

Abstract
Juvenile scleroderma (JS) represents a rarely seen group of connective tissue diseases with multiple organ involvement. Cardiac involvement in JSS is well known and, although rare in children, it may be an important cause of mortality and morbidity. Therefore, an early determination of cardio-vascular and pulmonary involvement is of the most relevance to reduce the mortality in patients with juvenile scleroderma. The aim of the study was to explore the non-invasive methods (Doppler echocardiography, pulmonary function tests), Forced vital capacity (FVC) and Carbon monoxide diffusion capacity (DLCO) in the assessment of the cardiopulmonary involvement in patients with JS. The assessment of pulmonary arterial pressure (PAP) and risk factors for pulmonary arterial hypertension (PAH) were made by the measurement of maximum tricuspid insufficiency (TI), end-diastolic pulmonary insufficiency (PI), ratio of acceleration time (AT) to ejection time (ET) (AT/ET), right atrial pressure (RAP) and contraction of vena cava inferior during inspiration. Thirty-five patients with confirmed JS were included in the study. The mean age of onset of the disease was 9.57 years (median 10 years, range 2-18 years). The mean disease duration and follow-up time was 2 years (median 1 year, range 0.5-8 years) and 3.57 years (median 2 years, range 0.5-14.5 years), respectively. The values of all the analyzed parameters including TI, PI, AT/ET, PAP, FVC and DLCO were found to be within normal ranges in all the patients tested, confirming an uncommonness of cardiopulmonary involvement in patients with juvenile scleroderma.
KEY WORDS: Juvenile systemic sclerosis, juvenile localized scleroderma, pulmonary hypertension
INTRODUCTION
Juvenile scleroderma (JS) is a rare connective tissue disease affecting multiple organs [1]. The published data on JS are sparse [2]. Its clinical presentation differs from the adult form, typically manifesting as either juvenile localized scleroderma (JLS) or juvenile systemic sclerosis (JSS). Compared with JSS, JLS is an indolent, self-limiting form of the disease, associated with better prognosis that can occasionally progress into a systemic form. JLS is the most frequent form of scleroderma in childhood [3]. It is estimated that 10% of all patients with scleroderma develop the disease before the age of 8 years, with the mean age of onset at 8.8 years [1,3].
Cardiac involvement in JSS is well known and, although rare in children, it may be an important cause of mortality and morbidity [4]. Pulmonary arterial hypertension (PAH) represents the most pertinent form of pulmonary involvement. The presence of PAH in scleroderma patient has a devastating impact on survival. Prior to the availability of PAH-specific therapies, the 5-year survival rate was only 10% for scleroderma patients with PAH, compared with 80% for scleroderma patients without PAH [5]. The previous reports showed that the connective tissue disease-associated PAH cohort accounts for up to 30% of all cases of PAH, and the vast majority of these cases reflect scleroderma-associated PAH [6]. Among adults patients with scleroderma, the PAH prevalence is estimated to be 10-15%, whereas in children it was reported to be 7% [2]. The studies of PAH among juvenile scleroderma patients are insufficient and there is a need for further investigations.
PAH is defined by a mean pulmonary arterial pressure (MPAP) of 25 mmHg or greater at rest. The mean pulmonary arterial pressure of 30-40 mmHg produces clinical symptoms; when the MPAP reaches the value of 50-70 mmHg, a clinical worsening and a reduced heart output are observed [7]. Since the clinical symptoms are seen in the advanced disease, it is of crucial importance to determine the cardio-vascular involvement in its early stage, when the patients are still asymptomatic. Although the right-sided heart catheterization (RHC) remains the gold standard for diagnosis of PAH, the Doppler echocardiographic examination (Doppler-echo) is used as a reliable screening tool in patients with scleroderma [8]. Pulmonary function tests (PFT) are helpful in the investigation of PAH. The measurement of Functional vital capacity (FVC) and Carbon monoxide diffusion capacity (DLCO) is useful in determination of the interstitial lung disease, which could lead to the PAH development. The ratio of FVC/DLCO >1.6 is shown to be indicative for PAH [9]. The early diagnosis of cardio-vascular involvement influences the clinical course and prognosis of the disease. The aim of our study was to use the non-invasive methods (echocardiography, pulmonary function tests) to examine the cardiopulmonary involvement in the patients with juvenile scleroderma.
