

Abstract
The role of exhibition swine in influenza A virus transmission was recently demonstrated by >300 infections with influenza A(H3N2) variant viruses among individuals who attended agricultural fairs. Through active influenza A virus surveillance in US exhibition swine and whole-genome sequencing of 380 isolates, we demonstrate that exhibition swine are actively involved in the evolution of influenza A viruses, including zoonotic strains. First, frequent introduction of influenza A viruses from commercial swine populations provides new genetic diversity in exhibition pigs each year locally. Second, genomic reassortment between viruses cocirculating in exhibition swine increases viral diversity. Third, viral migration between exhibition swine in neighboring states demonstrates that movements of exhibition pigs contributes to the spread of genetic diversity. The unexpected frequency of viral exchange between commercial and exhibition swine raises questions about the understudied interface between these populations. Overall, the complexity of viral evolution in exhibition swine indicates that novel viruses are likely to continually reemerge, presenting threats to humans.
Keywords: influenza A virus, swine, human-animal interface, evolution, genomics, gene flow, United States, exhibition animals
(See the editorial commentary by Baranovich and Bahl on pages 169–70.)
Swine harbor a diverse population of influenza A viruses, which occasionally transmit from swine to humans and represent an important threat to public health. Swine-origin influenza A viruses (IAV-S) that transmit zoonotically to humans are referred to as variants [1]. Since 2005, there have been 351, 18, and 5 confirmed cases of variant H3N2 (H3N2v), H1N1 (H1N1v), and H1N2 (H1N2v) influenza A virus infection, respectively, in the United States; 343 of the cases due to H3N2v occurred during 2011–2014 [2]. Human H3N2v infections have predominantly occurred among children at US agricultural fairs, where exhibition swine come into contact with humans [3]. To date, there have been 18 hospitalizations and 1 death associated with H3N2v infections. While H3N2v cases have occurred in every year during 2011–2014, the majority were documented during the summer of 2012 in Indiana (138 cases) and Ohio (107 cases). Confirmed cases also were identified during these 4 years in 11 other states, including Hawaii, Illinois, Iowa, Maine, Maryland, Michigan, Minnesota, Pennsylvania, Utah, West Virginia, and Wisconsin.
The influenza A virus genome comprises 8 viral RNA segments (polymerase basic 2 [PB2], PB1, polymerase acidic [PA], hemagglutinin [HA], nucleoprotein [NP], neuraminidase [NA], matrix protein [MP], and nonstructural protein [NS]) that can be exchanged through reassortment. The 2009 pandemic influenza A(H1N1) virus (A[H1N1]pdm09) is a reassortant swine virus containing both North American triple-reassortant IAV-S and Eurasian avian-like IAV-S segments [4, 5]. A(H1N1)pdm09 also has reassorted repeatedly with IAV-S in multiple countries, generating diverse reassorted genotypes [6–8], including the 7:1 reassortant virus associated with the cases of H3N2v infection during 2011 and 2012 in humans [9, 10]. This genotype (referred to as H3-G1 [10]) contains 7 triple-reassortant IAV-S H3N2 segments and a A(H1N1)pdm09 MP segment. Prior to 2009, the evolutionary dynamics and characterization of IAV-S in pigs exhibited at fairs and exhibitions were generally overlooked, despite their high human contact rates. As a result, it remains unclear why H3N2v cases surged in 2012 and then decreased in 2013–2014. The decrease in cases may be related to recent education and/or to interventions initiated at agricultural fairs, including limiting the time that pigs are on exhibition grounds to 72 hours [11]. Potentially, swine exhibitors exposed in 2012 carried forward immunity into 2013 and 2014. Alternatively, differences between years may be related to population dynamics in swine: the H3-G1 genotype may have circulated in exhibition pigs at a higher frequency in 2012 than in 2013 or 2014. The US Department of Agriculture conducts year-round surveillance of IAV-S in commercial swine [12], but exhibition swine (which represent approximately 1.5% of the US swine herd) are distinct from commercial swine and are primarily raised in backyard or small-farm settings for the purposes of agricultural education and/or competition.
Further knowledge of the frequency of contact and viral gene flow between commercial swine and exhibition is key to understanding the role of exhibition swine in the evolution of H3-G1 viruses (Figure 1). To investigate this question, we performed a large-scale phylogenetic analysis using genome sequences from 380 influenza A viruses collected from exhibition swine in 5 US states via active surveillance during 2009–2013 (Supplementary Table 1). Specifically, we aimed to determine whether H3-G1 viruses are primarily generated within the larger commercial swine population, with exhibition swine serving merely as the conduit due to their high human contact rates during fair events (Figure 1A), or whether exhibition swine are active breeding grounds for the generation of H3-G1 and other reassortant viruses with zoonotic potential (Figure 1B).
