).
Fig 1. Inhibition of RyR2 phosphorylation and oxidation both inhibit Ca sparks in mdx mice.
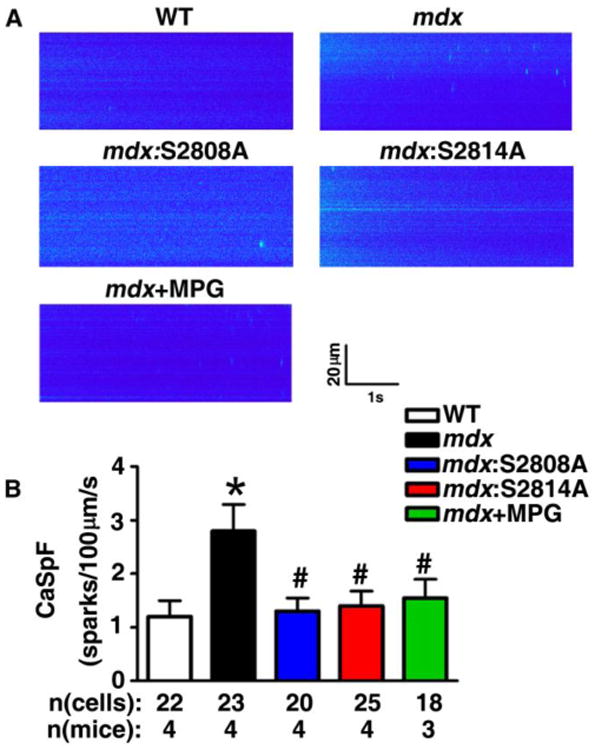
(A) Representative line scan images were recorded in ventricular myocytes from WT, mdx, mdx:S2808A, mdx:S2814A, and mdx mice treated with ROS scavenger MPG. (B) Bar graph of summary data showing Ca spark frequencies following 1 Hz pacing. *P<0.05 vs. WT; #P<0.05 vs. mdx.
Although not statistically significant, the amplitude of the calcium transients of mdx mice showed a trend toward a decrease probably due to the higher calcium leak in mdx cardiomyocytes, ROS scavenger MPG and genetic inhibition of RyR2 phosphorylation has similar amplitude with WT (Supplemental Fig 1A). In contrast to the Ca2+ sparks frequency, there were no significant difference between WT and mdx for sparks properties including Ca2+ sparks amplitude, the full-duration of half-maximum (FDHM) and the full-width of half-maximum (FWHM) (Supplemental Fig. 1, 2). Genetic inhibition of RyR2 phosphorylation and MPG did not significantly change sparks properties in mdx mice (Supplemental Fig. 1, 2).
Inhibition of RyR2 phosphorylation and oxidation both inhibit RyR2 activity in mdx mice
To directly assess the effects of post-translational modifications on RyR2 channel activity, single channel recordings were performed in planar lipid bilayers (Fig. 2A). RyR2 channels from 3-5 month-old mdx mice showed a higher open probability (0.084±0.017) compared to WT (0.013±0.004; P<0.05). In contrast, genetic inhibition of RyR2 phosphorylation normalized open probability in hearts of mdx:S2808A (0.016±0.009; P<0.05 vs mdx), and mdx:S2814A mice (0.015±0.011(P<0.05 vs mdx) (Fig. 2B). In addition, ROS scavenger MPG also normalized the open probability in mdx mice (0.02±0.011; P<0.05 vs mdx). Other channel properties were also affected in the hearts of mdx mice.
Fig 2. Inhibition of RyR2 phosphorylation and oxidation both inhibit RyR2 open probability in mdx mice.
