

Data indicate a compelling relationship between inflammation and depression that may have profound clinical relevance (1). A significant proportion of patients with depression reliably exhibit increased inflammatory markers, and exposure of humans to stress, a well-known precipitant of depression, is associated with activation of inflammatory responses. Nevertheless, an important unresolved question is whether inflammation is a cause or a consequence of depression. For example, if inflammation causes depression, strategies to reduce inflammation would be a rational approach to reduce the risk of the disease and ultimately to treat it. Alternatively, if increased inflammation is a consequence of the many biologic alterations that occur as a result of stress or depression including changes in the regulation of the hypothalamicpituitary-adrenal axis and autonomic nervous system, both of which have potent immunomodulatory effects, increased inflammation in depression, while representing a significant deleterious repercussion of the disorder, may have limited relevance to its pathogenesis. Thus, teasing apart this issue of cause or consequence becomes essential for understanding the relative strategy for addressing inflammation and determining its relevance to the risk for depression and other stress-related disorders.
To get a better grasp of this issue of cause and effect, investigators have conducted longitudinal studies in large population-based samples to determine whether increased inflammation is a risk factor for depression or vice versa. Several epidemiologic studies have provided data suggesting a one-way relationship between inflammation and depression. For example, in the Whitehall II study of >3000 participants, baseline concentrations of the inflammatory markers C-reactive protein and interleukin (IL)-6 predicted depressive symptoms over an approximately 10-year follow-up period, whereas baseline depressive symptoms did not predict inflammatory markers (2). Psychosocial stressors that are well known to be associated with increased inflammation including childhood maltreatment also have been found to be risk factors for the later development of depression (3). Moreover, inflammatory markers have been shown to predict development of posttraumatic stress disorder (4). In addition, administration of inflammatory stimuli leads to depressive symptoms in otherwise nondepressed subjects, further supporting the notion that inflammation can cause depression. Finally, a small literature has provided evidence that blockade of inflammation may reduce depressive symptoms, especially in patients with increased inflammation (5). Taken together, longitudinal epidemiologic studies and small clinical trials using proinflammatory or anti-inflammatory interventions indicate that inflammation may be a risk factor for depression. However, the results are not definitive, and cause and effect has yet to be established.
Another strategy to address the relationship between inflammation and depression is to take advantage of well-controlled studies using laboratory animals. Rodent models of stress-induced depression have provided key insights into how stress can activate the immune system to lead to depressive symptoms. Such models have included chronic social defeat, social disruption, predator stress, and resident-intruder paradigms, all of which reliably cause depressive-like behaviors, including anhedonia and social withdrawal. These studies also have demonstrated that stress independently induces “sterile inflammation” in the brain as reflected by activation of the inflammasome to produce IL-1β by danger-associated molecular pattern molecules such as adenosine triphosphate released during stress and ultimately leading to depressive-like behaviors (6). Activated peripheral monocytes/macrophages that invade the brain also have been shown to play a significant role in stress-induced behavioral alterations and may confer prolonged immunologic memory and sensitization to stress (7). Animal models have further demonstrated the importance of activation of relevant inflammatory signaling cascades (e.g., nuclear factor κB and p38 mitogen-activated protein kinase) as well as the role of inflammation in stimulating indoleamine 2,3-dioxygenase and the kynurenine pathway, inhibiting brain-derived neurotrophic factor and neurogenesis and inhibiting monoamine neurotransmission while stimulating extracellular glutamate (1). Together with data demonstrating that blockade of inflammation in the brain and the periphery can reverse stress-induced depressive-like behavior, these laboratory animal studies strongly support the notion that inflammation plays a significant causal role in stress-induced depression.
