

Abstract
During the last decade the use of ω-transaminases has been identified as a very powerful method for the preparation of optically pure amines from the corresponding ketones. Their immense potential for the preparation of chiral amines, together with their ease of use in combination with existing biocatalytic methods, have made these biocatalysts a competitor to any chemical methodology for (asymmetric) amination. An increasing number of examples, especially from industry, shows that this biocatalytic technology outmaneuvers existing chemical processes by its simple and flexible nature. In the last few years numerous publications and patents on synthetic routes, mainly to pharmaceuticals, involving ω-transaminases have been published. The review gives an overview of the application of ω-transaminases in organic synthesis with a focus on active pharmaceutical ingredients (APIs) and the developments during the last few years.
Keywords: Asymmetric synthesis, Biocatalysis, Enzyme catalysis, Transamination, Amines
1. Introduction
An ω-transaminase catalyzes the deamination of a primary amine (amine donor) and the concomitant amination of a ketone or aldehyde (amine acceptor), which results in a reaction in which an amino moiety is transferred between two molecules (Scheme 1).[1] Unlike α-transaminases, ω-transaminases are not restricted to α-amino and α-keto acids but can aminate virtually any ketone or aldehyde functionality and deaminate in theory any prim-amine; consequently they are also referred to as amine transaminases.[2] For the transamination reaction the enzyme requires pyridoxal 5′-phosphate (PLP, 1) as cofactor, acting as an intermediate amine acceptor and electron sink (see mechanism, Section 3). Because the amination/deamination is reversible, ω-transaminases can be used for the preparation of optically pure amines in two fashions: either (a) by kinetic resolution, in which one enantiomer of a racemic amine is converted into the corresponding ketone, leaving the desired amine enantiomer untouched (Scheme 1, a), or, more preferred, (b) in an asymmetric synthesis starting from the prochiral ketone (Scheme 1, b). The substrates and products of the reaction (amine donor and acceptor, product, co-product) exist in equilibrium which each other, which – depending on the substrates' and products' natures – might require reaction engineering or follow-up reactions to shift the reaction to the product side. Because, from a thermodynamic point of view, pyruvate is an ideal amine acceptor, ω-transaminases were at first mainly employed in kinetic resolution. The development of process-adapted enzymes or methods to shift the equilibrium launched the success of this biocatalytic method for the synthesis of amines. The obtained (chiral) amines represent highly valuable target molecules, because they are precursors for numerous synthetic targets, variously for the pharmaceutical industry (alkaloids etc.), the polymer area (e.g., carbamates, polyurethanes), or for plant protection [e.g., (+)-dihydropinidine]. It is worth noting that chiral amines can alternatively be prepared by employing other type of biocatalysts such as monoamine oxidases, amine dehydrogenases, and imine reductases; this has been the subject of other recent reviews.[1c],[3]
Scheme 1.
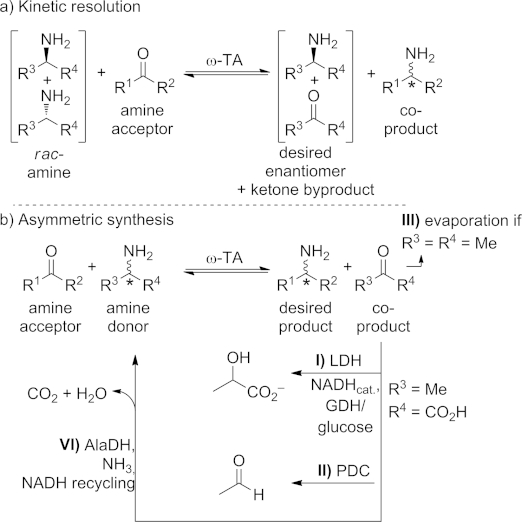
a) Kinetic resolution of a racemic primary amine by an ω-transaminase (ω-TA). b) ω-Transaminase-catalyzed asymmetric amine synthesis and common methods to shift the equilibrium to the product. PDC = pyruvate decarboxylase. LDH = lactate dehydrogenase. GDH = glucose dehydrogenase. AlaDH = alanine dehydrogenase. FDH = formate dehydrogenase.
2. The Early Days …
Although the first records concerning ω-transaminases date back about 50 years,[4] it took until the end of the last century for these enzymes to be spotted as possible catalysts for synthetic applications. The company Celgene demonstrated the first enantioselective production of chiral amines through kinetic resolution of a racemate on a 2.5 m3 scale.[5] At around the same time investigations into ω-transaminases for synthetic applications also started in academia, revealing that one of the major problems to overcome was shifting the equilibrium, especially in the case of asymmetric synthesis (Scheme 1, b), as well as inhibition by certain carbonyl compounds.[6] In order to circumvent inhibition, the kinetic resolution was performed in a biphasic system with removal of the inhibiting ketone byproduct obtained from the deaminated amine enantiomer from the aqueous reaction medium. Two years later, the energy barrier that had to be overcome in asymmetric synthesis was addressed (Scheme 1, b).[7] The target amine was only observed in <10 % yield with use of conventional amine donors, although up to 30 % conversion was reached when more elaborate or chiral amines were tested as donors. Subsequently a membrane reactor was employed to minimize inhibition by the ketone in a kinetic resolution.[8] However, none of these developments competed in any way with the – in those days dominant – ChiPros processes, which employ lipases and run at BASF.[9] Nevertheless, these initial studies marked a milestone in the biocatalytic approach to chiral amines and represent the finding and the identification of the hurdles that had to be jumped in order to achieve versatile and applicable synthetic processes.
3. General Developments and Challenges
At the beginning of the millennium, these initial studies began to attract the attention of several other researchers in the biocatalytic field. Various groups focused on the asymmetric synthesis of amines with the aid of ω-transaminases – 50 years after the first finding of this class of enzymes. The main challenge in the amination of ketones with alanine, an amine donor widely accepted by the enzymes, is the unfavorable equilibrium constant (e.g., for the transamination of acetophenone with alanine an equilibrium constant of 8.81 × 10–4 has been determined).[7],[10] This means that the equilibrium is far on side of the starting materials and only very small amounts of the desired amine product are obtained (usually <5 %).[7]
Using an excess of the amine donor was only effective for selected substrates. For instance, 4-methoxyphenylacetone was transformed into the desired amine with 94 % conversion with use of a 16-fold excess of alanine,[11] but most other substrates gave generally rather poor levels of conversion.[7] Using other amine donors (e.g., 1-aminotetralin) improved the levels of conversion slightly, but no general approach could be identified and most case studies reported highly substrate-dependent transamination reactions.
A first efficient method for equilibrium displacement was the removal of pyruvate – the co-product formed from the amine donor alanine – by use of a lactate dehydrogenase (LDH).[7] The LDH converts the pyruvate into lactic acid at the expense of NADH (nicotinamide adenine dinucleotide). For the recycling of NADH established methods can be exploited.[12] In most cases a glucose dehydrogenase (GDH) in combination with glucose is used nowadays (Scheme 1, b, pathway I).[13] The reaction conditions, such as the pH range of the transaminase and the recycling enzyme, must be compatible and a pH shift due to the formed byproduct (e.g., gluconic acid) has to be considered.
In another approach a pyruvate decarboxylase removes the pyruvate co-product (Scheme 1, b, pathway II).[10] The formed pyruvate is decarboxylated and the equilibrium is pulled to the product side, delivering the amine in average to good yields (results are comparable to those obtained with the LDH system). A significant drawback of this approach is the formation of acetaldehyde through the decarboxylation process, because acetaldehyde competes with the substrate to be aminated, leading to ethylamine as side product.
Other efforts focused on the physical removal of the formed co-product. By using 2-propylamine as amine donor and running the reaction at elevated temperatures, the co-product acetone can be removed by evaporation. High levels of conversion as well as perfect enantioselectivities can be obtained (Scheme 1, b, pathway III).[5a],[13b],[14]
To avoid high concentrations of possibly disruptive co-products, recycling of pyruvate to alanine with the aid of an alanine dehydrogenase was envisioned.[15] This recycles the formed pyruvate back to l-alanine at the expense of ammonia and NADH (see Scheme 1, b, pathway IV). Recycling of the nicotinamide cofactor was achieved by employing ammonium formate (which additionally also serves as a nitrogen source) and a formate dehydrogenase (FDH). If the complete reaction sequence is considered, this approach constitutes a formal asymmetric reductive amination at the expense of ammonia and formate, yielding chiral primary amines.[13b],[16] In the parlance of conventional organic chemistry, this approach represents a biocatalytic, asymmetric version of the Leuckart–Wallach reaction, a highly efficient reaction frequently used to access amines. It is worth noting that the transition-metal-catalyzed version of the same reaction uses precious rhodium catalysts that either give mixtures of the primary amine, the corresponding alcohol, and the carbamate,[17] or require expensive ligand systems.[18] The outlined biocatalytic version delivered the chiral amines in high yields (mainly >90 %) and with perfect stereoselectivies as the sole products after extraction. Very recently the amination of ketones just at the expense of NH3 and molecular hydrogen was described, with a related system in which FDH/formate were substituted by a hydrogenase and molecular hydrogen.[19] The [NiFe]-dehydrogenase reduces the oxidized NAD+ cofactor to NADH at the expense of H2 (2 bar). The S-selective biocatalysts gave reasonable results (77–98 % conv., ee 92–99) whereas the R-selective transaminations were more sensitive to the reaction conditions (conv. 32–94 conv., ee > 99); nevertheless, the [NiFe]-dehydrogenase is currently not competitive with established methods.
These reports brought ω-transaminases to the attention of the chemical community, because optically pure primary amines became accessible in a single reaction step from the corresponding ketones. However, one of the challenges for this enzyme-catalyzed amination was, and partially still remains, the amination of sterically demanding ketones possessing two bulky substituents (“bulky-bulky ketones”). Nevertheless, researchers at Merck/Codexis have subjected an R-selective transaminase from Arthrobacter sp. to directed evolution to provide a variant that accepts bulky-bulky substrates. They successfully applied it to an improved and more efficient production of sitagliptin (2, Figure1), and outperformed the previously established transition-metal-catalyzed process by significantly shortening the synthetic route to this important drug in diabetes treatment (for details see Section 5.2).[14]
Figure 1.
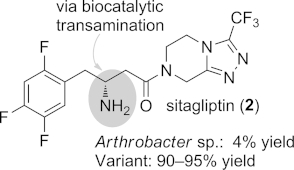
An ω-transaminase variant produced by directed evolution enabled the stereoselective amination of a bulky-bulky ketone for the production of sitagliptin (2), a drug used in the treatment of type II diabetes.
Unfortunately, no S-selective transaminase that generally accepts such bulky-bulky ketones has yet been described;[20] nevertheless, initial attempts have been made to engineer an S-selective transaminase, extending the tolerance from methyl to propyl and hydroxymethyl.[21] Therefore, further efforts to generate S-selective variants could have a significant impact, because a more general and broadly applicable concept for the biocatalytic asymmetric amination of ketones would be established.