MATERIALS AND METHODS
A total of 35 patients (31 female, 4 male) with diagnosis of juvenile scleroderma, followed up at Pedatric Rheumatology Department, Cerrahpasa Medical School were included in the study. The diagnosis of scleroderma was established according to 2013 updated classification criteria for systemic sclerosis, proposed by The American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) [9].
Doppler echocardiography was performed at Pediatric Cardiology Department.
Pulmonary function tests were performed at Laboratory for pulmonary function tests.
Criteria for inclusion in the study were: a) Diagnosis of juvenile scleroderma; b) Regular clinical follow-up at Pediatric Rheumatology Department, Cerrahpasa Medical Faculty; c) Performed Doppler echocardiography at Pediatric Cardiology Department; and d) Performed pulmonary function tests at the Laboratory for pulmonary function tests.
Patients with an irregular follow-up and those who were treatment non-compliant were excluded from the study (totally 5 patients: 4 with local scleroderma and one patient with systemic sclerosis).
Doppler echocardiographic examination
The assessment of pulmonary arterial pressure was made by measurement of maximum tricuspid insufficiency (TI), end-diastolic pulmonary insufficiency (PI), the ratio of acceleration time (AT) to ejection time (ET) (AT/ET), right atrial pressure (RAP) and contraction of vena cava inferior during inspiration [10]. Pulmonary arterial pressure was determined using three different methods: 1- Systolic pulmonary arterial pressure (SPAP) was estimated by measuring the maximal velocity of tricuspid insufficiency (TI)2- Diastolic pulmonary arterial pressure (DPAP) was estimated by measuring the velocity of pulmonary insufficiency at the end of diastole (PI)3- Mean pulmonary arterial pressure (MPAP) was determined by measuring the pulmonary insufficiency acceleration time (AT) and the ratio of AT to ejection time (ET).
Systolic pulmonary arterial pressure was calculated by modified Bernoulli equation, in which SPAP = 4 x TRVmax2 + right atrial pressure, where TRVmax was maximal velocity of tricuspid insufficiency. Right atrial pressure was evaluated by measuring the right atrial volume, the degree of tricuspid regurgitation and contraction of vena cava inferior during inspiration [11,12]. The volume of the right atrium was normal for all the patients, collapse of vena cava inferior during inspiration was more than 50% and none of the patients had a significant tricuspid regurgitation. Therefore, the right atrial pressure was supposed to be normal and was accepted to be 5 mmHg for all the patients. The cutoff value for the maximal velocity of tricuspid insufficiency was a 3 m/sec, while for calculated SPAP it was a 40 mmHg. All the values above the cutoff were considered as pathologic [4,5,11]. The diastolic pulmonary arterial pressure (DPAP) was estimated by measuring the velocity of pulmonary insufficiency at the end of diastole, with a cutoff value of 2m/s [12,13]. Mean pulmonary arterial pressure (MPAP) was determined by assessing the pulmonary insufficiency acceleration time (AT) and the ratio of AT to ejection time (ET); AT/ET < 0.3 was accepted as pathologic [11,13,14]. The same cardiologist performed all the measurements, repeating them for three times. The mean value was used in the statistical analysis.
Pulmonary function tests
Pulmonary function tests were performed in order to estimate the forced vital capacity and CO diffusion capacity. Results were adjusted to the age, gender, weight, height and hemoglobin levels of the patients. The previous studies showed that values of FVC < 80%, DLCO < 60% and FVC/DLCO > 1.6 imply a risk for PAH [4,10,11,12].
Statistical analysis
Patients were assigned into two groups: patients with JLS and those with JSS. Since the cardio-vascular involvement is not expected in the JLS patients, they were regarded as a control group. Statistical comparison was made between those two groups of patients. In the same time, all measurements were statistically analyzed according to the pathological values being established by the American Association of Pediatric Cardiology [13] and to the data from literature. Categorical variables were assessed by χ2 test and Fisher’s exact test. For the evaluation of the echocardiographic findings and pulmonary function tests results, we used a Mann-Whitney test. Reported p value was shown in 3 decimals. If the fourth decimal was found to be < 5, the p value is considered to be < 0.001. The p < 0.05 was considered significant. Statistical analyses were performed using SPSS software version 15.0 (Statistical Package for Social Sciences, IBM Corporation, USA).
RESULTS
The study was conducted between January 2013 and January 2014. A total of 35 patients with scleroderma (31 female, 4 male) were included in the study. 46% of the patients were diagnosed with a localized form of scleroderma, while 54% of patients had a systemic sclerosis. One patient was followed up with a diagnosis of mixed connective tissue disease, due to the symptoms of dermatomyositis concomitant with those of systemic sclerosis. This patient was included in systemic sclerosis patients group.