Figure 1.

The role of exhibition swine in the evolution of influenza A virus variants. There are 2 scenarios by which influenza A viruses (solid red circles) could evolve and be transmitted from exhibition swine to humans. In scenario A, the major transmission bottleneck occurs between commercial swine and exhibition swine: variant influenza A viruses initially evolve within the large and diverse influenza virus population found in commercial swine, are transmitted onward to exhibition swine in association with a major bottleneck in diversity, and are subsequently transmitted from exhibition swine to humans. In scenario B, the major transmission bottleneck occurs between exhibition swine and humans: multiple diverse influenza A viruses are transmitted from commercial swine to exhibition swine, where variant influenza A viruses evolve via reassortment and are transmitted onward to humans.
MATERIALS AND METHODS
Sample Collection, Influenza A Virus Detection and Recovery, and Genome Sequencing
Nasal swab or snout wipe specimens were collected from exhibition pigs at participating fairs and exhibitions [13, 14]. The Ohio State University Institutional Animal Care and Use Committee approved the use of animals in this study. Samples were screened for the influenza A virus MP gene with real-time revere transcription–polymerase chain reaction analysis, with positive samples inoculated into Madin-Darby canine kidney cells for virus isolation. From each fair event where influenza A virus was recovered from exhibition swine (n = 53), representative isolates were selected for genomic sequencing. The complete genomes of 380 influenza A virus isolates collected in exhibition swine (IAV-exS) during 2009–2013 were sequenced at the J. Craig Venter Institute, University of Minnesota, or the US Department of Agriculture National Veterinary Services Laboratories. Depending on the sequencing team, RNA extraction of the isolates was performed by either the ZR 96 viral RNA kit (Zymo Research, Irvine, California) or the MagMAX viral RNA isolation kit (Life Technologies, Carlsbad, California). Amplification of the influenza A virus genomic segments and preparation for next-generation sequencing was performed using previously described techniques [15, 16]. Next-generation sequencing was performed using the Ion Torrent PGM, Illumina HiSeq and MiSeq, or 454 GS FLX+. The reads from the sequencing instruments were sorted, trimmed, mapped to reference influenza A virus strains. All consensus sequences were deposited in GenBank (accession numbers are listed in Supplementary Table 1), and associated metadata were deposited in the Influenza Research Database [17].
Phylogenetic Analysis of Whole-Genome Sequence Data
In addition to the 380 IAV-exS isolates collected for this study, 470 whole-genome sequences from influenza A viruses collected from US commercial swine (IAV-comS) during 2009–2014 were downloaded from the Influenza Virus Resource at GenBank [18] and included as background data. Each segment was aligned separately using MUSCLE, version 3.8.1 [19], and separate alignments were made for H1, H3, N1, and N2. To broadly categorize the evolutionary origins of each segment, neighbor-joining trees were inferred using PAUP* [20]. Internal gene segments (PB2, PB1, PA, NP, MP, and NS) were categorized as triple-reassortant internal genes (TRIGs) or A(H1N1)pdm09 lineage (pdm09). H1 segments were categorized as H1δ1, H1δ2, H1γ, or H1-pdm09. N1 segments were categorized as N1-classical or N1-pdm09. N2 segments were categorized as N2-1998 or N2-2002. The evolutionary origins of each of the 8 genome segments were categorized for each virus (Supplementary Table 2).
Phylogenetic Analysis of Exhibition Swine in 2013
To study the spatial and evolutionary dynamics of IAV-exS in greater depth, we focused on the year 2013, for which the largest and most geographically diverse sampling was conducted in exhibition swine (149 viruses from 4 US states), although IAV-exS and IAV-comS sequences from 2009–2014 were retained as background. IAV-exS collected on the same day and location (ie, global positioning system [GPS] point) were subsampled down (2 viruses per day per location per genotype among closely related viruses) owing to the high numbers of identical sequences collected from Indiana and Ohio exhibitions. We also focused our analysis on the PB2 segment, which is the longest segment (2280 nucleotides) and provides the highest phylogenetic resolution. A key advantage of studying PB2 also is that all IAV-exS were TRIGs during 2009–2013, allowing for evolutionary relationships to be visualized on a single phylogenetic tree. A large proportion of IAV-comS in the United States during 2009–2013 also had TRIG PB2 segments, so focusing on TRIG PB2 did not remove as much background data as for other segments with more pdm09 versions [21]. Importantly, phylogenies inferred for other segments exhibited similar patterns as PB2 (provided in Supplementary Figures 1–12), and patterns of reassortment between segments were examined, as well (see below). The phylogenetic relationships of 411 PB2 TRIG sequences were inferred using the maximum likelihood method available in the program RAxML, version 7.2.6 [22], incorporating a general time-reversible model of nucleotide substitution with a gamma-distributed (Γ) rate variation among sites. To assess the robustness of each node, a bootstrap resampling process was performed (500 replicates), again using the maximum likelihood method available in RAxML, version 7.2.6. Pairwise genetic distances (Kimura 2-parameter) between 2013 exhibition swine viruses were estimated using MEGA6 [23] and plotted against pairwise geodesic distances, which were computed from GPS coordinates for each viral sample, using the R statistical software environment.