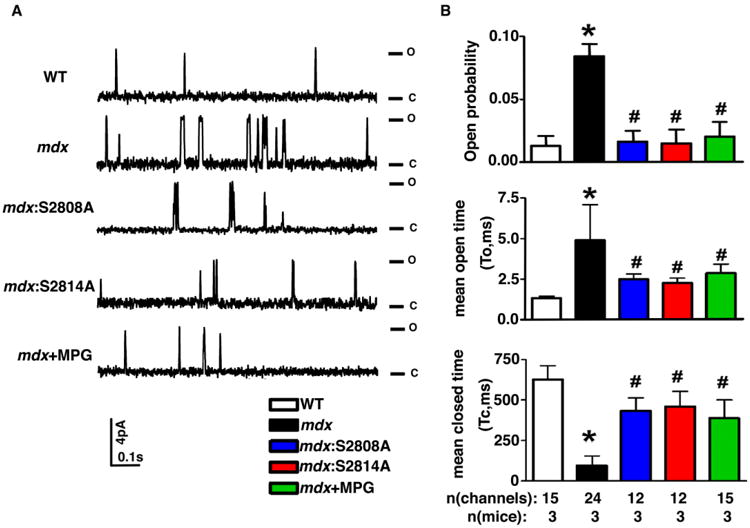
(A) Representative single channel recordings of RyR2 channels from WT, mdx, mdx:S2808A, mdx:S2814A, and mdx mice treated with ROS scavenger MPG. Channel openings (o) are shown as upward deflections from the closed (c) level. (B) Bar graphs showing averaged open probability, mean open time, and mean closed time. *P<0.05 vs. WT; #P<0.05 vs. mdx.
RyR2 channels from mdx mice exhibited a higher mean open time (To, 4.91±2.17ms) compared to WT mice (1.374±0.106ms; P<0.05), and a lower closed time in mdx hearts (Tc, 93.31±61.05ms) compared to WT (627.55±85.1ms; P<0.05). These RyR2 single channel properties were also mostly normalized by genetic inhibition of S2814 or S2808 phosphorylation on RyR2, compared to mdx mice (Fig. 2B). Finally, treatment with ROS scavenger MPG also normalized RyR2 channel abnormalities in mdx mice (Fig. 2B). The dwell time histograms of the open and close states of a single typical channel from WT, mdx and mdx with MPG were shown and fitted using an exponential probability distribution function (Supplemental Fig. 2). The lifetime of open states increased, and the lifetime of closed states decreased in mdx mice, and were normalized by MPG treatment (Supplemental Fig. 3). These data suggest that both phosphorylation and oxidation of RyR2 can contribute to elevated RyR2 activity in mdx mice.
Age-dependent changes in RyR2 oxidation and phosphorylation in mdx mice
We next assessed the level of RyR2 phosphorylation and oxidation in WT and mdx mice at 1-month and 3-months of age using Western blotting. Consistent with our previously published data, there was no difference in RyR2 phosphorylation at S2808 or S2814 at 1-month or 3-months of age between WT and mdx mice (Fig. 3A-B). In contrast, RyR2 oxidation – while similar at 1-month of age – was increased 1.6 fold in mdx mice compared to WT mice at 3 months (Fig. 3C).
Fig 3. Age-dependent changes in RyR2 oxidation and phosphorylation in mdx mice.
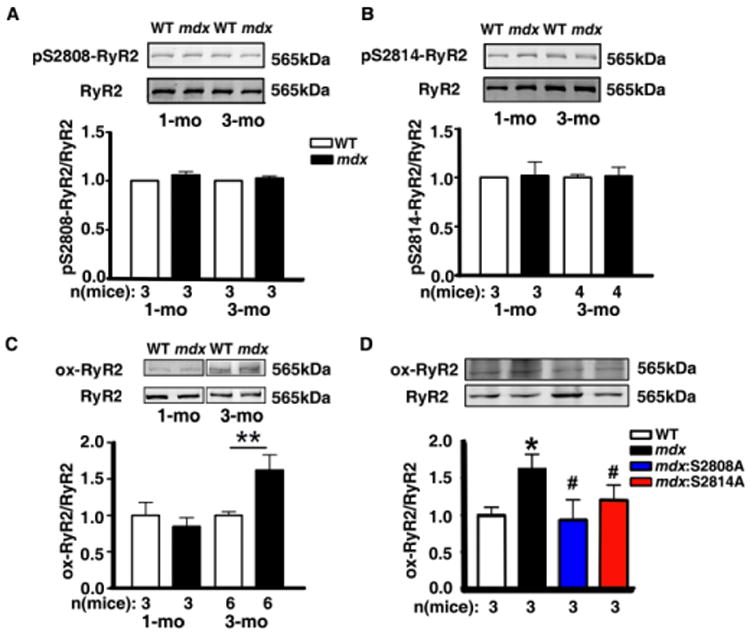
(A-B) Representative western blots and bar graphs showing unaltered phosphorylation level of RyR2 at the main PKA site S2808 and CaMKII site S2814 at 1 and 3 months in mdx mice. (C-D) Representative western blots and bar graphs showing increased RyR2 oxidation at 3 months in mdx mice, and inhibition of RyR2 oxidation due to genetic inhibition of RyR2 phosphorylation sites S2808 and S2814, respectively. *P<0.05 vs. WT; #P<0.05 vs. mdx.