The mean age of onset of the disease was 9.57 years (median 10 years, range 2-18 years). The mean disease duration and follow-up time was 2 years (median 1 year, range 0.5-8 years) and 3.57 years (median 2 years, range 0.5-14.5 years), respectively. The patients’ demographic characteristics and their clinical characteristics are summarized in the Table 1. There were no statistically significant differences between the two observed groups of patients in regard to demographic characteristics (p > 0.05). In contrary, laboratory investigations showed that there was a significant difference regarding serological tests between patients with JSS and those with JLS (ANA: p = 0.001, Anti Scl-70: p = 0.049, respectively). Analysis of the clinical features of the patients showed that there was also a significant difference in terms of the skin and musculoskeletal involvement (Table 1) (Figure 1).
TABLE 1.
General demographic characteristics of patients
FIGURE 1.
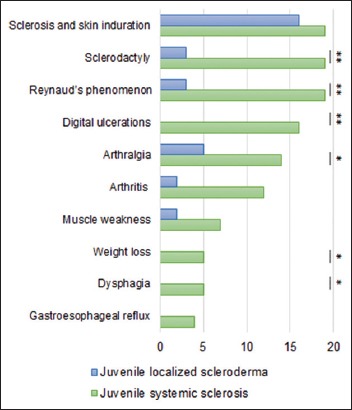
Clinical features in patients with JS. Skin, vascular and musculoscletal involvement were the most common findings. Gastrointestinal symptoms were not present among the patients with localized scleroderma. None of the patients had cardiological involvement. Statistical significance : ** : p < 0.01; * : p < 0.05
Results of the echocardiographic measurements and laboratory function tests are shown in the Table 2. The mean values of the pulmonary insufficiency at the end of diastole (PI) were 1.05 ± 0.24 m/sec and 1.00 ± 0.27 m/sec for patients with systemic sclerosis and those diagnosed with local scleroderma, respectively. The mean value of PI was statistically different from its upper referent value (2 m/sec) in the both groups of patients (p < 0.001). However, there was no significant difference in PI values between the two groups of patients (p = 0.64). The mean values of the maximal velocity of tricuspid insufficiency (TI) were 2.14 ± 0.21 m/sec and 2.07 ± 0.45 m/sec for the patients with systemic sclerosis and those with local scleroderma, respectively. The mean value of TI was significantly different from its upper referent value (3 m/sec) in the both groups (p < 0.001), while there was no significant difference for TI between the two groups of patients (p = 0.70). Systolic pulmonary arterial pressure was calculated by modified Bernoulli equation using the measurement of maximal velocity of tricuspid regurgitation. As it was mentioned previously, the systolic pulmonary arterial pressure above 40 mmHg was considered pathologic and indicative for diagnosis of PAH. The calculated MPAP was normal in both groups. Additionally, there was no significant difference for the value of MPAP between the two patients groups (p = 0.78). The mean values of the ratio of pulmonary insufficiency AT to ET (AT/ET) were found to be 0.46 ± 0.05 and 0.44 ± 0.05 for the patients with systemic sclerosis and those with local scleroderma, respectively. The both values were significantly different from the lower referent value of the AT/ET ratio (p < 0.001). Those echocardiographic measurement results indicate the absence of PAH in both groups of studied patients.
TABLE 2.
Results of echocardiographic measurements and pulmonary function tests
The mean values of FVC were 96.94 ± 16.67% and 102.12 ± 12.82% for patients with systemic sclerosis and those with local scleroderma, respectively. The results were significantly different from the lower referent value (70%) (p < 0.001). The difference in FVC value for patients with local scleroderma and those with systemic sclerosis was not statistically significant (p = 0.17).
Mean DLCO was 84.89 ± 21.57% and 94.46 ± 10.23% for JSS and JLS patients, respectively. Result for both groups of patients was significantly different from its pathologic value (p < 0.001). However, DLCO was not statistically different between two groups of patients (p = 0.16).
DISCUSSION
In this study, that represents the largest single-center collection of data on patients with juvenile scleroderma, we examined the cardio-vascular involvement by echocardiographic measurements and pulmonary function tests.
