

Abstract
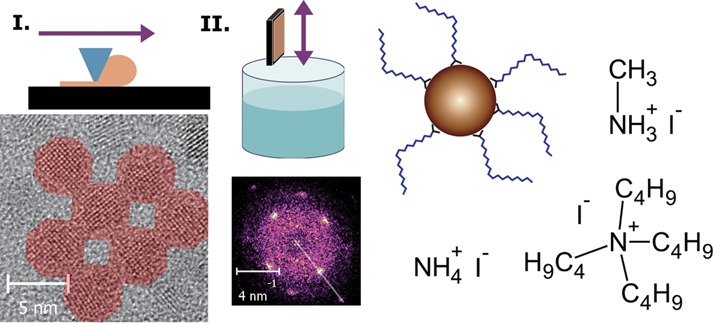
In the past years, halide capping became one of the most promising strategies to passivate the surface of colloidal quantum dots (CQDs) in thin films to be used for electronic and optoelectronic device fabrication. This is due to the convenient processing, strong n-type characteristics, and ambient stability of the devices. Here, we investigate the effect of three counterions (ammonium, methylammonium, and tetrabutylammonium) in iodide salts used for treating CQD thin films and shed light on the mechanism of the ligand exchange. We obtain two- and three-dimensional square-packed PbS CQD superlattices with epitaxial merging of nearest neighbor CQDs as a direct outcome of the ligand-exchange reaction and show that the order in the layer can be controlled by the nature of the counterion. Furthermore, we demonstrate that the acidity of the environment plays an important role in the substitution of the carboxylates by iodide ions at the surface of lead chalcogenide quantum dots. Tetrabutylammonium iodide shows lower reactivity compared to methylammonium and ammonium iodide due to the nonacidity of the cation, which eventually leads to higher order but also poorer carrier transport due to incomplete removal of the pristine ligands in the QD thin film. Finally, we show that single-step blade-coating and immersion in a ligand exchange solution such as the one containing methylammonium iodide can be used to fabricate well performing bottom-gate/bottom-contact PbS CQD field effect transistors with record subthreshold swing.
Keywords: colloidal quantum dots, lead sulfide, iodide-capped, transport, ligand exchange
In the past decade, colloidal quantum dot (CQDs) solids have attracted much interest for their promising applicability in solution-processed transistors, solar cells, and other optoelectronic devices.1−4 These materials are built up by forcing the individual quantum dots into proximity with each other by replacing the long capping ligands with shorter ones. This ligand exchange (LE) simultaneously affects the electronic coupling and the surface composition, altogether determining the electronic properties of the material; hence, a detailed understanding of the nature of the ligands, the LE process, and careful design of the procedure is required.
The most widely used ligands are mono- or bifunctional thiols (e.g., ethanedithiol (EDT), 3-mercaptopropionic acid (MPA), or benzenedithiol (BDT)), which bind strongly to the surface of the (usually lead- or cadmium-terminated) quantum dots.5−7 Often, however, the inherently high sensitivity of these organic ligands to the oxygen and moisture imparts electronic instability in the whole CQD solid. The very high ambient sensitivity of the ligands has even misled researchers when determining the dominant charge carrier in these solids.8
Recently, the use of halides during the CQD synthesis was suggested to passivate surface traps.9,10 Subsequently, these halides were found to replace the original ligands, resulting in n-type CQD solids.11 This partial halide shell provides a very good passivation of the CQD surface resulting in CQD solids with excellent air stability; the effectiveness of such treatment was demonstrated with the fabrication of highly efficient (8.5%) and ambient-stable solar cells.12−14 The insertion of heteroatoms onto the surface of the CQDs modifies their electronic structure and can be considered a sort of “doping”.15 Such doping was also exploited to obtain increased thermoelectric effects using hydrochloric acid treated lead chalcogenide CQDs.16 Following these early successes, solution-phase LE to halides, pseudohalides, and halometallates has also been accomplished.17−20
Even in such relatively simple ligand types as atomic halides, a large range of possibilities is provided by the broad variability of the organic salts and possible solvents. So far, most works have been performed using tetrabutylammonium iodide (TBAI) dissolved in methanol. However, it was noticed that use of methanol or 2-propanol resulted in different transport properties, but no light was shed to the origin of this difference.21 However, the nontrivial general role of the solvent in the LE process has also been reported.22−24 Methanol (compared to other common solvents) has a superior ability to strip the CQD surface from weakly bound ligands, while acetonitrile in the same process is practically inactive.
Here we report on the efficiency of solid-state LE of PbS CQDs with three different iodide salts, namely, ammonium iodide (NH4I), methylammonium iodide (CH3NH3+I–, MAI), and tetrabutylammonium iodide ((C4H9)4N+I–, TBAI). The first two have been used so far only in solution-phase LE, with limited success.20,25 To prove the effectiveness of the LE, in addition to the standard analytical techniques such as infrared absorption and HRTEM, we fabricated field-effect transistors (FETs) and investigated the photoexcitation dynamics of the CQD solids. Here, we reveal the different degrees of ordering upon the use of different salts, prove that the counterions play a role in the LE, and show the connection between the reactivity difference and the actual device performance. Furthermore, we demonstrate that the LE is hindered in aprotic conditions, showing that the process is acid-catalyzed, explaining, in this way, the role of the counterions. Importantly, we achieve epitaxial fusion of the CQDs and growth of multilayer superlattices by exposing the as-deposited OA capped CQDs to the LE solutions and show that the order depends on the reaction conditions. The good superlattice organization obtained with MAI is also evident in the fabricated FETs, which display electron mobility up to 0.05 cm2/(V s), an n-channel on/off ratio up to 106, and subthreshold swing as low as 2.4 V/dec. These findings might ease the fabrication in the near future of cheap and efficient CQD solar cells.
