

Abstract
Background
Although autophagy is an essential cellular salvage process to maintain cellular homeostasis, pathological autophagy can lead to cardiac abnormalities and ultimately heart failure. Therefore, a tight regulation of autophagic process would be important to treat chronic heart failure. Previously, we have shown that IL-10 strongly inhibited pressure overload-induced hypertrophy and heart failure, but role of IL-10 in regulation of pathological autophagy is unknown. Here we tested the hypothesis that IL-10 inhibits angiotensin II-induced pathological autophagy and this process, in part, leads to improve cardiac function.
Methods and Results
Chronic Ang II strongly induced mortality, cardiac dysfunction in IL-10 Knockout mice. IL-10 deletion exaggerated pathological autophagy in response to Ang II treatment. In isolated cardiac myocytes, IL-10 attenuated Ang II-induced pathological autophagy and activated Akt/mTORC1 signaling. Pharmacological or molecular inhibition of Akt and mTORC1 signaling attenuated IL-10 effects on Ang II-induced pathological autophagy. Furthermore, lysosomal inhibition in autophagic flux experiments further confirmed that IL-10 inhibits pathological autophagy via mTORC1 signaling.
Conclusion
Our data demonstrate a novel role of IL-10 in regulation of pathological autophagy; thus can act as a potential therapeutic molecule for treatment of chronic heart disease.
Keywords: Autophagy, Signaling, Cardiomyocytes, PI3K/AKT signaling, mTORC1
1. Introduction
Autophagy is a highly conserved bulk protein degradation process responsible for the turnover of long-lived proteins, disposal of organelles and clearance of aggregate-prone proteins [1, 2]. It plays a major role in clearance of misfolded and aggregated proteins to maintain cellular homeostasis and to keep cells healthy [3]. There are at least three distinct autophagic pathways; macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). During CMA, the misfolded cytosolic proteins containing exposed KFERQ motifs get degraded, while during microautophagy, cytosolic cargo is directly sequestered in the lysosome through invagination of lysosomal membrane. On the other hand, macroautophagy (here after termed as autophagy) is a non-selective multistep process by which portions of cytoplasm and/or organelles are sequestered in a vesicular structure including mitochondria (during mitophagy) called autophagosomes [4]. Furthermore this autophagosome fuses with lysosomes and degrades the autophagosomal contents. In the heart, autophagy occurs at low-levels under normal conditions but excessive and/or defective autophagy may lead to severe cardiac pathology and ultimately heart failure [5]. Indeed, elevated autophagy-associated vacuoles are detected in cardiomyocyte from failing heart in different organisms including human [6–8]. Up-regulated autophagy, along with hypertrophic remodeling, has also been reported in cardiomyocytes after chronic angiotensin II treatment [9]. Overexpression of beclin1 exacerbates TAC-induced pathological cardiac remodeling and heart failure in mice [7]. The precise role of autophagy during heart failure, however, remains to be elucidated.
The regulation of autophagy in heart is a complex process and involves many different signaling cascades [4, 10]. The major signaling cascades that regulate progress of autophagy in heart are, 1) AMPK, the major energy sensor in mammalian cells, can regulate autophagy during starvation via inhibition of mTORC1. 2) The FOXO3 protein is the positive regulator of autophagy and activates the transcription of many autophagy-associated genes. Additionally, FOXO3 also regulates the transcription of BNIP3 and PINK1, the major proteins involved during mitophagy [11, 12]. 3) JNK-mediated phosphorylation of bcl2 can also paly important role in regulation of autophagy [13]. Previously it has shown that phosphorylation of bcl2 releases beclin1 and thus activated autophagy. 4) Finally, PI3K-Akt-mTORC1 pathway also play critical role in autophagic regulation [4, 10].
Akt, a serine-threonine kinase, regulates cellular metabolism, is a pro-survival signaling molecule. Among numerous signaling pathways involved in the regulation of cellular apoptosis and autophagy, Akt plays crucial roles [14]. Phosphotidylinositol 3-Kinsae (PI3K) activates Akt in response to phosphorylation of receptor tyrosine kinase. Akt dependent suppression of autophagy can be mediated by activation of mTORC1, which inhibits the initiation of autophagy process [15]. It has been shown that IL-10, a pleiotropic cytokine promotes survival of astrocytes by a mechanism that involves activation of PI3-kinase, an upstream activator of Akt [16]. In another report, it has been shown that IL-10 promotes survival of myeloid progenitors by activating Akt pathway through the involvement of ERK 1/2 [17] and tyrosine kinase [18]. Previously, we have shown that IL-10 improves heart function both in ischemic and hypertrophic myocardium [19, 20]. Indeed, we have shown that IL-10-mediated activation of VEGF, a downstream target of Akt, improved endothelial progenitor cells survival [21]. Furthermore, IL-10 strongly inhibits pressure overload-induced cardiac cell death [20]. Recent studies advocate a critical contribution of class I PI3K/Akt in regulation of autophagy. Selective inhibition of Akt markedly induced autophagy [22].
However, neither the contribution of dysregulated autophagy to worsened cardiac functions in IL-10 null mice nor the role of IL-10 –induced signaling in the inhibition of pathological autophagy in cardiac cells is known. In the present study we provide the novel finding that IL-10 is a critical regulator of cardiac autophagy and that loss of IL-10 leads to exaggerated autophagy in response to Angiotensin II induced pressure overload. We also show that IL-10-mediated activation of PI3K/Akt/mTORC1 pathway inhibits Ang II-mediated pathological autophagy and improves cardiac function.
2.1. Animals and treatments
All animal experiments performed in this study adhered to the protocols approved by the Institutional Animal Care and Use Committee of the Temple University. Eight to ten-weeks-old wild-type (WT) and Interleukin-10-Knockout (IL-10 KO) mice of C57BL/6J background were procured from Jackson Research Laboratory (Bar Harbor, ME). Before treatments all animals were screened for baseline echocardiography. Left ventricular hypertrophy and heart failure was induced in these mice with Angiotensin II (1.6 mg/kg/day for 28 days) infusion using mini-osmotic pumps as described previously [20]. Twenty-eight days after treatment, mice were euthanized after final functional assessments and left ventricular tissue samples were collected for electron microscopy, qPCR and western blot analysis for hypertrophy and autophagy-associated protein as described previously [20, 23].
2.2. Isolation of neonatal rat ventricular cardiomyocytes (NRCM) and treatments
NRCM were prepared by enzymatic digestion of hearts obtained from newborn (0–2 day old) Sprague–Dawley rat pups using percoll gradient centrifugation and plated on six-well cell culture grade plates (coated with collagen IV) at a density of 0.85 × 106 cells/well in DMEM/M199 medium and maintained at 37°C in humid air with 5% CO2 [20, 23]. For treatments, NRCM were cultured in low serum media (2.5% fetal bovine serum and 2.5% horse serum; LS media) for 8 hours. NRCM were pre-treated with IL-10 (20 ng/ml) along with inhibitors (PI3K class I inhibitor, 30 μM; rapamycin, 50 nM and chloroquine 30 μM) for one hour followed by Angiotensin II (Ang II, 1 μM) for 48 hours. After treatments, cells were harvested and used for immunofluorescence staining and western blotting [20, 23]. Adenovirus transfection was performed in complete serum media for 36 hours as described previously.