Evolution of Genotype H3-G1 Viruses, Using Markov Jumps
To more quantitatively determine the evolutionary processes that generate H3-G1 viruses in exhibition swine, we performed a time-scaled Bayesian analysis using the Markov chain Monte Carlo (MCMC) method available via the BEAST, version 1.8.0, package [24] and the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health (Bethesda, Maryland; available at: http://biowulf.nih.gov). A relaxed uncorrelated log-normal molecular clock was used, with a flexible Bayesian skyline plot demographic model (10 piecewise constant groups) and a general-time reversible model of nucleotide substitution with Γ rate variation among sites. The MCMC was run separately 3 times for each of the data sets for at least 100 million iterations, with subsampling every 10 000 iterations, using the BEAGLE library to improve computational performance [25]. All parameters reached convergence, as assessed visually, using Tracer, version 1.6, with statistical uncertainty reflected in values of the 95% highest posterior density. At least 10% of the chain was removed as burn-in, and runs for the same segment were combined using LogCombiner, version 1.8.0, and down-sampled to generate a final posterior distribution of 1000 trees that was used in subsequent analyses.
To quantify gene flow between H3-G1 viruses and non–H3-G1 viruses in commercial and exhibition swine, we assigned each virus to one of 4 categories: commercial swine/non–H3-G1, commercial swine/H3-G1, exhibition swine/non–H3-G1, and exhibition swine/H3-G1. The specification of these categories (location states) allowed the expected number of location state transitions in the ancestral history, conditional on the data observed at the tree tips, to be estimated by means of Markov jump counts, using an asymmetrical model [26]. For computational efficiency the phylogeographic analysis was run using an empirical distribution of 1000 trees, allowing the MCMC chain to be run for 25 million iterations, with sampling performed every 1000 iterations. A Bayesian stochastic search variable selection was used to improve statistical efficiency and reveal epidemiological linkages, which were computed using SPREAD [27]. Maximum clade credibility trees were summarized using TreeAnnotator, version 1.8.0, and the trees were visualized in FigTree, version 1.4.2. The Markov jump analysis was repeated with the NP segment to ascertain that the PB2 segment was representative of patterns observed for the genome, even in the context of frequent reassortment (Supplementary Figure 9). To examine the frequency of reassortment of individual segments in greater detail, we again repeated the Markov jumps analysis, in this case assigning categories based on the evolutionary lineage of each segment (pdm09 or TRIG for internal gene segments; H1-β, H1-δ1, H1-δ2, H1-human, H1-γ, or H3 for HA; and N1-classical, N2-2002, or N2-1998 for NA) on the PB2 phylogeny. To compare the frequency of reassortment in commercial versus exhibition swine populations, we again categorized each virus as having an exhibition or commercial swine source, as well.
RESULTS
Frequent Transmission of Influenza A Viruses From Commercial Swine to Exhibition Swine
The 2009–2013 IAV-exS population did not form a discrete clade separate from the US IAV-comS population (Figure 2; phylogenies for other segments also exhibited similar patterns [Supplementary Figures 1–12]). Little linkage was observed between the viral clades found in exhibition pigs from year to year, which suggests that new viral diversity is introduced from commercial pigs into exhibition pigs annually (Figure 2). Limited persistence of viruses across years within exhibition pigs could not be ruled out, particularly during 2012–2013, but this was difficult to verify owing to high admixture between commercial and exhibition swine viruses. In 2013, the year for which our sampling of exhibition swine was most geographically diverse, IAV-exS was clustered into 12 highly supported, genetically distinct phylogenetic clades (bootstrap value, ≥70) and 4 singletons, representing 16 viral introductions into exhibition swine in a single year (Supplementary Figure 13). These clades also were evident on the phylogenies inferred for the HA and NA segment, although in some cases with lower bootstrap support (Supplementary Figures 1–5).
Figure 2.