Results and Discussion
First, the influence of different cations in iodide salts used for the LE on the nanostructure of the layer is investigated. Transmission electron microscopy (TEM) images of films prepared with the three different salts are reported in Figure 1a–c. Here, large domains of QDs organized in square-packed lattices can be observed. Since the oleic acid capped particles arrange in a hexagonal close-packed fashion, as can be seen in Figure S1 of the Supporting Information, this square-lattice arrangement is clearly induced during the LE process. Interestingly, the domain size appears to depend on the applied counterion in the LE process (Figure 1a–c). Domains of only a few CQDs and a single layer are observed for the sample fabricated with NH4I (Figure 1a), extended lattices and simultaneous formation of double layers are found in MAI (Figure 1b, see the darker areas on the right side of the image), and even larger domains and multiple layers are present in the case of TBAI-treated samples (Figure 1c).
Figure 1.

(a–c) TEM micrographs of PbS CQD solids formed via exposure to (a) NH4I, (b) MAI, and (c) TBAI; (d, e) real-space and Fourier-transformed HRTEM images, and (f) an extracted profile from the FT image of a square domain showing that the superlattice consists of CQDs oriented the same direction after treatment with MAI; (g) high-resolution image on the oriented assembly showing epitaxially connected CQDs; (h) schematic structure with the main facets of a PbS CQD.
A high-resolution transmission electron microscopy (HRTEM) image of the QDs assembled with MAI and its 2D Fourier transformation (FT) are displayed in parts d and e, respectively, of Figure 1. The real-space image shows that the CQDs are oriented in the same crystallographic direction. The combination of two patterns is observed on the FT image; in addition to the broad first- and second-order pattern of the rock-salt PbS structure, the smaller features related to the larger spacing of the superlattice are also visible around zero order (since the unit vectors of the superlattice are roughly 15 times longer than the PbS lattice vectors). The profile extracted from the FT image is shown on panel f, demonstrating that the directions of the two diffraction patterns coincide and that the primitive vectors of the superlattice lie in the <100> directions of the PbS rock-salt lattice (see also Figure 1g). Analogous self-organization along <100> axes has been observed previously for square26−30 and honeycomb31 PbSe CQD superlattices. Both PbS and PbSe CQDs are usually approximated as truncated octahedron or rhombicuboctahedron with three types of facets: {100}, {110}, and {111} (Figure 1h). Among those, {100} is the least acidic with Pb coordination number 5, whereas Pb atoms on {110} and {111} facets have coordination numbers 4 and 3, respectively. This should lead to a higher affinity of anionic ligand species to the latter facets leaving {100} facets more opened. Zherebetskyy et al. calculated that unpassivated {100} facets of PbS CQDs have approximately 2 times lower surface energy than the {111} facets, and ligands can be easily removed from the {100} surface.32 These facts together explain the observed self-organization of lead chalcogenide CQDs along <100> axes upon the exchange of bulky organic ligands with small inorganic anions.
Furthermore, the honeycomb PbSe superlattices reported in ref (31) fit these observation as well, since they can be considered as formed by the same primitive cubic superlattice viewed from the <111> direction. This orientation is formed and stabilized by the affinity difference of the OA to the different facets and the preferred exposure of the {111} facets to the surrounding medium due to the higher OA coverage. In our case, the reactive in situ LE to small anions allows a minimum amount of bulky organic leftovers, and the substrate used in these experiments has no preference toward the capped facets, leading to a denser structure, where the <100> axis is perpendicular to the surface. Similarly, drying of colloidal solutions of PbS QDs with undetectable amounts of bulky organic ligands (prepared by solution-phase LE to PbI3– anions, with MA+ as counterion)18 also forms square superlattices with CQD orientation along the <100> axes (Figure S2).
The higher resolution image (Figure 1g) reveals that the CQDs’ crystal lattice is continuous at the necking points; in other words, the CQDs are epitaxially fused together along the {100} facets. The superlattice spacing is found to be about 4.5 nm for iodide-capped CQDs of 3.8 nm size (Figure S3) and does not depend on the cation employed in the case of ordered domains. An increase of bond length in CQD superlattices with face-to-face attachment was observed and discussed also in refs (31) and (33); briefly, after atomic orientation of the CQDs, a gradual removal of remaining ligands occurs, which is followed by diffusion of Pb and S atoms into the interdot gap. Obviously, the interparticle distance right before necking depends on the original ligand size. This explains why the observed bond lengths (neck stretching) are constant for all iodide-capped CQDs and gives a convenient tool for further controlling the nanostructure of the layer.
As mentioned above, the CQDs appear to have reoriented during the LE process in order to interact between each other with the same facets, minimizing in this way the overall surface area, analogously to the “oriented attachment” occurring in solution.33 This implies that the self-orientation of the single QDs induces a macroscopic orientation at the superlattice level. The orientation is so powerful that it occurs even during a rapid LE employed in this research. It is important to notice that this assembly occurs in three dimensions, as seen from the multilayer structures on Figure 1b,c, opening up exciting possibilities in designing new QD-based metamaterials.
Absorbance, steady-state, and time-resolved photoluminescence of the three samples prepared with different iodide salts were recorded to understand if the observed differences in the nanostructures affect the electronic configuration and therefore the photophysical properties. Figure S4a reports the absorption spectra displaying a well-pronounced excitonic peak for all three samples and identical peak position (1070 nm). The photoluminescence spectra (Figure S4b) of the different samples are also practically identical with peaks around 1240 nm. Furthermore, the time-resolved measurements (Figure S4c) performed on the samples in inert atmosphere also display very similar lifetimes. The same peak position of the absorption and the photoluminesce spectra and the similar excitation lifetimes suggest that the samples are electronically identical (the emission originates from the same state) and that the degree of electronic wave function delocalization; thus, the average electronic coupling is also identical.
Recently, a dependence of the PL lifetime on the TBAI concentration was reported by Bawendi et al. for PbS CQDs.34 For concentrations similar to what is used in this work, much slower decay was observed. Besides their instrument having a worse resolution time, the higher coupling achieved here through longer exposure to the LE solution and more careful washing may be the origin of the difference.