2.3. Antibodies and reagents
Antibodies against beclin1, bcl2, pAkt, tAkt ser473, p4EBP1, t4EBP1, pmTORC, tmTORC, pJNK1/2, tJNK1/2, β-actin and GAPDH were purchased from Cell Signaling Inc. (Boston, MA). Antibody against LC3 (1/2) was purchased from MBL International Corporation. Recombinant murine IL-10 was obtained from R&D Systems (Minneapolis, MN). LY2940002 was obtained from Calbiochem. (San Diego, CA). Angiotensin II and chloroquine were purchased from Sigma (San Diego, CA). mRFP-GFP-LC3 adenovirus construct was a kind gift from Professor Joseph Rabinowits, Temple University. Rapamycin and Akt siRNA I were purchased from cell signaling technology.
2.4. Cardiac Imaging
Trans-thoracic 2-dimensional M-mode echocardiography was performed using Vevo 770 (VisualSonics, Toronto, Canada) equipped with a 30 MHz transducer. Echocardiographic studies were performed before (baseline) and 15 and 28 days post-osmotic pump implantation in mice. M-mode tracings were used to measure LV wall thickness, LV dimensions, LV volume and LV mass. The mean values of at least 3–5 cardiac cycles were used to determine the measurements for each animal. Percent fractional shortening (% LVFS) and ejection fraction (% LVEF) were calculated as described previously [20].
2.5. Electron Microscopy
Left ventricular tissues for electron microscopic examination were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1M sodium cacodylate buffer, pH7.4, overnight at 4°C. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide for 1 hour at room temperature, and then washed with buffer followed by distilled H2O. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Finally, thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software (Imaging department, University of Pennsylvania, PA). The number of autophagosomes was calculated in equal area of 4–5 sections of each mouse and 3–4 mice per group.
2.6. Preparation of Tissue/Cell Lysates, Immunoprecipitation and Western Blotting
Cells/tissue were lysed in RIPA cell lysis buffer (Cell Signaling, Boston, MA) supplemented with protease inhibitors pellets (Amarsham Biosciences, Piscataway, NJ). Insoluble debris was removed by centrifugation (12000 g) for 15 min at 4°C and cell lysates were boiled with Laemmle sample buffer (0.5 mol/L Tris-HCl [pH 6.8], 10% SDS, 10% glycerol, 4% β-merceptoethenol and 0.05% bromophenol blue). For immunoprecipitation experiments, cell lysates were immunoprecipitated using Peirce IP kit before boiling with Laemmli buffer. Equal amounts of protein (20–40 μg) were separated by SDS-PAGE and assessed by western blotting in indicated protein(s). GAPDH/β-actin was used as a loading control. The bands of interest were quantified using scanning densitometry using Image-quant software as described previously [23].
2.7. Immunoflourescent staining
Immunofluorescent staining for LC3, LAMP 1 and α-SA was performed in tissue sections or H9C2 cells plated onto chamber slides. After treatments and prior to immunostaining, cells were washed with PBS and fixed with 4% PFA for 10 min and permeabilized for 10 min using 0.05% Triton® 100. Cells were incubated for 1 h (22°C) with blocking solution (3% bovine serum albumin, 10% Horse serum and 0.2% Triton X-100) to block nonspecific binding. Cells were then incubated at 37°C for 2 h with primary antibodies and 45 min with the appropriate secondary antibody conjugated to Alexa-488 or Alexa 594 (Invitrogen). DAPI was used to counterstain the nuclei. Cells were covered with mounting media (Invitrogen), overlaid with a coverslips and examined under a fluorescence microscope with original magnification of 400X using oil emulsion. For tissue sections all the steps are same after de-parafinization steps and these section were imaged at 200X magnification.
2.8. Autophagic flux
Autophagic flux is a method to determine the defects in progress of autophagy. To measure autophagic flux we infect NRVM with tandem length mRFP-GFP-LC3 adenovirus for 40 hrs. After different treatment schedule cells were fixed in PFA as described above and stained with only DAPI. Cells were covered with mounting media (Invitrogen), overlaid with a coverslips and examined under a fluorescence microscope with original magnification of 400X using oil emulsion. Under fluorescence microscope, autophagosome, which express both GFP and RFP fluorescence, appears yellow and autolysosome appears red as acidic medium of lysosome quench GFP [24].
2.9. Transient transfection of siRNA and plasmid
Transient transfection of siRNA in cardiomyocytes was performed using Lipofectamine RNAiMAX (Life Technologies). Cells were plated in six-well plates the day before transfection at 70 to 80% confluency. Transfection reagent and siRNA were mixed in low glucose DMEM (Life Technologies) without serum and antibiotic/antimicotic. After 48 hours, the medium was changed to Low glucose DMEM medium and the cells were cultured overnight before drugs treatment. The transfection efficiency of Lipofectamine RNAiMax is consistently high and has been reported in our previous manuscript [25].
2.10. Statistical Analysis
Results are expressed as the mean ± standard error of the mean (SEM), computed from separate experiments. Comparisons between control and experimental groups were performed using the unpaired t test. Multiple comparisons among treatment groups were performed using one-way or repeated two way analysis of variance (ANOVA) and levels of significance were determined using the Tukey-Kramer multiple comparison post-hoc test if overall comparison across group was statistically significant (GraphPad prism 6 Software Inc., San Diego, CA). P values <0.05 were considered to be statistically significant.
3. Results
3.1. IL-10 deletion exacerbates Ang II-induced hypertrophic response in IL-10 KO mice
Ang II treatment significantly increased mortality among IL-10 KO mice compare to WT mice (Figure 1A). M-mode echocardiography showed no baseline differences in left ventricular (LV) function between WT and IL-KO mice. Similarly, saline treatment did not affect the cardiac function of WT and IL-10 KO mice. Significant impairment in LV function was observed from day 14 after Ang II treatment, as evidenced by reduced % ejection fraction (%EF Day 14: 50% and Day 28: 26% vs Baseline: 76%) and % fractional shortening (%FS Day 14: 29% and Day 28: 14% vs Baseline: 43%) (Figure 1B). Ang II-induced impairment in heart function was further aggravated in IL-10 KO mice compared to WT mice (Figure 1B). As hemodynamic ratio and fetal gene reprogramming are considered as hallmarks for stress-induced pathological remodeling thus next we measured both heart weight tibia length ratio and ANP and BNP genes expression. Ang II significantly increased heart weight/tibia length ratio both in WT and IL-10 KO mice compared to respective controls. Hemodynamic ratio was accentuated in IL-10 KO mice as compared to WT Ang II treated mice (Fig. 1C). Furthermore, chronic Ang II infusion increased ANP and BNP fetal gene expression (Figure 1D), which was further exaggerated in IL-10 KO mice (Figure 1D).
Figure 1. IL-10 deletion exaggerates Ang II-induced hypertrophy and heart failure.