In order to determine systolic pulmonary arterial pressure, we measured a maximal velocity of tricuspid insufficiency in patients with systemic sclerosis and those with the local disease. Results were found to be normal and significantly different from the pathologic values. There was no significant difference in the maximal velocity of tricuspid insufficiency between patients with JSS and those with JLS. Additionally, echocardiographic examination was used to assess the diastolic and mean pulmonary arterial pressure. The values for both measurements were found to be normal in both groups of patients, differing significantly from the pathological value. In the other words, none of the patients had an elevated pulmonary arterial pressure. In addition, the comparison of the two patient groups showed no difference in diastolic and mean pulmonary arterial pressure between patients with systemic sclerosis and those with local scleroderma.
Although detailed and repeated measurements were performed for all the patients, pulmonary atrial pressure was found to be within normal ranges. The previous reports showed that the connective tissue disease-associated PAH cohort accounts for up to 30% of all cases of PAH, and the vast majority of these cases reflect scleroderma-associated PAH [6]. Among adults patients with scleroderma, the PAH prevalence is estimated to be 10-15%, whereas in children it was reported to be 7% [2,15]. The main demographic and clinical features of the disease found in our study are similar to those reported by Martini G et al. in children with systemic sclerosis. The mentioned study reports a pulmonary hypertension in 7% of a total of 153 SS patients from 55 different rheumatologic centers, in the period of January 2002- June 2003. The mean duration of their patients’ follow up was 3.9 years, similarly to our results (3.5 years). Additionally, the mean time between the first signs of the disease and the diagnosis was reported as being 1.9 years; in our study the mean disease duration at diagnosis was found to be 2 years (median 1 year, range 0.5-8 years) [2]. However, studies on pulmonary hypertension in juvenile scleroderma patients are lacking [16-19].
Pulmonary fibrosis, the lung involvement of the disease, contributes the development of the PAH in patients with systemic scleroderma. We examined the presence of pulmonary fibrosis by providing the lung functional tests [20-23]. Pulmonary function test revealed a normal value of FVC and DLCO for all of the studied patients. The measured values were significantly different from those defined as pathological. The comparison of FVC and DLCO between JSS and JLS patients showed no differences between those two groups. Namely, our patients had neither clinical nor the functional signs of pulmonary fibrosis. In the medical literature, pulmonary function test is described as an essential tool for PAH screening in patients with scleroderma. A declining DLCO and elevated FVC% / DLCO% ratio are very useful as predictors of the presence of the PAH or its advanced development [10]. A study performed by Borowiec at al. among patients with juvenile scleroderma showed decreased FVC in 60% of patients with JSS [4].
There are several possible reasons explaining the absence of PAH findings in our patients. The majority of patients were diagnosed shortly after the beginning of the symptoms (mean time to diagnosis was found to be 2 years). The early diagnoses lead to an early treatment possibly influencing the progression of the disease. Martini et al. showed that a short time between disease onset and the diagnosis is a protective factor in patients with juvenile scleroderma [16]. Additionally, all the studied patients were taking the regular treatment with regular annual follow-up. Patients with irregular follow-up and those who had been non-compliant were excluded from the study. Early diagnosis, a regular follow up and a proper therapy are thought to be important in reduction of cardio-vascular complications of the disease. Normal results of the cardio-vascular assessment for all of the studied patients as well as their significant difference from the proposed pathological values imply that the treatment was appropriate and important in prevention of disease complications.
At the time we performed the study, the mean age of patients was 14.5 ± 3.3 years and 13.3 ± 2.7 years for systemic sclerosis and local scleroderma group, respectively. The mean follow up was 2 years for both groups of patients. The age of the patients also possibly influences the development of the PAH. Although we could not find the exact data about the time of PAH development in patients with juvenile scleroderma in the literature, the risk possibly increases with the progression of the disease. A multicenter study, provided by Martini et al. showed a mortality rate of 11.9%, with a cardiac failure as the most important cause of mortality. The death occurred after a mean 4 years from diagnosis, with median 2.8 years and a range from 13 days to 18.1 years. [2]. In our study, duration of disease was considered to be relatively short for the PAH development. The long-standing follow-up of patients would give more information about the progression of the disease and its possible complications. There was no fatal outcome in our study. This finding could be possibly explained by the absence of the major mortality risk factor (cardio-vascular involvement).
The advantages of our study are repeated and detailed echocardiographic examinations and pulmonary tests for all of patients. However, one of the limitations of our study is the small number of patients. Furthermore, our study is a cross-sectional investigation. A further multicenter, prospective studies would give a better insight into the development of PAH at juvenile scleroderma.