More recently, animal studies have begun to address more nuanced issues inherent in the cause-and-effect discussion, shedding more light on exactly what role inflammation plays as a risk factor in the complex pathway from stress to depression and the multiplicity of individual differences observed. The study in this issue of Biological Psychiatry by Wood et al. (8) corroborates the idea that an exaggerated inflammatory response to stress confers risk for developing depressive symptoms and lends insight into the role of individual differences in stress coping mechanisms. Wood et al. examined gene transcripts in the brains of outbred rats previously shown to exhibit either an active or a passive coping strategy in the face of social defeat stress, based on the latency to exhibit a submissive posture. Of a panel of 88 genes related to G protein–coupled receptor signaling, IL-1β was the only transcript that was found to be upregulated in passive coping animals and downregulated in active coping animals in the locus coeruleus and dorsal raphe (compared with control animals). Increases in IL-1β messenger RNA in passive coping animals also corresponded with an increase in IL-1β protein in the locus coeruleus and a shift toward proinflammatory cytokines in the plasma, including an increased IL-6-to-IL-10 (inflammatory to anti-inflammatory) ratio and increased monocyte chemoattractant protein-1. In the dorsal raphe of active coping animals, IL-1β protein was decreased 24 hours after final stress exposure. In addition, the passive coping strategy was associated with the induction of anhedonia as reflected by reduced sucrose preference following stress. Of considerable interest to our discussion of cause and effect and the role of inflammation in the risk for depression, intracerebroventricular administration of IL-1 receptor antagonist before each session of social defeat completely reversed depressive-like behavior in animals that employed the passive coping strategy. However, IL-1 blockade had no effect on whether animals employed passive or active coping. These findings indicate that passive coping, which serves as a risk factor for stress-induced depression, is independent of IL-1β, yet the increased IL-1β in the brain that resulted from this coping strategy led to anhedonia. Although individual differences in coping strategies conferred a risk for depressive-like behavior, increased inflammation in the brain was the proximate cause of individual differences in the development of anhedonia.
This work by Wood et al. is complemented by a recent study by Hodes et al. (9), which found that increased stress-induced plasma IL-6 responses and increased ex vivo production of stimulated IL-6 predicted subsequent development of social avoidance after repeated social defeat. Similar to the results reported by Wood et al., blockade of IL-6 with a peripherally administered monoclonal antibody before social defeat prevented stress-induced social avoidance. Transfer of bone marrow hematopoietic progenitor cells from mice producing high IL-6 to mice producing low IL-6 also was able to confer risk to stress-induced social withdrawal (9). These findings indicate that inflammatory responses serve not only as a factor that determines risk but also play a central role in the causation of stress-induced behavioral changes, accounting for the individual differences in the development of depressive-like behavior in these animals.
Wood et al. also found that animals with active coping strategies exhibited a resilient phenotype characterized by reduced IL-1β production in the brain, less monocyte chemoattractant protein-1 induction, and less of an increase in the IL-6-to-IL-10 ratio compared with passive coping animals. These findings are consistent with the work by Hodes et al. (9), who demonstrated that mice producing low IL-6 were resilient to the effects of stress on social withdrawal. Relevant to potential mechanisms of stress resilience, Brachman et al. (10) recently observed that transfer of T cells from mice chronically exposed to social defeat to lymphocyte-deficient, stress-naïve animals (Rag2−/− mice) conferred an anti-inflammatory and antidepressant phenotype. Indeed, animals that received adoptive transfer of T cells from stressed mice exhibited reduced serum inflammatory cytokines, increased progenitor cells in the dentate gyrus of the hippocampus, increased microglia with a protective M2-like phenotype, and reduced depressive and anxiety-like behaviors (10). These findings highlight the role of T cells in shifting the inflammatory tone of monocytes/macrophages and microglia to a protective M2 phenotype that supports neurogenesis and stress resilience, an effect that has been shown to be mediated in part by T-cell production of IL-4 (1).
Taken together, findings from animal models provide compelling data that inflammation plays a key causative role in the development of depressive-like behavior and serves as a lynchpin in the elaboration of phenotypes of risk and resilience to the behavioral effects of stress (Figure 1). Moreover, as indicated by the study of Wood et al., the inflammatory response can interact with other environmental and psychosocial variables including coping styles to lead to individual differences in the development of depressive-like behavior. Collectively, these studies provide the conceptual framework to further explore the role of inflammation as a mutable risk factor in clinical populations with susceptibilities to depression and other stress-related disorders. Just as in other medical specialties, increased inflammation may become a target for both the prevention and treatment of psychiatric disease.
Figure 1.