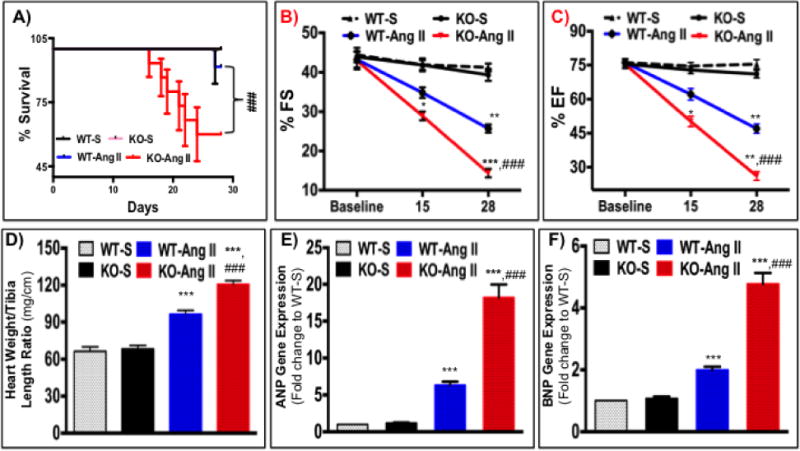
After series of echocardiography mice were euthanized the left ventricular tissue was used for biochemical analysis. A) Kaplan-Meier plot to estimate survival after Angiotensin II infusion in wild-type and IL-10 KO (N=10–12). Ang II infusion strongly increased mortality only in IL-10 KO mice. B&C) % Ejection fraction (%EF) and % Fractional shortening (%FS) were calculated by M-mode echocardiography in IL-10 KO and WT mice. Cardiac function of IL-10 KO mice was severely impaired after Ang II treatment (N= 6–8). D) Heart weight/tibia length was calculated in IL-10 KO mice as compared to WT after Ang II treatment. IL-10 KO mice showed significant increase in HW after Ang II treatment. E&F) ANP and BNP gene expression was measured in heart tissue isolated from WT and IL-10 KO mice by quantitative PCR. Relative mRNA expression of target genes was normalized with endogenous 18S and represented as fold-change vs. WT-saline. *** p<0.001 vs. WT-saline. **p<0.01 vs. WT-saline, *p<0.05 vs. WT-saline, ###p<0.001 vs. WT-Ang II. N=4–5
3.2. IL-10 KO mice showed enhanced autophagic response after Ang II infusion
To determine if increased mortality and depressed function in IL-10 KO mice might reflect enhance pathological autophagy, we performed immunohistochemical analysis for autophagic marker protein LC3 (1/2) in paraffin embedded left ventricular sections of WT and IL-10 KO mice. Ang II strongly activated LC3 staining both in WT and IL-10 KO cardiomyocytes of mice heart (Figure 2A). Strikingly, LC3 staining was further increased in IL-10 KO mice heart sections compared to WT (Figure 2A). Furthermore, western blots in left ventricular tissue lysate for autophagy-associated markers protein (beclin1 and LC3 1/2) confirmed that Ang II-treatment significantly increased both beclin 1 and LC3 protein expression in WT mice (Figure 2B and 2C) and Ang II-infused IL-10 KO mice showed an enhanced expression of these proteins (Figure 2B and 2C). Next we measured the number of autophagic vesicles in left ventricular sections of WT and IL-10 KO by transmission electron microscopy. Ang II significantly increased autophagosomal vesicles both in WT and IL-10 KO mice (Figure 2D and 2E). Interestingly, Ang II-induced autophagosomes were markedly increased in IL-10 KO mice (Figure 2D and 2E).
Figure 2. IL-10 deletion exaggerates Ang II-induced pathological autophagy.
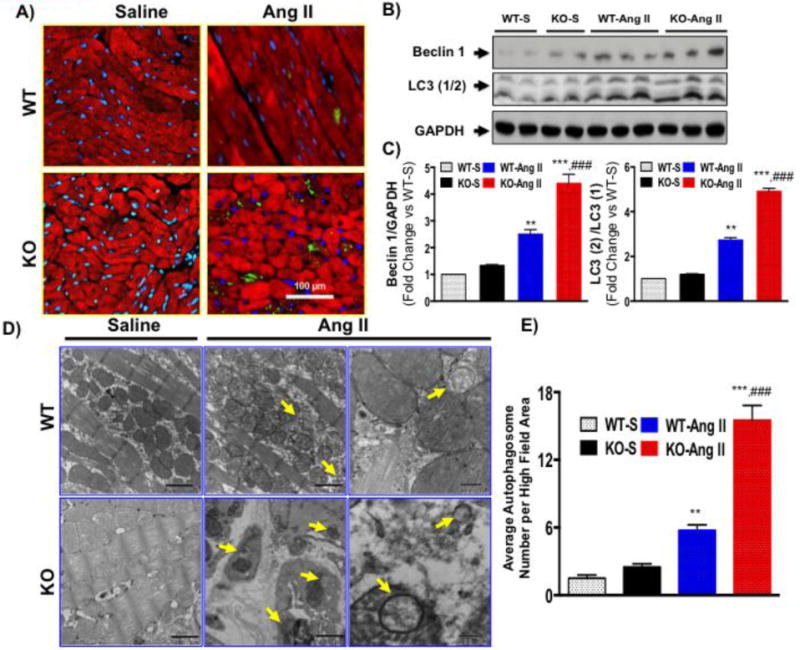
At the end of treatment schedule mice were euthanized and left ventricular tissue was harvested and used for biochemical analysis. A) Paraffin embedded sections were stained for LC3 (Green) and α-sarcomeric actin (Red) to identify autophagosomes in cardiomyocytes and visualized by fluorescence microscope at 400X magnification. More LC3 positive cardiomyocytes were detected in IL-10 KO mice compared to WT after Ang II treatment. B&C) Autophagy-associated proteins (beclin 1 and LC3 (1/2)) were measured in WT and IL-10 KO mice after Ang II treatment by western blot in whole tissue lysate. IL-10-KO mice showed enhanced expression of autophagic modulators after Ang II treatment. D&E) Electron microscopic images of LV sections in WT and IL-10 KO mice after saline control or Ang II treatment to measure autophagosomes. Bar graph indicates mean number of autophagosomes/high field area. Enhance autophagosome in IL-10 KO mice further corroborate our immunofluorescence data. (EM magnification was a) 500 nm = 20000X and b) 100 nm = 75000X) *** p<0.001 vs. WT-saline. **p<0.01 vs. WT-saline, *p<0.05 vs. WT-saline, ###p<0.001 vs. WT-Ang II. N=4–5
3.3. Acute long-term Ang II treatment induces pathological autophagy in neonatal rat ventricular cardiomyocytes (NRVM)
To test whether acute long-term treatment of Ang II induces pathological autophagy, we performed Ang II dose and time response study in NRVM. Cells were treated with two different doses of Ang II for three different time points as described in method section. Expression of autophagic marker proteins (beclin1 and LC3 (1/2)) was measured by western blot analysis. Ang II treatment significantly increased expression of autophagic markers within 8 hrs at both doses and then declined at 24 hrs. Interestingly, chronic long-term treatment of Ang II (for 48 hrs) exaggerated autophagic markers levels (supplementary Figure S1). Therefore in rest of study we used higher dose (1 μM) of Ang II for 48 hrs.