Notably, ω-transaminases can be exploited for regioselective conversion of a single ketone moiety in a di- or even triketone system in a more complex molecule. This regioselective approach has been applied to the asymmetric synthesis of all four diastereoisomers of dihydropinidine (Scheme 2, a).[22] The diastereoselective reduction of the imine precursor can subsequently be achieved by use either of Pd/C in the presence of hydrogen (cis diastereoisomers) or of LiAlH4 in the presence of a Lewis acid (e.g., Et3Al).
Scheme 2.
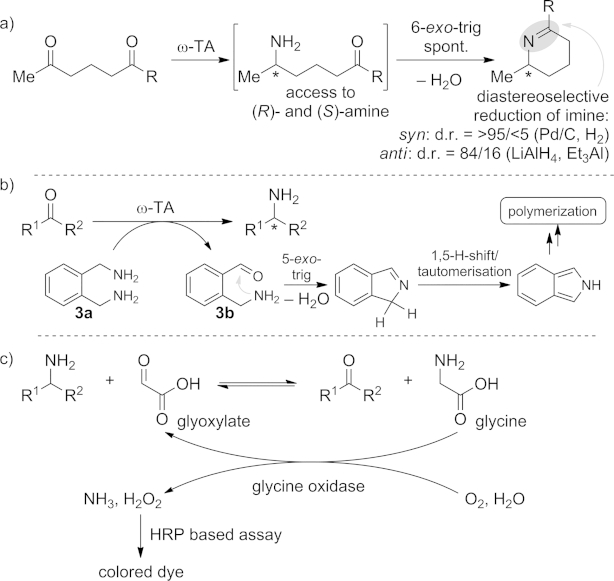
a) Regioselective monoamination of diketones for the diastereoselective synthesis of dihydropinidines. b) Screening assay with ortho-xylylenediamine as amine donor. c) Screening method with a glycine oxidase coupled with a HRP-based colorimetric assay.
Another important aspect – aside from the shift of the equilibrium – is the identification of the appropriate catalyst. Especially when it comes to the creation of variant libraries, fast and reliable detection methods to identify the best catalyst are essential.
Alongside other protocols, a protocol involving the lactate dehydrogenase/glucose dehydrogenase system for rapid identification of potential hits among the transaminase enzymes has been reported recently.[23] Glucose is converted into gluconic acid during recycling of the NADH cofactor. The change in the pH – due to the acidic coproduct – can be monitored with an appropriate pH-sensitive dye (e.g., phenol red). Even quantitative results can be read out from 96-well microtiter plates with a plate reader.
In another approach the amine donor ortho-xylylenediamine (3a) gives the corresponding aminoaldehyde 3b, which spontaneously undergoes a 5-exo-trig cyclization (Scheme 2, b).[24] The formed byproduct rearranges to the energetically more favored aromatic isoindole. The isoindole polymerizes, resulting in colored derivatives and thus simplifying the identification of positive hits in the tested library. It is noteworthy that, because the amine donor spontaneously undergoes the outlined “degradation” after reaction, no other system is required to shift the equilibrium.
In another approach a direct UV-based assay for the detection of acetophenone (ε = 12 mm–1 cm–1 at 245 nm) was envisaged. With this assay, reaction conditions can be optimized very quickly. Nevertheless, the method is substrate-dependent and suffers from interference with the aromatic amino acid residues of the protein.[25]
In another assay using glyoxylate as the amine acceptor, the formed glycine was oxidized back to glyoxylate by a glycine oxidase (Scheme 2, c). The formed hydrogen peroxide was detected by a colorimetric assay based on horseradish peroxidase (HRP). This approach allows the quick identification of active enzymes, especially because it can be employed as a solid-phase test by use of nitrocellulose membranes.[26]
The first R-selective ω-transaminases described by academia were identified by a computational approach. The crucial amino acids for catalysis having been determined, the Brookhaven protein database (PDB) was screened electronically.[27] By that subtle approach seventeen R-selective ω-transaminases were identified, representing a milestone for the identification of R-selective enzymes,[28] and were then successfully applied for asymmetric synthesis.[29]
In another approach to identifying a suitable transaminase quickly, a mix of eight amine donors and seven acceptors was subjected to enzyme transformation in a one-pot procedure. Analysis of the ketone composition after incubation gave a simple overview of which compounds were transformed. This allowed the affinities of the different substrates to the enzymes to be determined quickly, and the catalysts could be arrayed more rapidly with respect to their substrate scope.[30]
Another important feature of ω-transaminases is that they do not only work in buffer but also in organic solvents.[31],[32] For instance, transaminases work in organic solvents such as methyl tert-butyl ether (MTBE) and isopropyl acetate, with the water activity of the organic solvent used being of high importance for the reaction outcome.[32] In particular, substrates leading to amines, which can form intramolecular hydrogen bonds to a neighboring group, were transformed with high conversion employing a crude enzyme preparation (not immobilized). Other substrates were converted at slower rates. The stereopreference and -selectivity were not altered in organic media in relation to the results obtained in buffer. Using an immobilized crude enzyme preparation improved the conversion significantly: 1-phenylethylamine, for example, was formed in 87 % yield, whereas the system without immobilization gave roughly 40 %, even after extended reaction times.[31] Although organic solvents seem to be rather appealing, the formation of the imine between, for example, the substrate ketone and amine donor in an additional equilibrium reaction has to be considered.
Aldehydes are superb substrates too and have in particular been aminated in cascade reactions, in which they were formed as reactive intermediates by another biocatalyst through oxidation of a primary alcohol.[33] For example, galactose oxidase from Fusarium NRRL 2903 was used to oxidize allylic and benzylic alcohols to the corresponding aldehydes at the expense of innocent molecular oxygen (Scheme 3, a).[33a] The reaction sequence was completed by the transamination of the intermediate, yielding the primary amine. All reactions were run simultaneously in one-pot fashion, so all biocatalysts were mutually compatible. In a “borrowing hydrogen” approach the abstracted hydride of the oxidation step is recycled in the subsequent reductive amination. Thus, an alcohol dehydrogenase (ADH) is used to oxidize the primary alcohol to the aldehyde at the expense of NAD+ to give NADH (Scheme 3, b).[33c] The formed NADH is reused for the subsequent transamination reaction (recycling of the alanine cofactor), giving back NAD+ for the next oxidation. The result is a redox-self-sufficient process, which formally aminates the alcohol only at the expense of ammonium chloride, circumventing the need for any additional redox reagents.
Scheme 3.
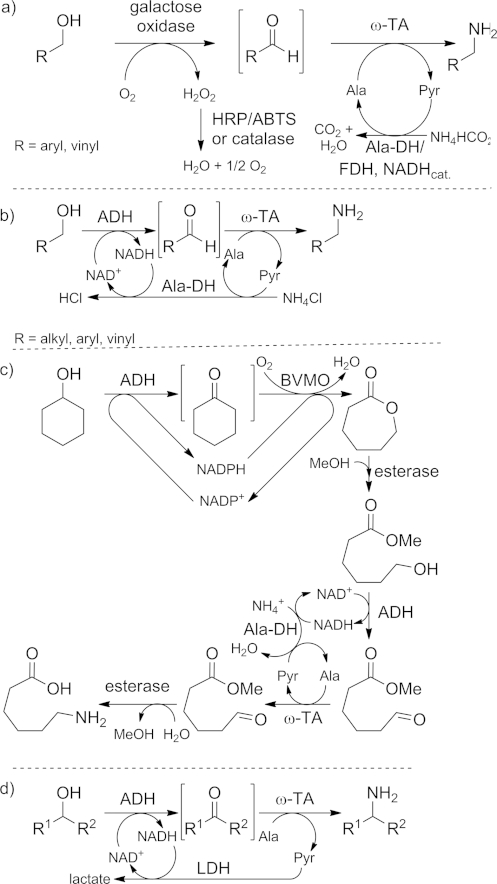
Oxidation/transamination cascade reactions. HRP = horse radish peroxidase. Ala = alanine. Pyr = pyruvate. Ala-DH = alanine dehydrogenase. ADH = alcohol dehydrogenase. NAD+/NADH = nicotinamide adenine dinucleotide cofactor (oxidized/reduced form). BVMO = Baeyer–Villiger monooxygenase. NADP+/NADPH = nicotinamide adenine dinucleotide phosphate cofactor (oxidized/reduced form). LDH = lactate dehydrogenase.
This approach has been further extended to a biocatalytic system for the preparation of hydrolyzed ε-caprolactam, the monomer of nylon (Scheme 3, c).[33b] This system additionally contained a Baeyer–Villiger monooxygenase and an esterase. Again it was shown that all of these biocatalysts function in each other's presence. Comparable chemical approaches require sophisticated transition-metal catalysts, and high temperature and pressure are unavoidable.[34] It is notable that no comparable one-pot route to ε-caprolactam by chemical approaches has been reported.
The biocatalytic sequence to aminate primary alcohols was also extended to secondary alcohols (Scheme 3, d, Table 1). However, the lower reactivity of the transaminase towards the ketone, as well as the additional complexity due to a chiral starting material, required more elaborate enzymatic systems in order to achieve good levels of conversion.[35] Either a NADH oxidase (NOX) or a lactate dehydrogenase system was required for an efficient amination.
Table 1.
ADH/ω-TA-catalyzed amination of sec-alcohols
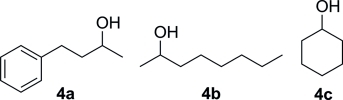 | ||||
|---|---|---|---|---|
| Entry | System | Substrate | Conv. [%][a] | ee [%] |
| 1 | NOX | 4a | 45 | n.d. |
| 2[b] | LDH | 4a | 64 | 88 |
| 3[b] | LDH | 4b | 64 | 96 |
| 4[c] | LDH | 4c | 91 | n.a. |
Conversion was determined by GC-FID.
ω-TA from Vibrio fluvialis was used.
ω-TA from Bacillus megaterium was used; n.d.: not determined; n.a.: not applicable.
Recent studies have established the potential of ω-transaminases for the dynamic kinetic resolution of α-chiral 2-phenylpropanals, which are converted into the corresponding β-chiral amines. The stereocenter of the aldehyde precursor is prone to racemization under the reaction conditions, so up to full conversion can be obtained with ee values up to 98 %[36] (Scheme 4).
Scheme 4.
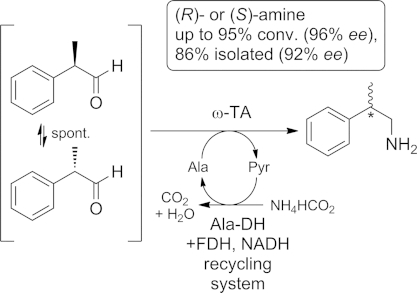
Dynamic kinetic resolution of α-chiral aldehydes. Ala-DH = alanine dehydrogenase. NADH = nicotinamide adenine dinucleotide cofactor (reduced form). FDH = formate dehydrogenase. Ala = alanine. Pyr = pyruvate.