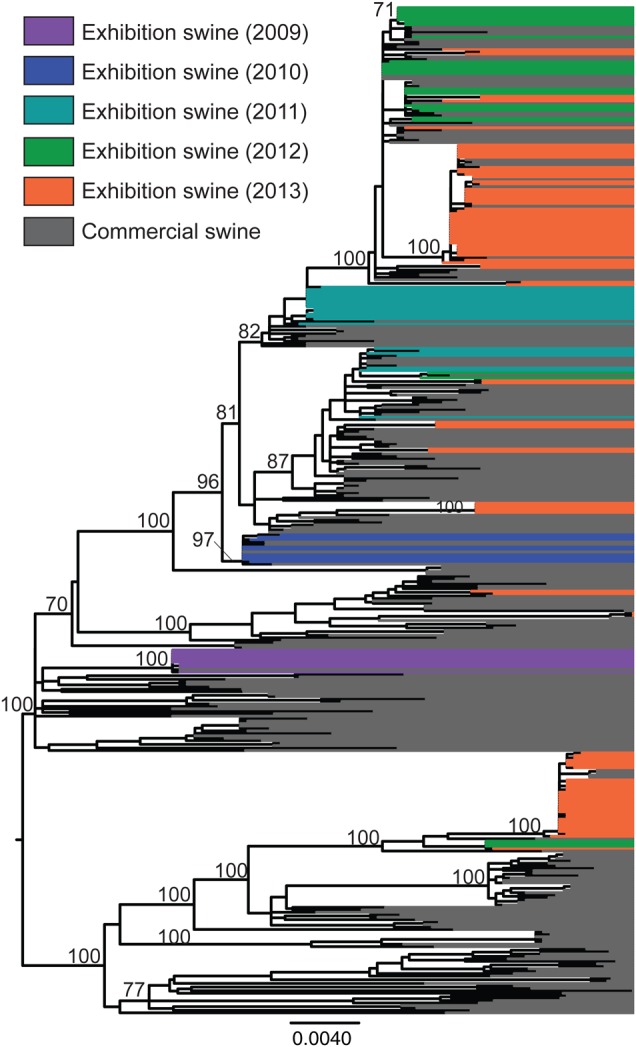
Phylogenetic relationships between influenza A viruses in exhibition swine. Phylogenetic relationships of 411 TRIG PB2 sequences collected from commercial swine and exhibition swine in the United States during 2009–2014, inferred using maximum likelihood methods. Branch lengths represent genetic distance and are drawn to scale. Bootstrap values for key nodes >70 are provided. Tip labels have been aligned and shaded to highlight the phylogenetic relationships between viruses collected from exhibition swine in different years; IAV-S collected from exhibition swine in 2009, purple; IAV-S collected from exhibition swine in 2010, blue; IAV-S collected from exhibition swine in 2011, turquoise; IAV-S collected from exhibition swine in 2012, green; IAV-S collected from exhibition swine in 2013, orange; IAV-S collected from commercial swine during 2009–2013, grey. A phylogenetic tree with tip labels is available in Supplementary Figure 13.
Diverse Genotypes Identified in Exhibition Swine
At a complete-genome level, 12 distinct influenza A virus genotypes were identified in exhibition swine in 2013 (Figure 3A). These genotypes included 4 antigenically distinct HA segment lineages (H1δ1, H1δ2, H1γ, and H3), 2 NA lineages (classical N1 and N2-2002), and 2 types of internal gene segments (TRIG and pdm09). In 2013 exhibition swine, both TRIG and pdm09 versions of the PA and NP were observed, all MP segments were pdm09, and all PB2, PB1, and NS segments were TRIG. The 3 H3 genotypes are numbered according to nomenclature used previously [10], with H3-G1 referring to the genotype that has been associated with the human H3N2v cases from 2011–2012. Viruses with the same genotype did not necessarily cluster together on the phylogeny, indicating that genotypes evolved multiple times independently (Supplementary Figure 14).
Figure 3.

Twelve influenza A virus genotypes identified in exhibition swine in 2013. A, The evolutionary origins of each of the 6 internal gene segments of the influenza A virus genome (polymerase basic 2 [PB2], PB1, polymerase acidic [PA], nucleoprotein [NP], matrix protein [MP], and nonstructural protein [NS]) are classified as triple-reassortant internal genes (red) or 2009 pandemic influenza A(H1N1) (blue). The hemagglutinin (HA) segments are classified as H1δ1 (H1d1; orange), H1δ2 (H1d2, shaded beige), H1γ (H1 g, shaded green), or H3 (shaded purple). The neuraminidase (NA) segments are classified as classical N1 (N1C; green) or N2-2002 (N2-02; purple). The A(H1N1)/A(H1N2) genotypes are numbered H1-G1 to H1-G9. The A(H3N2) genotypes are numbered H3-G1 to H3-G3, consistent with genotype nomenclature used previously [10]. The asterisk indicates that genotype H3-G1 viruses represents the genotype responsible for variant A(H3N2) cases in humans that occurred during 2011–2012. The US states in which each genotype was identified in exhibition swine in 2013 are specified. B, The global positioning system–based location where viruses of each genotype were identified in exhibition swine in 2013 is provided, with the shading of solid circles corresponding to those provided in panel A. C, Plot of geodesic distances (km) vs genetic distances (nucleotide substitutions per site) between influenza viruses collected from exhibition swine in 2013.