For the prospective applications, we sought to shed light onto the influence of the counterions on the charge-transport properties. Field-effect transistors (FETs) are exceptionally sensitive to the material’s quality; therefore they can be used as a general testing method, although one has to consider numerous factors that influence the measured current. We fabricated FETs of PbS CQDs using blade-coating, which is known to be an efficient and up-scalable method for the preparation of solution-based thin films. This one-step deposition allows simplification of the fabrication of CQDs transistors, going away from the material-costly spin-coating method and from the time-consuming layer-by-layer approach.8,35 Details of the fabrication are reported in the Experimental Section; briefly, we deposit the desired thickness on a prepatterned Si/SiO2 substrate, which also acts as the gate electrode, and the CQD layer is then dipped into the LE solution.
Figure 2a shows the transfer curves of FETs fabricated with 30 nm PbS films treated with the three different solutions. We compare the subthreshold and the linear regime behavior, using the subthreshold swing and mobility values extracted from the curves in Figure 2b. All transfer curves show similar main characteristics, electron-dominated weak ambipolarity, with a substantially different quality of the transport for the different ligands. In contrast to the CQD organization trend observed on the TEM images, the maximum currents show a TBAI < NH4I ≈ MAI trend, which is confirmed by the extracted mobility values. The subthreshold swing was found to be the lowest for the FET prepared using MAI and was the highest for the one with NH4I. There is only a marginal difference between the “on” state behaviors of the NH4I- and MAI-treated samples, but their “off” currents are considerably different. The FET prepared with TBAI showed instrument-limited “off” current, likely hiding the real subthreshold region, which might be due to the overall lower conductivity. One and 3 orders of magnitude higher “off” currents were measured in the MAI and NH4I samples, respectively; the increase may be explained with the generation (or leftover) of charged species in the channel during the LE.
Figure 2.

(a) ID–VG curves of PbS CQD FETs fabricated using different LE solutions; (b) extracted linear regime electron mobility and n-channel subthreshold swing values for the different ligands; (c) FTIR spectra of the corresponding samples and the untreated films showing traces of oleic acid.
The subthreshold swing is a direct measure of the sub-bandgap trap density in the thin film and at its interface with the dielectric.36 Since the SiO2 surface is the same in all fabricated devices, the bulk properties of the films account for the difference in the swing. The higher swing (lower logarithmic slope in the subthreshold region) means higher trap density or, in other words, more states to be filled before the Fermi level reaches the conduction band. Based on the TEM results (Figure 1), the increased energy disorder due to positional disorder may also account for the different trap density, resulting in a wider and deeper distribution of energy levels. However, the significance of this influence is minimal, since the transport in disordered systems generally occurs through states below the conduction band (above the valence band), leading to an apparent shallower band tail.37 Comparing the hysteresis of the MAI- and TBAI-treated samples (Figure S5), it is clear that the subthreshold behavior is similar in the forward and reverse scans for both samples (ruling out large difference in the sub-bandgap energy distribution and significant filling of these states). However, the difference between the forward and reverse slopes is significant, which correlates with filling the trap states below the transport edge, causing a mobility increase.7 This shift is higher in the TBAI-based FET (showing 10-fold mobility increase compared to the 2.7-fold in the MAI-based FET); this fact together with the minimal difference in the sub-bandgap trap density suggests the presence of deep trap states. A difference in the doping levels may also explain the overall higher conductivity achieved with MAI treatment compared to the TBAI one. However, a change in the dopant concentration is expected to shift the ratio of the hole and electron mobilities, which is roughly the same for TBAI and MAI. Consequently, the low conductivity of the TBAI sample seems to be related to a higher amount of traps. In the case of NH4I, the electron/hole balance changes significantly respect to the other two ligands (see the p-channel transfer curves and the mobility ratios on Figure S6), indicating a probable extrinsic doping (leftover of charge species) which is also in agreement with the high off current.
FTIR spectra of the films were taken (Figure 2c) to check the completeness of the LE. The peaks around 2900 and 1400–1600 cm–1 are signatures of the remaining oleic acid after LE for the same 1 min exposure time; the significant presence of ammonium ions can be ruled out since the NH or CN signatures are missing. Remains of OA are seen in the NH4I-treated and in increased amount in the TBAI-treated samples, suggesting incomplete LE, while no traces of oleic acid can be detected in samples prepared using MAI.
Altogether, these findings suggest that MAI is superior to both NH4I and TBAI for the fabrication of well performing CQD FETs. Therefore, the deposition process was further optimized for high mobility and low subthreshold swing (Figure S7). In Figure 3 are reported the output and transfer characteristics of the best device. A n-channel on/off ratio around 106 and subthreshold swing as low as 2.4 V/dec are the most relevant features. The latter is the lowest value reported for CQD transistors using SiO2 gating. In these samples, the electron mobility is around 0.5 cm2/(V s), while the hole mobility is much lower (3 × 10–5 cm2/(V s), Figure S8).
Figure 3.