Additionally, ω-transaminases have been successfully employed in flow chemistry. The enzyme – immobilized on polymeric methacrylate beads – was shown to be a perfect catalytic system: pumping the substrate, dissolved in MTBE, over the immobilized catalyst gave good yields of the corresponding enantiopure amine. The levels of conversion were almost constant, even over a reaction time of 10 days. However, the long residence times (tR = 30–60 min) and the limited cartridge size (0.5 mL volume) at substrate concentrations of 25–50 mm resulted in a productivity of the process of only 50 μmol h–1 (corresponds to 12 mmol within 10 d). Nevertheless, the proof of the concept and the successful implementation of ω-transaminases into a flow process have been provided.[37]
4. Mechanism and Active Site
To allow a general understanding of the mode of the transamination, the mechanism is discussed below.[38] The crucial molecule for the reaction is the cofactor PLP (1), together with its reductively aminated form PMP (5, pyridoxamine 5′-phosphate). In its resting state, PLP (1) forms a Schiff base with a lysine residue in the enzyme backbone (A in Scheme 5). The C=N bond of the Schiff base is attacked by the substrate amine, leading to imine formation between substrate amine and PLP (B). This attack is facilitated by a water molecule in the active site and the phenolic oxygen of the PLP (1) molecule.[39] The phenolic hydroxy group in the ortho-position stabilizes the Schiff base in both cases through a hydrogen bond. A suitably orientated lysine residue then abstracts the proton at the amine carbon, which has become more acidic due to the imine formation, formally creating a carbanion. The proton abstraction initiates isomerization to the resonance methide form (D), which is quenched and rearomatized by the proton of the base from the first deprotonation step (structure E). The newly formed imine is hydrolyzed, releasing the ketone product and an enzyme-PMP complex (F), which is ready for transferring the amino group to another ketone substrate by the reverse pathway, because all steps in the mechanism are reversible. The outlined mechanism has recently been investigated by DFT calculations on an active site model, which led to similar conclusions.[39]
Scheme 5.
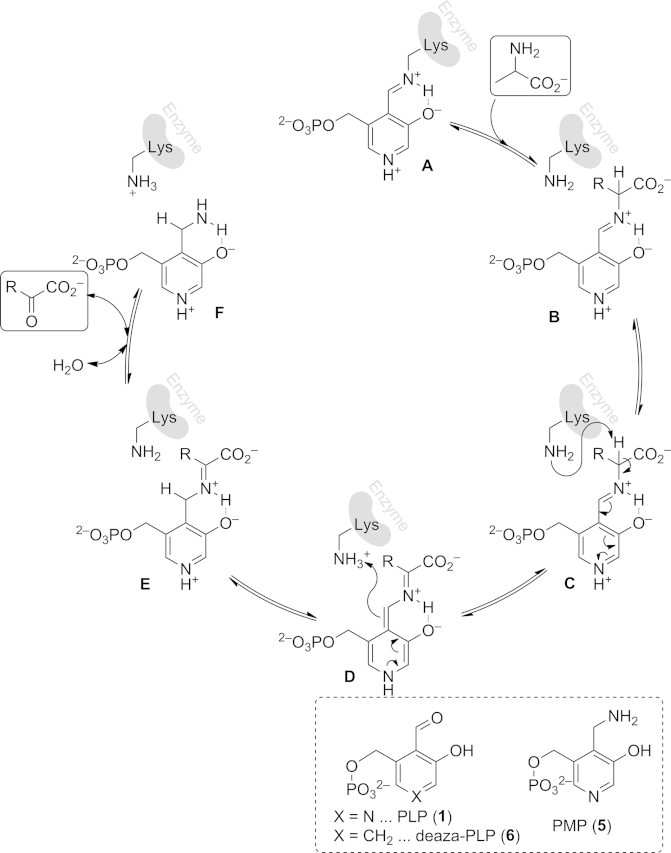
Mechanism of the deamination (clockwise) and amination (anticlockwise) process with alanine as amine donor and pyruvate as amine acceptor, respectively. Lys = lysine.
A very important feature of the cofactor is its pyridine nitrogen, because the heterocycle, especially in its protonated form, can stabilize the postulated methide form (D) of the cofactor, which is required for efficient catalysis. For example, if deaza-PLP (6) is used as cofactor, the enzymatic activity drops by four to nine orders of magnitude. This also reveals another mechanistic detail, because the reaction can be tuned by the strength of the corresponding acid, which protonates the cofactor, allowing nature to adjust the reactivity of the enzyme in question.[40],[41]
The substrate binding – in terms of chiral information transfer – is based on a large (L) pocket and a small (S) pocket in the active site (see Figure2). The basic residue that assists the 1,3-prototropic shift is usually the amine arm of a lysine, which also binds the PLP cofactor prior to the entry of the substrate into the active site.[42]
Figure 2.
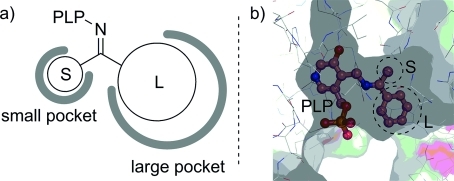
a) Schematic view of the binding site of an ω-TA. b) Active site of the ω-TA from Pseudomonas putida with pre-optimized, docked substrate-PLP complex [PDB entry 3a8u, picture was prepared with Pymol (vs. 0.99rc6)].
These mechanistic investigations have also triggered interest in biomimetic catalytic systems, leading to investigation of an asymmetric transamination catalyzed by chiral Lewis acids (e.g., compound 7), albeit with low enantioselectivities (<46 % ee, Scheme 6).[43]
Scheme 6.
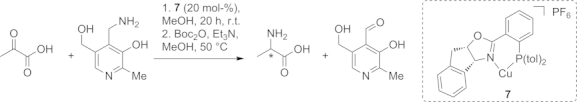
Mimicking transamination with a PLP-like amine donor and chiral Lewis acid catalyst 7.
Recent X-ray structures of various transaminases[44] and the understanding of the substrate binding have opened the door for rational protein engineering. For example, two variants of the V. fluvialis transaminase that can convert bulky-bulky ketones were developed (although only a narrow substrate spectrum was presented). The key mutations were two amino acid residues in the small pocket of the active site: a valine was changed to an alanine and a tyrosine was mutated to phenylalanine. Through these mutations the small pocket chamber was extended in order to tolerate bulkier residues.[21]
The same enzyme was used to convert 3-methylcyclohexan-1-one into the corresponding amines (for details see Scheme 7 and Section 5.1). The wild-type enzyme showed only modest diastereoselectivity (14 % de), whereas the L56V variant of the enzyme gave the (R)-amine with 66 % de. Even more interesting was the fact that changing the same leucine for isoleucine (L56I) inverted the stereopreference towards the (S)-amine, with 70 % de.[45]
Scheme 7.
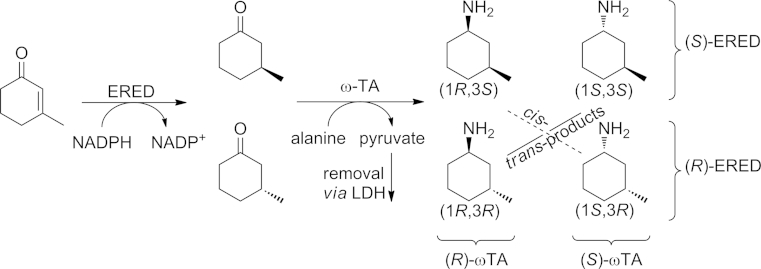
Combination of an enoate reductase (ERED) and an ω-TA for the preparation of all stereoisomers of 1-amino-3-methylcyclohexanes.
5. Applications
Thanks to the broad substrate scope of ω-transaminases (ω-TAs), their usually high turnover rate, and their remarkable stereoselectivity, along with their environmentally friendly reaction setups, ω-TAs are considered a convenient alternative to classical chemical approaches for the asymmetric synthesis of valuable amine building blocks and bioactive compounds. ω-TA-catalyzed reactions represent key steps in a wide range of chemo-enzymatic syntheses and, thanks to their compatibility with other enzymes, they are often employed in multiple-enzyme cascade reactions.
5.1. Multiple-Enzyme Cascade Reactions Involving ω-TAs
In the last few years, ω-TA-catalyzed reactions have been combined with other enzymatic transformations in concurrent one-pot processes for the preparation of highly functionalized molecules.[1b],[46] Such multi-enzymatic cascades offer various advantages, such as shortened reaction times and a reduced number of workup steps, allowing high atom-efficiency.
ω-TAs have been combined, for example, with an enoate reductase for the synthesis of 3-methyl-cyclohexylamines amines containing two stereocenters.[47] Through the employment of an engineered ω-TA in the presence of an organic co-solvent (30 % DMSO) a diastereoselective reduction/transamination sequence was successfully performed in one-pot fashion (89 % de max., Scheme 7,b).
ω-TAs have also been successfully combined with other biocatalysts such as transketolases, lyases, and hydrolases with particular regard to the formation and cleavage of C–C bonds. In a recent example, an ω-TA was applied together with a thiamine-diphosphate-dependent (ThDP-dependent) transketolase in two sequential steps for the production of various amino alcohols on a preparative scale (100 mL, 300 mm substrate concentration).[48] Vicinal amino alcohols are highly valuable intermediates for various pharmaceutical compounds as well as synthons in organic syntheses.[49] Therefore, this cascade was employed for the preparation of the drugs (1R,2R)-norpseudoephedrine (NPE, 10) and (1R,2S)-norephedrine (NE, 10) by starting from the inexpensive substrates pyruvate and benzaldehyde (Scheme 8, a).[50] In the first step these two molecules were coupled together by the ThDP-dependent lyase acetohydroxyacid synthase I (AHAS-I) to yield (R)-phenylcarbinol (11). This intermediate keto alcohol was then directly aminated by one of two stereocomplementary ω-TAs, giving access either to (1R,2R)-NPE or to (1R,2S)-NE in high yields and with perfect optical purity. The coproduct pyruvate – which is produced in the transamination reaction – was removed through its consumption as the substrate in the first reaction step.
Scheme 8.
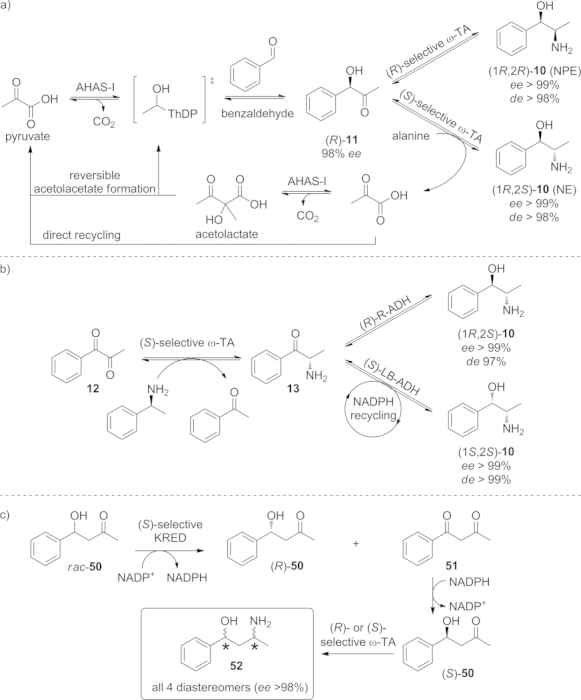
One-pot, two-step cascade for the synthesis of various diastereomers of norpseudoephedrine (NPE) and norephedrine (NE) by combining either a) a ThDP-dependent lyase (AHAS-I) with stereocomplementary ω-TAs, or b) an S-selective ω-TA with stereocomplementary ADHs. c) Preparation of all four diastereoisomers of 1,3-amino alcohol 52.
