

Abstract
Drug addiction is driven, in part, by powerful and enduring memories of sensory cues associated with drug intake. As such, relapse to drug use during abstinence is frequently triggered by an encounter with drug-associated cues, including the drug itself. L-type Ca2+ channels (LTCCs) are known to regulate different forms of synaptic plasticity, the major neural substrate for learning and memory, in various brain areas. Long-term potentiation (LTP) of NMDA receptor (NMDAR)-mediated glutamatergic transmission in the ventral tegmental area (VTA) may contribute to the increased motivational valence of drug-associated cues triggering relapse. In this study, using rat brain slices, we found that isradipine, a general LTCC antagonist used as antihypertensive medication, not only blocks the induction of NMDAR LTP but also promotes the reversal of previously induced LTP in the VTA. In behaving rats, isradipine injected into the VTA suppressed the acquisition of cocaine-paired contextual cue memory assessed using a conditioned place preference (CPP) paradigm. Furthermore, administration of isradipine or a CaV1.3 subtype-selective LTCC antagonist (systemic or intra-VTA) before a single extinction or reinstatement session, while having no immediate effect at the time of administration, abolished previously acquired cocaine and alcohol (ethanol) CPP on subsequent days. Notably, CPP thus extinguished cannot be reinstated by drug re-exposure, even after 2 weeks of withdrawal. These results suggest that LTCC blockade during exposure to drug-associated cues may cause unlearning of the increased valence of those cues, presumably via reversal of glutamatergic synaptic plasticity in the VTA.
Introduction
Addiction is a chronic, relapsing disorder driven in part by strong associations formed between drugs and sensory cues experienced during drug intake, such as places, people, and interoceptive drug cues, i.e., subjective effects caused by drugs themselves1-3. Addictive drugs are thought to hijack synaptic plasticity mechanisms in key brain circuits involved in reward learning, especially the mesolimbic dopaminergic system comprising the ventral tegmental area (VTA) and its projections to the nucleus accumbens and other limbic structures4-6. As such, powerful and enduring memories of drug-related cues are formed, overshadowing other cues associated with non-drug rewards and driving continued drug use as well as relapse after a period of abstinence. Therefore, reducing the strength of drug cue memories by manipulating the underlying synaptic plasticity mechanisms has received particular attention.
During cue-reward conditioning, dopamine neuron burst responses [2–10 action potentials (APs) at 10–50 Hz] shift in time from the reward to the cue. As a consequence, the reward-associated cue acquires positive valence and triggers approach behavior7. Glutamatergic inputs activating NMDA receptors (NMDARs) play a critical role in driving burst firing8-11, while the role AMPA receptors (AMPARs) in burst generation remains controversial12, 13. In addition to different forms of synaptic plasticity of AMPARs in dopamine neurons5, 6, NMDAR-mediated transmission also undergoes long-term potentiation (LTP) following repeated pairing of glutamatergic input stimulation with postsynaptic burst firing14, an activity pattern that may be experienced during cue-reward pairing15. Hence, this form of glutamatergic synaptic plasticity may contribute, at least partially, to the acquisition of cue-induced burst responses. Induction of LTP requires AP-evoked Ca2+ signals amplified by preceding activation of metabotropic glutamate receptors (mGluRs, more specifically mGluR1), in addition to the activation of NMDARs themselves, presumably at the glutamatergic inputs to be potentiated16. In contrast, previously induced LTP can be reversed when potentiated inputs are repeatedly stimulated in the absence of postsynaptic APs, raising the possibility that cue memory, or learned valence of the cue, could be unlearned under certain conditions.
Voltage-gated Ca2+ channels are the major source of activity-dependent Ca2+ influx. Dihydropyridine-sensitive L-type Ca2+ channels (LTCCs) are a well-established target for antihypertensive medication because of their involvement in excitation-contraction coupling in the cardiovascular system17. LTCCs are also widely expressed in the CNS and regulate diverse neuronal processes, such as gene expression, cell survival, and synaptic plasticity18. Dopamine neurons in both the VTA and substantia nigra express LTCCs19, 20. In the substantia nigra, these channels, particularly the low-threshold CaV1.3 subtype, have been implicated in driving tonic pacemaker firing and, more recently, in neuronal death associated with Parkinson's disease20-22; however, the pathophysiological roles of LTCCs in the VTA remain unclear. A number of studies have reported that systemic administration of LTCC antagonists blocks the acquisition of drug-induced conditioned place preference (CPP)23-26, a form of Pavlovian contextual cue learning dependent on NMDAR-mediated transmission in the VTA9, 27-29 (but also see30). Our previous study has shown that acquisition of psychostimulant CPP is inhibited by mGluR1 or NMDAR antagonist in the VTA, while CPP expression is attenuated by NMDAR antagonist, but not by mGluR1 antagonist, in the VTA31, supporting the potential contribution of NMDAR LTP in driving CPP. Here, mGluR1/NMDAR blockade would suppress CPP acquisition via inhibiting LTP induction at glutamatergic inputs activated by contextual cues of the CPP box, while blocking potentiated NMDAR-mediated excitation at those inputs would interfere with CPP expression. In this study, we examined how LTCC blockade in the VTA affects NMDAR LTP in ex vivo brain slices and drug (cocaine/ethanol)-induced CPP in behaving rats.
Materials and Methods
Animals
Male Sprague-Dawley rats (3–10 weeks old; Harlan Laboratories) were housed in groups of three and maintained on a 12h light/dark cycle with food and water available ad libitum. All animal procedures were approved by the Universtiy of Texas Institutional Animal Care and Use Committee.
Electrophysiology
Horizontal midbrain slices (∼200 μm) were prepared from rats (3–7 weeks old) and recordings were made at 33–35°C in physiological saline, as in our previous studies14, 16, 31. Recordings were performed in the lateral VTA located 50–150 μm from the medial border of the medial terminal nucleus of the accessory optic tract. Internal solution contained (in mM): 115 K-methylsulfate, 20 KCl, 1.5 MgCl2, 10 HEPES, 0.025 EGTA, 2 Mg-ATP, 0.2 Na2-GTP, and 10 Na2-phosphocreatine (pH ∼7.25, ∼285 mOsm/kg). Putative dopamine neurons were identified by spontaneous firing (1–5 Hz) with broad APs (>1.2 ms) in cell-attached configuration and large Ih currents (>200 pA; evoked by a 1.5 s hyperpolarizing step of 50 mV) in whole-cell configuration. Voltage-clamp recordings were made at a holding potential of –62 mV, corrected for a liquid junction potential of –7 mV. Recordings were discarded if the series resistance increased above 16 MΩ or the input resistance dropped below 200 MΩ.