We next sought to explore the effects of genetic inhibition of RyR2 phosphorylation on the level of RyR2 oxidation, since our work shows that inhibition of RyR2 phosphorylation can reduce SR Ca2+ leak despite unaltered RyR2 phosphorylation levels at 3-months of age. Genetic inhibition of RyR2 phosphorylation at S2808 and S2814 in mdx mice blunted the elevated oxidation level of RyR2 in mdx mice at 3 months-of-age (Fig. 3D). These data suggested functional interplay between RyR2 phosphorylation and oxidation in the hearts of mdx mice.
Inhibition of RyR2 phosphorylation reduces ROS production in mdx mice
To assess the level of oxidative stress and production of reactive oxygen species (ROS), ventricular myocytes were incubated with ROS indicator CM-H2DCFDA (Fig. 4). The rate of ROS production was found to be significantly higher in cardiomyocytes from 3-month-old mdx mice compared to WT controls (Fig. 4B-C). In contrast, treatment with ROS scavenger MPG reduced the ROS production in mdx mice to levels similar to those seen in WT myocytes (Fig. 4B-C). Genetic inhibition of RyR2 phosphorylation in mdx mice also reduced ROS production compared to mdx mice at 3-months of age (Fig. 4B-C). A second assay was used to quantify ROS production using DHE staining, which confirmed the aforementioned results (Supplemental Fig 4). Finally, the changes in ROS production were not present at the age of 1-month, suggesting that cardiac remodeling occurs between the 1-to- 3 months of age time period (Supplemental Fig. 5).
Fig 4. Inhibition of RyR2 phosphorylation reduces ROS production in mdx mice.
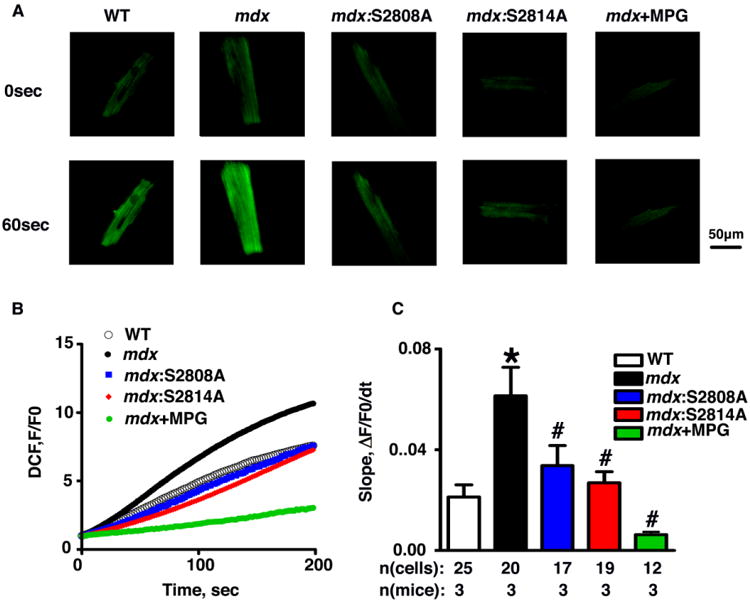
(A) Representative DCF fluorescence images of ROS in ventricular myocytes from WT, mdx, mdx:S2808A, and mdx:S2814A mice. (B) Time course of DCF fluorescence and (C) quantification of the time course slope in myocytes from WT, mdx, mdx:S2808A, and mdx:S2814A mice. *P<0.05 vs. WT; #P<0.05 vs. mdx.