Spatial Dynamics of Influenza Viruses in Exhibition Swine
Extensive IAV-exS diversity also was observed in 2013 at the state level (Figure 3B), with multiple genotypes observed in Indiana (n = 7), Ohio (n = 7), and Texas (n = 5) (Figure 3A). Frequent viral exchange between exhibition swine in Indiana and Ohio was evident during 2013 (eg, clade 3, clade 9, and clade 12; Table 1). All samples were geocoded by GPS, allowing for investigation into the extent of IAV-exS diversity at the level of individual agricultural fairs. For example, 4 genotypes (H1-G2, H1-G4, H1-G6, and H3-G3) were identified at a single fair in Texas within the same month (February 2013; Figure 3A), each associated with a separate viral introduction, most likely from commercial swine (singleton 2, clade 10, singleton 4, and singleton 3, respectively; Supplementary Figure 1 and Table 1). The IAV-exS identified in Texas was genetically distinct from IAV-exS identified in other US states in 2013. However, A/swine/Texas/13TOSU0050/2013(H1N1) (singleton 4; Supplementary Figure 1) was closely related to IAV-exS identified in Kentucky 3 months earlier (eg, A/swine/Kentucky/12TOSU1053/2012[H1N2], collected November 2012), forming a discrete clade and possibly representing interstate transmission (Supplementary Figure 13). IAV-exS in Iowa formed an independent clade that also likely represented a separate introduction from commercial swine (clade 7; Table 1 and Supplementary Figures 2, 4, and 13). Consistent with independent introductions of genetically divergent viruses from commercial swine to exhibition in multiple US states, there was little association between geographical distance and genetic distance between US exhibition swine viruses in 2013 (Figure 3C). A strong spatial pattern would have been more likely in the case of a single viral lineage diffusing through the exhibition swine population during this year.
Table 1.
Diversity of Influenza Viruses in Exhibition Swine in Indiana, Iowa, Ohio, and Texas During 2013
| Clade | Genotype |
State(s) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H1 |
H3 |
||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 1 | 2 | 3 | ||
| Singleton 1 | … | … | … | … | … | … | … | … | … | × | … | … | IN |
| Singleton 2 | … | × | … | … | … | … | … | … | … | … | … | … | TX |
| Singleton 3 | … | … | … | … | … | … | … | … | … | × | … | … | TX |
| Singleton 4 | … | … | … | … | … | × | … | … | … | … | … | TX | |
| Clade 1 | … | … | … | … | … | … | … | … | … | × | … | … | OH |
| Clade 2 | … | … | … | … | … | … | … | … | … | × | … | … | OH |
| Clade 3 | … | … | … | × | × | … | × | × | × | × | × | × | IN, OH |
| Clade 4 | … | … | … | × | … | … | … | … | … | … | … | … | IN |
| Clade 5 | … | … | … | … | … | … | … | … | … | × | … | … | IN |
| Clade 6 | … | … | … | … | × | … | … | … | … | … | … | … | IN |
| Clade 7 | … | … | … | × | … | … | … | … | … | … | … | … | IA |
| Clade 8 | … | … | × | … | … | … | … | … | … | … | … | … | OH |
| Clade 9 | … | … | … | … | … | … | … | … | … | … | × | … | IN, OH |
| Clade 10 | … | … | … | × | … | … | … | … | … | … | … | … | TX |
| Clade 11 | × | … | … | … | … | … | … | … | … | … | … | … | TX |
| Clade 12 | … | … | … | × | … | … | … | × | … | × | … | … | IN, OH |
Clades refer to those indicated in Figure 5 and Supplementary Figure 1. Genotypes refer to those listed in Figure 3A.
Transmission of Human Influenza Viruses to Exhibition Swine
The Kentucky 2012 and Texas 2013 viruses (described above) also were notable for their pdm09 MP segments, which provided the only possible evidence of direct transmission of a human influenza A virus to exhibition swine (Supplementary Figure 11). Whereas all other IAV-exS segments on the pdm09 MP tree (and phylogenies for other segments) were closely related to IAV-comS, a single highly supported (97% bootstrap) clade of 8 pdm09 MP segments from exhibition swine in Kentucky (November 2012) and Texas (February 2013) was more closely related to human pdm09 diversity from 2009 than to IAV-comS. Given the long branch length between the 2012/2013 IAV-exS clade and the 2009 human viruses, and the undersampling of IAV-comS in Kentucky and Texas, it is possible that the IAV-exS clade represented a spillover from an unsampled pdm09 lineage circulating among commercial swine, rather than a direct human-to-exhibition swine transmission.