A 2 ms depolarizing pulse of 55 mV was used to elicit an unclamped AP. Time integral of the outward tail current, termed IK(Ca), was calculated between 20 ms and 400–600 ms after the depolarizing pulse (expressed in pC). IK(Ca) thus measured is eliminated by TTX and by apamin, a selective antagonist of Ca2+-activated SK channels, and thus can be used as a readout of AP-induced Ca2+ transients16.
Loose-patch recordings (∼20 MΩ seal) were made using pipettes filled with 150 mM NaCl to monitor dopamine neuron firing. Aspartate iontopheresis (1 M L-aspartate in ∼100 MΩ pipette placed at ∼10–50 μm from the soma/proximal dendrites) was used to evoke NMDAR-dependent bursts10, 16. Amplitude (∼100–200 nA) and duration (∼50–150 ms) of the iontophoretic current was adjusted to produce a burst of 5-10 spikes with a minimum instantaneous frequency of 15 Hz.
NMDAR LTP experiments
Synaptic stimuli were applied every 30 s using a bipolar tungsten electrode (∼300 μm tip separation) placed rostral to the recorded neuron. To isolate NMDAR EPSCs, recordings were performed in the presence of DNQX (10 μM), picrotoxin (100 μM), CGP55845 (50 nM), and eticlopride (100 nM) to block AMPA/kainate, GABAA, GABAB, and D2 dopamine receptors, and in glycine (20 μM) and low Mg2+ (0.1 mM) to enhance NMDAR activation. Stimulation intensity was adjusted after break-in (within ∼1 min) to obtain ∼100 pA EPSCs. Cells with baseline EPSC amplitude (averaged from 10 traces during 5 min window before LTP induction) outside the 90–110 pA range were excluded.
Following 10 min baseline EPSC recording, the effect of sustained synaptic stimulation (33 stimuli at 33 Hz) on IK(Ca) was assessed immediately before LTP induction. Here, IK(Ca) was evoked by a single AP alone and an AP with preceding synaptic stimulation (140 ms interval between the offset of synaptic stimulation and AP; each repeated twice). LTP was induced by pairing sustained synaptic stimulation (50 stimuli at 33 Hz) with a burst (5 APs at 20 Hz), where the burst onset was delayed by 1 s from the onset of synaptic stimulation. This synaptic stimulation-burst pairing was repeated 10 times every 20 s. In LTP reversal experiments, sustained synaptic stimulation alone or synaptic stimulation paired with a single AP (delayed by 1 s from the synaptic stimulation onset) was delivered repeatedly (10 or 30 times) 30 min after LTP induction. Magnitude of LTP and its reversal was determined by averaged EPSC amplitude from a 5 min window (10 traces) immediately before LTP induction and that from 5 min windows at 25–30 min after LTP induction/reversal. For AP5 experiments (Figure 3c), a 5 min window before AP5 perfusion (i.e., 20–25 min after LTP induction) was used.
Figure 3.
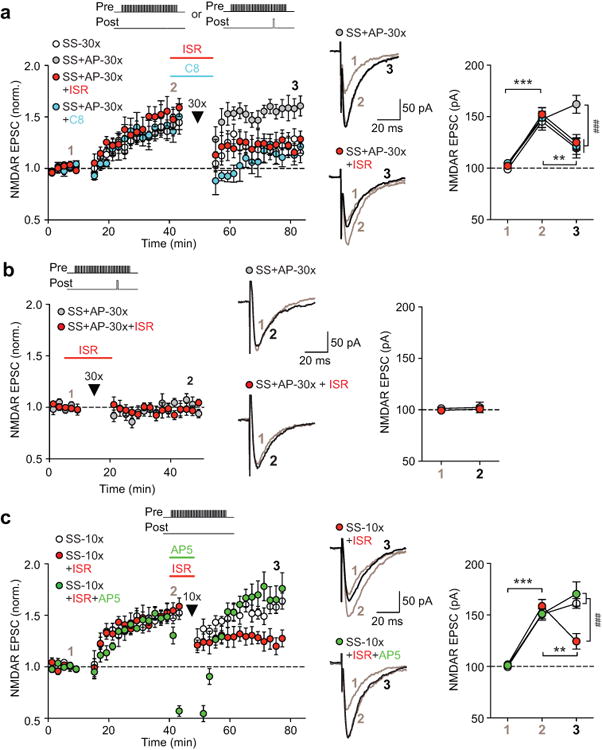
Isradipine and compound 8 promote reversal of previously induced NMDAR LTP. (a) After LTP induction and its development, LTP reversal protocol consisting of 1) sustained synaptic stimulation alone or 2) synaptic stimulation paired with a single AP was repeatedly delivered (30 times; arrowhead). In the third and fourth groups, the latter protocol (“SS+AP-30×”) was delivered in the presence of isradipine (ISR) or compound 8 (C8). (b) The “SS+AP-30×” protocol was delivered in control solution or in isradipine without prior LTP induction. (c) LTP reversal protocol consisting of synaptic stimulation alone was repeatedly delivered (10 times) in control solution, in isradipine, or in isradipine and AP5 (5 μM; produced 83 ± 4% peak inhibition of potentiated EPSCs, n = 6 cells). Summary time graphs of these experiments are shown on the left, while graphs depicting average EPSC amplitude during baseline, after LTP [except for (b)], and following delivery of LTP reversal protocol are shown on the right [(a) F6,40 = 4.85, p < 0.001, n = 5–7 cells/group; (b) F1,10 = 0.01, p = 0.94, n = 6 cells/group; (c) F4,32 = 10.49, p < 0.001, n = 6–7 cells/group; mixed two-way ANOVA). Example traces for the experiments indicated are shown in the middle. Synaptic stimulation intensity was adjusted to evoke ∼100 pA baseline EPSCs in each cell; thus the degree of synaptic facilitation of IK(Ca) was similar in different groups (Supplementary Figures S8d-f). **p < 0.01, ***p < 0.001 between two LTP stages; ###p < 0.001 between groups (Bonferroni post hoc test).
Place conditioning
A CPP box (Med Associates) consisting of two distinct compartments separated by a small middle chamber was used for conditioning. Rats (4–10 weeks old) were first subjected to a pretest, in which they explored the entire CPP box for 15 min. The percentage of time spent in each compartment was determined after excluding the time spent in the middle chamber. Rats with initial side preference >60% were excluded. During the next 6 days, rats were given saline injection (1 ml/kg) and confined to one compartment (days 1, 3, 5) or received cocaine injection (10 mg/kg, i.p.) and confined to the other compartment (days 2, 4, and 6; 15 min each). For ethanol CPP, rats were given saline (4.2 ml/kg) or ethanol (0.5 g/kg, 15% v/v, i.p.) injection and confined to one compartment for 7 min. Compartment assignment was counterbalanced such that animals had, on average, ∼50% initial preference for the drug-paired side in the pretest. A 15 min posttest was performed 1 day after the last conditioning session. In extinction experiments, animals underwent repeated posttests (once daily). For reinstatement, rats received priming injection of cocaine (10 mg/kg) or ethanol (0.5 g/kg) prior to the posttest. In some CPP experiments, rats received bilateral intra-VTA infusion (0.3 μl/side, 0.15 μl/min) of 1) isradipine (0.6 pmol) or vehicle [0.01% ethanol (=1.7 mM)], 2) compound 8 (6 pmol) or vehicle (0.02% DMSO), or 3) AP5 (6 nmol) or vehicle (PBS). Intra-VTA microinjection procedure is detailed in Supplementary Materials and Methods. Data from rats with injection sites outside the VTA were excluded from the analysis.