3.4. IL-10 treatment attenuates Ang II-induced pathological autophagy in NRVM
To understand the molecular mechanisms of IL-10 on pathological autophagy inhibition, NRVM were treated with Ang II and/or IL-10 (20 ng/ml) for 48 hrs and total cells lysates were prepared for proteins analysis. Western blots for autophagic indicators showed Ang II treatment significantly increased beclin1 and LC3 (2) proteins levels (Figure 3A & 3B). However, IL-10 co-treatment attenuated Ang II-induced autophagic indicators level (Figure 3A & 3B). Akt is a survival-signaling molecule important in cells proliferation and inhibition of apoptosis. Additionally, Akt tightly regulates progress of autophagy in many cells [22]. Previously we have shown that IL-10 regulates Akt activity in endothelial progenitor cells [25]. But the role of IL-10 on cardiomyocytes in autophagy regulation is not known. Therefore, to determine whether IL-10-mediated inhibition in pathological autophagy is dependent on Akt activation, we measured Akt (Akt-serine-473) and 4EBP1 phosphorylation in whole cells lysates. Chronic Ang II significantly inhibited both Akt and 4EBP1 phosphorylation and this inhibition was abolished by IL-10 treatment (Figure 3C & 3D). As JNK1/2 is known to be a critical regulator of autophagy, next we asked whether IL-10 alters Ang II-induced JNK1/2 activity. In alignment with our previous study [26], chronic long-term (48 hrs) Ang II treatment significantly inhibited JNK1/2 phosphorylation. Interestingly, Ang II-induced JNK1/2 inhibition was modestly rescued by IL-10; however, this affect was statistically insignificant (Suppl. Figure S2). This result suggests that Ang II-induced autophagy in this model is independent of JNK1/2 signaling pathway.
Figure 3. IL-10 treatment inhibits Ang II-induced pathological autophagy in neonatal rat ventricular cardiomyocytes (NRVM).
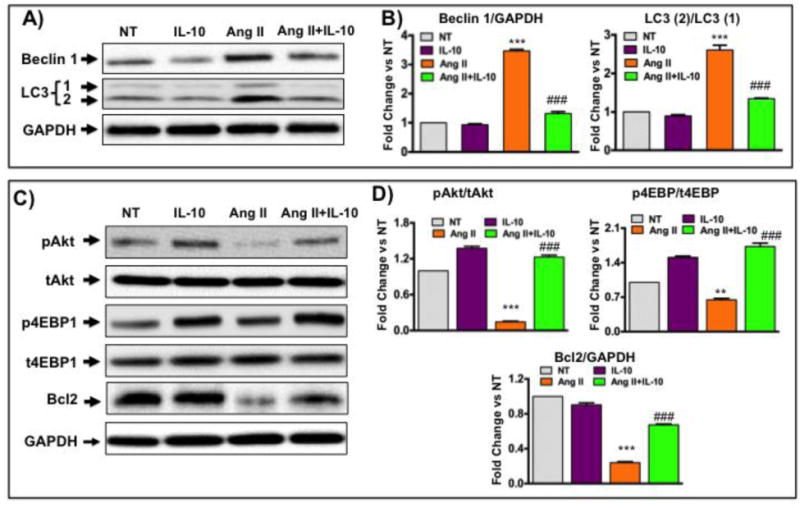
NRVM were isolated from 1–2 days-old rat pups. Cells were pretreated with IL-10 for 60 min followed by Ang II treatment for 48 hrs. A) Western blot analysis was performed to measure autophagy-associated proteins expression. IL-10 treatment significantly inhibited Ang II-induced beclin1 and LC3 (1/2) protein level. C) Western blot measured the effect of IL-10 and Ang II on Akt-mTORC1 (pAkt-ser473 and p4EBP1-thr37/46) signaling components and bcl2 levels in cells lysate. IL-10 significantly reestablished Ang II-mediated inhibition in Akt and 4EBP1 phosphorylation. Interestingly, Ang II-induced inhibition of bcl2 was rescued by IL-10. GAPDH were used as internal control in both Western blot experiments. B&D) Densitometric analysis was performed on the western blots using Image quant software. Bar graphs show fold change in respective proteins compared to NT group. *** p<0.001 vs. NT, ###p<0.001 vs. Ang II. N=3–4
3.5. IL-10-mediated PI3K/Akt signaling regulates Ang II-induced pathological autophagy
To confirm the role of PI3K/Akt signaling on IL-10-mediated regulation of pathological autophagy, we used phosphatidylinositol-3-kinase (PI3K) inhibitor LY294002 (LY02). LY02 compound significantly reduced both basal (Figure 4A lane 3 and Figure 4B) and IL-10-induced Akt and 4EBP1 phosphorylation (Fig. 4A and 4B). Interestingly, pharmacological inhibition of PI3 kinase resulted in robust activation of both basal and Ang II-mediated autophagy mediators (Fig. 4C and 4D). Furthermore, effect of IL-10 on Ang II-induced autophagic marker proteins expression was curbed by LY02 compound (Fig. 4C and 4D). These results suggest the important role of PI3K/Akt signaling on anti-autophagic responses of IL-10. Furthermore, to corroborate our pharmacological data, next we silenced the Akt expression in NRVM using Akt siRNA I. Akt siRNA I selectively inhibits expression of both Akt1 and Akt2, the major Akt isoforms present in cardiac cells. The efficacy of this siRNA was validated by western blot using total Akt antibody. Akt siRNA strongly inhibited both basal and Ang II-induced Akt levels and its phosphorylation (Figure 5A and 4B). Interestingly, IL-10 induced restoration of Akt phosphorylation was strongly inhibited by Akt siRNA (Figure 5A and 5B). In addition, Akt inhibition exaggerated Ang II-induced pathological autophagy (Figure 5A, 5C and 4D). Intriguingly, the inhibitory effect of IL-10 on pathological autophagy was diminished by Akt silencing. These results further validated our pharmacological inhibition data and strongly suggest that IL-10 mediated cardiac cell protection is mainly mediated by PI3K/Akt signaling pathway.
Figure 4. Akt activation is required for IL-10-mediated inhibition of autophagy in NRCM after chronic Ang II treatment.