However, this approach is not suitable for the preparation of the complementary (1S,2S)-10 or (1S,2R)-10, because of the lack of an enzyme that affords (S)-11 with sufficient optical purity. As a consequence, a recent report provides an alternative reaction sequence to overcome this limitation.[51] 1-Phenylpropane-1,2-dione (12) was used as starting material and was converted into all four N(P)E isomers in two sequential steps by combining an ω-TA and an ADH. Thereby, full stereochemical control was achieved through the stereopreferences of the two enzymes (Scheme 8, b).
Very recently, 1,3-amino alcohols were accessed by oxidative kinetic resolution of keto alcohol 50 by a ketoreductase in the first step (Scheme 8, c). The oxidized diketone 51 was separated and reduced to (S)-50. Subsequent transamination with the appropriate biocatalyst yielded each of the four diastereomers of 52 in optically pure form.[52]
Another concept involves the stereoselective hydrolytic C–C bond cleavage of prochiral bicyclic β-diketones by a hydrolase. After bond cleavage as the first step, the esterification of the newly formed free acid with MeOH is catalyzed by a CAL-B lipase. Subsequently, an ω-TA converts the ketone moiety into the corresponding amine with perfect diastereoselectivity in the final step of the three-step cascade. By this approach the corresponding cyclohexylamines could be obtained in either cis or trans configurations in moderate to high yields (Scheme 9).[53]
Scheme 9.
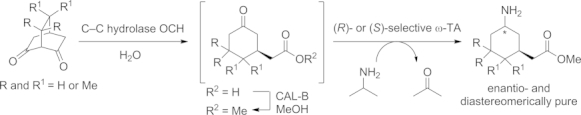
Diastereoselective synthesis of 3-substituted cyclohexylamines by a three-step cascade sequence employing two hydrolases and an ω-TA.
Multi-enzymatic cascades also play a significant role in the deracemization of rac-amines. One example of such a deracemization is the combination of two stereocomplementary ω-TAs in a one-pot, two-step fashion, which was successfully applied for the preparation of a broad range of α-chiral amines in optically pure form (Scheme 10, a).[54]
Scheme 10.
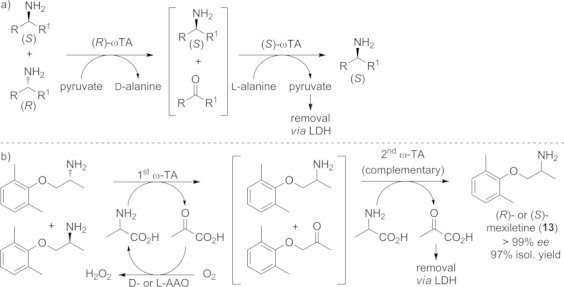
a) One-pot, two-step cascade employing two stereocomplementary ω-TAs for the deracemization of racemic amines. b) The procedure was successfully applied for the preparation of both enantiomers of mexiletine (13).
Firstly, an R-selective ω-TA transformed one enantiomer into the corresponding ketone, whereas the S enantiomer remained untouched. In this process pyruvate had to be employed in stoichiometric amounts, serving as an amine acceptor and forming D-alanine as byproduct. After 24 h a second, S-selective ω-TA was added in order to convert the obtained ketone into the (S)-amine, thus leading to a single enantiomer. Because the order of the ω-TAs can be inverted, both enantiomers can be prepared with high yields and perfect stereoselectivities. In initial studies, this one-pot stepwise deracemization methodology only yielded good results when the biocatalyst from the first reaction step was inactivated by heat treatment after the kinetic resolution had been completed. Otherwise it interfered with the stereocomplementary ω-TA in the second step and therefore the optical purity of the final amine product was decreased. The applicability of this procedure was demonstrated for the deracemization of the antiarrhythmic drug mexiletine (13) on preparative scale (100 mg).[55]
In order to circumvent the heat deactivation procedure, the first step of the cascade was performed with an immobilized ω-TA (solid support: sol-gel/celite matrix) in a subsequent study.[56] The first enzyme can then be removed by simple filtration after the first reaction step and the economically unviable decomposition of the catalyst can be avoided.
To reduce the amount of pyruvate required for the first step (kinetic resolution) an amino acid oxidase (l- or D-AAO) was introduced to catalyze the recycling of the formed l- or D-alanine to pyruvate.[23],[55] This minimizes the amount of the co-substrate to catalytic amounts and improves the overall efficiency of the reaction (Scheme 10, b).
Nevertheless, an approach that allows the interference of the stereocomplementary enzymes to be avoided relies on a correct and smart selection of the amine acceptor/donor system.[57] By using α-ketoglutarate instead of pyruvate as amine acceptor for the S-selective catalyst both the deamination and amination steps can be performed simultaneously, because α-ketoglutarate is not accepted by the R-selective ω-TA.
A different concept for deracemization of amines takes advantage of the hydrolysis of imines. The S enantiomer of the racemic amine is selectively oxidized by a monoamine oxidase to the corresponding imine. The imine spontaneously hydrolyzes to the ketone in the aqueous reaction medium. Subsequently, the ketone intermediate is converted into the (R)-amine by an ω-TA (Scheme 11, a).[58] Additionally, this cascade was employed for selective N-dealkylation of secondary amines. The benzylic position of the secondary amine was oxidized to produce the imine, which was again hydrolyzed to afford the corresponding carbonyl functionality. The latter is then “reaminated” by an appropriate ω-TA (Scheme 11, b).
Scheme 11.
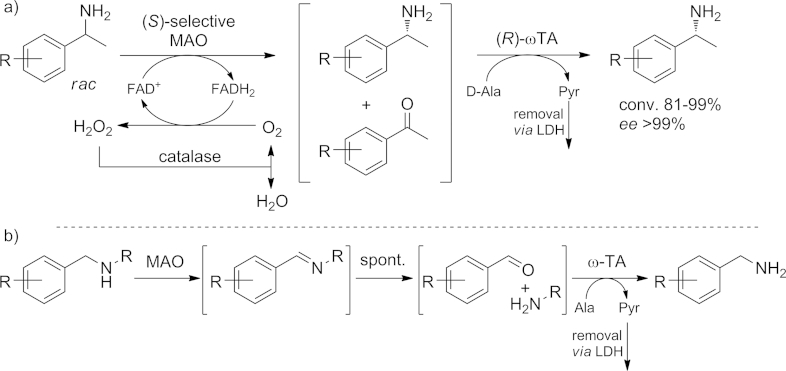
a) Cascade reaction combining a monoamine oxidase (MAO) and an ω-TA for the deracemization of 1-phenylethylamine derivatives, based on the rapid hydrolysis of imines. b) Dealkylation of sec-amines through the action of a MAO and an ω-TA. FAD+/FADH = flavin adenine dinucleotide (oxidized/reduced form). MAO = monoamine oxidase. Ala = alanine. Pyr = pyruvate. LDH = lactate dehydrogenase.
For the amination of alcohols, various cascades combining the oxidation of an alcohol with a subsequent ω-TA-catalyzed amination step have been developed. For example, an oxidation/transamination cascade via an aldehyde intermediate was developed for the preparation of benzylic amines (vide supra; see Scheme 3, a). Various benzylic alcohols (50 mm substrate concentration) were successfully transformed into the corresponding amines, and the practical applicability of this enzymatic cascade reaction was also shown in the synthesis of the antifungal compound naftifine (8, Figure3).[33a] The already mentioned “borrowing hydrogen” approach (Scheme 3, b) achieved in a redox-self-sufficient cascade is based on the oxidation of the alcohol by an ADH and has been employed, for example, for the diamination of 1,ω-diols.[33c] The amination of decane-1,10-diol was performed successfully on preparative scale (126 mg) in 70 % isolated yield. The corresponding 1,ω-diamines are versatile building blocks in polymer chemistry. Another successful application of an oxidation/transamination cascade has been reported for the conversion of the diol isosorbide (9, Figure3) originating from renewable resources.[59] Its transformation gave access to isosorbide monoamine, with 7 % conversion.
Figure 3.
Structures of naftifine (8) and isosorbide (9).
The redox-self-sufficient oxidation/transamination cascades have been improved by co-expression of the three required enzymes (ADH, ω-TA, and Ala-DH) in a single E. coli host.[60] The applicability of the single-cell catalyst was demonstrated in the amination of aliphatic and aromatic mono- and dialcohols. Because this concept simplifies the catalyst preparation, it represents a more practicable and economic approach. A single-cell catalyst has also been used for the ω-functionalization of fatty acids in an in vivo one-pot, three-step cascade (Scheme 12).[61] The ω-position of the fatty acid was hydroxylated by a monooxygenase (AlkBGT), which also oxidized the primary alcohol to the corresponding aldehyde, but also to the corresponding acid. In the presence of an ω-TA, the aldehyde was aminated before it could be oxidized to the acid. This process was successfully transferred to a pilot plant by Evonik Industries in 2013; it yielded ω-aminolauric acid, which is the monomer of a sustainable high-performance plastic.[62]
Scheme 12.
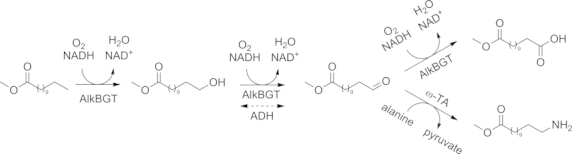
In vivo three-step cascade for the terminal amino-functionalization of fatty acids by an alkane monooxygenase (AlkBGT) and an ω-TA.
4.2. Chemo-Enzymatic Processes
A cascade process can also be based on a subsequent spontaneous chemical reaction that shifts the thermodynamically disfavored amination reaction to completion by removing the product(s) from the reaction mixture. In this context, a transamination/lactamization cascade has been successfully applied for the synthesis of various chiral heterocycles. The initial reductive amination of carbonyl compounds is followed by spontaneous ring closure to afford the corresponding lactams (Scheme 13). By this approach, the amination of ethyl 4-acetylbutyrate (14) was accomplished at a concentration of 50 g L–1, and after cyclization of the intermediate amino ester the product 6-methylpiperidin-2-one (15) was obtained optically pure in >90 % isolated yield.[63]
Scheme 13.
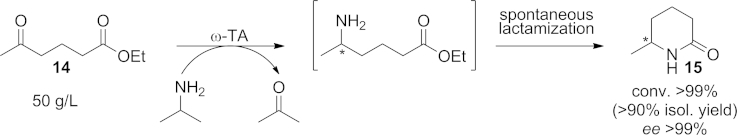
Amination/lactamization cascade for the preparation of 6-methylpiperidin-2-one (15) in high isolated yield and in enantiopure form.
These cascades based on the spontaneous subsequent cyclization of the amine product represent highly efficient and scalable methodologies because the equilibrium is driven towards the product side without a significant excess of the amine donor.
The combination of biocatalytic steps with common chemical transformations has been increasingly considered for manufacturing routes to various building blocks[64] and pharmaceuticals.[1c],[65] Often these chemo-enzymatic routes represent scalable, cost-efficient, and green alternatives, with fewer synthetic steps, reduced waste production, and an improved overall synthetic efficiency. Consequently, ω-TA-catalyzed reactions have already been integrated into classical synthetic routes, resulting in highly successful chemo-enzymatic processes for the preparation of several chiral intermediates and active pharmaceutical ingredients (APIs).