Data Analysis
No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in previous publications14, 16, 31-33. Group assignment was mostly done in a random fashion, except for certain CPP experiments (Figures 4 and 5; Supplementary Figures S11 and S12), where rats were assigned to treatment groups in a counterbalanced manner based on the first posttest data. Data acquisition and analysis was not blinded. Data are expressed as mean SEM with the sample size in each group indicated. Data distribution was assumed to be normal, but this was not formally tested. Statistical significance was determined by two-tailed t test or ANOVA followed by Bonferroni post hoc test using GraphPad Prism (significant at p < 0.05; details provided in figure legends).
Figure 4.
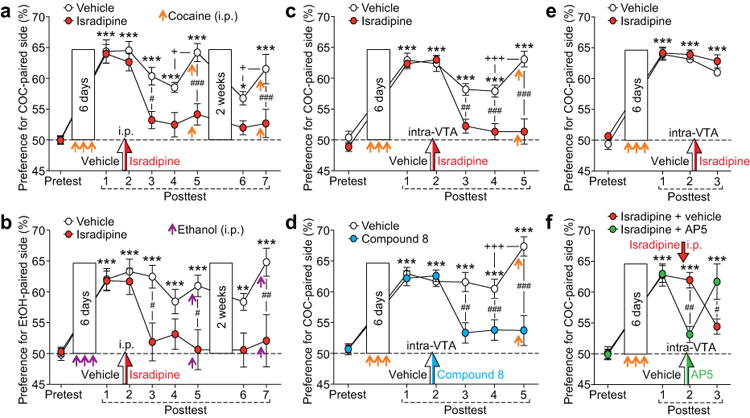
Isradipine and compound 8 promote extinction of cocaine/ethanol CPP and prevent future reinstatement. (a and b) Summary graphs depicting the effects of systemic isradipine administration (i.p.) on the expression and extinction of CPP previously induced with cocaine (a) or ethanol (b) (three conditioning sessions for both). A single injection of isradipine (1.2 mg/kg) or vehicle [1 ml/kg of 16% ethanol (0.13 g/kg)] was made prior to the second posttest, while cocaine/ethanol injections were made immediately before the fifth and seventh posttests to trigger reinstatement (2-week interval between fifth and sixth posttests) [(a) F7,126 = 3.40, p < 0.001, n = 10 rats/group; (b) F7,91 = 3.21, p < 0.01, n = 7–8 rats/group; mixed two-way ANOVA). (c and d) Summary graphs showing the effects of intra-VTA injection of isradipine (c) or compound 8 (d) made before the second posttest following cocaine CPP acquisition. Cocaine-induced reinstatement was tested on the fifth posttest [(c) F5,80 = 13.82, p < 0.001, n = 9 rats/group; (d) F5,70 = 9.62, p < 0.001, n = 7–9 rats/group; mixed two-way ANOVA). (e) Intra-VTA isradipine injection had no effect when done immediately after the second posttest (F3,42 = 0.45, p = 0.72, n = 7–9 rats/group; mixed two-way ANOVA). (f) Systemic isradipine injection (i.p.) followed by intra-VTA injection of AP5 or vehicle was made before the second posttest (F3,30 = 9.71, p < 0.001, n = 6 rats/group; mixed two-way ANOVA). *p < 0.05, **p < 0.01, ***p < 0.001 vs. pretest; +p < 0.05, +++p < 0.001 between two successive posttests; #p < 0.05, ##p < 0.01, ###p < 0.001 between groups (Bonferroni post hoc test).
Figure 5.
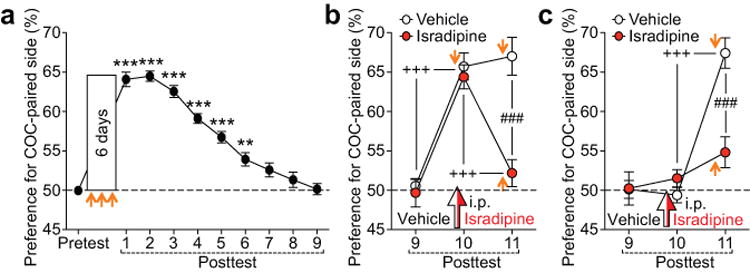
Isradipine prevents future reinstatement when administered before the posttest following complete extinction. (a) Average time course of cocaine CPP extinction during repeated posttests over 9 days (F9,288 = 77.30, p < 0.001, n = 33 rats; repeated measures one-way ANOVA). (b and c) A single systemic injection of isradipine or vehicle was made before the tenth posttest performed with (b) or without (c) cocaine injection (orange arrow), while the eleventh posttest was always done with cocaine injection [(b) F2,32 = 16.54, p < 0.001, n = 9 rats/group; (c) F2,26 = 30.33, p < 0.001, n = 7–8 rats/group; mixed two-way ANOVA]. **p < 0.01, ***p < 0.001 vs. pretest; +++p < 0.001 between two successive posttests; ###p < 0.001 between groups (Bonferroni post hoc test).
Results
Isradipine inhibits induction of NMDAR LTP
In order to gain insight into the LTCC-dependent mechanisms in the VTA, we performed electrophysiological recordings in ex vivo VTA slices to examine the effects of isradipine, a dihydropyridine LTCC antagonist used as antihypertensive medication in humans. Isradipine was first tested on NMDAR-dependent dopamine neuron excitation/bursting, which likely plays an important role in the acquisition of CPP, as well as its expression9, 27-29, 31. Bath application of isradipine (2 μM) had no effect on NMDAR-mediated EPSCs elicited by local synaptic stimulation (Figure 1a) or on NMDAR-dependent burst firing evoked by aspartate iontophoresis10 (Figure 1b). Furthermore, isradipine had no effect on tonic firing (Figure 1b), consistent with an LTCC-independent mechanism of pacemaker activity of dopamine neurons in the VTA20, 21, 34.
Figure 1.