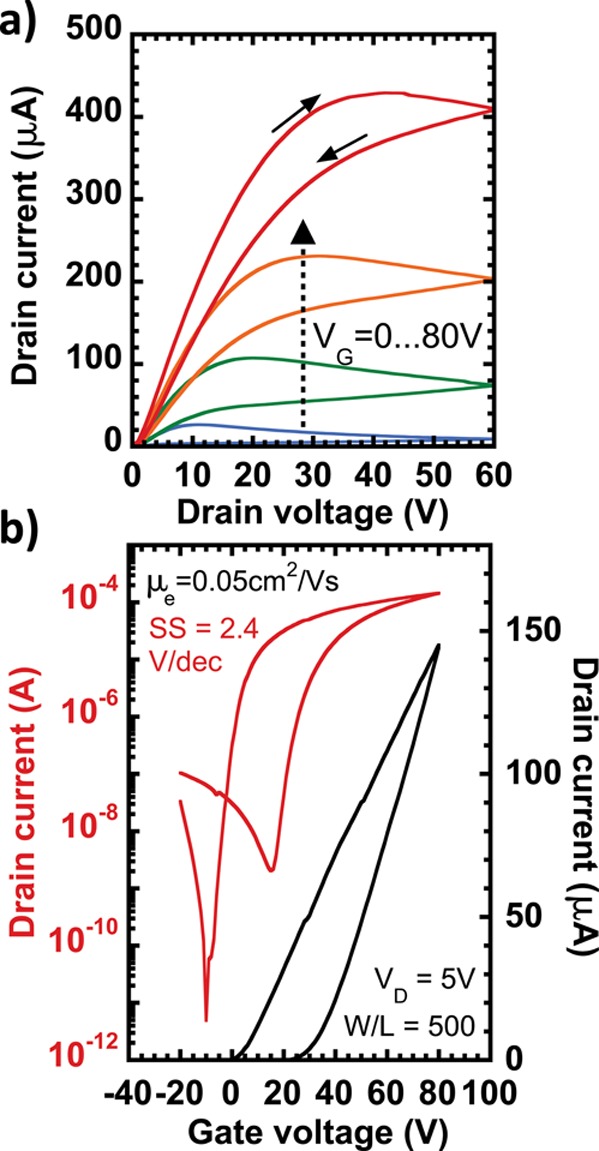
(a) ID–VD (output) and (b) ID–VG (transfer) characteristics measured in one of the champion MAI-treated devices. The average electron mobility is 0.043 ± 0.011 cm2/(V s) and the average subthreshold swing is 4.0 ± 0.9 V/dec calculated with 95% confidence from 14 samples.
As observed already with the HRTEM reported in Figure 1 treatments of the same duration result in films with different disorder depending on the couterion used. Also, partial delamination and cracking of the films is observed frequently using NH4I and only very occasionally using MAI. These phenomena suggest that the LE for NH4I is too fast to leave sufficient time for a full reorganization of the CQDs; at the same time, the LE using TBAI appears to be mild, but incomplete, resulting in many defect states and large oleic acid leftover. These observations could indicate a reactivity difference between the three salts; we therefore investigated if the LE is acid catalyzed and if the acidity of the cations influences the exchange rate.
To test this hypothesis, we performed experiments where no protic species are present (namely TBAI, the nonprotic salt was dissolved in aprotic acetonitrile). By measuring the transistor fabricated in this way, we found that the LE does not take place in fully aprotic conditions, even up to 10 min exposure, as shown in Figure 4a. A large amount of oleic acid was found in the film (see the bound COO– peak at around 1530 cm–1 and the CH stretching peaks around 2900 cm–1 (Figure 4b), and the film remained soluble in apolar solvents. We observed the same inactivity using other polar aprotic solvents, such as propylene carbonate and γ-butyrolactone (Figure S9). However, upon addition of formic acid to the acetonitrile solution, the LE takes place as in methanol, and the film turns conductive; the conductivity in both n- and p-channels depends on the acid concentration (see Figure 4a). Importantly, the sign of the charge carriers in these experiments is significantly different from that of the formic acid only treatment, which suggests that the CQDs are not capped by formate instead of iodide, but the former assists the attachment of the latter. Proof for the removal of the oleic acid and the absence of bound formate species is obtained by FTIR measurements showing no signs of either CH or bound COO– peaks (Figure 4b).
Figure 4.

(a) ID–VG (transfer) curves of FETs fabricated using acetonitrile solutions containing 10 mM TBAI and 0, 1, or 10 mM HCOOH; (b) corresponding FTIR spectra showing the removal of oleic acid and the lack of carboxylate groups in the iodide-treated sample in the presence of acid; (c) ID–VG (transfer) curves measured in layers treated with acetonitrile solutions containing 10 mM MAI and 1, 10, or 100 mM TEA; (d) corresponding FTIR spectra showing different amounts of remaining oleic acid at different pH’s. All samples were fabricated from the same solution and had the same layer thickness treated for the exact same time, for perfect comparison. All curves were measured at VD = 5 V.
The fast oleic acid–formic acid replacement is driven by the higher adsorption energy of the shorter aliphatic ligand in polar solvent, since the functional groups are the same.38 Energetically, the same stands for the oleate–iodide couple (given the high lattice energy of lead iodide), but the lack of acidic species in the mixture seems to hinder the reaction. Based on this difference, we suggest that the presence of acidic species in the solution can stabilize ligand removal analogously to the equilibrium ligand stripping achieved in ref (30). The surface is then partially “naked”, and the equilibrium will be shifted toward capping with the strongest binding ligand present in the solution in the order: oleate < formate < iodide. Whether the stabilization is of protic or Lewis acidic origin remains a topic of future investigations.
Furthermore, we found that LE occurs when the PbS CQD film is exposed to MAI dissolved in acetonitrile (Figure S10), likely through the mediation of the acidic counterion. If the reaction is indeed happening through the contribution of acidic species, addition of a base (with low affinity to the surface) will decrease the LE reaction rate. To demonstrate the deactivation of the LE solution, we added different concentrations of triethylamine (TEA) in an acetonitrile solution of MAI. With this, we expect that the acidity of the MA+ will be masked by its interaction with the strongest base present, the TEA. We found that films treated at higher pH show lower conductivity after the same exposure time (Figure 4c), and the amount of oleic acid remaining in the film increases accordingly (Figure 4d). The simultaneous variation of the p-channel and n-channel currents in the transistor suggests increasing resistance in the layer, and not doping or trapping effects, which would influence only one of the channels. Moreover, the CH2 and CH3 peak ratios, the similar trend in the COO– peak, and the lack of CN vibration also rule out TEA being adsorbed on the surface; therefore the incomplete exchange of the OA is the cause of the lower current.