In conclusion, PAH is an uncommon finding among patients with juvenile scleroderma. Early diagnosis, regular follow-up and proper therapy are thought to be important factors in reducing the cardio-vascular complications of the disease. In addition, this study points out the importance of annual cardio-vascular screening for patients with juvenile sclerosis.
CONCLUSIONS
Although quite rare in juvenile scleroderma, cardio-vascular and pulmonary involvement is the most important factor for the prognosis of the disease. Early diagnosis, regular follow-up and appropriate treatment are important factors in reducing the cardiovascular and pulmonary complications of the disease.
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
- [1].Foeldvari I. Update on juvenile systemic sclerosis. Curr Rheumatol Rep. 2015;17(3):17–18. doi: 10.1007/s11926-014-0491-y. Doi: http://dx.doi.org/10.1007/s11926-014-0491-y . [DOI] [PubMed] [Google Scholar]
- [2].Martini G, Foeldvari I, Russo R, Cuttica R, Eberhard A, Ravelli A, et al. Systemic Sclerosis in Childhood. Clinical and Immunologic Features of 153 Patients in an International Database. Arthritis & Rheumatism. 2006;54(12):3971–3978. doi: 10.1002/art.22207. Doi: http://dx.doi.org/10.1002/art.22207 . [DOI] [PubMed] [Google Scholar]
- [3].Zulian F, Cuffaro G, Spretto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25(5):643–650. doi: 10.1097/BOR.0b013e3283641f61. Doi: http://dx.doi.org/10.1097/BOR.0b013e3283641f61 . [DOI] [PubMed] [Google Scholar]
- [4].Borowiec A, Dabrowski R, Wozniak J, Jasek S, Chwyczko T, Kowalik I, et al. Cardiovascular assessment of asymptomatic patients with juvenile-onset localized and systemic scleroderma:10 years prospective observation. Scand J Rheumatol. 2012;41(1):33–38. doi: 10.3109/03009742.2011.609489. Doi: http://dx.doi.org/10.3109/03009742.2011.609489 . [DOI] [PubMed] [Google Scholar]
- [5].Fischer A, Bull TM, Steen VD. Practical Approach to Screening for Scleroderma-Associated Pulmonary Arterial Hypertension. Arthritis Care & Research. 2012;64(3):303–310. doi: 10.1002/acr.20693. Doi: http://dx.doi.org/10.1002/acr.20693 . [DOI] [PubMed] [Google Scholar]
- [6].Coghlan JG, Handler C. Connective tissue associated pulmonary arterial hypertension. Lupus. 2006;15(3):138–142. doi: 10.1191/0961203306lu2280rr. Doi: http://dx.doi.org/10.1191/0961203306lu2280rr . [DOI] [PubMed] [Google Scholar]
- [7].Bossone E1, D’Andrea A, D’Alto M, Citro R, Argiento P, Ferrara F, et al. Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr. 2013;26(1):1–14. doi: 10.1016/j.echo.2012.10.009. Doi: http://dx.doi.org/10.1016/j.echo.2012.10.009 . [DOI] [PubMed] [Google Scholar]
- [8].Mukerjee D, St George D, Coleiro B, Knight C, Denton CP, Davar J, et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis. 2003;62(11):1088–1093. doi: 10.1136/ard.62.11.1088. Doi: http://dx.doi.org/10.1136/ard.62.11.1088 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–2747. doi: 10.1002/art.38098. Doi: http://dx.doi.org/10.1002/art.38098 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fischer A, Bull TM, Steen VD. Practical Approach to Screening for Scleroderma-Associated Pulmonary Arterial Hypertension. Arthritis Care Res. 2012;64(3):303–310. doi: 10.1002/acr.20693. Doi: http://dx.doi.org/10.1002/acr.20693 . [DOI] [PubMed] [Google Scholar]
- [11].Mukerjee D, St George D, Knight C, Davar J, Wells AU, Du Bois RM. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology. 2004;43(4):461–466. doi: 10.1093/rheumatology/keh067. Doi: http://dx.doi.org/10.1093/rheumatology/keh067 . [DOI] [PubMed] [Google Scholar]