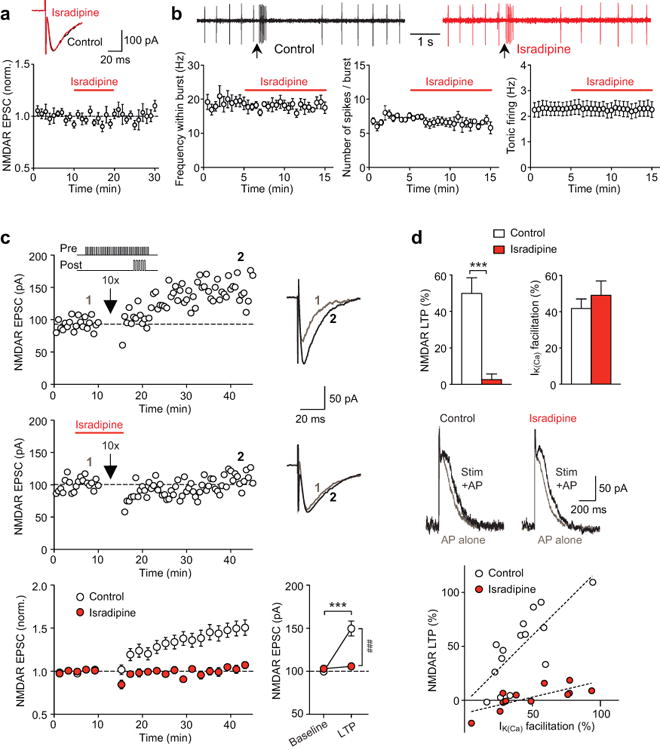
Isradipine blocks NMDAR LTP induction in the VTA. (a) Isradipine (2 μM) had no effect on NMDAR EPSCs (n = 6 cells; example EPSC traces before and after isradipine application). (b) Example traces (top; aspartate iontophoresis at arrows) and summary time graphs (bottom) illustrating that isradipine had no effect on the frequency/number of spikes within the burst (n = 5 cells) or tonic firing (n = 8 cells). (c) Example experiments (EPSC traces at the times indicated) and summary time graph showing that isradipine blocked the induction of NMDAR LTP. Graph at the bottom right depicts average EPSC amplitude during baseline and after LTP (F1,25 = 21.89, p < 0.001, n = 12–15 cells/group; mixed two-way ANOVA). ***p < 0.001 vs. baseline; ###p < 0.001 between groups (Bonferroni post hoc test). (d) Isradipine blocked LTP induction without affecting synaptic facilitation of IK(Ca). Data are from the same cells shown in (c) (LTP: t25 = 4.71, p < 0.001; IK(Ca) facilitation: t25 = 0.79, p = 0.43; unpaired t test). Example traces depict IK(Ca) evoked by a single AP alone and with preceding synaptic stimulation. Bottom graph illustrates the relationship between LTP magnitude and the degree of IK(Ca) facilitation obtained from each cell (dashed lines: linear fit to all data points in each group).
Next we asked if isradipine interferes with the mGluR1-dependent induction of NMDAR LTP. LTP was induced using a synaptic stimulation-burst pairing protocol (see Materials and Methods). Here, sustained glutamatergic input stimulation leads to mGluR1-dependent production of the cytosolic messenger inositol 1,4,5-trisphosphate (IP3), which amplifies AP-evoked Ca2+ signals via Ca2+-induced Ca2+ release from intracellular stores14. Baseline NMDAR EPSC amplitude, recorded in low Mg2+ (0.1 mM), was set at ∼100 pA to control for synaptic stimulation intensity and thus for the degree of synaptic activation of the mGluR-IP3 pathway. Under these conditions, repeated synaptic stimulation-burst pairing (10 times) produced LTP of NMDAR EPSCs that gradually developed over ∼30 min (Figure 1c). As in previous studies14, 32, 33, LTP magnitude was positively correlated with the degree of synaptic mGluR-induced facilitation of AP-evoked Ca2+ signals [assessed immediately before LTP induction using the size of Ca2+-activated K+ (SK) currents, termed IK(Ca)] (Figure 1d). In contrast, LTP was virtually abolished when isradipine was applied 5 min before and during the delivery of LTP induction protocol, although synaptic facilitation of IK(Ca) at the time of induction was comparable to that observed in control solution. In separate experiments, we confirmed that isradipine, which did suppress Ca2+ currents evoked by small depolarizations (10–15 mV) from –62 mV (Supplementary Figure S1), had no effect on the size of burst-evoked IK(Ca) or the magnitude of IK(Ca) facilitation produced by photolytic application of IP3 (Supplementary Figure S2). Therefore, LTCC inhibition with isradipine suppresses LTP induction without affecting burst-evoked Ca2+ signals or the mGluR/IP3-dependent amplification machinery [e.g., the size of IP3-sensitive Ca2+ stores (Supplementary Figure S3)].
We further examined if enhancing LTCC activation with the LTCC agonist S(-)-Bay K 8644 promotes NMDAR LTP induction. The magnitude of LTP induced in the presence of Bay K 8644 (1 μM) was comparable to that in control (Supplementary Figure S4). Furthermore, inhibiting NMDARs with AP5 blocked LTP induced in Bay K 8644, as has been reported for LTP in control solution14. Hence, increasing LTCC activation appears to have no significant effect on NMDAR LTP induction, in contrast to the major suppression of LTP observed with LTCC inhibition.
LTCC blockade in the VTA inhibits acquisition of cocaine CPP
Systemic injection (i.p.) of isradipine has been shown to suppress the acquisition of psychostimulant (cocaine and amphetamine) CPP23, 24. Blockade of NMDAR LTP induction in the VTA might contribute to CPP suppression. Thus we sought to determine if isradipine affects acquisition of cocaine CPP via its effect in the VTA. We found that bilateral intra-VTA injection of isradipine [0.6 pmol/0.3 μl (= 2 μM) in each side] 5 min before each of the three cocaine conditioning sessions completely blocked CPP acquisition, as was observed with systemic isradipine injection (1.2 mg/kg, i.p.) made 10 min prior to each conditioning session (Figure 2a). Isradipine (both systemic and intra-VTA) had no significant effect on the overall activity during the conditioning sessions (Supplementary Figure S5). Furthermore, CPP was partially suppressed when intra-VTA isradipine injection was made immediately after each cocaine conditioning session (Figure 2b) [Note that the burst-inducing effect of cocaine (10 mg/kg, i.p.) persists >45 min35 and thus should be still robust after the 15 min conditioning session]. Intra-VTA injection of isradipine by itself did not affect side preference (Supplementary Figure S6). These results are consistent with the involvement of LTCC-dependent plasticity processes in the VTA in the acquisition of appetitive cocaine cue memory and, likely, its consolidation.
Figure 2.