ROS source in mdx mice
To identify the potential source of ROS generation in mdx mice, we measured expression levels of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits in the hearts of WT, mdx, mdx:S2808A and mdx:S2814A mice. Previous studies suggest that NADPH oxidase 2 (Nox2) and NADPH oxidase 4 (Nox4) are major contributors to elevated ROS production in dystrophic hearts [9, 10]. Western blot analysis revealed that protein levels of Nox2 and Nox4 were increased by 20-30% in mdx mice compared to WT at 3-5 months (Fig. 5A, 5B). Genetic inhibition of RyR2 phosphorylation did not affect the increased Nox2 and Nox4 expression levels in mdx mice. Since activation of Nox2 requires translocation of cytosolic factors (p47-phox, p67-phox and p40-phox) to the NoX2/p22phox complex by the “organizer subunit” p47-phox,[10] and p47-phox was shown to colocalize with RyR1 in skeletal muscle,[11] we also measured p47-phox expression levels (Fig. 5C). There were no significant changes in p47-phox expression level in mdx mice compared to WT mice (Fig. 5C).
Fig 5. ROS sources in mdx mice.
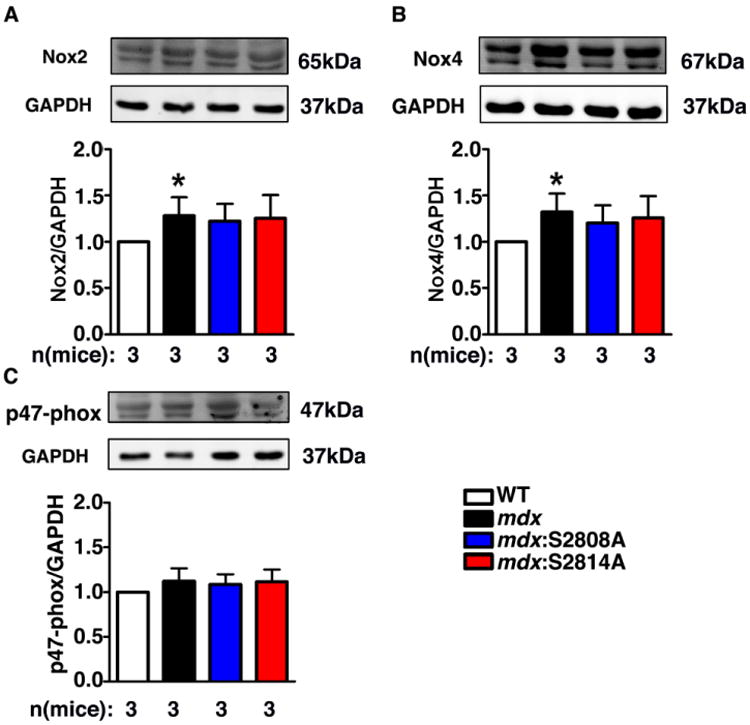
Representative Western blots and bar graphs showing (A) increased NADPH oxidase 2 (Nox2) expression, (B) increased NADPH oxidase 4 (Nox4) expression and (C) unaltered Nox2 subunit p47-phox expression in mdx mice. *P<0.05 vs. WT.
ROS production in mdx mice is RyR2-dependent
Finally, we set out to investigate whether the production of ROS was dependent on RyR2-mediated SR Ca2+ release. Caffeine was used to activate RyR2,[12] whereas RyR2 stabilizer JTV519 (JTV, also known as K201)[13] was applied to inhibit RyR2, and ROS production was measured in ventricular myocytes from mdx mice (Fig. 6A, 6B). RyR2 blockade by JTV519 decreased ROS production (slope: 0.015±0.004) compared to buffer-treated mdx myocytes (0.06±0.008; P<0.05) (Fig. 6B). On the other hand, caffeine activated ROS production (slope increased to 0.09±0.01; P<0.05 vs mdx) (Fig. 6B). These data suggest that SR Ca2+ release through RyR2 is an important mediator of ROS production. Finally, to determine the contribution of Nox2 to ROS production, Nox2 inhibitor gp91ds-tat was applied and ROS production was measured. After treatment with gp91ds-tat, ROS production in mdx mice was significantly decreased (0.013±0.003 vs 0.06±0.008 P<0.05), confirming Nox2 as a ROS source in mdx mice (Fig. 6).
Fig 6. ROS production in mdx mice is RyR2-dependent.