One of the first published examples of a chemo-enzymatic route including an ω-TA-catalyzed amination step was the preparation of the anticholinergic agent (S)-rivastigmine (16), a potent cholinesterase inhibitor used for the treatment of Alzheimer's or Parkinson's disease.[66] The drug was prepared and isolated in 71 % overall yield through a short reaction sequence including the ω-TA-catalyzed amination of the ketone precursor 17 (45 mm substrate concentration, 100 mg scale) to afford the chiral amine moiety 18, representing the synthetic key step (Scheme 14, a).[67]
Scheme 14.
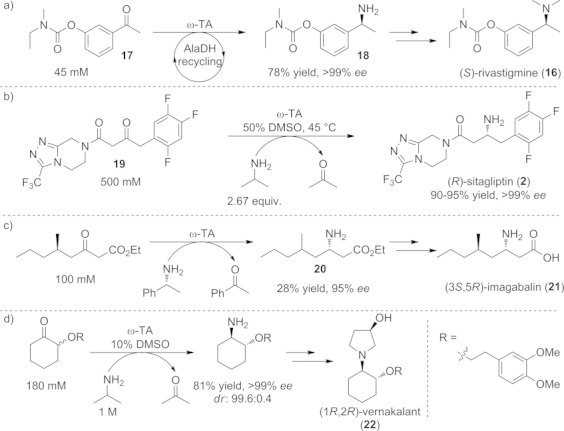
Chemo-enzymatic routes involving ω-TA-catalyzed key steps for the synthesis of a) the anticholinergic agent (S)-rivastigmine, b) the antidiabetic therapeutic sitagliptin, c) imagabalin, and d) the antiarrhythmic drug vernakalant.
The most prominent transamination-based process implemented on industrial scale, and hence a benchmark in the biocatalytic research field, was the synthesis of the antidiabetic drug sitagliptin (2).[14] Directed evolution provided an enzyme that was able to accept the sterically demanding ketone and to tolerate increased concentrations of organic co-solvents (e.g., DMSO) and higher temperatures. The combination of these improvements led to a reductive amination of 19, a prositagliptin ketone precursor, at elevated substrate concentration (200 g L–1) with 2-propylamine as amine donor. The target amine 2 was obtained in 90–95 % overall yield and in optically pure form (ee >99 %, Scheme 14, b).
An ω-TA was also engineered in order to adapt it to the demand for a process for the preparation of (3S,5R)-ethyl 3-amino-5-methyloctanoate (20), a precursor of imagabalin (21, Scheme 14, c)[68] and the antiarrhythmic compound (1R,2R)-vernakalant (22, Scheme 14, d).[69] In the latter example, directed evolution was used to provide an ω-TA with inverted diastereoselectivity, and a high selectivity for the recognition of the adjacent chiral center was achieved. Careful choice of the right reaction conditions enabled the racemization of the chiral center α to the ketone, leading to a dynamic kinetic resolution and the formation of the desired trans isomer with excellent enantio- and diastereoselectivity (>99:1 dr, >99 % ee). Consequently, this transamination represents the asymmetric key reaction in a five-step chemo-enzymatic route leading to vernakalant (22, 56 % overall yield). Identically to the sitagliptin (2) process, 2-propylamine turned out to be the ideal amine donor.
However, in some cases the basicity of 2-propylamine has led to side reactions. For instance, a careful choice of amine donor was necessary for the asymmetric key step in the chemo-enzymatic synthesis of the antiallergic drug ramatroban (23, Scheme 15, a). Use of the basic 2-propylamine led to enol/enamine formation, followed by oxidation in air to afford the corresponding energetically favored aromatic system.[70] However, by employing the sterically more demanding (R)-1-phenylethylamine (24) as amine donor, the formation of side products could be decreased dramatically and, moreover, the thermodynamic equilibrium was shifted towards the product side even with smaller amounts of amine donor. By this approach the amine intermediate 25 on the synthetic path to ramatroban (23) was prepared successfully on preparative scale (0.5 g) in 96 % isolated yield and enantiopure form. This substituted three steps of the previous chemo-enzymatic synthesis, which used lipase- or oxidoreductase-based techniques.[71]
Scheme 15.
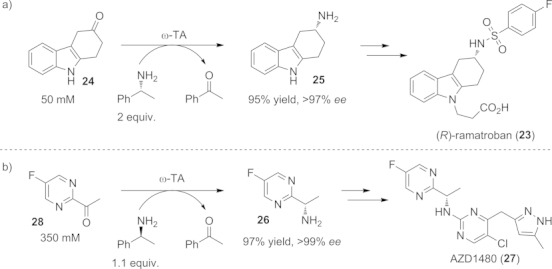
Reductive amination of the ketone precursors to a) the antiallergic drug ramatroban (23), and b) the JAK2 kinase inhibitor AZD1480 (27) by employing ω-TAs and the sterically more demanding 1-phenylethylamine as amine donor.
The excellent thermodynamic properties of 1-phenylethylamine (24) as amine donor were also exploited for the multigram preparation of (S)-1-(5-fluoropyrimidin-2-yl)ethanamine (26, 500 g, 20 L scale), a valuable precursor for the synthesis of the Janus kinase 2 (JAK2) inhibitor AZD1480 (27, Scheme 15, b).[72] The ketone precursor 28 was aminated by use of an S-selective ω-TA from V. fluvialis. In order to avoid inhibition due to the high substrate concentration (0.35 m), toluene was added as a co-solvent, resulting in a 97 % yield of the target amine 26. This ω-TA-catalyzed transformation was integrated into a recently proposed chemo-enzymatic long-term manufacturing route to the JAK2 kinase inhibitor AZD1480 (27) as a promising alternative to the classical route, showing again the great potential of the combination of bio- and chemocatalysis.[72a]
As already mentioned, amination/lactamization cascades – or, more generally, amination/cyclization cascades initiated by an ω-TA-catalyzed step – are quite efficient and have therefore been used in various chemo-enzymatic synthetic routes. For instance, biologically active γ-aminobutyric acid (GABA) derivatives such as 29, which play an important role in nervous system functions, were prepared by this approach (Scheme 16, a).[73]
Scheme 16.
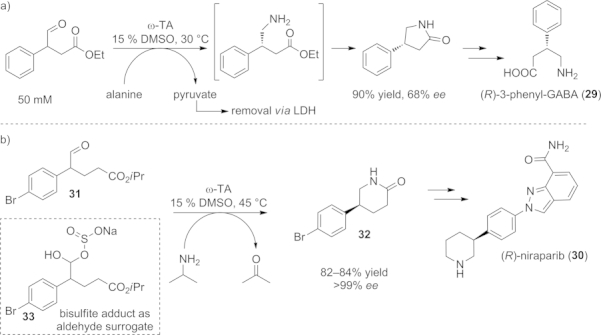
a) Amination/lactamization cascade initiated by an ω-TA-catalyzed step giving access to valuable lactams such as 4-phenylpyrrolidin-2-one, a precursor of GABA derivatives. b) The practical applicability of this cascade was shown in the chemo-enzymatic synthesis of the API niraparib.
Another application of that strategy has been reported for the preparation of niraparib (30), an inhibitor of the poly(ADP-ribose)polymerase involved in numerous cellular processes.[74] Thorough the employment of an ω-TA-catalyzed dynamic kinetic resolution starting from the racemic open-chain aldehyde 31, the (R)-lactam 32 [chiral building block for niraparib (30)] was obtained on preparative scale (35 g) with high yields (82 %) and stereoselectivity (Scheme 16, b). Because of the low stability of aldehyde 31 during the preliminary experiments, the transamination step was alternatively performed by starting from bisulfite adduct 33. This aldehyde surrogate is more stable and liberates the corresponding aldehyde 31 in situ under the basic conditions of the transaminase reaction, resulting in an 84 % yield of (R)-lactam 32. Using the aldehyde surrogate 33 improved the robustness and reliability of the process, making it suitable for large-scale applications.
A similar amination/lactamization concept was exploited for the dual orexin receptor antagonist MK-6096 (34), used for the treatment of insomnia (Scheme 17, a).[75] The preparation of this therapeutic agent was accomplished on a kilogram scale, leading to 1.2 kg of MK-6096 (34) in nine steps with an overall yield of 13 %. In the asymmetric key step, which builds up the α-methylpiperidine core 35, the keto diester substrate 36 is converted through an ω-TA-catalyzed transamination reaction into the corresponding amino diester 37 (50–57 g L–1). Compound 37 spontaneously cyclizes to the optically pure (R)-lactam 35. In addition to the already favorable thermodynamic equilibrium due to the lactamization step, product formation was further supported by removing the pyruvate byproduct through the action of the LDH/GDH system.
Scheme 17.
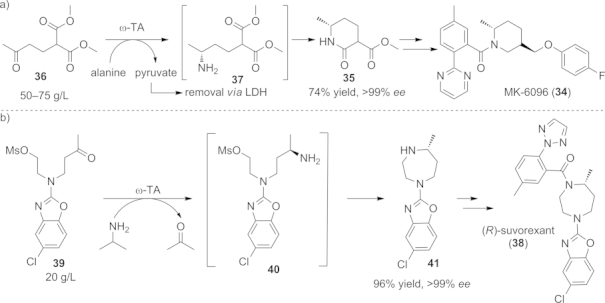
Chemo-enzymatic syntheses for the preparation of the dual orexin receptor antagonists a) MK-6096 (34), and b) suvorexant (38), each involving an ω-TA-catalyzed amination/cyclization step.
Suvorexant (38), another dual orexin receptor antagonist already in phase III clinical trials for the therapy of primary insomnia, has also been produced on a multigram scale by a chemo-enzymatic route including a similar amination/cyclization step (Scheme 17, b).[76] A key feature of this compound is the core chiral diazepane ring, which was obtained through the stereoselective amination of ketone precursor 39 by use of the ω-TA variant from Merck/Codexis[14] followed by a spontaneous cyclization of the unstable amino mesylate intermediate 40. In order to avoid the formation of various side products in the cyclization step, a thorough optimization of the pH and the leaving group had to be performed, ultimately giving the 1,4-diazepane ring 41 in good yield and perfect enantiopurity.
In this context the amination/imination cascade sequence already described above (see Scheme 2, a) leading to dihydropiperidines should be mentioned, because it represents a similar amination/cyclization approach.[22b] Nevertheless, standard reactions conditions were used, and no investigations of a potential beneficial effect of the spontaneous cyclization were reported.
Besides the synthesis of natural alkaloids, ω-TAs have also proven to be suitable catalysts for the enzymatic synthesis of 17α-aminosteroids (Scheme 18), which are used as intermediates in the preparation of biologically active steroidal derivatives.[77]
Scheme 18.
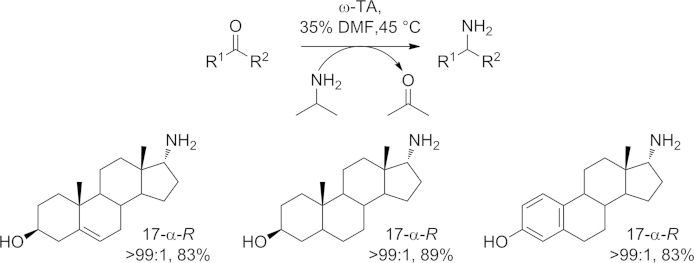
Biocatalytic route for the synthesis of 17α-aminosteroids with the aid of ω-TAs.