Evolution of H3-G1 in Exhibition Swine
Since genotype H3-G1 is associated with the vast majority of human cases of H3N2v infection, their evolutionary dynamics were explored in finer detail. H3-G1 viruses emerged in exhibition swine at least 7 independent times in 2013, either via reassortment or introduction from commercial swine, based on the number of discrete clades containing H3-G1 viruses or H3-G1 singletons (clade 1, clade 2, clade 3, clade 5, clade 12, singleton 1, and singleton 3; Table 1 and Supplementary Figure 1). To distinguish between these evolutionary processes (in situ reassortment in exhibition swine vs introduction of H3-G1 from commercial swine), we used Markov jump counts of the expected number of state transitions over the posterior distribution of trees of the PB2 segment (represented by the maximum clade credibility tree in Figure 4C). A heat map depicting Markov jump counts indicates that the H3-G1 viruses identified in exhibition swine emerged by both evolutionary mechanisms, which approximately 11.8 direct introductions of H3-G1s from commercial swine (Figure 4A) and approximately 6.1 in situ reassortment events between non–H3-G1 viruses circulating in exhibition swine (Figure 4A). High rates of gene flow also were observed between commercial and exhibition swine for non–H3-G1 viruses in both directions: approximately 11.1 introductions were from commercial to exhibition swine, and approximately 9.8 introductions were from exhibition to commercial swine. These linkages were also supported by high Bayes factors (Figure 4B).
Figure 4.

Evolution of H3-G1 viruses. Time-scaled maximum clade credibility phylogeny of 306 influenza viruses (TRIG PB2 segment) collected from commercial and exhibition swine during 2009–2013 (C). Branches are shaded by 4 categories: commercial swine/non–H3-G1, red; commercial swine/H3-G1, green; exhibition swine/non–H3-G1, blue; and exhibition swine/H3-G1, purple. Posterior probabilities of >0.90 are provided for key nodes. Viruses representing the 12 clades and 4 singletons identified in exhibition swine in 2013 depicted in Supplementary Figure 13 are indicated. A heat map depicts viral transmissions between commercial and exhibition swine (A). The intensity of the color (red, high; white, low) reflects the number of Markov jump counts from one swine population (y-axis) to another (x-axis), a measure of the number of inferred location state transitions, modeled by a continuous-time Markov chain process, that occur along the branches of the phylogeny. To further understand the evolution of H3-G1 viruses, viral populations from commercial and exhibition swine have both been differentiated as H3-G1 and other/non–H3-G1. For clarity, the heat map has been divided into 4 sections that represent transmission events (I) between commercial swine, (II) from commercial swine to exhibition swine, (III) from exhibition swine to commercial swine, and (IV) between exhibition swine. Well-supported rates were estimated using the Bayes factors (BF) test (B). The colors of the circles are identical to those in panels A and C, and the width of the lines is proportional to the log BF.
Frequency of Reassortment of Influenza Virus Segments
We extended our Markov jump approach to quantify the frequency of reassortment of individual segments of the influenza A virus genome within the commercial and exhibition swine populations (Figure 5). The highest frequency of reassortment in both commercial and exhibition swine was observed in the HA and NA segments (Figure 5B). This higher frequency at least in part relates to the greater number of genetically distinct HA and NA lineages that circulate in US swine, compared with the internal genes. Among the internal gene segments, the highest frequency of reassortment was observed in the MP segment, owing to the large number of transitions from TRIG to PDM in both the commercial and exhibition swine populations (21.4 and 9.4, respectively; Figure 5A). Frequent reassortment of the pdm09 MP segment in commercial swine was consistent with prior findings [9]; our results indicated that the emergence of human H3N2v cases was also likely related to frequent reassortment of pdm09 MP within exhibition swine. High levels of bidirectional transmission between commercial and exhibitor swine were observed for the H3 segment, the N2-2002 segment, and TRIG segments but the MP TRIG segment. Only in the case of MP was bidirectional transmission between commercial and exhibition swine higher for pdm09 (approximately 32.3 introductions) than for TRIG (approximately 7.0 transmissions; Figure 5A).
Figure 5.

Heat map of reassortment frequency. A heat map (similar to that in Figure 4A) depicts reassortment between polymerase basic 2 (PB2) and other internal gene segments (A) and the hemagglutinin (HA) and neuraminidase (NA) segments (B). For clarity, the heat map has been divided into 4 sections that represent (I) reassortment events within the commercial swine population, (II) transmission from commercial swine to exhibition swine, (III) transmission from exhibition swine to commercial swine, and (IV) reassortment events within the exhibition swine population. Direct viral transmission events from exhibition to commercial swine (or vice versa) that do not involve reassortment are differentiated by the symbol §. The high frequency of reassortment involving the pdm09 matrix protein (MP) in commercial swine is indicated with an asterisk. For each internal gene segment, the total frequency of reassortment in exhibition swine, commercial swine, and both swine populations is provided in brackets (A) and plotted, along with HA and NA frequencies, in panel B. Estimates for exhibition swine (EX) are blue, those for commercial swine (COM) are orange, and those for the entire population (ALL) are green.