- [12].Shah AA, Chung SE, Wigley FM, Wise RA, Hummers LK. Changed in estimated right ventricular systolic pressure predict mortality and pulmonary hypertension in a cohort of scleroderma patients. Ann Rheum Dis. 2013;72(7):1136–1140. doi: 10.1136/annrheumdis-2012-201861. Doi: http://dx.doi.org/10.1136/annrheumdis-2012-201861 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Armstrong WF, Ryan T. Feigenbaum’s Echocardiography. seventh ed. Philadelphia: Lippincott, Williams & Wilkins; 2003. [Google Scholar]
- [14].Litwin SE. Noninvasive assessment of pulmonary artery pressures. Moving beyond tricuspid regurgitation velocities. Circ Cardiovasc Imaging. 2010;3(2):132–133. doi: 10.1161/CIRCIMAGING.110.945121. Doi: http://dx.doi.org/10.1161/CIRCIMAGING.110.945121 . [DOI] [PubMed] [Google Scholar]
- [15].Sweiss NJ, Hushaw L, Thenappan T, Sawaqed R, Machado RF, Patel AR, et al. Diagnosis and management of pulmonary hypertension in systemic sclerosis. Curr Rheumatol Rep. 2010;12(1):8–18. doi: 10.1007/s11926-009-0078-1. Doi: http://dx.doi.org/10.1007/s11926-009-0078-1 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Martini G, Vittadello F, Kasapçopur O, Magni MS, Corona F, Duarte-Salazar C, et al. Factors affecting survival in juvenile systemic sclerosis. Rheumatology (Oxford) 2009;48(2):119–122. doi: 10.1093/rheumatology/ken388. Doi: http://dx.doi.org/10.1093/rheumatology/ken388 . [DOI] [PubMed] [Google Scholar]
- [17].Misra R, Singh G, Aggarwal P, Aggarwal A. Juvenile onset systemic sclerosis: a single center experience of 23 cases from Asia. Clin Rheumatol. 2007;26(8):1259–1262. doi: 10.1007/s10067-006-0483-z. Doi: http://dx.doi.org/10.1007/s10067-006-0483-z . [DOI] [PubMed] [Google Scholar]
- [18].Valerio CJ, Schreiber BE, Handler CE, Denton CP, Coghlan JG. Borderline mean pulmonary artery pressure in systemic sclerosis patients: trans-pulmonary gradient predicts risk of developing pulmonary hypertension. Arthritis Rheum. 2013;65(4):1074–1084. doi: 10.1002/art.37838. Doi: http://dx.doi.org/10.1002/art.37838 . [DOI] [PubMed] [Google Scholar]
- [19].Quartier P, Bonnet D, Fournet JC, Bodemer C, Acar P, Ouachée-Chardin M, et al. Severe cardiac involvement in children with systemic sclerosis and myositis. J Rheumatol. 2002;29(8):1767–1773. [PubMed] [Google Scholar]
- [20].Foeldvari I. New developments in juvenile systemic and localized scleroderma. Rheum Dis Clin North Am. 2013;39(4):905–920. doi: 10.1016/j.rdc.2013.05.003. Doi: http://dx.doi.org/10.1016/j.rdc.2013.05.003 . [DOI] [PubMed] [Google Scholar]
- [21].Fraisse A, Jais X, Schleich JM, di Filippo S, Maragnes P, Beghetti M, et al. Characteristics and prospective 2-year-follow – up of children with pulmonary arterial hypertension in France. Arch Cardiovasc Dis. 2010;103(2):66–74. doi: 10.1016/j.acvd.2009.12.001. Doi: http://dx.doi.org/10.1016/j.acvd.2009.12.001 . [DOI] [PubMed] [Google Scholar]
- [22].Langleben D, Orfanos SE, Givinazzo M, Hirsch A, Baron M, Senécal JL, et al. Pulmonary capillary endothelial metabolic dysfunction: Severity in pulmonary arterial hypertension related to connective tissue disease versus idiopathic pulmonary arterial hypertension. Arthritis Rheum. 2008;58(4):1156–1164. doi: 10.1002/art.23405. Doi: http://dx.doi.org/10.1002/art.23405 . [DOI] [PubMed] [Google Scholar]
- [23].Le Pavec J, Humbert M, Mouthon L, Hasooun PM. Systemic sclerosis associated pulmonary hypertension. Am J Respir Crit Care Med. 2010;181(12):1285–1293. doi: 10.1164/rccm.200909-1331PP. Doi: http://dx.doi.org/10.1164/rccm.200909-1331PP . [DOI] [PMC free article] [PubMed] [Google Scholar]