Another example showing the potential of combining classical chemical transformations with biocatalytic steps for the development of more scalable and cost-efficient manufacturing routes is the chemo-enzymatic synthesis of the CRTH2 antagonist MK-7246 (42, Scheme 19, a).[78] This compound was reported to be a potential therapeutic agent for the treatment of respiratory diseases.[79] The previous chemical route, consisting of eighteen steps with a 10 % overall yield, was optimized by introducing a direct transformation of the ketone precursor 43 into the desired chiral amine 44 by an ω-TA with excellent stereoselectivity. Consequently, the number of required reaction steps was reduced to nine, starting from commercially available materials. Without the need for any chromatographic purifications the product was obtained in 49 % overall yield, thus representing a quite efficient process optimization amenable to pilot-plant scale production.[78]
Scheme 19.
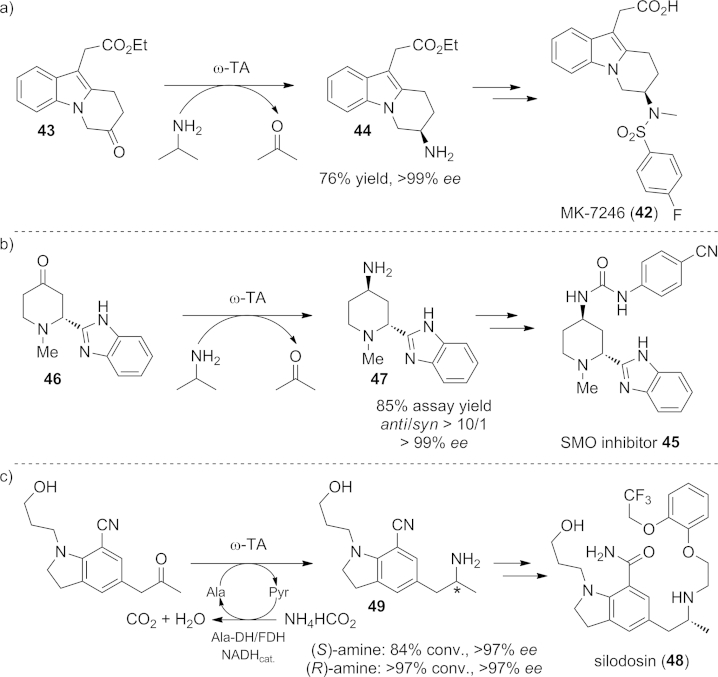
ω-TA-catalyzed key steps of chemo-enzymatic routes for the manufacture of a) the antiallergic CRTH2 antagonist MK-7246 (42), b) the anticarcinogenic smoothened receptor (SMO) inhibitor 45, and c) the silodosin intermediate 49.
Another concise asymmetric synthesis involving a transamination step has been developed recently for the large-scale commercial supply of compound 45, a smoothened receptor (SMO) inhibitor and a new therapeutic for the treatment of a broad range of human cancers.[80] The challenging key step of the synthesis was the establishment of the anti stereocenters on the 2- and 4-positions of the piperidine ring. By means of an ω-TA-catalyzed transamination of the 4-piperidone precursor 46, both centers – the required C-4 amino functionality as well as the anti-configured C-2 stereocenter – were generated simultaneously through a dynamic kinetic resolution process. The targeted anti-amine intermediate 47 was obtained in >10:1 dr and 99 % ee in a single step, and therefore the SMO inhibitor 45 was prepared in only five steps with 40 % overall yield, representing a highly attractive alternative route (Scheme 18, b). An enzyme-catalyzed transamination was also considered for the preparation of the amine 49, an intermediate on the pathway to the pro-drug silodosin (48), which is used for the treatment of dysuria and urinary disturbance (Scheme 18, c).[81]
Conclusions
Various examples, which have been provided to a significant extent by companies, demonstrate the applicability of ω-transaminases for the synthesis of valuable targets. Detailed investigation of the substrate scope, as well as a well-understood mechanism and detailed crystallographic analysis of the catalysts' protein structures, have built up a consolidated base for the application of transaminase enzymes on a laboratory scale, as well as on a pilot-plant scale. The interest results from the high demand for chiral amines as building blocks or intermediates within synthetic routes. The broad range of accepted ketone and aldehyde precursors, the robust reaction conditions, and the relatively easy preparation of the catalysts represent a highly attractive alternative to recent amination methodologies. Therefore the increasing number of laboratories focusing on the implementation of this biocatalytic technology is due to the fact that alternative methods from other fields of catalysis are less attractive or lack the robustness of the biocatalytic equivalent. Thus, ω-transaminases show great potential to become one of the big players in the preparation of (chiral) amines.
Michael Fuchs graduated from the university of Graz under the supervision of Prof. Kurt Faber. After his PhD in the same group – applying enzymes for the preparation of complex organic molecules – he spent two years at the MPI in Mülheim an der Ruhr with Prof. Alois Fürstner, where he worked on natural product synthesis and mechanistic studies involving ruthenium-catalyzed hydrogenations. Since 2015 he has been a postdoc in the working group of Prof. Wolfgang Kroutil, employing ω-transaminases for cascade processes and efficient synthesis of APIs.

Judith E. Farnberger did her bachelor's studies in environmental system sciences with focus on chemistry at the University of Graz. For her master's studies she switched to the TU Graz, where she focused on biotechnology and biocatalysis. She did her master's thesis under the supervision of Prof. W. Kroutil on ω-transaminases. After completing her master's studies in 2014 she is now working on her PhD in the same group.

Wolfgang Kroutil is professor at the University of Graz, Austria. He did his master's studies at the TU Graz and his doctoral studies at the University of Exeter (S. M. Roberts) and TU Graz (Kurt Faber). After a postdoc in industry (Syngenta) he worked as an R&D manager (Krems Chemie). In 2000 he became an assistant prof. at the University of Graz, in 2003 associate and in 2013 full professor. His research interests are in shortening chemical syntheses or in making them more efficient by using biocatalysts. Bio-oxidation and -reduction, C–C bond formation, cascades, and systems biocatalysis are some of his research topics.

Acknowledgments
M.F. is grateful for an Erwin-Schrödinger fellowship of the Austrian Science Fund (FWF, J3466-N28). J.F. has been supported by the Austrian BMWFJ, BMVIT, SFG, Standortagentur Tirol and ZIT through the Austrian FFG-COMET- Funding Program.
References
- 1a.Ghislieri D, Turner NJ. Top. Catal. 2014;57:284–300. [Google Scholar]
- 1b.Simon RC, Richter N, Busto E, Kroutil W. ACS Catal. 2014;4:129–143. [Google Scholar]
- 1c.Kohls H, Steffen-Munsberg F, Höhne M. Curr. Opin. Chem. Biol. 2014;19:180–192. doi: 10.1016/j.cbpa.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 1d.Kroutil W, Fischereder EM, Fuchs CS, Lechner H, Mutti FG, Pressnitz D, Rajagopalan A, Sattler JH, Simon RC, Siirola E. Org. Process Res. Dev. 2013;17:751–759. doi: 10.1021/op4000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e.Mathew S, Yun H. ACS Catal. 2012;2:993–1001. [Google Scholar]
- 1f.Malik MS, Park ES, Shin JS. Appl. Microbiol. Biotechnol. 2012;94:1163–1171. doi: 10.1007/s00253-012-4103-3. [DOI] [PubMed] [Google Scholar]
- 1g.Rudat J, Brucher BR, Syldatk C. AMB Express. 2012;2:11. doi: 10.1186/2191-0855-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h.Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W, Woodley JM. Biotechnol. Bioeng. 2011;108:1479–1493. doi: 10.1002/bit.23154. [DOI] [PubMed] [Google Scholar]
- 2.Yamada H, Kimura T, Tanaka A, Ogata K. Agric. Biol. Chem. 1964;28:443–450. [Google Scholar]
- 3a.Nestl BM, Hammer SC, Nebel BA, Hauer B. Angew. Chem. Int. Ed. 2014;53:3070–3095. doi: 10.1002/anie.201302195. Angew. Chem.2014, 126, 3132. [DOI] [PubMed] [Google Scholar]
- 3b.Ghislieri D, Turner NJ. Top. Catal. 2014;57:284–300. [Google Scholar]
- 4a.Kim K-H. J. Biol. Chem. 1964;239:783–786. [PubMed] [Google Scholar]
- 4b.Noe FF, Nickerson WJ. J. Bacteriol. 1958;75:674–681. doi: 10.1128/jb.75.6.674-681.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5a.Matcham G, Bhatia M, Lang W, Lewis C, Nelson R, Wang A, Wu W. Chimia. 1999;53:584–589. [Google Scholar]
- 5b.Stirling DI, Zeitlin AL, Matcham GW. 1990. US Patent, US 4,950,606.