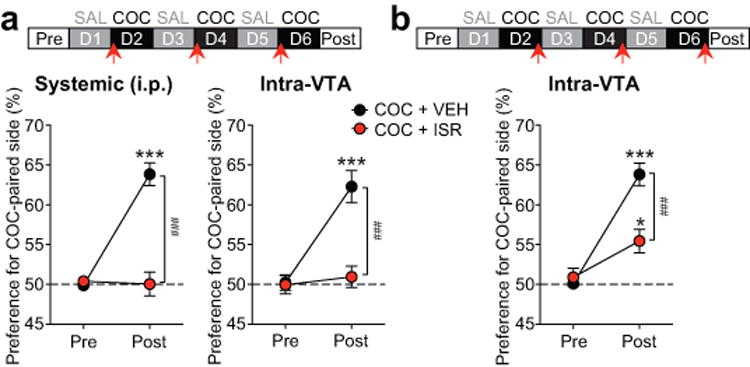
Isradipine in the VTA blocks cocaine CPP acquisition. (a and b) Timeline of experiments [top; systemic (i.p.) or intra-VTA injection of isradipine (ISR)/vehicle (VEH) made at arrows] and summary graphs depicting changes in the preference for the cocaine (COC)-paired compartment after three conditioning sessions [(a) systemic: F1,30 = 41.08, p < 0.001, n = 16 rats/group; intra-VTA: F1,12 = 26.71, p < 0.001, n = 6–8 rats/group; (b) F1,12 = 17.92, p < 0.01, n = 7 rats/group; mixed two-way ANOVA). *p < 0.05, ***p < 0.001 vs. pretest; ###p < 0.001 between groups (Bonferroni post hoc test).
Next we examined if cocaine conditioning alters NMDAR-mediated excitation in the VTA. In these experiments, we used rats that had undergone cocaine conditioning with systemic injection of isradipine or vehicle (Figure 2a, left panel; recordings made one day after the posttest), and the data were compared to those from control rats with no cocaine conditioning experience. There was no change in overall NMDAR-dependent excitation assessed with bath application of NMDA (10 μM; Supplementary Figure S7a), as has been reported36, 37. A recent study in mice has shown that in vivo cocaine experience induces synaptic insertion of GluN3A-containing NMDARs, which display reduced Mg2+ blockade at hyperpolarized potentials38. However, NMDAR EPSCs measured in normal Mg2+ (1.2 mM) displayed similar voltage dependence in the three groups of rats (Supplementary Figure S7b). Finally, comparable NMDAR LTP magnitude was observed in these groups (Supplementary Figure S7c), as opposed to dramatic alterations in synaptic plasticity of AMPARs produced by cocaine experience36-41. Altogether, these data suggest that cocaine conditioning, with or without isradipine, caused no significant changes in global NMDAR-mediated excitation in the VTA (see Discussion for further details on this issue).
Isradipine promotes reversal of NMDAR LTP
Previously induced NMDAR LTP can be reversed by repeated stimulation of the potentiated inputs without postsynaptic firing14, which may resemble the activity pattern during extinction training where the cue is repeatedly presented without the reward/drug7. Indeed, 30 min after inducing LTP, repeated delivery of the synaptic stimulation train (30 times) (SS-30×; Figure 3a and Supplementary Figure S8a) caused persistent depression of potentiated EPSC amplitude toward the baseline level. In contrast, only transient depression was observed and EPSC amplitude returned to the LTP level in ∼5–10 min when 1) a single AP was paired with the synaptic stimulation train (SS+AP-30×; Figure 3a and Supplementary Figure S8a) or 2) the number of repetition of the synaptic stimulation train was reduced to 10 times (SS-10×; Figure 3c and Supplementary Figure S8c). We found that delivery of these protocols in the presence of isradipine invariably caused persistent depression of potentiated EPSCs. Notably, delivery of the “SS+AP-30×” protocol in isradipine failed to significantly depress baseline EPSCs without prior LTP induction (Figure 3b and Supplementary Figure S8b). These data demonstrate that isradipine facilitates the reversal, or depotentiation, of previously induced LTP. Application of AP5, which inhibited potentiated NMDAR EPSCs, together with isradipine during the delivery of the “SS-10×” protocol prevented LTP depotentiation (Figure 3c and Supplementary Figure S8c). Therefore, NMDAR activation at the potentiated inputs is likely required to reverse LTP.
CaV1.3 is the major subtype of LTCCs expressed in dopamine neurons19, 20. A CaV1.3-selective LTCC antagonist 1-(3-chlorophenethyl)-3-cyclopentylpyrimidine-2,4,6-(1H,3H,5H)-trione, termed compound 8, was recently developed42. Indeed, compound 8 (20 μM) also enabled the “SS+AP-30×” protocol to cause depotentiation of potentiated EPSCs (Figure 3a and Supplementary Figure S8a).
LTCC blockade in the VTA promotes extinction of cocaine and ethanol CPP
Previously acquired drug CPP gradually diminishes, i.e., undergoes extinction, with repeated expression tests (posttests; once daily), where rats are exposed to CPP compartments without the drug. To test if LTCCs are involved in the expression and extinction of cocaine CPP, a single systemic injection of isradipine (1.2 mg/kg, i.p.) was made 10 min before the second posttest. Isradipine failed to affect CPP expression on the day of injection, in line with the lack of effect of isradipine on tonic and burst firing in VTA slices from rats that had undergone cocaine conditioning (Supplementary Figure S9). However, no significant CPP was observed on the following 2 days (third and fourth posttests, performed without isradipine injection), while the vehicle-injected control rats still displayed robust CPP (Figure 4a). Importantly, priming injection of cocaine before subsequent posttests, which significantly increased CPP in control rats, failed to reinstate CPP previously extinguished in the presence of isradipine, even after 2 weeks of withdrawal in the home cage (i.e., no exposure to the CPP box or cocaine). Systemic isradipine injection also promoted extinction of CPP acquired with alcohol (ethanol), a different class of addictive drug, in a similar manner and prevented subsequent ethanol-induced reinstatement (Figure 4b).
Next, we wished to determine if LTCC blockade in the VTA affects CPP extinction. In cocaine-conditioned animals, we made bilateral intra-VTA injection of isradipine or compound 8 [6 pmol/0.3 μl (= 20 μM) in each side] 5 min before the second posttest, which produced no immediate effect on CPP expression. However, as with systemic isradipine injection, CPP was virtually abolished on subsequent days, and CPP thus extinguished was resistant to cocaine-induced reinstatement (Figures 4c and d). Isradipine/compound 8 injection (systemic or intra-VTA) had no effect on the overall activity during the posttest (Supplementary Figure S10).