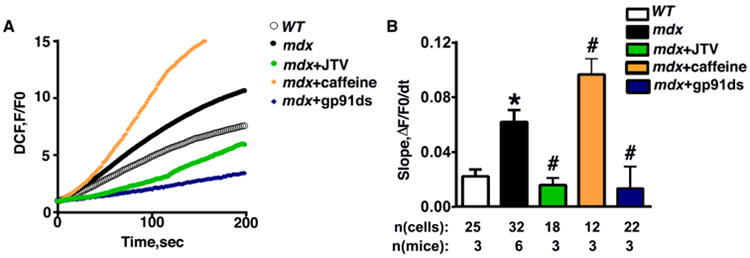
(A) DCF fluorescence time course showing increased ROS production in mdx mice, which is reduced by Nox2 inhibitor gp91ds-tat and RyR2-inhibitor JTV519 (JTV) and enhanced by RyR2 activator caffeine. (B) Quantification of normalized DCF slope in ventricular myocytes from WT mice, mdx mice, mdx with JTV, mdx with caffeine cardiomyocytes and mdx with gp91ds-tat. *P<0.05 vs. WT. #P<0.05 vs. mdx.
Discussion
Previous studies have shown that alterations in either the phosphorylation status or the oxidation state of RyR2 lead to abnormal intracellular Ca2+ release during the progression of cardiac disease in mdx mice, a commonly used animal model of DMD [4, 14]. Here we show that these two post-translation modifications are intimately connected, regulating RyR2 function and intracellular Ca2+ homeostasis in mdx mice. In 3 month-old mdx mice, prior to the development of structural heart disease, we found increased oxidation of RyR2 yet unaltered phosphorylation of RyR2 at both PKA and CaMKII phosphorylation sites. Intriguingly, genetic inhibition of RyR2 phosphorylation decreased RyR2 activity, reduced ROS production, and decreased the oxidation state of RyR2. These data suggest a cross-talk between oxidation and phosphorylation, resulting in increased SR Ca2+ leak and enhanced ROS production, both of which lead to the pathogenesis of heart disease in dystrophic hearts.
Role of RyR2 dysfunction in cardiac remodeling in mdx mice
Mdx mice develop progressive cardiac dysfunction with age. At 2 months of age, mdx mice have normal ventricular function based on echocardiography, whereas at 8 months of age they typically exhibit evidence of dilated hypertrophy, fibrosis, and alterations of contractility [2, 15]. However, at the cellular level, cardiomyocytes from young (3 months) mdx mice already show increased diastolic Ca2+ leak, increased Ca2+ spark frequency, and increased RyR2 single channel open probability. Therefore, alterations in RyR2 function may represent early events that drive the progression of dystrophic cardiomyopathy.
Several studies - including the present paper - have reported that RyR2 phosphorylation at the S2808 phosphorylation site is not elevated in mdx mice up to about 6 months of age [3, 6]. At an older age of 15-months, however, phosphorylation at this S2808 site does become elevated as cardiac dysfunction manifests [3]. Despite the lack of elevated S2808 phosphorylation in young mdx mice, genetic modification of this site (S2808A) can normalize aberrant SR Ca2+ release in ventricular myocytes [3]. For example, we have previously shown that genetic or pharmacological inhibition of RyR2 phosphorylation at the S2808 phosphorylation sites restored Ca2+ handling by reducing diastolic Ca2+ leak in 3 month old mdx mice[3]. Similar findings were obtained for the S2814 phosphorylation site of RyR2,[4] which is believed to be the primary CaMKII phosphorylation site on RyR2 [16].
This apparent discrepancy between unchanged phosphorylation levels in mdx mice and a protective effect of genetic ablation of the same phosphorylation site might be explained by crosstalk with another posttranslational modification of RyR2 (oxidation), which does occur by 3-months of age in the hearts of mdx mice (Fig. 3C-D). In cardiomyocytes isolated from mdx mice, ROS production has been shown to be elevated and ROS scavengers or Nox2 inhibition was able to normalize alterations in calcium homeostasisis [9, 14, 17]. Interestingly, the oxidation level of RyR2 was significantly reduced in RyR2-S2808A mice (with genetic ablation of PKA phosphorylation of RyR2) after isoproterenol treatment[18], while both Cysteine-nitrosylation and oxidation of RyR2 were increased in RyR2-S2808D (constitutive PKA hyper-phosphorylation of RyR2) mice [19]. In addition, inhibition of neuronal nitric oxide synthase (NOS1) decreased RyR2 phosphorylation at S2814 site (CaMKII phosphorylation site) [20]. These studies suggest a functional cross-talk between ROS/RNS modifications and phosphorylation of RyR2.