- 6.Shin J-S, Kim B-G. Biotechnol. Bioeng. 1997;55:348–358. doi: 10.1002/(SICI)1097-0290(19970720)55:2<348::AID-BIT12>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 7.Shin J-S, Kim B-G. Biotechnol. Bioeng. 1999;65:206–211. [PubMed] [Google Scholar]
- 8.Shin J-S, Kim B-G, Liese A, Wandrey C. Biotechnol. Bioeng. 2001;73:179–187. doi: 10.1002/bit.1050. [DOI] [PubMed] [Google Scholar]
- 9a.Balkenhohl F, Ditrich K, Nübling C. 2001. US Patent, US 6214608(A1)
- 9b.Balkenhohl F, Hauer B, Ladner W, Nuebling C, Pressler U. 1995. PCT Int. Appl, WO 9508636(A1)
- 10.Höhne M, Kühl S, Robins K, Bornscheuer UT. ChemBioChem. 2008;9:363–365. doi: 10.1002/cbic.200700601. [DOI] [PubMed] [Google Scholar]
- 11.Nakamichi K. Appl. Microbiol. 1990;33:637–640. [Google Scholar]
- 12a.Kara S, Schrittwieser JH, Hollmann F, Ansorge-Schumacher MB. Appl. Microbiol. Biotechnol. 2014;98:1517–1529. doi: 10.1007/s00253-013-5441-5. [DOI] [PubMed] [Google Scholar]
- 12b.Weckbecker A, Groger H, Hummel W. Biosystems Engineering I: Creating Superior Biocatalysts. 2010;120:195–242. doi: 10.1007/10_2009_55. [DOI] [PubMed] [Google Scholar]
- 12c.Berenguer-Murcia A, Fernandez-Lafuente R. Curr. Org. Chem. 2010;14:1000–1021. [Google Scholar]
- 12d.Liu WF, Wang P. Biotechnol. Adv. 2007;25:369–384. doi: 10.1016/j.biotechadv.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 13a.Koszelewski D, Lavandera I, Clay D, Rozzell D, Kroutil W. Adv. Synth. Catal. 2008;350:2761–2766. [Google Scholar]
- 13b.Truppo MD, Rozzell JD, Moore JC, Turner NJ. Org. Biomol. Chem. 2009;7:395–398. doi: 10.1039/b817730a. [DOI] [PubMed] [Google Scholar]
- 14.Savile CK, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ. Science. 2010;329:305–309. doi: 10.1126/science.1188934. [DOI] [PubMed] [Google Scholar]
- 15a.Galkin A, Kulakova L, Yamamoto H, Tanizawa K, Tanaka H, Esaki N, Soda K. J. Ferment. Bioeng. 1997;83:299–300. [Google Scholar]
- 15b.Richter N, Farnberger JE, Pressnitz D, Lechner H, Zepeck F, Kroutil W. Green Chem. 2015;17:2952–2958. [Google Scholar]
- 16.Koszelewski D, Lavandera I, Clay D, Guebitz GM, Rozzell D, Kroutil W. Angew. Chem. Int. Ed. 2008;47:9337–9340. doi: 10.1002/anie.200803763. Angew. Chem.2008, 120, 9477. [DOI] [PubMed] [Google Scholar]
- 17.Kadyrov R, Riermeier TH. Angew. Chem. Int. Ed. 2003;42:5472–5474. doi: 10.1002/anie.200352503. Angew. Chem.2003, 115, 5630. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Q, Wen J, Tan R, Huang K, Metola P, Wang R, Anslyn EV, Zhang X. Angew. Chem. Int. Ed. 2014;53:8467–8470. doi: 10.1002/anie.201404570. Angew. Chem.2014, 126, 8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzer AK, Hiebler K, Mutti FG, Simon RC, Lauterbach L, Lenz O, Kroutil W. Org. Lett. 2015;17:2431–2433. doi: 10.1021/acs.orglett.5b01154. [DOI] [PubMed] [Google Scholar]
- 20.Park E-S, Park S-R, Han S-W, Dong J-Y, Shin J-S. Adv. Synth. Catal. 2014;356:212–220. [Google Scholar]
- 21.Nobili A, Steffen-Munsberg F, Kohls H, Trentin I, Schulzke C, Höhne M, Bornscheuer UT. ChemCatChem. 2015;7:757–760. [Google Scholar]
- 22a.Simon RC, Grischek B, Zepeck F, Steinreiber A, Belaj F, Kroutil W. Angew. Chem. Int. Ed. 2012;51:6713–6716. doi: 10.1002/anie.201202375. Angew. Chem.2012, 124, 6817. [DOI] [PubMed] [Google Scholar]
- 22b.Simon RC, Zepeck F, Kroutil W. Chem. Eur. J. 2013;19:2859–2865. doi: 10.1002/chem.201202793. [DOI] [PubMed] [Google Scholar]
- 23.Truppo MD, Turner NJ, Rozzell JD. Chem. Commun. 2009:2127–2129. doi: 10.1039/b902995h. [DOI] [PubMed] [Google Scholar]
- 24.Green AP, Turner NJ, O'Reilly E. Angew. Chem. Int. Ed. 2014;53:10714–10717. doi: 10.1002/anie.201406571. Angew. Chem.2014, 126, 10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schätzle S, Höhne M, Redestad E, Robins K, Bornscheuer UT. Anal. Chem. 2009;81:8244–8248. doi: 10.1021/ac901640q. [DOI] [PubMed] [Google Scholar]
- 26.Weiß MS, Pavlidis IV, Vickers C, Höhne M, Bornscheuer UT. Anal. Chem. 2014;86:11847–11853. doi: 10.1021/ac503445y. [DOI] [PubMed] [Google Scholar]
- 27.Höhne M, Schätzle S, Jochens H, Robins K, Bornscheuer UT. Nat. Chem. Biol. 2010;6:807–813. doi: 10.1038/nchembio.447. [DOI] [PubMed] [Google Scholar]
- 28.Jiang JJ, Chen X, Zhang DL, Wu QQ, Zhu DM. Appl. Microbiol. Biotechnol. 2015;99:2613–2621. doi: 10.1007/s00253-014-6056-1. [DOI] [PubMed] [Google Scholar]
- 29.Schätzle S, Steffen-Munsberg F, Thontowi A, Höhne M, Robins K, Bornscheuer UT. Adv. Synth. Catal. 2011;353:2439–2445. [Google Scholar]
- 30.Steffen-Munsberg F, Vickers C, Thontowi A, Schätzle S, Tumlirsch T, Svedendahl Humble M, Land H, Berglund P, Bornscheuer UT, Höhne M. ChemCatChem. 2013;5:150–153. [Google Scholar]
- 31.Truppo MD, Strotman H, Hughes G. ChemCatChem. 2012;4:1071–1074. [Google Scholar]
- 32.Mutti FG, Kroutil W. Adv. Synth. Catal. 2012;354:3409–3413. [Google Scholar]
- 33a.Fuchs M, Tauber K, Sattler J, Lechner H, Pfeffer J, Kroutil W, Faber K. RSC Adv. 2012;2:6262–6265. [Google Scholar]
- 33b.Sattler JH, Fuchs M, Mutti FG, Grischek B, Engel P, Pfeffer J, Woodley JM, Kroutil W. Angew. Chem. Int. Ed. 2014;53:14153–14157. doi: 10.1002/anie.201409227. Angew. Chem.2014, 126, 14377. [DOI] [PubMed] [Google Scholar]
- 33c.Sattler JH, Fuchs M, Tauber K, Mutti FG, Faber K, Pfeffer J, Haas T, Kroutil W. Angew. Chem. Int. Ed. 2012;51:9156–9159. doi: 10.1002/anie.201204683. Angew. Chem.2012, 124, 9290. [DOI] [PubMed] [Google Scholar]
- 34a.Gunanathan C, Milstein D. Angew. Chem. Int. Ed. 2008;47:8661–8664. doi: 10.1002/anie.200803229. Angew. Chem.2008, 120, 8789. [DOI] [PubMed] [Google Scholar]
- 34b.Imm S, Bähn S, Neubert L, Neumann H, Beller M. Angew. Chem. Int. Ed. 2010;49:8126–8129. doi: 10.1002/anie.201002576. Angew. Chem.2010, 122, 8303. [DOI] [PubMed] [Google Scholar]
- 35.Tauber K, Fuchs M, Sattler JH, Pitzer J, Pressnitz D, Koszelewski D, Faber K, Pfeffer J, Haas T, Kroutil W. Chem. Eur. J. 2013;19:4030–4035. doi: 10.1002/chem.201202666. [DOI] [PubMed] [Google Scholar]
- 36.Fuchs CS, Hollauf M, Meissner M, Simon RC, Besset T, Reek JNH, Riethorst W, Zepeck F, Kroutil W. Adv. Synth. Catal. 2014;356:2257–2265. [Google Scholar]
- 37.Andrade LH, Kroutil W, Jamison TF. Org. Lett. 2014;16:6092–6095. doi: 10.1021/ol502712v. [DOI] [PubMed] [Google Scholar]
- 38a.Soda K, Yoshimura T, Esaki N. Chem. Rec. 2001;1:373–384. doi: 10.1002/tcr.1021. [DOI] [PubMed] [Google Scholar]
- 38b.Cassimjee KE, Humble MS, Miceli V, Colomina CG, Berglund P. ACS Catal. 2011;1:1051–1055. [Google Scholar]
- 39.Cassimjee KE, Manta B, Himo F. Org. Biomol. Chem. 2015;13:8453–8464. doi: 10.1039/c5ob00690b. [DOI] [PubMed] [Google Scholar]
- 40.Griswold WR, Toney MD. J. Am. Chem. Soc. 2011;133:14823–14830. doi: 10.1021/ja2061006. [DOI] [PubMed] [Google Scholar]
- 41a.Stryer L. In: Biochemistry. Freeman and Company WE, editor. New York: 1999. p. 633. [Google Scholar]
- 41b.Schiroli D, Peracchi A. Biochim. Biophys. Acta - Prot. Proteom. 2015;1854:1200–1211. doi: 10.1016/j.bbapap.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 41c.Steffen-Munsberg F, Vickers C, Kohls H, Land H, Mallin H, Nobili A, Skalden L, van den Bergh T, Joosten H-J, Berglund P, Höhne M, Bornscheuer UT. Biotechnol. Adv. 2015;33:566–604. doi: 10.1016/j.biotechadv.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 42a.Humble MS, Cassimjee KE, Håkansson M, Kimbung YR, Walse B, Abedi V, Federsel H-J, Berglund P, Logan DT. FEBS J. 2012;279:779–792. doi: 10.1111/j.1742-4658.2012.08468.x. [DOI] [PubMed] [Google Scholar]
- 42b.Łyskowski A, Gruber C, Steinkellner G, Schürmann M, Schwab H, Gruber K, Steiner K. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0087350. e87350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42c.Park E-S, Dong J-Y, Shin J-S. Appl. Microbiol. Biotechnol. 2014;98:651–660. doi: 10.1007/s00253-013-4846-5. [DOI] [PubMed] [Google Scholar]
- 42d.Park E-S, Kim M, Shin J-S. Appl. Microbiol. Biotechnol. 2012;93:2425–2435. doi: 10.1007/s00253-011-3584-9. [DOI] [PubMed] [Google Scholar]
- 42e.Sayer C, Martinez-Torres RJ, Richter N, Isupov MN, Hailes HC, Littlechild JA, Ward JM. FEBS J. 2014;281:2240–2253. doi: 10.1111/febs.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43a.Bachmann S, Knudsen KR, Jorgensen KA. Org. Biomol. Chem. 2004;2:2044–2049. doi: 10.1039/b404381b. [DOI] [PubMed] [Google Scholar]
- 43b.Knudsen KR, Bachmann S, Jorgensen KA. Chem. Commun. 2003:2602–2603. doi: 10.1039/b308395k. [DOI] [PubMed] [Google Scholar]
- 44a.Łyskowski A, Gruber C, Steinkellner G, Schürmann M, Schwab H, Gruber K, Steiner K. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0087350. e87350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b.Thomsen M, Skalden L, Palm GJ, Höhne M, Bornscheuer UT, Hinrichs W. Acta Crystallogr., Sect. D. 2014;70:1086–1093. doi: 10.1107/S1399004714001084. [DOI] [PubMed] [Google Scholar]