Certain manipulations disrupt previously acquired CPP when performed shortly after the posttest, via interacting with the memory reconsolidation process43-45, or even without a posttest, via non-specific memory ablation46, 47. However, intra-VTA injection of isradipine immediately after the second posttest or without a posttest, i.e., while rats stayed in the home cage, had no effect on CPP expression the following day (Figure 4e and Supplementary Figure S11). Thus, in order to promote CPP extinction, isradipine needs to be present in the VTA during the posttest when rats are exposed to cocaine-associated contextual cues. Accordingly, intra-VTA injection of the NMDAR antagonist AP5, which suppressed CPP expression likely by blocking NMDARs at glutamatergic inputs activated by cocaine-associated cues, prevented CPP extinction induced by systemic isradipine injection (Figure 4f and Supplementary Figure S12).
Repeated posttests over 8–9 consecutive days, during which rats are repeatedly exposed to the CPP compartments without cocaine, resulted in complete extinction of cocaine CPP (Figure 5a). In contrast to extinction induced in the presence of isradipine, CPP simply extinguished with repeated posttests was robustly reinstated by priming injection of cocaine (Figure 5b). However, systemic injection of isradipine, made 10 min before the cocaine-induced reinstatement session (Figure 5b) or before the tenth posttest without cocaine injection (Figure 5c), led to suppression of cocaine-induced reinstatement on the following day.
Altogether, these results show that LTCC blockade during exposure to cocaine/ethanol-paired contextual cues, and to interoceptive cocaine cues during cocaine-induced reinstatement, may cause persistent disruption of appetitive cue memory.
Discussion
Isradipine, a dihydropyridine LTCC antagonist that crosses the blood-brain barrier, is currently undergoing clinical trials to test if daily isradipine slows neurodegeneration in Parkinson's disease48, 49. Our study suggests that isradipine may also be used to treat a critical component of addiction, i.e., increased motivational valence of drug-associated cues triggering craving and relapse.
LTCCs are involved in the induction of synaptic plasticity in different brain areas, such as the hippocampus, cerebral cortex, and striatum50-53, where LTCC activation associated with postsynaptic depolarization is thought to drive synaptic plasticity. In VTA dopamine neurons, our data suggest that basal Ca2+ levels maintained by constant LTCC-mediated Ca2+ influx are essential not only for the induction of NMDAR LTP but also for its maintenance, i.e., in preventing LTP reversal triggered by glutamatergic input activity. Here, LTCCs do not contribute to burst-evoked Ca2+ signals or the mGluR/IP3-dependent amplification mechanism necessary for LTP induction14. CaV1.3 LTCCs, which activate at relatively hyperpolarized membrane potentials18, are suited for providing tonic Ca2+ influx, even though they do not drive subthreshold oscillations underlying pacemaker activity in VTA dopamine neurons20, 21, 34. The cellular machinery sensing basal Ca2+ levels (LTCC-dependent) together with transient AP/burst-evoked Ca2+signals (LTCC-independent) during the delivery of LTP induction and depotentiation protocols remains to be determined. Protein kinase C, which mediates the induction of mGluR/Ca2+-dependent LTP of NMDAR EPSCs at hippocampal mossy fiber synapses54, 55, has been ruled out in dopamine neurons14.
In the lateral amygdala, LTCC inhibition by antagonists suppresses the induction of AMPAR LTP and impairs aversive Pavlovian conditioning (i.e., fear conditioning)56, 57, in a manner analogous to the effects of isradipine in the VTA on NMDAR LTP induction and CPP acquisition. Interestingly, LTCC inhibition in the lateral amygdala during extinction training blocks the extinction of conditioned responses58, presumably by interfering with the induction of certain forms of synaptic plasticity within the lateral amygdala underlying inhibitory learning59. In contrast, LTCC inhibition in the VTA facilitates the reversal of NMDAR LTP. This may cause unlearning of cue memory by reversing the synaptic plasticity induced during CPP acquisition, thereby promoting CPP extinction and preventing future reinstatement. Hence, LTCC blockade timed with cue exposure would allow for the manipulation of specific cue memory by controlling LTP induction and reversal60.
In the present study, no global alterations in NMDAR-dependent excitation were found in the VTA after cocaine conditioning. It remains to be determined if NMDAR potentiation can be observed specifically at those inputs activated by cocaine-associated cues, as has been demonstrated recently in the lateral amygdala following fear conditioning, where only those inputs paired with a foot shock during conditioning display AMPAR potentiation60. Therefore, firm evidence for the role of NMDAR LTP in CPP, or more generally in reward-associated cue learning, is lacking at the moment. Interestingly, a reduction in unitary NMDAR EPSCs at individual glutamatergic synapses has been reported after in vivo cocaine experience39. This may represent redistribution of NMDARs from synaptic to extrasynaptic sites even if the overall number of NMDARs (synaptic and extrasynaptic) is not changed, as assessed with bath application of NMDA61. Alternatively, although speculative, this might represent a form of homeostatic synaptic plasticity62, in which NMDAR transmission at inputs not activated by cocaine-associated cues (e.g., interoceptive cocaine cues or the experimenter performing injection40) are scaled down in response to LTP induced in the presumably small subset of glutamatergic inputs encoding cocaine cues, thereby maintaining the overall strength of NMDAR-dependent excitatory transmission in each neuron. It should be noted these two possibilities are not necessarily mutually exclusive.
It has been shown that NMDAR blockade in the VTA inhibits the induction of NMDAR LTP and acquisition of psychostimulant CPP14, 31. In the present study, we found that NMDAR blockade in the VTA also prevents the reversal of NMDAR LTP and extinction of cocaine CPP enabled by isradipine. These results are consistent with the idea that NMDAR activation at specific glutamatergic inputs activated by contextual cues of the CPP box is required for both the learning and unlearning of those cues. In this regard, it is of note that isradipine was effective in preventing future CPP reinstatement, i.e., in disrupting cue memory, even when administered before the tenth posttest without CPP expression, where CPP had been completely extinguished during repeated posttests. This suggests that contextual cue inputs were still active during the tenth posttest, thus enabling isradipine to reverse NMDAR LTP at those inputs, but were not capable of supporting CPP expression as a consequence of inhibitory learning during extinction training that would suppress the learned response43, 44, 63. Isradipine administration prior to cocaine-induced reinstatement might further reverse LTP at glutamatergic inputs activated by interoceptive cocaine cues.
Mouse studies with genetic deletion of NMDARs selectively in dopamine neurons reported impaired drug (cocaine/nicotine) and food CPP9, 27, 28, while another study observed normal cocaine CPP with impaired reinstatement of extinguished CPP30. Although this discrepancy may be due to differences in the CPP protocol (e.g., the number and duration of conditioning sessions), these studies overall support the role of NMDARs in dopamine neurons in CPP acquisition, expression, and/or reinstatement.