DISCUSSION
The identification of influenza A(H3N2) viruses associated with human illness in 2012 within the context of agricultural fairs emphasized the importance of studying viral dynamics in exhibition swine, about which little was known until recently [13, 28, 29]. By conducting several years of intensive surveillance in exhibition swine in multiple US states, our study demonstrates that the exhibition swine population is likely to be an active, dynamic breeding ground for new viral diversity, similar to the much larger commercial swine production systems. One may have expected commercial and exhibition swine to host separate influenza A virus populations that reflect differences in their genetics and production. Rather, our study demonstrates that frequent transmission of influenza A viruses between commercial and exhibition swine is central to the evolution of IAV-exS, seeding diversity that has the opportunity to reassort and generate new genotypes, including H3-G1. Extensive genetic diversity, reassortment, and multiple viral introductions also were observed even in much less intensively sampled states, such as Texas. However, because the interface between commercial swine and exhibition swine remains poorly understood, further understanding of when and how contact occurs between these populations is needed.
Another outstanding question that could not be addressed fully is the extent to which influenza A viruses transmit spatially within the US exhibition swine population. Exhibition pigs have the ability to attend several livestock exhibitions and fairs in multiple states each year, providing the opportunity for intrastate and interstate spread of viruses. However, in contrast to well-documented commercial swineflows [30], the interstate movements of exhibition swine are not systematically tracked. Extensive viral gene flow was observed in our study between Indiana and Ohio exhibition pigs. The lack of viral migration observed between Indiana/Ohio and Iowa, Kentucky, and Texas could be related to the very low sampling of IAV-exS in these 3 states, and further surveillance beyond Indiana and Ohio is needed. It is also important to note that the spatial unevenness of sampling of IAV-exS in our study also suggests that our analysis detected only a fraction of the total number of viral transmission events between commercial and exhibition swine during 2009–2013 and that it likely underestimated the frequency of reassortment within exhibition swine, as well.
Ideally, knowledge of IAV-exS dynamics would inform annual predictions of the threat of zoonotic influenza A viruses for humans. An observable increase in the proportion of H3-G1 viruses identified in exhibition swine did occur at the time of H3N2v emergence in humans in 2011, consistent with prior findings that the presence of the virus in exhibition swine is associated with human infections [13, 29]. However, variations in sampling in exhibition swine over this period make it difficult to compare genotype frequencies between years. IAV-comS surveillance also is rarely population based and is complicated by selection biases, undermining efforts to track changes in the prevalence of particular genotypes. Risk assessment is further complicated by the local variability of IAV-exS in the United States and the rarity of a single virus dominating each year at a national scale (Figure 1A). Instead, multiple diverse influenza A virus lineages cocirculate in exhibition swine each year, vary from state to state, and undergo further reassortment. Efforts to mitigate risk are likewise complicated, as IAV-exS is derived from multiple geographically diverse commercial swine sources, and it is not possible to target a single key point of entry. However, our findings provide support for one prediction: given the high rate of detection and reassortment of the pdm09 MP segment in both commercial and exhibition swine, it is likely that H3-G1 and other viruses with zoonotic potential will continue to circulate, reemerge, and transmit between commercial and exhibition swine, presenting a long-term threat to high-risk groups, including children, and requiring continued vigilance.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank all our participating agricultural fairs; the field staff who tirelessly collected these samples; Sarah Nelson and Jody Edwards, for their laboratory expertise and technical abilities; Aaron Schwartzbard, for his technical assistance in making figures in R.
Disclaimer. The opinions expressed in this article are the authors' own and do not reflect the views of the Centers for Disease Control, the Department of Health and Human Services, or the US government. Any mention of trade names or commercial products is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Financial support. This work was supported by the Centers of Excellence for Influenza Research and Surveillance, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Department of Health and Human Services (DHHS; contracts HHSN266200700007C and HHSN272201400006C); the NIAID, NIH, DHHS (contract HHSN272200900007C); the Multinational Influenza Seasonal Mortality Study, an ongoing international collaborative effort to understand influenza epidemiology and evolution, led by the Fogarty International Center, NIH, with funding from the Office of Global Affairs at the Department of Health and Human Services; and the National Veterinary Services Laboratories, Animal and Plant Health Inspection Service, US Department of Agriculture.