- 44c.Humble MS, Cassimjee KE, Håkansson M, Kimbung YR, Walse B, Abedi V, Federsel H-J, Berglund P, Logan DT. FEBS J. 2012;279:779–792. doi: 10.1111/j.1742-4658.2012.08468.x. [DOI] [PubMed] [Google Scholar]
- 44d.Rausch C, Lerchner A, Schiefner A, Skerra A. Proteins Struct., Funct., Bioinf. 2013;81:774–787. doi: 10.1002/prot.24233. [DOI] [PubMed] [Google Scholar]
- 44e.Sayer C, Isupov MN, Littlechild JA. Acta Crystallogr., Sect. F. 2007;63:117–119. doi: 10.1107/S1744309107000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skalden L, Peters C, Dickerhoff J, Nobili A, Joosten H-J, Weisz K, Höhne M, Bornscheuer UT. ChemBioChem. 2015;16:1041–1045. doi: 10.1002/cbic.201500074. [DOI] [PubMed] [Google Scholar]
- 46a.Oroz-Guinea I, Garcia-Junceda E. Curr. Opin. Chem. Biol. 2013;17:236–249. doi: 10.1016/j.cbpa.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 46b.Ricca E, Brucher B, Schrittwieser JH. Adv. Synth. Catal. 2011;353:2239–2262. [Google Scholar]
- 46c.O'Reilly E, Turner NJ. Perspectives Sci. 2015;4:55–61. [Google Scholar]
- 47.Skalden L, Peters C, Dickerhoff J, Nobili A, Joosten H-J, Weisz K, Höhne M, Bornscheuer UT. ChemBioChem. 2015;16:1041–1045. doi: 10.1002/cbic.201500074. [DOI] [PubMed] [Google Scholar]
- 48a.Ingram CU, Bommer M, Smith MEB, Dalby PA, Ward JM, Hailes HC, Lye GJ. Biotechnol. Bioeng. 2007;96:559–569. doi: 10.1002/bit.21125. [DOI] [PubMed] [Google Scholar]
- 48b.Smith MEB, Chen BH, Hibbert EG, Kaulmann U, Smithies K, Galman JL, Baganz F, Dalby PA, Hailes HC, Lye GJ, Ward JM, Woodley JM, Micheletti M. Org. Process Res. Dev. 2010;14:99–107. [Google Scholar]
- 49a.Bergmeier SC. Tetrahedron. 2000;56:2561–2576. [Google Scholar]
- 49b.Karjalainen OK, Koskinen AMP. Org. Biomol. Chem. 2012;10:4311–4326. doi: 10.1039/c2ob25357g. [DOI] [PubMed] [Google Scholar]
- 49c.Lait SM, Rankic DA, Keay BA. Chem. Rev. 2007;107:767–796. doi: 10.1021/cr050065q. [DOI] [PubMed] [Google Scholar]
- 50.Sehl T, Hailes HC, Ward JM, Wardenga R, von Lieres E, Offermann H, Westphal R, Pohl M, Rother D. Angew. Chem. Int. Ed. 2013;52:6772–6775. doi: 10.1002/anie.201300718. Angew. Chem.2013, 125, 6904. [DOI] [PubMed] [Google Scholar]
- 51.Sehl T, Hailes HC, Ward JM, Menyes U, Pohl M, Rother D. Green Chem. 2014;16:3341–3348. [Google Scholar]
- 52.Kohls H, Anderson M, Dickerhoff J, Weisz K, Córdova A, Berglund P, Brundiek H, Bornscheuer UT, Höhne M. Adv. Synth. Catal. 2015;357:1808–1814. [Google Scholar]
- 53.Siirola E, Mutti FG, Grischek B, Hoefler SF, Fabian WMF, Grogan G, Kroutil W. Adv. Synth. Catal. 2013;355:1703–1708. [Google Scholar]
- 54a.Koszelewski D, Clay D, Rozzell D, Kroutil W. Eur. J. Org. Chem. 2009:2289–2292. [Google Scholar]
- 54b.Koszelewski D, Grischek B, Glueck SM, Kroutil W, Faber K. Chem. Eur. J. 2011;17:378–383. doi: 10.1002/chem.201001602. [DOI] [PubMed] [Google Scholar]
- 55.Koszelewski D, Pressnitz D, Clay D, Kroutil W. Org. Lett. 2009;11:4810–4812. doi: 10.1021/ol901834x. [DOI] [PubMed] [Google Scholar]
- 56.Koszelewski D, Müller N, Schrittwieser JH, Faber K, Kroutil W. J. Mol. Catal. B. 2010;63:39–44. [Google Scholar]
- 57.Shin G, Mathew S, Shon M, Kim BG, Yun H. Chem. Commun. 2013;49:8629–8631. doi: 10.1039/c3cc43348j. [DOI] [PubMed] [Google Scholar]
- 58.O'Reilly E, Iglesias C, Turner NJ. ChemCatChem. 2014;6:992–995. [Google Scholar]
- 59.Lerchner A, Achatz S, Rausch C, Haas T, Skerra A. ChemCatChem. 2013;5:3374–3383. [Google Scholar]
- 60.Klatte S, Wendisch VF. Bioorg. Med. Chem. 2014;22:5578–5585. doi: 10.1016/j.bmc.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 61.Schrewe M, Ladkau N, Buhler B, Schmid A. Adv. Synth. Catal. 2013;355:1693–1697. [Google Scholar]
- 62.Karau A, Sieber V, Haas T, Heager H, Grammann K, Buehler B, Blank L, Schmid A, Jach G, Lalla B, Mueller A, Schullehner K, Welters P, Eggert T, Weckbecker A. 2010. US Patent, US20100324257 A1.
- 63.Truppo MD, Rozzell JD, Turner NJ. Org. Process Res. Dev. 2010;14:234–237. [Google Scholar]
- 64a.Schmidt NG, Simon RC, Kroutil W. Adv. Synth. Catal. 2015;357:1815–1821. [Google Scholar]
- 64b.Andrade LH, Silva AV, Milani P, Koszelewski D, Kroutil W. Org. Biomol. Chem. 2010;8:2043–2051. doi: 10.1039/b920946h. [DOI] [PubMed] [Google Scholar]
- 64c.Pressnitz D, Fuchs CS, Sattler JH, Knaus T, Macheroux P, Mutti FG, Kroutil W. ACS Catal. 2013;3:555–559. [Google Scholar]
- 64d.Cuetos A, Lavandera I, Gotor V. Chem. Commun. 2013;49:10688–10690. doi: 10.1039/c3cc46760k. [DOI] [PubMed] [Google Scholar]
- 64e.Paul CE, Rodriguez-Mata M, Busto E, Lavandera I, Gotor-Fernandez V, Gotor V, Garcia-Cerrada S, Mendiola J, de Frutos O, Collado I. Org. Process Res. Dev. 2014;18:788–792. [Google Scholar]
- 64f.Esparza-Isunza T, Gonzalez-Brambila M, Gani R, Woodley JM, Lopez-Isunza F. Chem. Eng. J. 2015;259:221–231. [Google Scholar]
- 65a.Gröger H, Hummel W. Curr. Opin. Chem. Biol. 2014;19:171–179. doi: 10.1016/j.cbpa.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 65b.Huisman GW, Collier SJ. Curr. Opin. Chem. Biol. 2013;17:284–292. doi: 10.1016/j.cbpa.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 65c.Tao JH, Zhao LS, Ran NQ. Org. Process Res. Dev. 2007;11:259–267. [Google Scholar]
- 66.Emre M. CNS Drugs. 2006;20:748–748. [Google Scholar]
- 67a.Cabirol FL, Gohel A, Oh SH, Smith DJ, Wong B, Lalonde JJ. 2015. US Patent, US 08932838.
- 67b.Fuchs M, Koszelewski D, Tauber K, Kroutil W, Faber K. Chem. Commun. 2010;46:5500–5502. doi: 10.1039/c0cc00585a. [DOI] [PubMed] [Google Scholar]
- 67c.Fuchs M, Koszelewski D, Tauber K, Sattler J, Banko W, Holzer AK, Pickl M, Kroutil W, Faber K. Tetrahedron. 2012;68:7691–7694. [Google Scholar]
- 68.Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K, Gehlhaar DK, Mistry A, Chang JS, Anderson M, Villalobos A, Minshull J, Govindarajan S, Wong JW. Protein Eng. Des. Sel. 2013;26:25–33. doi: 10.1093/protein/gzs065. [DOI] [PubMed] [Google Scholar]
- 69.Limanto J, Ashley ER, Yin JJ, Beutner GL, Grau BT, Kassim AM, Kim MM, Klapars A, Liu ZJ, Strotman HR, Truppo MD. Org. Lett. 2014;16:2716–2719. doi: 10.1021/ol501002a. [DOI] [PubMed] [Google Scholar]
- 70.Busto E, Simon RC, Grischek B, Gotor-Fernández V, Kroutil W. Adv. Synth. Catal. 2014;356:1937–1942. [Google Scholar]
- 71.Busto E, Gotor-Fernandez V, Gotor V. J. Org. Chem. 2012;77:4842–4848. doi: 10.1021/jo300552v. [DOI] [PubMed] [Google Scholar]
- 72a.Frodsham L, Golden M, Hard S, Kenworthy MN, Klauber DJ, Leslie K, Macleod C, Meadows RE, Mulholland KR, Reilly J, Squire C, Tomasi S, Watt D, Wells AS. Org. Process Res. Dev. 2013;17:1123–1130. [Google Scholar]
- 72b.Meadows RE, Mulholland KR, Schurmann M, Golden M, Kierkels H, Meulenbroeks E, Mink D, May O, Squire C, Straatman H, Wells AS. Org. Process Res. Dev. 2013;17:1117–1122. [Google Scholar]
- 73.Koszelewski D, Clay D, Faber K, Kroutil W. J. Mol. Catal. B. 2009;60:191–194. [Google Scholar]
- 74.Chung CK, Bulger PG, Kosjek B, Belyk KM, Rivera N, Scott ME, Humphrey GR, Limanto J, Bachert DC, Emerson KM. Org. Process Res. Dev. 2014;18:215–227. [Google Scholar]
- 75.Girardin M, Ouellet SG, Gauvreau D, Moore JC, Hughes G, Devine PN, O'Shea PD, Campeau LC. Org. Process Res. Dev. 2013;17:61–68. [Google Scholar]
- 76.Mangion IK, Sherry BD, Yin JJ, Fleitz FJ. Org. Lett. 2012;14:3458–3461. doi: 10.1021/ol3014123. [DOI] [PubMed] [Google Scholar]
- 77.Richter N, Simon RC, Kroutil W, Ward JM, Hailes HC. Chem. Commun. 2014;50:6098–6100. doi: 10.1039/c3cc49080g. [DOI] [PubMed] [Google Scholar]
- 78.Molinaro C, Bulger PG, Lee EE, Kosjek B, Lau S, Gauvreau D, Howard ME, Wallace DJ, O'Shea PD. J. Org. Chem. 2012;77:2299–2309. doi: 10.1021/jo202620r. [DOI] [PubMed] [Google Scholar]
- 79.Gallant M, Beaulieu C, Berthelette C, Colucci J, Crackower MA, Dalton C, Denis D, Ducharme Y, Friesen RW, Guay D, Gervais FG, Hamel M, Houle R, Krawczyk CM, Kosjek B, Lau S, Leblanc Y, Lee EE, Levesque JF, Mellon C, Molinaro C, Mullet W, O'Neill GP, O'Shea P, Sawyer N, Sillaots S, Simard D, Slipetz D, Stocco R, Sorensen D, Truong VL, Wong E, Wu J, Zaghdane H, Wang ZY. Bioorg. Med. Chem. Lett. 2011;21:288–293. doi: 10.1016/j.bmcl.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 80.Peng ZH, Wong JW, Hansen EC, Puchlopek-Dermenci ALA, Clarke HJ. Org. Lett. 2014;16:860–863. doi: 10.1021/ol403630g. [DOI] [PubMed] [Google Scholar]
- 81.Simon RC, Sattler JH, Farnberger JE, Fuchs CS, Richter N, Zepeck F, Kroutil W. Tetrahedron: Asymmetry. 2014;25:284–288. [Google Scholar]