The same consideration about the pH effect on ligand removal is relevant not only for the original organic ligands, but also for the new inorganic anionic species (e.g., PbI3–). Since ligand removal can be promoted/hindered by the presence/absence of acidic species, one can control the rate of the CQDs’ “oriented attachment” by solvent acidity. In order to prove it, we used two solutions of MAPbI3-capped PbS CQDs: the first in net aprotic protophobic propylene carbonate (PC) and the second in PC with addition of N-methylformamide (MFA/PC = 1:100), which is a strong H-bond donor solvent (one of the strongest among the nonalcohols). We observed the previously described square-ordered superlattices in both cases, but the level of ordering was noticeably different (Figure S2a,b). The sample deposited from pure PC demonstrated a relatively low level of ordering, whereas the MFA additive allowed notable extension of the ordered domains of QDs. In this case, the nonacidic PC does not provide a driving force for the ligand removal in solution; thus, only a low number of CQDs have ligandless, reactive facets that can participate in the “oriented attachment”. On the other hand, PbI3– ions are stabilized in solution in the presence of MFA,18 increasing the number of “naked” CQDs, which is a prerequisite for the “oriented attachment” and the formation of large domains.
Clearly, the reactivity of the environment affects the CQDs ordering during in situ LE as well as the transport in the layer. The use of TBAI, where only the weak acid solvent catalyzes the LE, leads to extended superlattice domains, since more time is available for the “oriented attachment”. However, when considering the electronic properties, the lower reactivity is less attractive, since low conductivity and worse material quality (in sense of high trap density) are achieved. The lower acidity of 2-propanol than methanol can also account for the increased trap density reported in the literature.21 Longer (e.g., 10 min) exposure to the TBAI–methanol solution leads to improvement in the transport (Figure S8), but the mobility is still below that of in the MAI sample. This suggests that there may be other factors causing the dissimilar conductivity, such as the TBA+ ions being unable to diffuse fast through the layer. If the solubilized oleates are not replaced by iodide ions due to diffusion problems, the unbalanced surface chemistry will likely cause charge trapping. Consequently, if one aims for one-step deposition of thicker layers (e.g., hundreds of nanometers for a solar cell) at a reasonable time scale, TBAI has to be dropped and a different salt has to be chosen.
The acidic ammonium and methylammonium cations increase the reactivity of the LE solution, leading to higher disorder, with FET mobilities invariant with the exposure time within the measurement reproducibility limits. However, the “off” current is very high in the NH4I-treated sample, which is either result of the presence of mobile ionic species or stems from the increased n-type doping (see Figure S6). As we have demonstrated here a fine-tuning of the reactivity of the ligands may be obtain by tuning the acidity of the solution.
Conclusions
To conclude, we have shown that epitaxial growth of extended CQD superlattices can be obtained simply by immersing a PbS CQD film into a solution of an iodine salt and that these ordered regions extend in 3D leading to largely improved FETs subthreshold behavior. We found that TBAI dissolved in methanol shows low reactivity, not providing complete LE in a short time, while both MAI and NH4I are capable thereof. We delivered proof for an acidic catalysis mechanism of the exchange of carboxylates on the surface of lead chalcogenides to iodide ions, likely through improved organic ligand displacement. Furthermore, the higher reactivity of NH4I compared to MAI is due to the higher acidity of the cation which is responsible for a higher disorder and worse carrier transport in the layers. Finally, we showed that single-step blade-coating and immersion in a LE solution can be used to fabricate well performing bottom-gate/bottom-contact PbS CQD field effect transistors.
Experimental Section
Materials
The reagents and solvents mentioned below were used as received. Methylammonium iodide was prepared following a literature procedure.39 Briefly, 24 mL of methylamine (33% in ethanol) and 10 mL of HI (57% in water) were mixed in 100 mL of ethanol at 0 °C with constant stirring for 2 h. The salt was formed upon evaporation of the solvent at 60 °C on a hot plate, and the product was washed three times with ethyl ether and dried in vacuum at 60 °C overnight. PbS quantum dots of 3.8 nm diameter capped with oleic acid and dispersed in chloroform were synthesized as reported previously using a hot-injection method.40 A lead precursor solution consisting of 1.516 g of PbAc2·H2O in 50 mL of ODE and 4.5 mL of OA was vacuum dried at 120 °C in a three-neck reaction flask. The temperature was subsequently raised to 145 °C, after which a sulfur precursor of 0.420 mL of TMS2S in 10 mL of ODE was quickly injected and the flask cooled in a water bath. Toluene and ethanol were added to the solution followed by centrifugation to separate the QDs. Two more washing steps were performed by redispersion in toluene and precipitation by ethanol, and then the particles were dried. Redispersion in anhydrous chloroform and precipitation with methanol resulted in the desired cleanness, followed by redispersion in anhydrous chloroform. Anhydrous solvents were also used for the fabrication of samples for spectroscopy and transistors.
PbS CQDs for LE with MAPbI3 were prepared in the same way except that 30 mL of ODE and 20 mL of OA were used, whereas injection was carried out at 120 °C. After washing, CQDs were redissolved in hexane. LE was performed according to the literature procedure.18 MAI (1.5 mmol) and PbI2 (1.5 mmol) were dissolved in 30 mL of MFA. The solution of organically capped PbS CQDs in hexane (150 mg/30 mL) was added to the ligand solution. The biphasic system was stirred vigorously for 12 h until the CQDs were completely transferred from the nonpolar to the polar organic phase. The hexane layer was removed, and the MFA solution was rinsed three times with pure hexane. To remove the excess of metal halide ligands, the particles were precipitated from MFA by acetone, centrifuged, and redispersed in 1.5 mL of MFA.
TEM Characterization
TEM micrographs were taken using a FEI Tecnai G2, a JEOL JEM-2010, or a Hitachi HD2700CS (STEM) instrument; Fourier-transform images were calculated from selected areas of the micrographs. OA-capped PbS CQDs were deposited onto carbon grids by drop-casting from a dilute solution, followed by immersion in the LE solution and solvent. The MFA solution was diluted 100 times by propylene carbonate, and the samples were dried at 80 °C in vacuo. For PC solutions of CQDs, we additionally precipitated MAPbI3-capped particles from MFA by toluene and redissolved them in pure PC. Samples from PC have been dried on TEM grids at 85 °C in vacuo.