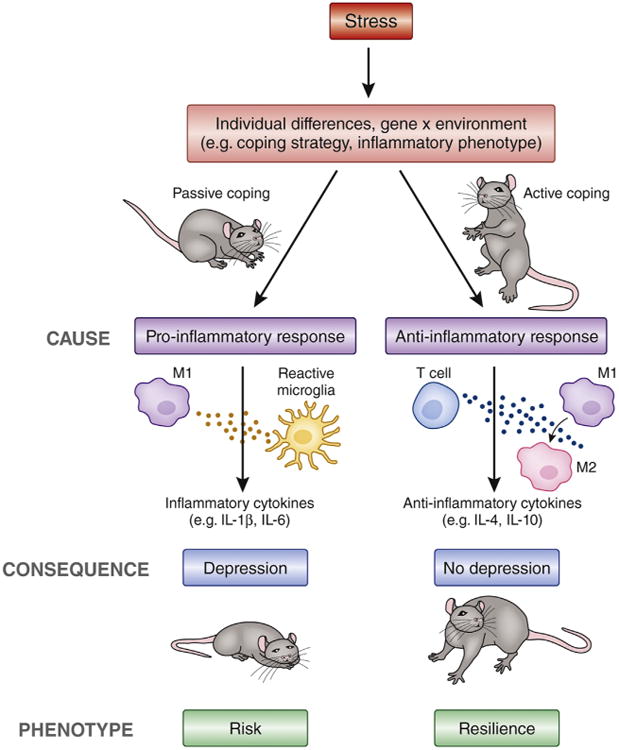
Inflammation mediates individual differences in risk and resilience to stress-induced depression. Individual differences in the response to stress (via gene × environment interactions) predict risk or resilience to the development of stress-induced behavioral changes. Inflammation has been shown in animal studies to be a pivotal causative factor of these individual differences. Animals who exhibit poor coping strategies exhibit exaggerated inflammatory responses (release of inflammatory cytokines, shift toward M1 macrophage phenotype, and activation of microglia) and are at risk for the development of depressive-like behavior. The causal role of inflammation in development of stress-induced depression was highlighted in the study by Wood et al. (8), which demonstrated that blocking interleukin-1β before stress had no effect on individual differences in coping strategy but prevented individual differences in the induction of depressive-like behavior. Furthermore, resilient animals displayed anti-inflammatory responses to stress, consistent with a previously established protective role of the acquired immune system (T cells) and anti-inflammatory cytokines in promoting a shift toward an M2-like macrophage phenotype, reduced inflammation in the brain, and prevention of stress-induced depression. IL, interleukin.
Acknowledgments
This work was supported by U.S. Department of Health and Human Services Public Health Service Grant Nos. UL1TR000454 and KL2TR000455 from the National Center for Advancing Translational Sciences of the National Institutes of Health and Grant Nos. R01MH087604 and R03MH100273 from the National Institute of Mental Health.
Footnotes
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Haroon E, Raison CL, Miller AH. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology. 2012;37:137–162. doi: 10.1038/npp.2011.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gimeno D, Kivimaki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol Med. 2009;39:413–423. doi: 10.1017/S0033291708003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry. 2008;65:409–415. doi: 10.1001/archpsyc.65.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eraly SA, Nievergelt CM, Maihofer AX, Barkauskas DA, Biswas N, Agorastos A, et al. Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry. 2014;71:423–431. doi: 10.1001/jamapsychiatry.2013.4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: The role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwata M, Ota KT, Duman RS. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav Immun. 2013;31:105–114. doi: 10.1016/j.bbi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry. 2014;75:970–981. doi: 10.1016/j.biopsych.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood SK, Wood CS, Lombard CM, Lee CS, Zhang XY, Finnell JE, et al. Inflammatory factors mediate vulnerability to a social stress-induced depressive-like phenotype in passive coping rats. Biol Psychiatry. 2015;78:38–48. doi: 10.1016/j.biopsych.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci U S A. 2014;111:16136–16141. doi: 10.1073/pnas.1415191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brachman RA, Lehmann ML, Maric D, Herkenham M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J Neurosci. 2015;35:1530–1538. doi: 10.1523/JNEUROSCI.2278-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]