Mechanisms of altered RyR2 oxidation in mdx mice
Consistent with previous work [9, 21], we found increased expression of Nox2 and Nox4 and increased ROS production in hearts from mdx mice. Increased Nox2-dependent ROS production has been shown to promote RyR2 Ca2+ leak in response to mechanical stretch [9, 14]. In skeletal muscle, Nox2 was previously shown to be the primary source of ROS in young mdx mice (2-6 weeks of age) [9, 22, 23].
In this study, we found that genetic inhibition of RyR2 phosphorylation reduced the overall ROS production as well as the oxidation state of RyR2. In addition, caffeine, which increases RyR2 dependent Ca2+ release, increased ROS production in WT mice and blockade of RyR2 leak with the RyR2 inhibitor JTV519 (or K201) decreased ROS production in mdx mice. While Nox2 and Nox4 are not directly activated by Ca2+, increased RyR2 leak could activate Ca2+ sensitive PKC, leading to activation of Nox [24, 25].
Our data also revealed that Nox2 inhibitor gp91ds-tat blunted ROS production in ventricular myocytes from mdx mice. This is consistent with evidence in the literature that Nox2 plays a major role in increased ROS production in dystrophic cardiomyocytes [9, 21]. In contrast, much less is known about the potential role of Nox4, which is localized to the endo/sarcoplasmic reticulum [5, 21]. Nox4 is found in an endoplasmic reticulum (ER)-related perinuclear location [26], and prior studies have shown that Nox4 co-immunoprecipitates with RyR1 in skeletal muscle [27]. Future studies are required to investigate the potential contribution of Nox4 to abnormal RyR2 function in cardiomyocytes of mdx mice.
Potential limitations
In this paper, we focused on RyR2 regulation by oxidation and phosphorylation. Prior work has revealed that other post-translational modifications such as S-nitrosylation and S-glutathionylation also modify RyR2 in muscular dystrophy [6]. Future work is needed to investigate whether modifications of the RyR2 phosphorylation sites also affect regulation by those pathways in hearts of mdx mice. Although mdx mice have led to important advances in our understanding of muscular dystrophy, the disease progression is slower than in most human patients. Moreover, therapeutic approaches that yielded promising results in mdx mice were often not effective in patients [28]. Therefore, future validations in large animal models or human-based clinical studies would be important to confirm the potential benefits of anti-oxidant or RyR2-modifying therapeutic strategies.
Conclusions
Our findings suggest that increased reactive oxygen species (ROS) production leads to oxidation of RyR2 and drives the progression of cardiac disease in mdx mice, and that cross-talk between oxidation and phosphorylation of RyR2 also contributes the cardiac disease progression. These studies have the potential to open new avenues for the development of drugs that modify Ca2+ handling by RyR2 directly or by targeting ROS production, which might prove to be beneficial not only for DMD patients but also for other causes of cardiomyopathy and sudden cardiac death.
Supplementary Material
Highlights.
Increased ROS production contributes to cardiac dysfunction in mdx mice
Inhibition of RyR2 phosphorylation suppresses SR calcium leak in mdx mouse hearts.
ROS production in mdx mice is proportional to RyR2-mediated SR calcium leak.
Acknowledgments
Funding Sources: This work was supported by grants from the Muscular Dystrophy Association (186530), the Mrs. Clifford Elder White Graham Endowed Research Fund, NIH-NHLBI (HL089598, HL091947, HL117641, HL129570), the American Heart Association (13EIA14560061), and the Juanita P. Quigley endowed chair in cardiology.