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.Standardization of terminology for the variant A(H3N2) virus recently infecting humans. Joint announcement of FAO, OIW, WHO. 23 December 2011. http://www.who.int/influenza/gisrs_laboratory/terminology_ah3n2v/en/. Accessed 10 August 2015.
- 2.Blanton L, Brammer L, Smith S et al. . Update: influenza activity—United States and worldwide, May 18–September 20, 2014. MMWR Morb Mortal Wkly Rep 2014; 63:861–4. [PMC free article] [PubMed] [Google Scholar]
- 3.Jhung MA, Epperson S, Biggerstaff M et al. . Outbreak of variant influenza A(H3N2) virus in the United States. Clin Infect Dis 2013; 57:1703–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garten RJ, Davis CT, Russell CA et al. . Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009; 325:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith GJD, Vijaykrishna D, Bahl J et al. . Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009; 459:1122–5. [DOI] [PubMed] [Google Scholar]
- 6.Vijaykrishna D, Poon LLM, Zhu HC et al. . Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 2010; 328:1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harder TC, Grosse Beilage E, Lange E et al. . Expanded cocirculation of stable subtypes, emerging lineages, and new sporadic reassortants of porcine influenza viruses in swine populations in Northwest Germany. J Virol 2013; 87:10460–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ducatez MF, Hause B, Stigger-Rosser E et al. . Multiple reassortment between pandemic (H1N1) 2009 and endemic influenza viruses in pigs, United States. Emerg Infect Dis 2011; 17:1624–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson MI, Vincent AL, Kitikoon P, Holmes EC, Gramer MR. Evolution of novel reassortant A/H3N2 influenza viruses in North American swine and humans, 2009–2011. J Virol 2012; 86:8872–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitikoon P, Nelson MI, Killian ML et al. . Genotype patterns of contemporary reassorted H3N2 virus in US swine. J Gen Virol 2013; 94(Pt 6):1236–41. [DOI] [PubMed] [Google Scholar]
- 11.Marsh B, Scheftel J, Bender J et al. . Measures to minimize influenza transmission at Swine exhibitions, 2014. 2014.
- 12.Anderson TK, Nelson MI, Kitikoon P, Swenson SL, Korslund JA, Vincent AL. Population dynamics of cocirculating swine influenza A viruses in the United States from 2009 to 2012. Influenza Other Respi Viruses 2013; 7(suppl 4):42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowman AS, Nelson SW, Page SL et al. . Swine-to-human transmission of influenza A(H3N2) virus at agricultural fairs, Ohio, USA, 2012. Emerg Infect Dis 2014; 20:1472–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowman AS, Nolting JM, Nelson SW, Slemons RD. Subclinical influenza virus A infections in pigs exhibited at agricultural fairs, Ohio, USA, 2009–2011. Emerg Infect Dis 2012; 18:1945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou B, Donnelly ME, Scholes DT et al. . Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol 2009; 83:10309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djikeng A, Halpin R, Kuzmickas R et al. . Viral genome sequencing by random priming methods. BMC Genomics 2008; 9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Squires RB, Noronha J, Hunt V et al. . Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respi Viruses 2012; 6:404–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bao Y, Bolotov P, Dernovoy D et al. . The influenza virus resource at the National Center for Biotechnology Information. J Virol 2008; 82:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Swofford DL. Phylogenetic analysis using parsimony (*and other methods). Sunderland, MA: Sinauer Associates, 2003. [Google Scholar]
- 21.Nelson MI, Stratton J, Killian ML, Janas-Martindale A, Vincent AL. Continual reintroduction of human pandemic H1N1 influenza A viruses into Swine in the United States, 2009 to 2014. J Virol 2015; 89:6218–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006; 22:2688–90. [DOI] [PubMed] [Google Scholar]
- 23.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28:2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012; 29:1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suchard MA, Rambaut A. Many-core algorithms for statistical phylogenetics. Bioinformatics 2009; 25:1370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minin VN, Suchard MA. Counting labeled transitions in continuous-time Markov models of evolution. J Math Biol 2008; 56:391–412. [DOI] [PubMed] [Google Scholar]
- 27.Bielejec F, Rambaut A, Suchard MA, Lemey P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011; 27:2910–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng Z, Gomez J, Bowman AS et al. . Antigenic characterization of H3N2 influenza A viruses from Ohio agricultural fairs. J Virol 2013; 87:7655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bowman AS, Sreevatsan S, Killian ML et al. . Molecular evidence for interspecies transmission of H3N2pM/H3N2v influenza A viruses at an Ohio agricultural fair, July 2012. Emerg Microbes Infect 2012; 1:e33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson MI, Lemey P, Tan Y et al. . Spatial dynamics of human-origin H1 influenza A virus in North American swine. PLoS Path 2011; 7:e1002077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.