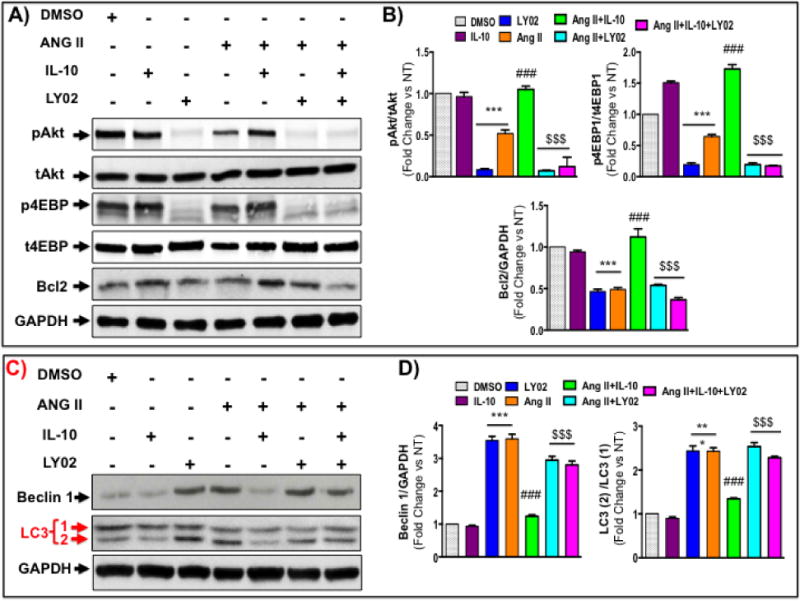
Cells were pretreated with PI3K inhibitor (LY02 compound; 30 μM) and IL-10 for 60 min followed by Ang II treatment for 48 hrs. A&B) Western blot measured pAkt, p4EBP1 and bcl2 in cells lysate after above treatments. PI3 kinase inhibition abolished IL-10 effect on Akt and 4EBP1 activation. LY02 compound also inhibited IL-10-mediated bcl2 levels. C&D) Western blot measured Akt and autophagy-associated proteins. IL-10-mediated inhibition of autophagy was abolished by LY02 compound. *** p<0.001 vs. NT, ###p<0.001 vs. Ang II, $$$p<0.001 vs. Ang II+IL-10, $$p<0.01 vs. Ang II+IL-10, $p<0.05 vs. Ang II+IL-10. N=3–4
Figure 5. Akt inhibition attenuates IL-10-mediated inhibition of autophagy in NRCM after chronic Ang II treatment.
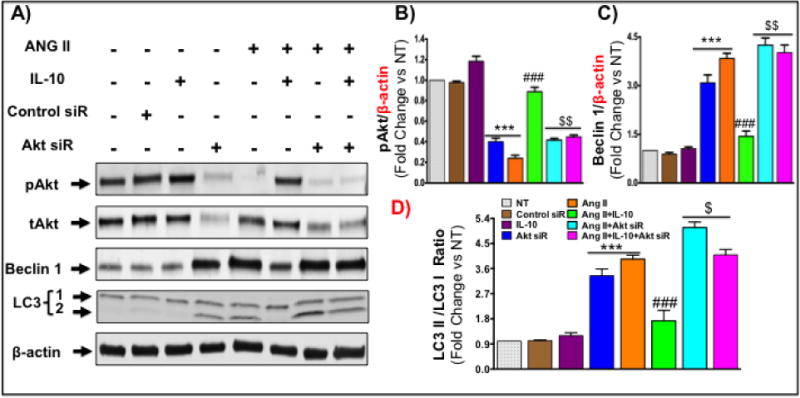
Cells were transfected with Akt 1 siRNA (100 nM; Cell signaling) using lipofectamin transfection reagent for 48 hrs. After siRNA transfection, transfected cells were pretreated with IL-10 for 60 min followed by Ang II treatment for 48 hrs. A–D) Western blot measured pAkt and p4EBP1, beclin1 and LC3 (1/2) in cells. Akt silencing abolished IL-10 effect on Akt and 4EBP1 activation. Akt siRNA I exaggerated the expression of Ang II-induced autophagic mediators and anti-autophagic effects of IL-10 was blocked by this siRNA. Bar graphs show fold change in respective proteins compared to NT group. *** p<0.001 vs. NT, ###p<0.001 vs. Ang II, $$p<0.01 vs. Ang II+IL-10. N=3–4
3.6. Down regulation of mTORC1 subsides IL-10 effects on Ang II-induced pathological autophagy
It is well established the mTORC1, a downstream target of Akt, acts as negative regulator of autophagy. To determine whether the IL-10-mediated inhibition of pathological autophagy is mediated by Akt-induced mTORC1 activation, we treated NRVM with mTORC1 specific inhibitor, rapamycin together with IL-10 and Ang II. Ang II alone or in combination with rapamycin strongly inhibit mTORC1 phosphorylation. However, IL-10 treatment significantly restored Ang II-induced mTORC1 inhibition. Interestingly, rapamycin attenuated IL-10 effect on Ang II-induced mTORC1 phosphorylation (Figure 6A–C). Furthermore, rapamycin strongly activate both basal as well as Ang II-induced autophagic markers protein levels. In agreement to our Akt silencing data, rapamycin diminished the effect of IL-10 on Ang II-induced pathological autophagy (Figure 6A, 6D and 6E). Furthermore, to explore whether Ang II caused some defect in autophagy process, we measured autophagic flux in NRVM. To measure autophagic flux, NRVM were co-transfected with adenovirus harboring tandem length mRFP-GFP-LC3 [24]. mRFP retains its fluorescence, whereas GFP loses its fluorescence in the acidic environment of lysosomes during fusion of autophagosome with lysosome revealing autophagosomes as green/yellow puncta and autolysosome as red puncta. Ang II treatment alone or in combination with rapamycin increased the red puncta indicating autophagic flux is maintained and form autolysosomes with high rate (Figure 6F panels v, vii and viii). IL-10 treatment significantly reduced the pace of autophagic flux (as indicated by less number of puncta both yellow and red) (Figure 6F panel vi). Intriguingly, IL-10-mediated inhibition of autophagic flux was restored by rapamycin (Figure 6F panel viii). To further conform that role of IL-10 on autophagic flux, we inhibited cellular lysosome using lysosome inhibitor (chloroquine which neutralizes the acidic environment of lysosome). Chloroquine treatment elevated autophagosome (more yellow puncta) in cells. Ang II treatment strongly activated autophagy as shown by red dots. Interestingly, IL-10 treatment significantly inhibited autophagosome accumulation (yellow puncta) after Ang II treatment even in the presence of chloroquine (Supplementary Figure S3 panels vi vs. iii). This result suggested that IL-10 treatment strongly inhibits autophagosome numbers and therefore chloroquine was not able to accumulate autophagosomes in presence of IL-10. Similar result was obtained in H9C2 myoblast cell lines using LC3 and lysosomal marker protein LAMP 1 immunocytochemistry (Supplementary Figure S4). These results confirm that Ang II strongly activates pathological autophagy and IL-10 inhibited it via PI3K/Akt/mTORC1 signaling.
Figure: 6. Inhibition of mTORC1 reversed IL-10 effects in Ang II-induced pathological autophagy in NRVM.