Non-standard Abbreviations and Acronyms
- CaMKII
Calcium/calmodulin-dependent protein kinase II
- CaSpF
Calcium sparks frequency
- DCFH-DA
5-(6)-chloromethyl-2,7-dichlorodihydrofluorescein diacetate
- DHE
Dihydroethidium
- DMD
Duchenne muscular dystrophy
- MPG
N-2-mercaptopropionyl glycine
- NOS
Nitric oxide synthase
- NOX
NADPH oxidase
- PKA
Protein kinase A
- PTM
Post-translational modification
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- RyR2
Ryanodine receptor type 2
- SR
Sarcoplasmic reticulum
- SERCA2a
Sarco/endoplasmic reticulum Ca2+-ATPase
Footnotes
Disclosures: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94:1023–31. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- 2.Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–6. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 3.Sarma S, Li N, van Oort RJ, Reynolds C, Skapura DG, Wehrens XH. Genetic inhibition of PKA phosphorylation of RyR2 prevents dystrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:13165–70. doi: 10.1073/pnas.1004509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ather S, Wang W, Wang Q, Li N, Anderson ME, Wehrens XH. Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm. 2013;10:592–9. doi: 10.1016/j.hrthm.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol. 2007;292:H846–55. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- 6.Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, et al. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2010;107:1559–64. doi: 10.1073/pnas.0908540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyrychenko S, Polakova E, Kang C, Pocsai K, Ullrich ND, Niggli E, et al. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc Res. 2013;97:666–75. doi: 10.1093/cvr/cvs425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009;458:915–28. doi: 10.1007/s00424-009-0670-2. [DOI] [PubMed] [Google Scholar]
- 10.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 11.Pal R, Basu Thakur P, Li S, Minard C, Rodney GG. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One. 2013;8:e63989. doi: 10.1371/journal.pone.0063989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Porta M, Zima AV, Nani A, Diaz-Sylvester PL, Copello JA, Ramos-Franco J, et al. Single ryanodine receptor channel basis of caffeine's action on Ca2+ sparks. Biophys J. 2011;100:931–8. doi: 10.1016/j.bpj.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304:292–6. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 14.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–5. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 15.Zhang W, ten Hove M, Schneider JE, Stuckey DJ, Sebag-Montefiore L, Bia BL, et al. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: an MRI and MRS study. J Mol Cell Cardiol. 2008;45:754–60. doi: 10.1016/j.yjmcc.2008.09.125. [DOI] [PubMed] [Google Scholar]
- 16.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ Res. 2004;94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 17.Shirokova N, Niggli E. Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies. J Mol Cell Cardiol. 2013;58:217–24. doi: 10.1016/j.yjmcc.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–98. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–87. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cutler MJ, Plummer BN, Wan X, Sun QA, Hess D, Liu H, et al. Aberrant S-nitrosylation mediates calcium-triggered ventricular arrhythmia in the intact heart. Proc Natl Acad Sci U S A. 2012;109:18186–91. doi: 10.1073/pnas.1210565109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spurney CF, Knoblach S, Pistilli EE, Nagaraju K, Martin GR, Hoffman EP. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul Disord. 2008;18:371–81. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pal R, Palmieri M, Loehr JA, Li S, Abo-Zahrah R, Monroe TO, et al. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nat Commun. 2014;5:5425. doi: 10.1038/ncomms5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One. 2010;5:e15354. doi: 10.1371/journal.pone.0015354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dekker LV, Leitges M, Altschuler G, Mistry N, McDermott A, Roes J, et al. Protein kinase C-beta contributes to NADPH oxidase activation in neutrophils. Biochem J. 2000;347(Pt 1):285–9. [PMC free article] [PubMed] [Google Scholar]
- 25.Raad H, Paclet MH, Boussetta T, Kroviarski Y, Morel F, Quinn MT, et al. Regulation of the phagocyte NADPH oxidase activity: phosphorylation of gp91phox/NOX2 by protein kinase C enhances its diaphorase activity and binding to Rac2, p67phox, and p47phox. FASEB J. 2009;23:1011–22. doi: 10.1096/fj.08-114553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun QA, Hess DT, Nogueira L, Yong S, Bowles DE, Eu J, et al. Oxygen-coupled redox regulation of the skeletal muscle ryanodine receptor-Ca2+ release channel by NADPH oxidase 4. Proc Natl Acad Sci U S A. 2011;108:16098–103. doi: 10.1073/pnas.1109546108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chamberlain JS. Duchenne muscular dystrophy models show their age. Cell. 2010;143:1040–2. doi: 10.1016/j.cell.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.