Isradipine, administered systemically or into the VTA, failed to affect the expression of cocaine CPP, including cocaine priming-induced reinstatement of extinguished CPP. This is in line with the lack of effect of isradipine on NMDAR-dependent excitation in the VTA that drives CPP expression29, 31. Dopamine D1 receptor-mediated activation of LTCCs in the nucleus accumbens has been implicated in the reinstatement of cocaine self-administration64. Thus, dopamine regulation of LTCCs in the nucleus accumbens appears to be selectively involved in the expression of operant, but not Pavlovian, drug-seeking behavior.
Current cue exposure-based strategies to treat addiction are aimed at facilitating inhibitory extinction learning (e.g., with the NMDAR partial agonist d-cycloserine) or disrupting memory reconsolidation following retrieval (e.g., with the β-adrenergic receptor antagonist propranolol)43, 44. Based on rodent studies demonstrating the effectiveness of isradipine and other LTCC antagonists on the acquisition of CPP induced by cocaine and other addictive drugs23-26, high dosage of isradipine has been tested in human cocaine addicts in a laboratory setting, which failed to reduce measures of cocaine-induced subjective euphoria with no effect on cognitive performance65-67. The present study suggests that isradipine, if taken prior to the retrieval of cue memory, as occurs upon an encounter with environmental cues (places, people, etc.) or with interoceptive drug cues during a relapse, would enable unlearning of the increased valence of those cues, thus preventing craving and relapse in the future.
Supplementary Material
Acknowledgments
We thank Dr. D. James Surmeier for compound 8 and Dr. Kamran Khodakhah for caged IP3. We also thank Garry Cooper for helpful discussions on the effect and usage of compound 8. This work was supported by NIH grants DA015687 and AA015521.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Supplementary information is available at Molecular Psychiatry's website.
References
- 1.Wise RA, Kiyatkin EA. Differentiating the rapid actions of cocaine. Nat Rev Neurosci. 2011;12:479–484. doi: 10.1038/nrn3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson TE, Yager LM, Cogan ES, Saunders BT. On the motivational properties of reward cues: Individual differences. Neuropharmacology. 2014;76 Pt B:450–459. doi: 10.1016/j.neuropharm.2013.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Volkow ND, Wang GJ, Fowler JS, Tomasi D, Telang F, Baler R. Addiction: decreased reward sensitivity and increased expectation sensitivity conspire to overwhelm the brain's control circuit. Bioessays. 2010;32:748–755. doi: 10.1002/bies.201000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- 5.Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- 6.Luthi A, Luscher C. Pathological circuit function underlying addiction and anxiety disorders. Nat Neurosci. 2014;17:1635–1643. doi: 10.1038/nn.3849. [DOI] [PubMed] [Google Scholar]
- 7.Schultz W. Predictive reward signal of dopamine neurons. J Neurophysiol. 1998;80:1–27. doi: 10.1152/jn.1998.80.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- 9.Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, et al. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc Natl Acad Sci U S A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morikawa H, Khodakhah K, Williams JT. Two intracellular pathways mediate metabotropic glutamate receptor-induced Ca2+ mobilization in dopamine neurons. J Neurosci. 2003;23:149–157. doi: 10.1523/JNEUROSCI.23-01-00149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang LP, Li F, Wang D, Xie K, Wang D, Shen X, et al. NMDA receptors in dopaminergic neurons are crucial for habit learning. Neuron. 2011;72:1055–1066. doi: 10.1016/j.neuron.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deister CA, Teagarden MA, Wilson CJ, Paladini CA. An intrinsic neuronal oscillator underlies dopaminergic neuron bursting. J Neurosci. 2009;29:15888–15897. doi: 10.1523/JNEUROSCI.4053-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blythe SN, Atherton JF, Bevan MD. Synaptic activation of dendritic AMPA and NMDA receptors generates transient high-frequency firing in substantia nigra dopamine neurons in vitro. J Neurophysiol. 2007;97:2837–2850. doi: 10.1152/jn.01157.2006. [DOI] [PubMed] [Google Scholar]
- 14.Harnett MT, Bernier BE, Ahn KC, Morikawa H. Burst-timing-dependent plasticity of NMDA receptor-mediated transmission in midbrain dopamine neurons. Neuron. 2009;62:826–838. doi: 10.1016/j.neuron.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown J, Bullock D, Grossberg S. How the basal ganglia use parallel excitatory and inhibitory learning pathways to selectively respond to unexpected rewarding cues. J Neurosci. 1999;19:10502–10511. doi: 10.1523/JNEUROSCI.19-23-10502.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui G, Bernier BE, Harnett MT, Morikawa H. Differential regulation of action potential- and metabotropic glutamate receptor-induced Ca2+ signals by inositol 1,4,5-trisphosphate in dopaminergic neurons. J Neurosci. 2007;27:4776–4785. doi: 10.1523/JNEUROSCI.0139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuccotti A, Clementi S, Reinbothe T, Torrente A, Vandael DH, Pirone A. Structural and functional differences between L-type calcium channels: crucial issues for future selective targeting. Trends in pharmacological sciences. 2011;32:366–375. doi: 10.1016/j.tips.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 18.Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. Journal of neurophysiology. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- 19.Rajadhyaksha A, Husson I, Satpute SS, Kuppenbender KD, Ren JQ, Guerriero RM, et al. L-type Ca2+ channels mediate adaptation of extracellular signal-regulated kinase 1/2 phosphorylation in the ventral tegmental area after chronic amphetamine treatment. J Neurosci. 2004;24:7464–7476. doi: 10.1523/JNEUROSCI.0612-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, et al. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- 21.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Branch SY, Sharma R, Beckstead MJ. Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:9310–9318. doi: 10.1523/JNEUROSCI.4228-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pani L, Kuzmin A, Martellotta MC, Gessa GL, Fratta W. The calcium antagonist PN 200-110 inhibits the reinforcing properties of cocaine. Brain research bulletin. 1991;26:445–447. doi: 10.1016/0361-9230(91)90022-c. [DOI] [PubMed] [Google Scholar]
- 24.Pucilowski O, Plaznik A, Overstreet DH. Isradipine suppresses amphetamine-induced conditioned place preference and locomotor stimulation in the rat. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 1995;12:239–244. doi: 10.1016/0893-133X(94)00080-J. [DOI] [PubMed] [Google Scholar]
- 25.Biala G, Langwinski R. Effects of calcium channel antagonists on the reinforcing properties of morphine, ethanol and cocaine as measured by place conditioning. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society. 1996;47:497–502. [PubMed] [Google Scholar]
- 26.Pucilowski O, Garges PL, Rezvani AH, Hutheson S, Janowsky DS. Verapamil suppresses d-amphetamine-induced place preference conditioning. European journal of pharmacology. 1993;240:89–92. doi: 10.1016/0014-2999(93)90551-r. [DOI] [PubMed] [Google Scholar]