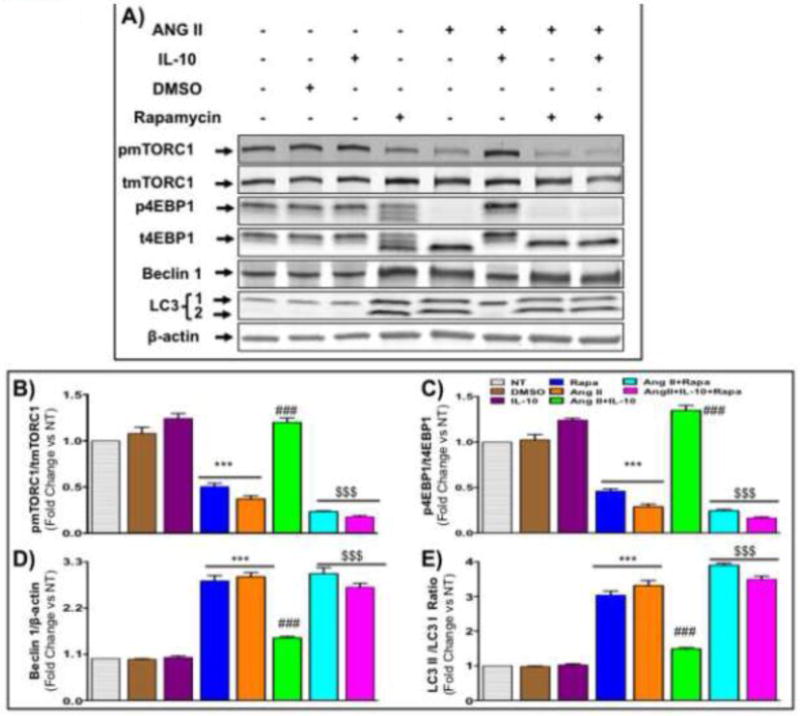
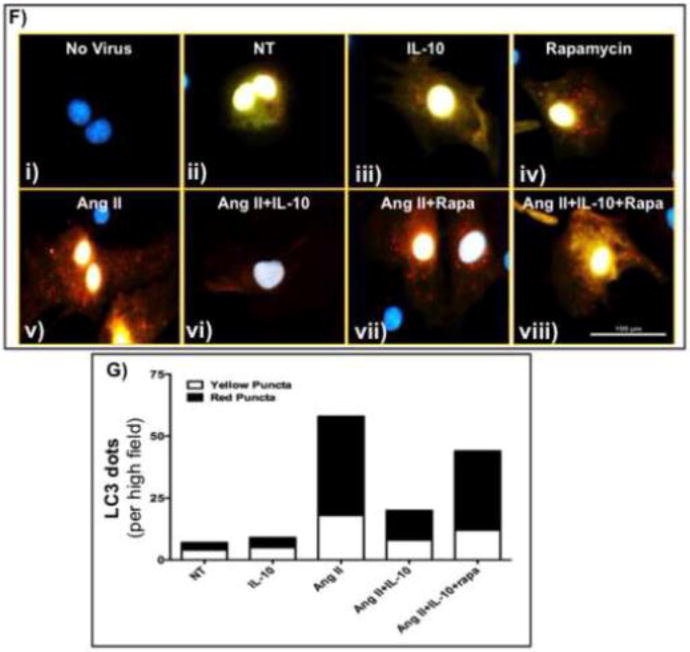
Cells were pretreated with mTORC1 inhibitor (rapamycin) and IL-10 for 60 min followed by Ang II treatment for 48 hrs in low serum medium. A&B) Western blot measured pmTORC1, beclin 1 and LC3 (1/2) in cells lysate. Rapamycin abolished IL-10 effect on mTORC1 activation. IL-10-mediated inhibition of autophagy and associated protein expression was abolished by rapamycin. C&D) Tandem length mRFP-GFP-LC3 adenovirus was infected in cardiac cells to measure autophagic flux. This adenovirus express yellow puncta during accumulation of autophagosomes but as acidic enzyme of lysosome inactivate GFP signal, a red puncta appears when autophagosome fuse with lysosome called autolysosome. Ang II treatment induced autolysosome formation. IL-10 strongly inhibited Ang II-induced both autophagosome and autolysosome. Rapamycin treatment attenuated IL-10 effects. Bar graph indicates mean number of autophagosome (empty box) and autolysosome (filled box) per cell. Scale bar, 100 um and original magnification was 400X with oil emulsion. GFP: green fluorescent protein, RFP: red fluorescent protein, Rapa: rapamycin. N=3–4
3.7. IL-10 regulates autophagy via Akt-mTORC1
Previously it was established that rapamycin treatment up-regulated autophagy in mouse [27, 28] suggesting a critical role of mTORC1 in the regulation of autophagy. Additionally, while systemic mTOR knockout mice die early after implantation, cardiac specific deletion of mTOR results in only 8% of live births with severely compromised cardiomyocyte proliferation and number and presenting a phenotype heart failure [29]. To investigate whether the Akt-mTORC1 signaling was involved in IL-10 mediated regulation of autophagy, we treated cardiomyocytes with rapamycin with/without IL-10 and without Ang II. Rapamycin alone activated autophagy. Rapamycin-induced autophagy was unaltered by IL-10. This result suggests that IL-10 is upstream regulator of autophagy and it is independent of Ang II treatment (Supplementary Figure S5).
3.8. IL-10 promotes Bcl2-beclin 1 interaction to regulate Ang II-induced pathological autophagy
Beclin1 was first discovered as a bcl2-interacting protein and this interaction is modulated during autophagy [30, 31]. Therefore, we investigated the role of IL-10 on interaction of bcl2 with beclin1 in NRCM after Ang II treatment. Beclin1 was co-immunoprecipitated using protein A-tagged bcl2 antibody. Ang II inhibited physical interaction of beclin1 with bcl2 and this interaction was reestablished by IL-10 (Figure S6). Thus in addition to direct regulation of Akt-mTORC1 signaling, IL-10 also promotes bcl2-beclin1 interaction to inhibit pathological autophagy.
3.9. IL-10 deletion exacerbates Ang II-induced Akt inhibition in IL-10 KO mice
Finally, to examine the status of Akt activation in myocardium after Ang II treatment, we measured Akt activation in whole tissue lysate 28 days after Ang II infusion. In agreement to our in vitro data, Ang II treatment negatively regulated Akt phosphorylation both in WT and IL-10 KO mice (Figure 7A and 6B). Interestingly, IL-10 deletion exaggerated Ang II-induced inhibition in Akt phosphorylation. Our results suggest that IL-10 KO mice are more susceptible for Ang II-induced pathological autophagy and restoration of this signaling axis using IL-10 would be beneficial to regulate transition of adaptive hypertrophy to heart failure. A novel signaling model of IL-10 effects is shown in figure 7C.
Figure 7. Ang II infusion abolished Akt activation in IL-10 KO mice.