FET Fabrication
For device fabrication, oleic acid capped quantum dots were deposited by blade-coating on HMDS-treated Si/SiO2 wafers with prepatterned gold electrodes.41 The optimal parameters for the deposition were found to be 10 mg/mL of CQD concentration, 25 mm/s blade speed, and 30 °C substrate temperature, resulting in 20–22 nm thickness. The films were immersed in a 10 mM solution of the ligand in methanol (or acetonitrile) for 1 min and then washed with clean solvent for another minute. The reaction time was measured precisely to get comparable results. These parameters were used for all devices unless specifically mentioned. The fabrication was completed by baking the samples at 120 °C for 20 min, which does not sinter the CQDs (Figure S12). All steps took place in a glovebox, as well as the transport measurements. For this, an Agilent E5262A semiconductor parameter analyzer was used. The mobility values were calculated from the linear regime transfer curves from the forward scan (|VD| increases in time) to avoid the screening effect of trapped carriers.42 Similar behavior observed in >10 devices confirmed the reproducibility.
FTIR measurements were performed on films deposited on double-polished silicon wafers following the protocol used for FET fabrication. The measurements were done using a Bruker IFS 66/v in transmission mode or with a Shimadzu IRTracer100-ATR averaging 50 scans.
Photoluminescence Spectroscopy
Photoluminescence measurements were performed on samples prepared on quartz substrates by repeating the deposition 4× to achieve ∼90 nm thickness. The second harmonic (400 nm) of a Ti:sapphire laser (Coherent, Mira 900, repetition rate 76 MHz) was used to excite the samples. The optical emission was spectrally dispersed in a single spectrometer and recorded by a cooled array detector (Andor, iDus 1.7 μm). Time-resolved PL spectra were detected using a Hamamatsu streak camera with a cathode sensitive to near-IR radiation. The excitation density was reduced to 7 μJ/cm2 by a neutral density filter, and the samples were encapsulated to prevent photodegradation during the experiment. All PL transients were fitted with biexponential functions.
Acknowledgments
We thank Arjen Kamp for technical support. The Groningen team is grateful for the financial support of the European Research Council (ERC Starting Grant “Hy-SPOD” No. 306983). M.V.K. acknowledges financial support from the Swiss National Science Foundation (Grant No. 200021_143638) and the European Research Council (ERC Starting Grant “NANOSOLID” No. 306733). D.N.D. acknowledges the European Union for a Marie Curie Fellowship (IIF-GA-2012-330524). We acknowledge Dr. Frank Krumeich for electron microscopy conducted at ETH Zurich (at the Scientific Center for Optical and Electron Microscopy) and Dr. Maryna Bodnarchuk for electron microscopy at the Empa Electron Microscopy Center.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b04547.
Additional TEM images, FET characteristics, and UV–vis absorption (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Talapin D. V.; Murray C. B. PbSe Nanocrystal Solids for n- and p-channel Thin Film Field-Effect Transistors. Science 2005, 310, 86–89 10.1126/science.1116703. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Law M.; Beard M. C.; Song Q.; Reese M. O.; Ellingson R. J.; Nozik A. J. Schottky Solar Cells Based on Colloidal Nanocrystal Films. Nano Lett. 2008, 8, 3488–3492 10.1021/nl802476m. [DOI] [PubMed] [Google Scholar]
- Szendrei K.; Cordella F.; Kovalenko M. V.; Böberl M.; Hesser G.; Yarema M.; Jarzab D.; Mikhnenko O. V.; Gocalinska A.; Saba M.; et al. Solution-Processable Near-IR Photodetectors Based on Electron Transfer from PbS Nanocrystals to Fullerene Derivatives. Adv. Mater. 2009, 21, 683–687 10.1002/adma.200801752. [DOI] [Google Scholar]
- Shirasaki Y.; Supran G. J.; Bawendi M. G.; Bulovic V. Emergence of Colloidal Quantum-dot Light-emitting Technologies. Nat. Photonics 2012, 7, 13–23 10.1038/nphoton.2012.328. [DOI] [Google Scholar]
- Luther J. M.; Law M.; Song Q.; Perkins C. L.; Beard M. C.; Nozik A. J. Structural, Optical and Electrical Properties of Self-assembled Films of PbSe Nanocrystals Treated with 1,2-Ethanedithiol. ACS Nano 2008, 2, 271–280 10.1021/nn7003348. [DOI] [PubMed] [Google Scholar]
- Barkhouse D. A. R.; Pattantyus-Abraham A. G.; Levina L.; Sargent E. H. Thiols Passivate Recombination Centers in Colloidal Quantum Dots Leading to Enhanced Photovoltaic Device Efficiency. ACS Nano 2008, 2, 2356–2362 10.1021/nn800471c. [DOI] [PubMed] [Google Scholar]