- 27.Wang LP, Li F, Shen X, Tsien JZ. Conditional knockout of NMDA receptors in dopamine neurons prevents nicotine-conditioned place preference. PLoS One. 2010;5:e8616. doi: 10.1371/journal.pone.0008616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462. doi: 10.1111/j.1369-1600.2007.00070.x. [DOI] [PubMed] [Google Scholar]
- 30.Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 31.Whitaker LR, Degoulet M, Morikawa H. Social deprivation enhances VTA synaptic plasticity and drug-induced contextual learning. Neuron. 2013;77:335–345. doi: 10.1016/j.neuron.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn KC, Bernier BE, Harnett MT, Morikawa H. IP3 receptor sensitization during in vivo amphetamine experience enhances NMDA receptor plasticity in dopamine neurons of the ventral tegmental area. J Neurosci. 2010;30:6689–6699. doi: 10.1523/JNEUROSCI.4453-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernier BE, Whitaker LR, Morikawa H. Previous ethanol experience enhances synaptic plasticity of NMDA receptors in the ventral tegmental area. J Neurosci. 2011;31:5205–5212. doi: 10.1523/JNEUROSCI.5282-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khaliq ZM, Bean BP. Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage-dependent sodium conductances. J Neurosci. 2010;30:7401–7413. doi: 10.1523/JNEUROSCI.0143-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koulchitsky S, De Backer B, Quertemont E, Charlier C, Seutin V. Differential effects of cocaine on dopamine neuron firing in awake and anesthetized rats. Neuropsychopharmacology. 2012;37:1559–1571. doi: 10.1038/npp.2011.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 37.Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan T, Mameli M, EC OC, Dey PN, Verpelli C, Sala C, et al. Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron. 2013;80:1025–1038. doi: 10.1016/j.neuron.2013.07.050. [DOI] [PubMed] [Google Scholar]
- 39.Mameli M, Bellone C, Brown MT, Luscher C. Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat Neurosci. 2011;14:414–416. doi: 10.1038/nn.2763. [DOI] [PubMed] [Google Scholar]
- 40.Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, et al. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 42.Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, et al. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nature communications. 2012;3:1146. doi: 10.1038/ncomms2149. [DOI] [PubMed] [Google Scholar]
- 43.Torregrossa MM, Taylor JR. Learning to forget: manipulating extinction and reconsolidation processes to treat addiction. Psychopharmacology (Berl) 2013;226:659–672. doi: 10.1007/s00213-012-2750-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Milton AL, Everitt BJ. The persistence of maladaptive memory: addiction, drug memories and anti-relapse treatments. Neurosci Biobehav Rev. 2012;36:1119–1139. doi: 10.1016/j.neubiorev.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Xue YX, Luo YX, Wu P, Shi HS, Xue LF, Chen C, et al. A memory retrieval-extinction procedure to prevent drug craving and relapse. Science. 2012;336:241–245. doi: 10.1126/science.1215070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee AM, Kanter BR, Wang D, Lim JP, Zou ME, Qiu C, et al. Prkcz null mice show normal learning and memory. Nature. 2013;493:416–419. doi: 10.1038/nature11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li YQ, Xue YX, He YY, Li FQ, Xue LF, Xu CM, et al. Inhibition of PKMzeta in nucleus accumbens core abolishes long-term drug reward memory. J Neurosci. 2011;31:5436–5446. doi: 10.1523/JNEUROSCI.5884-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Group PS. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson's disease (STEADY-PD) Movement disorders : official journal of the Movement Disorder Society. 2013;28:1823–1831. doi: 10.1002/mds.25639. [DOI] [PubMed] [Google Scholar]
- 49.Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiology of disease. 2011;43:364–371. doi: 10.1016/j.nbd.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seoane A, Massey PV, Keen H, Bashir ZI, Brown MW. L-type voltage-dependent calcium channel antagonists impair perirhinal long-term recognition memory and plasticity processes. J Neurosci. 2009;29:9534–9544. doi: 10.1523/JNEUROSCI.5199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adermark L, Lovinger DM. Combined activation of L-type Ca2+ channels and synaptic transmission is sufficient to induce striatal long-term depression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:6781–6787. doi: 10.1523/JNEUROSCI.0280-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 54.Hunt DL, Castillo PE. Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Curr Opin Neurobiol. 2012;22:496–508. doi: 10.1016/j.conb.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rebola N, Srikumar BN, Mulle C. Activity-dependent synaptic plasticity of NMDA receptors. J Physiol. 2010;588:93–99. doi: 10.1113/jphysiol.2009.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langwieser N, Christel CJ, Kleppisch T, Hofmann F, Wotjak CT, Moosmang S. Homeostatic switch in hebbian plasticity and fear learning after sustained loss of Cav1.2 calcium channels. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:8367–8375. doi: 10.1523/JNEUROSCI.4164-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davis SE, Bauer EP. L-type voltage-gated calcium channels in the basolateral amygdala are necessary for fear extinction. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:13582–13586. doi: 10.1523/JNEUROSCI.0809-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pare D, Duvarci S. Amygdala microcircuits mediating fear expression and extinction. Curr Opin Neurobiol. 2012;22:717–723. doi: 10.1016/j.conb.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, Malinow R. Engineering a memory with LTD and LTP. Nature. 2014;511:348–352. doi: 10.1038/nature13294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–696. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor perspectives in biology. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan WX, Schmidt R, Wickens JR, Hyland BI. Tripartite mechanism of extinction suggested by dopamine neuron activity and temporal difference model. J Neurosci. 2008;28:9619–9631. doi: 10.1523/JNEUROSCI.0255-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, et al. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nature neuroscience. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- 65.Johnson BA, Javors MA, Lam YW, Wells LT, Tiouririne M, Roache JD, et al. Kinetic and cardiovascular comparison of immediate-release isradipine and sustained-release isradipine among non-treatment-seeking, cocaine-dependent individuals. Progress in neuro-psychopharmacology & biological psychiatry. 2005;29:15–20. doi: 10.1016/j.pnpbp.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 66.Roache JD, Johnson BA, Ait-Daoud N, Mauldin JB, Thornton JE, Wells LT, et al. Effects of repeated-dose isradipine on the abuse liability of cocaine. Experimental and clinical psychopharmacology. 2005;13:319–326. doi: 10.1037/1064-1297.13.4.319. [DOI] [PubMed] [Google Scholar]
- 67.Johnson BA, Roache JD, Ait-Daoud N, Wallace CL, Wells LT, Wang Y, et al. Effects of isradipine on cocaine-induced changes in cognitive performance in recently abstinent cocaine-dependent individuals. The international journal of neuropsychopharmacology/official scientific journal of the Collegium Internationale Neuropsychopharmacologicum (CINP) 2005;8:549–556. doi: 10.1017/S1461145705005511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.