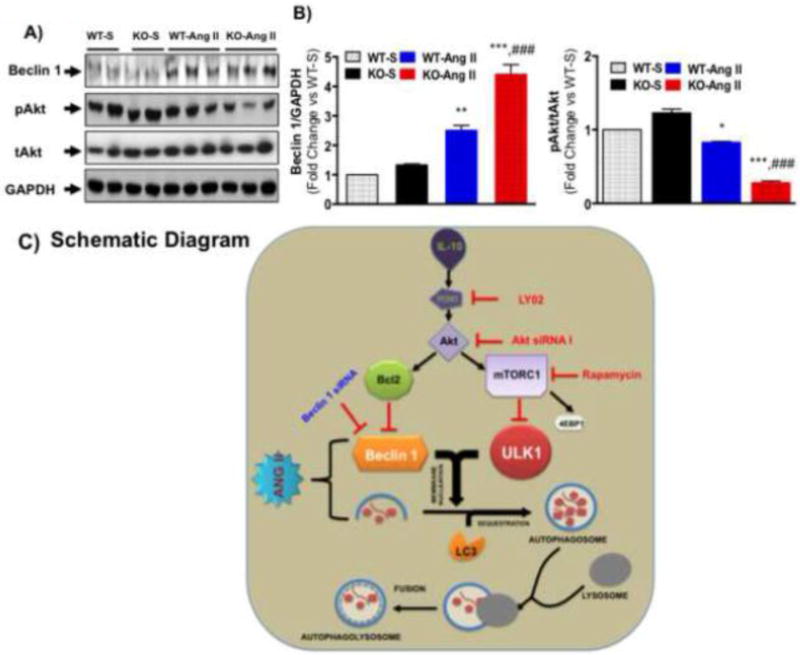
At the end of treatment schedule mice were euthanized and left ventricular tissue was harvested and used for biochemical analysis. A&B) Akt phosphorylation and Beclin 1 were measured in WT and IL-10 KO mice after Ang II treatment by western blot in whole tissue lysate. IL-10-KO mice showed enhanced expression of beclin 1 after Ang II treatment. Interestingly, drastic reduction in AKT phosphorylation was seen in IL-10 KO after Ang II treatment. (N=4–5) C) Schematic diagram: Under stimulation, beclin 1 and other autophagy-associated molecules (LC3) promote autophagosome formation. Later, autophagosomes fuse with lysosome and make autolysosomes. Our data suggest that Ang II exaggerates autophagosome formation. Ang II also disrupts the fusion of autophagosomes with lysosomes and induces pathological autophagy and cardiac remodeling. Conversely, IL-10 activates PI3K/Akt signaling to regulate mTORC1 and bcl2 and therefore, suppress pathological autophagy. Our data strongly suggest that IL-10 may inhibit pathological autophagy by regulating Akt-mTORC1 signaling components.
4. Discussion
Autophagy salvages the dysfunctional protein and organelles [32]. Defects in this process can lead to accumulation of proteins and organelles that can be toxic for cells and lead to autophagic cell death [5, 32]. Excessive autophagy in heart can cause non-selective degradation of proteins and organelles and thus functional defects in cardiac cells and ultimately heart failure [5, 32]. Therefore, tight regulation of autophagy is critical during disease condition. Here we showed that IL-10 acts to regulate the rate of autophagy and restrict the transition of physiological autophagy to the pathological or defective autophagy. The essential and clinically relevant finding of this study is that IL-10 deletion accentuates pathological autophagy and erodes cardiac function in the face of pressure overload stress. Most importantly, IL-10 treatment significantly inhibited the deleterious effects of Ang II and attenuated pathological autophagy in vitro. To the best of our knowledge, this is the first report showing direct regulation of pathological autophagy by IL-10 in diseased heart. The functional significance of IL-10 on Ang II-induced autophagy is further evident in that IL-10 KO mice exhibit an exaggerated response to angiotensin II compared to the WT mice. Mechanistically for the first time we have demonstrated that PI3K-Akt-mTORC1 signaling pathways mediate the beneficial effects of IL-10 in cardiac cell.
Autophagy has emerged as pivotal in cardiovascular pathogenesis. Although, autophagy is initially beneficial and contributes to cell survival, excessive autophagy leads to cell death and actively contributes to cardiomyocyte attrition exacerbating heart failure [3, 5]. Cardiac autophagy could be a maladaptive response to hemodynamic stress [7]. Zhu et.al. (2007) documented that beclin1 overexpression exaggerated autophagic activity and accentuated pathological remodeling. Heterozygous disruption of this gene decreased cardiomyocyte autophagy and diminished load-induced pathological remodeling. Consistent with these studies, our data suggests that long-term Ang II infusion in mice strongly activates autophagy. IL-10 deletion exaggerated Ang II-induced autophagic response in heart. Furthermore, excessive autophagic vesicles in IL-10 KO mouse heart confirmed that failing heart in IL-10 KO mice have acute autophagic activity as compared to WT control. Thus, IL-10 could be a negative regulator of pathological autophagy and cardiac remodeling.
The process that regulates autophagy is very complex and involved many signaling molecules. Among three types of PI3-kinases, class I PI3 kinase signaling molecule is known to tightly regulate the progress of autophagy [22, 33]. In general, Akt by activation of mTORC1 inhibits UNC-51-like kinase (ULK1-3), which is important for phagophore formation during initiation of autophagy [34, 35]. Additionally, PI3K/Akt signaling pathway is important for physiological growth of heart and regulates cardiac cell survival during pathological stress [36]. As transient Akt activation is beneficial for proper cardiac function, we wondered whether IL-10-mediated PI3K/Akt signaling negatively regulates chronic Ang II-induced pathological autophagy. Our data demonstrates that IL-10 treatment restores the effect of Ang II on Akt signaling. Loss-of-function experiments using PI3K inhibitor (LY2940002) or by direct silencing of Akt expression using Akt1 siRNA I show that IL-10 dependent autophagy inhibition is mediated by PI3K-Akt signaling pathway as these inhibitors mitigates IL-10 effects on pathological autophagy.
Several mechanisms may contribute in IL-10-mediated inhibition of pathological autophagy. Chronic stress-induced inhibition of Akt signaling can alter mTORC1 activities. Owing to its energy sensing functions, mTORC1 is considered the master regulator of autophagy [33]. To verify that IL-10 signaling targets mTORC1 via Akt to regulated pathological autophagy, we examined the phosphorylation of mTORC1 downstream target 4EBP1, after IL-10 treatment. IL-10 treatment strongly activated mTORC1 activity. Furthermore, direct inhibition of mTORC1 by rapamycin attenuates IL-10-mediated autophagy inhibition. Our results strongly conformed that anti-autophagic effect of IL-10 is mainly mediated by PI3 kinase dependent activation of mTORC1 via Akt in cardiomyocytes [34, 37].
Other signaling outputs of Akt, such as activation of bcl2, can also contribute to regulate autophagy independent of mTORC1 [30, 31, 38]. Bcl2 is an anti-apoptotic and anti-autophagic protein that inhibits autophagy through direct interaction with the BH3 domain of pro-autophagic protein beclin1 [30, 31]. The stability of the bcl2/beclin1complex is regulated by posttranslational modification of bcl2 and beclin1. Disruption of bcl2-beclin 1 interaction is crucial for stimulation of autophagy in mammalian cells [31]. Previously we have shown that exogenous treatment of IL-10 strongly increased bcl2 levels in mouse left ventricular tissue after TAC [20]. IL-10 mediated activation of bcl2 could be another potential mechanism to regulate pathological autophagy. Our data in this study also confirmed that Ang II negatively regulates bcl2 levels and IL-10 restored it. The bcl2-mediated regulation of autophagy by IL-10 may be important to maintain autophagy at levels compatible with cell survival, rather than cell death. Here it is important to note that the regulation of IL-10 on bcl2 is independent of JNK1/2, as IL-10 did not affect JNK activity.
5. Conclusion
In conclusion, this is the first study describing the novel role of IL-10 on pathological autophagy. A model depicting the possible mechanism of IL-10-mediated regulation of Ang II-induced autophagy is shown in Figure 6C. In sum, our studies suggest that IL-10 regulates pathological autophagy by activating PI3K/Akt/mTORC1 signaling pathway. As excessive or defective autophagy contributes to development of pathological remodeling and heart failure, selective agents that manipulate signaling pathways to regulate autophagy during pathological stress (such as IL-10) may represent potential therapeutic molecules to treat or prevent development of heart disease.