- Bisri S. Z.; Piliego C.; Yarema M.; Heiss W.; Loi M. A. Low Driving Voltage and High Mobility Ambipolar Field-Effect Transistors with PbS Colloidal Nanocrystals. Adv. Mater. 2013, 25, 4309–4314 10.1002/adma.201205041. [DOI] [PubMed] [Google Scholar]
- Balazs D. M.; Nugraha M. I.; Bisri S. Z.; Sytnyk M.; Heiss W.; Loi M. A. Reducing Charge Trapping in PbS Colloidal Quantum Dot Solids. Appl. Phys. Lett. 2014, 104, 112104. 10.1063/1.4869216. [DOI] [Google Scholar]
- Tang J.; Kemp K. W.; Hoogland S.; Jeong K. S.; Liu H.; Levina L.; Furukawa M.; Wang X.; Debnath R.; Cha D.; et al. Colloidal-Quantum-Dot Photovoltaics Using Atomic-Ligand Passivation. Nat. Mater. 2011, 10, 765–771 10.1038/nmat3118. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Gao J.; Miller E. M.; Luther J. M.; Beard M. C. Diffusion-Controlled Synthesis of PbS and PbSe Quantum Dots with in situ Halide Passivation for Quantum Dot Solar Cells. ACS Nano 2014, 8, 614–622 10.1021/nn405236k. [DOI] [PubMed] [Google Scholar]
- Zhitomirsky D.; Furukawa M.; Tang J.; Stadler P.; Hoogland S.; Voznyy O.; Liu H.; Sargent E. H. N-Type Colloidal-Quantum-Dot Solids for Photovoltaics. Adv. Mater. 2012, 24, 6181–6185 10.1002/adma.201202825. [DOI] [PubMed] [Google Scholar]
- Chuang C. H.; Brown P. R.; Bulovic V.; Bawendi M. G. Improved Performance and Stability in Quantum Dot Solar Cells Through Band Alignment Engineering. Nat. Mater. 2014, 13, 796–801 10.1038/nmat3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler P.; Mohamed S. A.; Gasiorowski J.; Sytnyk M.; Yakunin S.; Scharber M. C.; Enengl C.; Enengl S.; Egbe D. A. M.; El-Mansy M. K.; et al. Iodide-Capped PbS Quantum Dots: Full Optical Characterization of a Versatile Absorber. Adv. Mater. 2015, 27, 1533–1539 10.1002/adma.201404921. [DOI] [PubMed] [Google Scholar]
- Crisp R. W.; Kroupa D. M.; Marshall A. R.; Miller E. M.; Zhang J.; Beard M. C.; Luther J. M. Metal Halide Solid-State Surface Treatment for High Efficiency PbS and PbSe QD Solar Cells. Sci. Rep. 2015, 5, 9945. 10.1038/srep09945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voznyy O.; Zhitomirsky D.; Stadler P.; Ning Z.; Hoogland S.; Sargent E. H. A Charge-Orbital Balance Picture of Doping in Colloidal Quantum Dot Solids. ACS Nano 2012, 6, 8448–8455 10.1021/nn303364d. [DOI] [PubMed] [Google Scholar]
- Ibáñez M.; Korkosz R. J.; Luo Z.; Riba P.; Cadavid D.; Ortega S.; Cabot A.; Kanatzidis M. G. Electron Doping in Bottom-Up Engineered Thermoelectric Nanomaterials through HCl-Mediated Ligand Displacement. J. Am. Chem. Soc. 2015, 137, 4046–4049 10.1021/jacs.5b00091. [DOI] [PubMed] [Google Scholar]
- Fafarman A. T.; Koh W. K.; Diroll B. T.; Kim D. K.; Ko D. K.; Oh S. J.; Ye X.; Doan-Nguyen V.; Crump M. R.; Reifsnyder D. C.; et al. Thiocyanate-capped Nanocrystal Colloids: Vibrational Reporter of Surface Chemistry and Solution-based Route to Enhanced Coupling in Nanocrystal Solids. J. Am. Chem. Soc. 2011, 133, 15753–15761 10.1021/ja206303g. [DOI] [PubMed] [Google Scholar]
- Dirin D. N.; Dreyfuss S.; Bodnarchuk M. I.; Nedelcu G.; Papagiorgis P.; Itskos G.; Kovalenko M. V. Lead Halide Perovskites and Other Metal Halide Complexes As Inorganic Capping Ligands for Colloidal Nanocrystals. J. Am. Chem. Soc. 2014, 136, 6550–6553 10.1021/ja5006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Jang J.; Liu W.; Talapin D. V. Colloidal Nanocrystals with Inorganic Halide, Pseudohalide, and Halometallate Ligands. ACS Nano 2014, 8, 7359–7369 10.1021/nn502470v. [DOI] [PubMed] [Google Scholar]
- Ning Z.; Dong H.; Zhang Q.; Voznyy O.; Sargent E. H. Solar Cells Based on Inks of n-Type Colloidal Quantum Dots. ACS Nano 2014, 8, 10321–10327 10.1021/nn503569p. [DOI] [PubMed] [Google Scholar]
- Stadler P.; Sutherland B. R.; Ren Y.; Ning Z.; Simchi A.; Thon S. M.; Hoogland S.; Sargent E. H. Joint Mapping of Mobility and Trap Density in Colloidal Quantum Dot Solids. ACS Nano 2013, 7, 5757–5762 10.1021/nn401396y. [DOI] [PubMed] [Google Scholar]
- Law M.; Luther J. M.; Song O.; Hughes B. K.; Perkins C. L.; Nozik A. J. Structural, Optical, and Electrical Properties of PbSe Nanocrystal Solids Treated Thermally or with Simple Amines. J. Am. Chem. Soc. 2008, 130, 5974–5985 10.1021/ja800040c. [DOI] [PubMed] [Google Scholar]
- Hassinen A.; Moreels I.; De Nolf K.; Smet P. F.; Martins J. C.; Hens Z. Short-Chain Alcohols Strip X-type Ligands and Quench the Luminescence of PbSe and CdSe Quantum Dots, Acetonitrile Does Not. J. Am. Chem. Soc. 2012, 134, 20705–20712 10.1021/ja308861d. [DOI] [PubMed] [Google Scholar]
- Kirmani A. R.; Carey G. H.; Abdelsamie M.; Yan B.; Cha D.; Rollny L. R.; Cui X.; Sargent E. H.; Amassian A. Effect of Solvent Environment on Colloidal-Quantum-Dot Solar-Cell Manufacturability and Performance. Adv. Mater. 2014, 26, 4717–4723 10.1002/adma.201400577. [DOI] [PubMed] [Google Scholar]