Supplementary Material
Highlights.
IL-10 deletion exaggerates pathological myocardial autophagy in response to Ang II.
IL-10 treatment attenuates Ang II-induced pathological autophagy.
PI3K, Akt or mTORC1 inhibition mitigates IL-10 effect on Ang II-induced autophagy.
Lysosome inhibition does not alter autophagic flux in IL-10 treated cells.
Acknowledgments
Work described in this manuscript was in part supported by National Institute of Health grants HL091983, HL105597, HL126186, HL053354, HL108795 and HL108806 and American Heart Association-Scientist Development Grant 14SDG20480104.
Non-standard Abbreviation and Acronyms
- NRCM
Neonatal rat ventricular cardiomyocytes
- Class I PI3K
Class I phosphoinositide 3-Kinase
- 4EBP1
4E binding protein 1
- CHOP
C/EBP homologous protein
- EF
Ejection fraction
- FS
Fractional shortening
- LY02
LY2940002
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material.
Supplemental Information includes SIX figures (S1–S6) and can be found with this article as online-only supplement.
Disclosures
None
References
- 1.Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annual review of nutrition. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 2.Lee Y, Lee HY, Gustafsson AB. Regulation of autophagy by metabolic and stress signaling pathways in the heart. Journal of cardiovascular pharmacology. 2012;60:118–24. doi: 10.1097/FJC.0b013e318256cdd0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orogo AM, Gustafsson AB. Therapeutic targeting of autophagy: potential and concerns in treating cardiovascular disease. Circulation research. 2015;116:489–503. doi: 10.1161/CIRCRESAHA.116.303791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothermel BA, Hill JA. Myocyte autophagy in heart disease: friend or foe? Autophagy. 2007;3:632–4. doi: 10.4161/auto.4913. [DOI] [PubMed] [Google Scholar]
- 6.Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, et al. Autophagy in chronically ischemic myocardium. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13807–12. doi: 10.1073/pnas.0506843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. The Journal of clinical investigation. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shimomura H, Terasaki F, Hayashi T, Kitaura Y, Isomura T, Suma H. Autophagic degeneration as a possible mechanism of myocardial cell death in dilated cardiomyopathy. Japanese circulation journal. 2001;65:965–8. doi: 10.1253/jcj.65.965. [DOI] [PubMed] [Google Scholar]
- 9.Porrello ER, D’Amore A, Curl CL, Allen AM, Harrap SB, Thomas WG, et al. Angiotensin II type 2 receptor antagonizes angiotensin II type 1 receptor-mediated cardiomyocyte autophagy. Hypertension. 2009;53:1032–40. doi: 10.1161/HYPERTENSIONAHA.108.128488. [DOI] [PubMed] [Google Scholar]
- 10.Gatica D, Chiong M, Lavandero S, Klionsky DJ. Molecular mechanisms of autophagy in the cardiovascular system. Circulation research. 2015;116:456–67. doi: 10.1161/CIRCRESAHA.114.303788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 12.Mei Y, Zhang Y, Yamamoto K, Xie W, Mak TW, You H. FOXO3a-dependent regulation of Pink1 (Park6) mediates survival signaling in response to cytokine deprivation. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5153–8. doi: 10.1073/pnas.0901104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–88. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. The Journal of clinical investigation. 2005;115:2618–24. doi: 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pahan K, Khan M, Singh I. Interleukin-10 and interleukin-13 inhibit proinflammatory cytokine-induced ceramide production through the activation of phosphatidylinositol 3-kinase. Journal of neurochemistry. 2000;75:576–82. doi: 10.1046/j.1471-4159.2000.0750576.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhou JH, Broussard SR, Strle K, Freund GG, Johnson RW, Dantzer R, et al. IL-10 inhibits apoptosis of promyeloid cells by activating insulin receptor substrate-2 and phosphatidylinositol 3′-kinase. Journal of immunology. 2001;167:4436–42. doi: 10.4049/jimmunol.167.8.4436. [DOI] [PubMed] [Google Scholar]
- 18.Makuta Y, Sonoda Y, Yamamoto D, Funakoshi-Tago M, Aizu-Yokota E, Takebe Y, et al. Interleukin-10-induced CCR5 expression in macrophage like HL-60 cells: involvement of Erk1/2 and STAT-3. Biological & pharmaceutical bulletin. 2003;26:1076–81. doi: 10.1248/bpb.26.1076. [DOI] [PubMed] [Google Scholar]
- 19.Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, et al. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:2484–94. doi: 10.1096/fj.09-149815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation. 2012;126:418–29. doi: 10.1161/CIRCULATIONAHA.112.112185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnamurthy P, Thal M, Verma S, Hoxha E, Lambers E, Ramirez V, et al. Interleukin-10 deficiency impairs bone marrow-derived endothelial progenitor cell survival and function in ischemic myocardium. Circulation research. 2011;109:1280–9. doi: 10.1161/CIRCRESAHA.111.248369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hua Y, Zhang Y, Ceylan-Isik AF, Wold LE, Nunn JM, Ren J. Chronic Akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic research in cardiology. 2011;106:1173–91. doi: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 23.Verma SK, Lal H, Golden HB, Gerilechaogetu F, Smith M, Guleria RS, et al. Rac1 and RhoA differentially regulate angiotensinogen gene expression in stretched cardiac fibroblasts. Cardiovascular research. 2011;90:88–96. doi: 10.1093/cvr/cvq385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circulation research. 2015;116:264–78. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 25.Garikipati VN, Krishnamurthy P, Verma SK, Khan M, Abramova T, Mackie AR, et al. Negative Regulation of miR-375 by Interleukin-10 Enhances Bone Marrow-Derived Progenitor Cell-Mediated Myocardial Repair and Function After Myocardial Infarction. Stem Cells. 2015 doi: 10.1002/stem.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lal H, Verma SK, Golden HB, Foster DM, Smith M, Dostal DE. Stretch-induced regulation of angiotensinogen gene expression in cardiac myocytes and fibroblasts: opposing roles of JNK1/2 and p38alpha MAP kinases. J Mol Cell Cardiol. 2008;45:770–8. doi: 10.1016/j.yjmcc.2008.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu L, Feng Z, Cui S, Hou K, Tang L, Zhou J, et al. Rapamycin upregulates autophagy by inhibiting the mTOR-ULK1 pathway, resulting in reduced podocyte injury. PLoS One. 2013;8:e63799. doi: 10.1371/journal.pone.0063799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim YC, Guan KL. mTOR: a pharmacologic target for autophagy regulation. The Journal of clinical investigation. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circulation research. 2014;114:549–64. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Decuypere JP, Parys JB, Bultynck G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells. 2012;1:284–312. doi: 10.3390/cells1030284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–5. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao DJ, Gillette TG, Hill JA. Cardiomyocyte autophagy: remodeling, repairing, and reconstructing the heart. Current hypertension reports. 2009;11:406–11. doi: 10.1007/s11906-009-0070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. The Journal of cell biology. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Molecular and cellular biology. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nature reviews Molecular cell biology. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 36.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovacic S, Soltys CL, Barr AJ, Shiojima I, Walsh K, Dyck JR. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. The Journal of biological chemistry. 2003;278:39422–7. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 38.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.