- Kim S.; Noh J.; Choi H.; Ha H.; Song J. H.; Shim H. C.; Jang J.; Beard M. C.; Jeong S. One-Step Deposition of Photovoltaic Layers Using Iodide Terminated PbS Quantum Dots. J. Phys. Chem. Lett. 2014, 5, 4002–4007 10.1021/jz502092x. [DOI] [PubMed] [Google Scholar]
- Oh S. J.; Wang Z.; Berry N. E.; Choi J. H.; Zhao T.; Gaulding E. A.; Paik T.; Lai Y.; Murray C. B.; Kagan C. R. Engineering Charge Injection and Charge Transport for High Performance PbSe Nanocrystal Thin Film Devices and Circuits. Nano Lett. 2014, 14, 6210–6216 10.1021/nl502491d. [DOI] [PubMed] [Google Scholar]
- Koh W. K.; Saudari S. R.; Fafarman A. T.; Kagan C. R.; Murray C. B. Thiocyanate-capped PbS Nanocubes: Ambipolar Transport Enables Quantum Dot Based Circuits on a Flexible Substrate. Nano Lett. 2011, 11, 4764–4767 10.1021/nl202578g. [DOI] [PubMed] [Google Scholar]
- Baumgardner W. J.; Whitham K.; Hanrath T. Confined-but-connected Quantum Solids via Controlled Ligand Displacement. Nano Lett. 2013, 13, 3225–3231 10.1021/nl401298s. [DOI] [PubMed] [Google Scholar]
- Oh S. J.; Berry N. E.; Choi J. H.; Gaulding E. A.; Lin H.; Paik T.; Diroll B. T.; Muramoto S.; Murray C. B.; Kagan C. R. Designing High-Performance PbS and PbSe Nanocrystal Electronic Devices Through Stepwise, Post-Synthesis, Colloidal Atomic Layer Deposition. Nano Lett. 2014, 14, 1559–1566 10.1021/nl404818z. [DOI] [PubMed] [Google Scholar]
- Doris S. E.; Lynch J. J.; Li C.; Wills A. W.; Urban J. J.; Helms B. A. Mechanistic Insight into the Formation of Cationic Naked Nanocrystals Generated under Equilibrium Control. J. Am. Chem. Soc. 2014, 136, 15702–15710 10.1021/ja508675t. [DOI] [PubMed] [Google Scholar]
- Boneschanscher M. P.; Evers W. H.; Geuchies J. J.; Altantzis T.; Goris B.; Rabouw F. T.; van Rossum S. A.; van der Zant H. S.; Siebbeles L. D.; Van Tendeloo G.; et al. Long-Range Orientation and Atomic Attachment of Nanocrystals in 2D Honeycomb Superlattices. Science 2014, 344, 1377–1380 10.1126/science.1252642. [DOI] [PubMed] [Google Scholar]
- Zherebetskyy D.; Scheele M.; Zhang Y.; Bronstein N.; Thompson C.; Britt D.; Salmeron M.; Alivisatos P.; Wang L. W. Hydroxylation of the Surface of PbS Nanocrystals Passivated with Oleic Acid. Science 2014, 344, 1380–1384 10.1126/science.1252727. [DOI] [PubMed] [Google Scholar]
- Li D.; Nielsen M. H.; Lee J. R.; Frandsen C.; Banfield J. F.; De Yoreo J. J. Direction-Specific Interactions Control Crystal Growth by Oriented Attachment. Science 2012, 336, 1014–1018 10.1126/science.1219643. [DOI] [PubMed] [Google Scholar]
- Wanger D. D.; Correa R. E.; Dauler E. A.; Bawendi M. G. The Dominant Role of Exciton Quenching in PbS Quantum-Dot-Based Photovoltaic Devices. Nano Lett. 2013, 13, 5907–5912 10.1021/nl402886j. [DOI] [PubMed] [Google Scholar]
- Speirs M. J.; Balazs D. M.; Fang H. H.; Lai L. H.; Protesescu L.; Kovalenko M. V.; Loi M. A. Origin of the Increased Open Circuit Voltage in PbS–CdS Core–Shell Quantum Dot Solar Cells. J. Mater. Chem. A 2015, 3, 1450–1457 10.1039/C4TA04785K. [DOI] [Google Scholar]
- Sze S. M.; Ng K. K.. Physics of Semiconductor Devices; Wiley-Interscience: Hoboken, NJ, 2007. [Google Scholar]
- Erslev P. T.; Chen H.; Gao J.; Beard M. C.; Frank A. J.; van de Lagemaat J.; Johnson J. C.; Luther J. M. Sharp exponential band tails in highly disordered lead sulfide quantum dot arrays. Phys. Rev. B: Condens. Matter Mater. Phys. 2012, 86, 155313. 10.1103/PhysRevB.86.155313. [DOI] [Google Scholar]
- You H.; Wang W.; Yang S. A Universal Rule for Organic Ligand Exchange. ACS Appl. Mater. Interfaces 2014, 6, 19035–19040 10.1021/am504918z. [DOI] [PubMed] [Google Scholar]
- Fang H.; Raissa R.; Abdu-Aguye M.; Adjokatse S.; Blake G. R.; Even J.; Loi M. A. Photophysics of Organic-Inorganic Hybrid Lead Iodide Perovskite Single Crystals. Adv. Funct. Mater. 2015, 25, 2378–2385 10.1002/adfm.201404421. [DOI] [Google Scholar]
- Piliego C.; Protesescu L.; Bisri S. Z.; Kovalenko M. V.; Loi M. A. 5.2% Efficient PbS Nanocrystal Schottky Solar Cells. Energy Environ. Sci. 2013, 6, 3054–3059 10.1039/c3ee41479e. [DOI] [Google Scholar]
- Nugraha M. I.; Häusermann R.; Bisri S. Z.; Matsui H.; Sytnyk M.; Heiss W.; Takeya J.; Loi M. A. High Mobility and Low Density of Trap States in Dual-Solid-Gated PbS Nanocrystal Field-Effect Transistors. Adv. Mater. 2015, 27, 2107–2112 10.1002/adma.201404495. [DOI] [PubMed] [Google Scholar]
- Kang M. S.; Sahu A.; Norris D. J.; Frisbie C. D. Size- and Temperature-Dependent Charge Transport in PbSe Nanocrystal Thin Films. Nano Lett. 2011, 11, 3887–3892 10.1021/nl2020153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.